
Abstract
Proteins derived from microorganisms that survive in the harshest environments on Earth have stable activity under extreme conditions, providing rich resources for industrial applications and enzyme engineering. Due to the time-consuming nature of experimental determinations, it is imperative to develop computational models for fast and accurate prediction of protein optimal conditions. Previous studies were limited by the scarcity of data and the neglect of protein structures. To solve these problems, we constructed an up-to-date dataset with 175,905 non-redundant proteins and proposed a new model GeoPoc based on geometric graph learning for the protein optimal temperature, pH, and salt concentration prediction. GeoPoc leverages protein structures and sequence embeddings extracted from pre-trained language model, and further employs a geometric graph transformer network to capture the sequence and spatial information. We first focused on in-house validation for optimal temperature prediction for robustness assessment, and achieved a PCC of 0.78. The algorithm is further confirmed in an independent test set, where GeoPoc surpasses the state-of-the-art method by 2.3% in AUC. Additionally, GeoPoc was extended to pH and salt concentration prediction, and obtained AUC scores of 0.78 and 0.77, respectively. Through further interpretable analysis, GeoPoc elucidates the critical physicochemical properties that contribute to enhancing protein thermostability.
Similar content being viewed by others


Introduction
Extremophiles, organisms that can survive in the harshest environments, demonstrate remarkable survival strategies on Earth. The proteins in these organisms display exceptional endurance to extreme temperatures, acidic or alkaline conditions, high salinity, and other harsh conditions1. Since these proteins can remain stable and active under extreme environmental conditions, they have various applications in biotechnology2, pharmaceuticals, chemical industry3, and environmental science4. Moreover, these proteins are crucial for understanding the adaptability mechanisms of life and provide a perspective for exploring the evolutionary patterns of life5. Therefore, it’s significant to develop a tool that accurately predicts the optimal temperature, pH, and salt concentration for extremophilic proteins from microorganisms.
Thermophilic proteins can maintain their structural integrity and function at high temperatures, with some variants showing stability at temperatures up to 100 °C6. Its unique thermal stability is closely related to biological characteristics such as amino acid composition, hydrogen bonds, salt bridges, and disulfide bonds7. Leveraging these characteristics, some thermophilic protein predictors have been developed by traditional machine learning methods, such as logistic model trees8, support vector machines9, ensemble learning10, and other classical algorithms11. In recent years, deep learning technology has also been used to predict thermophilic proteins, with a model called iThermo12 using a multilayer perceptron for thermophilic protein prediction. DeepTP13, another deep learning model, employs Convolutional Neural Networks to extract critical local information from protein sequences and then uses Bidirectional Long Short-Term Memory Networks to capture long-range dependency features. Besides the thermophilic proteins, proteins that maintain structural and functional stability under extreme pH and salt concentration play a crucial role in industrial production14,15. However, these proteins are relatively under-researched. For pH, the enzymes produced by acidophilic and alkaliphilic microorganisms are widely used in various fields, including food processing, biofuel production, and wastewater treatment16. In a previous study, EpHod was designed to predict enzyme optimal pH directly from protein sequences17. Halophilic proteins produced by microorganisms that survived in high salinity are positioned as promising candidates for biocatalytic applications18.
Despite previous methods that have already been developed for protein optimal condition prediction, their overall utility was limited by the scarcity of data, time-consuming features, and the lack of consideration of protein structures. The scarce samples and excessive homologous proteins impair the generalization ability13. This leaves a certain amount of room for constructing a reliable large-scale benchmark dataset. Furthermore, some methods10 that rely on sequence-based evolutional features are time-consuming. Considering the linguistic characteristics of protein sequences, unsupervised pre-training with language models has recently been applied to protein sequence representation19. Many methods have replaced these evolutional features with pre-trained embeddings20,21. In addition, previous studies have rarely utilized the structural information to predict protein optimal conditions. However, the three-dimensional structure of proteins serves as a crucial part of exploring the stability and functionality of proteins in extreme conditions22. With the development of deep learning techniques, significant breakthroughs have been achieved in protein structure prediction. The representative approach, AlphaFold223, has exhibited a degree of accuracy that is comparable to experimental results in most cases and provided accessible databases. Such breakthroughs will undoubtedly benefit downstream protein studies24, including protein optimal condition prediction. To fully leverage protein structure, various studies have represented proteins as graph data and utilized graph convolutional networks to extract sequence and structural features25.
In this study, we constructed an up-to-date and non-redundant data set for the research of protein optimal condition. We developed GeoPoc (Geometry-aware Protein Optimal Condition predictor), which applies a graph transformer network and AlphaFold-predicted protein structures for protein optimal condition prediction. Specifically, the structures were utilized to construct the protein graph and obtain the geometric features. We integrate sequence embeddings from a pre-trained language model with the geometric features to construct residual features. Then, a geometric graph transformer network is applied to capture the complex interactions and structural features. Next, we use the attention pooling layer to perform weighted pooling of information from nodes in the graph, aiming to enhance the model’s perception of the graph structure. Various evaluations have shown that GeoPoc accurately predicts the optimal protein temperature and surpasses state-of-the-art thermophilic protein predictors. At species-level validation, GeoPoc is robust for unseen species. Extending GeoPoc to predict pH and salt concentration also demonstrates accurate classification for each task. Furthermore, the model interpretability analysis indicates that our model captures the underlying mechanisms related to proteins adapted to extreme conditions and evolutionary patterns.
Results
Overview of GeoPoc
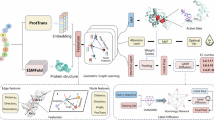
We set out to construct an up-to-date microorganism protein data set with GeoPoc tailored for protein optimal condition prediction, consisting of two major components: protein representation as graph and GeoFormer block with graph transformer network, as shown in Fig. 1 and Materials and Methods.

a The data collection and dataset preparation process. b The overall architecture of the GeoPoc model. ESM2.0 is used to extract the sequence embedding from the sequence, and the protein structure is taken from the AlphaFold2 database. After featuring these as protein graphs, the graph is input to the GeoFormer module to get hidden embeddings. Finally, the hidden embeddings are pooled by the self-attention pooling layer, which is input to the output MLP to predict the temperature, pH, and salt concentration. Note: SaltConc denotes salt concentration.
As shown in Fig. 1a, organism optimal culture condition data were retrieved from two prominent bacterial databases (BacDive26, HaloDom27) for 7766 entries on November 7, 2023, and protein sequences were downloaded from Uniport28 for 6,595,396 of these entries. While preserving species diversity, redundant proteins were removed to form the final data set.
The data set described above together with the corresponding structure and pre-trained language model embedding were represented as graph, which served as the input for the GeoFormer (Fig. 1b). The GeoFormer module is a geometry-aware versatile network consisting of norm layer and message passing. The message passing layer adopts the multi-head attention in the graph transformer network enhanced by edge features. We validated the performance of GeoPoc on protein optimal temperature prediction in our data set and external data, and further performed optimal pH and salt concentration prediction. The results demonstrated our framework to be accurate and robust. We then conducted several interpretability analyses and confirmed that GeoPoc is effective for capturing protein multidimensional structure and evolution patterns.
The up-to-date protein optimal condition datasets
In previous research, various data sets were used in thermophilic protein prediction (Table. S1). However, the lack of a comprehensive public benchmark data set prompted us to construct a reliable and extensive benchmark data set. The BacDive and HaloDom databases contain extensive optimal environmental conditions for various species without protein information, while UniProt has abundant protein sequences. Therefore, we linked BacDive and HaloDom to the UniProt database using species names to create a data set of optimal conditions for proteins. Our data set stands out as the largest and most diverse data set of thermophilic proteins, spanning a wide range of protein sequences from various protein families and species. Moreover, previous studies mainly focused on classifying thermophilic proteins, while our study represents the first accurate prediction of protein optimal temperatures. To enhance the overall quality and reliability of our data set, we removed redundant proteins with a sequence identity <25%, while the other data sets identity cutoff is 40% or more.
Our data set is comprised of three tasks: predicting protein optimal temperature, pH, and salt concentration. The data distribution of each task is shown in Fig. 2. The temperature data set we collected ranges from 4 °C to 105 °C, with 10,743 proteins above 50 °C from 157 species. The pH data set ranges from 1.1 to 12, with 8837 proteins from 100 species in the pH <5 and >9 ranges. The salt data set ranges from 0% to 37%, with 25,542 proteins above 4% from 535 species. The broader range of data distribution and abundance of extreme samples help us explore the intrinsic mechanisms of proteins adapted to extreme conditions.

a Optimal temperature distribution for species and proteins, with a temperature range from 4 °C to 105 °C. b Optimal pH distribution, ranging from 1.1 to 12. c Optimal salt concentration distribution, ranging from 0% to 37%. In each figure, blue bars represent species, and yellow bars represent proteins. Source data are provided as a Source Data file.
GeoPoc for optimal temperature prediction
We first focused on the task of predicting protein optimal temperature, and evaluated the performance of GeoPoc by PCC using 5-fold cross-validation (CV) and test set. As shown in Fig. 3a, b, GeoPoc achieved PCC values of 0.758 ± 0.005 and 0.779 for the 5-fold CV and test set, respectively. The consistent performances on the cross-validation and test set indicate the robustness of our model. We also extracted points in the dense red regions of Fig. 3b and the PCC is 0.777 (Fig. S1), indicating the robustness of our model. In addition, the model accuracy across different temperature ranges is primarily correlated with the size of the training set, with no clear positive correlation to structural accuracy, as shown in Fig. S2. We also trained models only using thermophilic proteins, but the model doesn’t work well on the test set (MAE of 6.975 vs 6.083 by the original model). To demonstrate the relative importance of each part we adopted, we conducted several ablation experiments. Compared with GeoPoc, GeoPoc (w/o attention pooling) removes the attention pooling layer, and GeoPoc (w/o geometric features) excludes the geometric information in the graph. We also included a naive baseline that uses ESM2.0 embeddings processed by a single transformer block to predict protein conditions. To assess the impact of protein structure, we also established a baseline method LSTM, and applied different feature inputs including ESM2.0 embedding and one-hot embedding. Additionally, we introduced another baseline with GNN using only one-hot embeddings for direct comparison against LSTM (one-hot).

The regression correlation between prediction and ground truth values of optimal temperature for all proteins in 5-fold CV (a) and test (b). For the color bar in the (a)/(b), we used a density-based color scale computed via Gaussian kde, and the square root transformation was applied to enhance visual clarity in regions with high data density. The MAE for the 5-fold CV is 6.402 ± 0.116 (a), while the MAE for the test set is 6.083 (b). c Performance comparison between GeoPoc and ablation methods in the test set using PCC. d Receiver Operating Characteristic curves of GeoPoc and comparison methods on the independent test set. e Comparison of GeoPoc and comparison methods on threshold-dependent metrics. f The regression correlation between ground truth and predicted values of optimal temperature (°C) for all species in the test set. Note: w/o denotes without. Source data are provided as a Source Data file.
As shown in Fig. 3c, the LSTM model utilizing one-hot embedding inputs exhibited the poorest performance with a PCC of 0.419. When the pre-trained language model embedding was used as the node feature, the LSTM model yielded a PCC of 0.680. This result confirms that a model that learns solely based on one-hot embedding inputs would be unable to yield any valuable predictions. Moreover, GeoPoc (without geometric features) which utilizes protein structures outperforms LSTM (ESM embeddings) and Transformer (ESM embeddings), indicating that structural information is more directly relevant to the protein optimal temperature. Moreover, the GNN using only one-hot features outperformed LSTM (one-hot) by 0.096 PCC, further demonstrating the importance of incorporating protein structure. When we enhance the node and edge features with the geometric information, the GeoPoc yields the highest PCC (0.779). This further demonstrates that geometric information provides insights into the protein optimal temperature, such as well-packed hydrophobic cores and optimized hydrogen bonding patterns, which can contribute to a protein’s ability to maintain its structure and function at a particular temperature. On the other hand, the removal of the attention pooling layer has the slightest impact on GeoPoc, with the drop of PCC from 0.779 to 0.764. This indicates that attention pooling can assist the network in capturing key features across the sequence more effectively, but its impact on improving our model’s performance is limited. The performance of GeoPoc and baselines is shown in Table. S2.
Since our model uses structures predicted by AF2, we also tried to incorporate structural accuracy information into the model, but the model performance slightly decreased (PCC from 0.779 to 0.770). Additionally, due to the limited accuracy of AF2 in predicting side chains, we also evaluated the impact of side chains on our model’s performance. When removing side chain-related features, the achieved PCC slightly decreased to 0.771 in the test set, indicating slight but positive contributions from the predicted side chains. We also explored the impact of different TransGCN depths on our model’s performance, with GeoPoc achieving the best results at a depth of 2, as shown in Table. S6.
To demonstrate the effectiveness of our method, we compared GPSite with DeepTP, TMPpred9, SCMTPP29, iThermo, and SAPPHIRE10 on an independent test set collected by Zhao13. The independent test set consisted of 817 thermophilic and 817 mesophilic proteins. We removed 1677 proteins from our train data set to ensure that the protein identity between the independent test set and our train set was <25%. Thermophiles are defined as organisms with optimal growth temperatures exceeding 45 °C, while mesophiles are those with optimal growth temperatures ranging between 15 °C and 45 °C30. Thus, GeoPoc was retrained and used the 45 °C cutoff to classify thermophilic and mesophilic proteins (Fig. S3). The baseline LSTM (ESM embedding) was also considered here.
Figure 3d shows that GeoPoc outperforms existing thermophilic protein predictors on the independent test set, with the highest AUC (0.966). In threshold-dependent metrics (Fig. 3e), GeoPoc achieved a MCC value of 0.823, which was 10.3% better than DeepTP (0.746) and 25.3% better than SAPPHIRE (0.657), and an accuracy value of 0.905, which was 3.7% better than DeepTP (0.873) and 10.2% better than SAPPHIRE (0.821). The performance of the other models is shown in Table. S3. Moreover, GeoPoc obtained the highest AUPR of 0.973 compared with the other predictors, as shown in Fig. S4. The comparable performance of LSTM (EMS embedding) and DeepTP suggests that the pre-trained language model can capture richer semantic and evolutionary information of proteins, leading to improved predictions of optimal temperature. The outperformance of GeoPoc indicates that structure and geometric features are an effective part of learning the patterns for temperature prediction. These patterns are likely the mechanisms by which proteins maintain functionality and structural stability at specific temperatures.
At the species-level validation, GeoPoc obtained a PCC of 0.780 for proteins from unknown species(Fig. S5). Moreover, we determined the predicted optimal temperature for each species by averaging the predicted temperatures of the proteins associated with that species, and we achieved a PCC of 0.805, as shown in Fig. 3f. The performance in the species-level validation indicates that our model has the potential to predict species optimal temperature through the proteins of the species.
GeoPoc for optimal pH and salt concentration prediction
We next considered extending GeoPoc to predict protein optimal pH and salt concentration, and evaluated the performance of GeoPoc by AUC, AUPR, F1, and MCC using a 5-fold CV and test set. Similar to the previous section, we perform an ablation study to evaluate the effect of different parts of GeoPoc, including GeoPoc (w/o attention pooling), GeoPoc (w/o geometric features), and LSTM (ESM embedding). As a result, GeoPoc outperforms all baselines with the highest AUPR of 0.56 and 0.59, AUC of 0.79 and 0.79 on the pH and salt concentration test set, respectively. Other performance metrics and 5-fold CV results are detailed in Table. S4. Although GeoPoc yields the highest MCC (0.32,0.40) in the test sets, there is still room for improvement in its performance. Figure 4c and Fig. S6a show that our model misclassifies some samples of extremely acidic or alkaline categories as neutral. Figure 4e and Fig. S6b show that our model incorrectly classifies some proteins with the highest salt concentrations as those originating from freshwater environments, reflecting the challenges in accurately predicting low salt concentrations. As a result, the model achieved a precision of only 0.41 for this class, indicating significant room for improvement. While stratified splitting was used to balance the dataset, the relatively small number of proteins with optimal salt concentrations below 0.05% likely contributed to the reduced predictive performance in this class. Furthermore, microorganism responses to salinity involve a wide array of genetic and metabolic pathways, including osmoregulation and ion transport mechanisms, which vary significantly between species and environmental conditions31. This complexity makes it harder to capture the relationship between a protein’s sequence and its optimal salt concentration.

Receiver Operating Characteristic curves of GeoPoc and the ablation methods of GeoPoc on the pH (a) and salt concentration (b) test sets. Sankey diagram visualizing the flow between ground truth and prediction of GeoPoc (c–e) and GeoPoc (w/o geometric features) (d–f) on the pH and salt concentration test sets. Source data are provided as a Source Data file.
As shown in Fig. 4a and b, the ROC curves of LSTM (ESM embedding) are closer to the diagonal, indicating that the prediction of the LSTM model is similar to random choice. However, GeoPoc (w/o attention pooling) and GeoPoc (w/o geometric features) performed better benefiting from the constructed graph based on the protein structures. This indicates that protein structures can reveal the physicochemical properties of proteins, which helps the model deeply understand the intrinsic mechanisms behind protein optimal pH and salt concentrations. Figure 4d, f show the predicted Sankey diagram for the three categories of pH and salt concentration after GeoPoc removes geometric features. GeoPoc (w/o geometric features) shows a 0.09 decrease in MCC, a 0.10 decrease in f1, and a 0.04 decrease in AUPR compared to GeoPoc, in the pH prediction task. GeoPoc (w/o geometrical features) demonstrates a 0.05 decrease in f1 and a 0.03 decrease in both MCC and AUPR in the salt concentration task. This indicates that geometric information can better capture the interactions between residues, such as charge regulation, salt bridges, and hydration effects, which are crucial for proteins to maintain their activity under extreme pH or salt concentrations.
GeoPoc is effective for capturing multidimensional structure and evolution patterns
We further conducted model interpretability analysis to obtain insights into the underlying mechanisms utilized by the model during prediction. The importance scores of secondary structures and 20 amino acids were analyzed to provide a multidimensional understanding of the physicochemical properties of the thermophilic protein. Fig. S7 shows the importance distribution of secondary structure among thermophilic and mesophilic proteins from the test set. After conducting a T-test with P < 0.0001, it’s convinced that the helix is more critical than other secondary structures. In the previous research32, the analysis of helix and intrahelical interactions within the protein led to the conclusion that maintaining high helical stability is essential for protein thermostability. Moreover, compared to mesophilic proteins, the importance of sheet secondary structures increased in thermophilic proteins. Figure 5a shows the importance of per-residue in protein M0HVG5, and Fig. S8 shows two other examples. In 73.6% of the thermophilic proteins, the average importance of helices exceeds that of other secondary structures. This confirmed the discriminative power of GeoPoc to capture the key information in protein optimal temperature prediction.

a Protein secondary structure with the color-coded importance level of residues in one example (UID: M0HVG5). The color indicates importance, where red represents higher importance, and blue indicates lower importance. b The amino acid’s frequency and the average importance in the thermophilic proteins. c Plot the A0A1I7KCI7 protein residue conservation score and its importance for each position in the aligned protein families (DNA_helicase_UvrDPEP).
As shown in Fig. 5b, the ranks of the top four important amino acids are Glutamic acid (E, 0.594), lysine (K, 0.593), isoleucine (I, 0.589), and tyrosine (Y, 0.584). Other thermophilic protein predictors also reveal that lysine12, glutamic acid29, isoleucine, and tyrosine are often among the most important amino acids10. Lysine and glutamic acid, due to their side chains carrying positive and negative charges, can form strong electrostatic interactions11. Their interactions can lead to the formation of salt bridges, further stabilizing the protein structure22. Furthermore, glutamic acid and lysine are known for their strong helix-forming tendencies due to their long side chains33. This is consistent with their role in stabilizing helices, which are essential for the thermal stability of protein structures, as previously analyzed. In contrast, amino acids such as glycine and proline, which are known as helix breakers due to their conformational flexibility and rigid ring structures, exhibit the lowest importance. Additionally, tyrosine contains a large hydrophobic phenyl ring, and isoleucine is a hydrophobic amino acid, which promotes the formation of a tight hydrophobic core within the protein34. The hydrophobic core enhances protein stability by compacting its interior, preserving the tertiary structure, and preventing structural disintegration under high temperatures35. These results are consistent with previous studies36,37. Moreover, we find that the importance and frequency of amino acids do not demonstrate a clear correlation, suggesting that the GeoPoc predicts optimal temperature without relying on the frequency of amino acid occurrence. The importance and occurrence of each amino acid in the thermophilic proteins are shown in Table S5.
To explore protein evolutionary patterns, we analyzed the conservation and importance scores of protein regions from two protein families, including DNA_helicase_UvrD/PEP and ABC_transpr_LoID_like. We calculated Spearman’s rank correlation coefficient between the conservation and importance of the 52 protein sequence regions from two protein families as shown in Fig. S9, and the average of Spearman’s rank correlation is 0.35. Figure 5c shows that conserved regions of the sequence are often more important for predicting the protein optimal temperature. These conserved regions are especially crucial for the stability and functionality of proteins during their evolution, including their optimal activity at specific temperatures. For future work, we can induce mutations in these essential regions and then utilize our model to screen for proteins possessing desired characteristics, such as improved stability or activity under specific temperatures in protein design.
Discussion and conclusion
The stability of extremophile proteins under extreme conditions is crucial for industrial applications and enzyme engineering. However experimental determination of the optimal temperature, pH, and salt concentration for specific proteins is time-consuming and expensive. Moreover, rapid advancements in sequencing technologies have led to the generation of a vast amount of protein sequence data. Therefore, computational models are urgently needed to screen and annotate extremophilic proteins. Previous studies were limited by scarce sample sizes in datasets and a lack of consideration for protein structural information.
In this study, we propose GeoPoc, a novel deep-learning model designed to accurately predict the optimal conditions for extremophile proteins. To develop GeoPoc, an up-to-date and large-scale data set has been constructed for the research of extremophile proteins. Our data set incorporates the latest data on protein sequences and optimal conditions measured in experiments from three databases (BacDive, HaloDom, UniProt), ensuring that our model is trained on the most relevant and reliable data. GeoPoc introduces several innovative features and geometric graph neural networks that enhance its predictive capabilities compared to existing methods. What sets GeoPoc apart from all existing state-of-the-art methods is its incorporation of protein structural information. By leveraging the predicted protein structure, GeoPoc can capture the intricate geometric features and spatial arrangements that influence protein stability and function under extreme conditions. We employ the pre-trained language model ESM to efficiently extract informative sequence embeddings, thus bypassing expensive sequence-based evolutional features. Moreover, GeoPoc achieved similar performance using ESM embeddings and evoformer embeddings in a 5-fold CV on a subset of 200 proteins, as shown in Table. S7. This is consistent with the previous observation38, where evoformer was shown not better than ESM for protein function predictions.
GeoPoc shows promising performance and generalization capabilities for protein optimal temperature in our data set, both at the protein and species levels. In independent testing, GeoPoc demonstrates superior performance compared to other existing methods for thermophilic protein prediction. Additionally, GeoPoc is extended to protein optimal pH and salt concentration prediction, exhibiting accurate classification for the pH task, while the performance for salt concentration, particularly in the low salt range, still leaves room for improvement. In the future, we will incorporate additional biochemical or environmental features to improve the performance of optimal salt concentration prediction. We also tried to directly predict the optimal pH and salt concentration for proteins, but the performances are low (the PCC is 0.480 and 0.558). This is likely because cells could regulate ion concentrations through membranes, and the working environments of proteins inside cells don’t exactly match the external optimal environment. Relatively, the multiple type classifications are less affected than real value predictions.
The model’s interpretability analysis reveals that GeoPoc elucidates the critical physicochemical properties that contribute to enhancing protein thermostability. Our model still has some limitations. Our current dataset comprises only single-chain proteins, which limits the extension of our model to predicting conditions for multi-chain proteins. We will further enhance the generalizability of our model in the future as we acquire multi-chain protein data. In the future, we will also explore fine-tuning AlphaFold2 to predict protein the optimal conditions and structures. This will enable us to better understand how proteins behave under different conditions and allow us to integrate these findings into protein design efforts, potentially leading to the development of proteins with tailored properties for specific industrial or biomedical applications.
Method
Data preparation
In this study, we created an up-to-date data set from two bacteria databases: BacDive26 and HaloDom27. The bacterial diversity metadatabase BacDive is the worldwide largest database for standardized bacterial phenotypic information. HaloDom has collected data on 1,268 halophilic organisms across all life domains, covering aspects from genome and sequence data to environmental details. We downloaded each organism’s species name and corresponding optimal culture conditions from these databases. Due to the optimal growth condition of organisms commonly used to estimate the stability of proteins encoded in their genomes39, we annotate the proteins with the optimal condition of the organism from which they originate. To get protein sequences, we downloaded data from UniProt28 by NCBI tax ID or species name of the organism. We retain protein sequences with a Protein Existence (PE) value of 3 or lower, which represents the evidence supporting the existence of the protein and is defined in the UniProt FASTA headers’ PE field. If the optimal condition is represented as a range, we will replace it with its average. Moreover, the unit of salt concentration is standardized to x%, which means xg/100 ml. The data collection process is shown in Fig. 1a. To this end, there are 6,595,396 reliable protein sequences from 4948 species spanning 39 different phyla, as shown in Fig. S10 and Fig. S11. To examine the generalization ability of our method, we removed redundant proteins sharing sequence identity >25 within each data set using MMSeq240. Finally, we formed a temp data set with 102,615 proteins, a pH data set with 84,646 proteins, and a salt data set with 66,618 proteins.
Due to microorganisms having mechanisms to regulate their intracellular pH and salt concentration41, we conducted different tasks at optimal condition prediction. For example, Natronomonas moolapensis was identified as a highly halophilic archaeon with an optimal growth salt concentration of 18–20% in the BacDive, belonging to the Halobacteriaceae family42. For adaptation to extreme saline environments, the Halobacteriaceae family commonly uses the ”salt-in” effect43 and consists of compensating ion levels: KCl usually provides an osmotic balance31. Microorganisms also have various regulatory mechanisms to adapt to acidic44 or alkaline45 environments. While bacteria possess the ability to regulate their internal pH and salt concentrations, it should be noted that this range of regulation is limited46. Therefore, the protein optimal pH and salt concentration prediction problem were converted into three-class classification tasks. We set up three categories for pH and salt concentration, with pH values split by 5 and 9 and salt concentrations split by 0.05% and 4%. Where 0.05% is the dividing line between freshwater and seawater and 4% is the maximum salt concentration of seawater. As for temperature, it is consistent inside and outside the microorganism, so we perform regression tasks for temperature prediction. Then, we remove the proteins with lengths greater than 1023 due to EMS2.0’s input limitation. Furthermore, the optimal temperature of Balnearium lithotrophicum was found to be between 70 and 75 °C47. Therefore, we adjusted the optimal temperature for the Balnearium lithotrophicum from 55 °C, as reported by BacDive, to 72.5 °C. Then the dataset was divided into training and test sets in a ratio of 8:2 using stratified sampling, the training set sizes for temperature, pH, and salt concentration were 79,272, 65,364, and 51,624, and the test set sizes were 19,817, 16,339, and 12,904, respectively. For the protein optimal pH prediction task, the range of 0–5 contains 3261 training samples and 787 test samples; the 5–9 range contains 57,578 training samples and 14,409 test samples; the 9–14 range contains 4525 training samples and 1143 test samples. For the salt concentration task, the training set contains 2988 samples, and the test set 746 samples for the 0 –0.05% range; the 0.05–4% range contains 27,396 training samples and 6849 test samples; the 4–100% range, there are 21,240 training samples and 5309 test samples. We perform five-fold cross-validation on the training set.
Protein representation as a graph
AlphaFold2 (AF2) has demonstrated remarkable accuracy in predicting protein structures, and we downloaded the predicted protein structures for our dataset from the AF2 database. To assess structural accuracy, AF2 uses the pLDDT (predicted Local Distance Difference Test) score, which estimates the model’s confidence in its predictions. For the proteins in our data set, the average pLDDT score is 81.76, greater than 70 for a correct backbone prediction as indicated by AF2. Furthermore, there is no decline in the accuracy of structures predicted by AF2 at extreme temperatures, as shown in Fig. S1a. Since protein structure represents a balance between environment and evolution, and AF2 utilizes evolutionary information through MSA, it can accurately predict protein structures in extreme environments. According to the protein structure, we represent protein as a graph G = (V, E), with V denoting the amino acids (nodes) and E denoting the contacts of amino acids pairwise distances (edges). Considering the crucial role of the geometric information of residues and the interactions between these residues. We derived protein graph representations from a 3D protein structure referring to48. Specifically, the node of residue i consists of sequence features from pre-trained language model, geometric node features as Xi = (Xe,i, XG,i), and the edge (i, j) contains geometric edge features Ej,k between residue j and k.
Node features Xe,i derived from the embeddings of a pre-trained 36-layer ESM2.0 model named ’esm2_t36_3B_UR50D’, which provides a rich and information-dense evolutionary representation for protein sequences. We extracted the output from the last layer of the encoder part of ESM2.0 as the embedding. The geometric node feature XG,i was derived from the 3D structure by taking angles and distances between nodes and the direction of local atoms relative to the Cα atom. Through the coordinates of three atoms, N − Cα − C, we calculated distances and angles consisting of dihedral angles and bond angles. The direction is calculated by computing the direction of each atom within an amino acid residue relative to the Cα atom and then projecting them onto the local coordinate system defined by N − Cα − C.
The geometric edge feature Ej,k comprises positional embeddings, edge orientation, edge distance, and edge direction. Positional embeddings calculate the frequency of the distances between nodes in the sequence and encode them, helping the model understand the order of the sequence or the relative positions of nodes in the graph structure. Edge direction is a directional vector calculated based on the two residue nodes of an edge relative to their respective local coordinate systems. It describes the relative directional relationship between these two nodes in space. Edge orientation is obtained by comparing the N − Cα − C coordinate systems of the two nodes, specifically by calculating the rotation between the two local coordinate systems, represented as quaternions, to describe the relative difference in the local spatial orientation of the two nodes.
The architecture of GeoPoc
Figure 1b shows GeoPoc represents protein structures and sequence embeddings as protein graphs. Subsequently, GeoPoc employs a GeoFormer block to learn the intricate interactions between nodes and the overall characteristics of the graph structure, generating encoded information that reflects the molecular features of the proteins.
In the GeoFormer module, we apply a norm layer and an MLP to project the initial node and edge features to facilitate better processing and learning in subsequent graph neural network layers. After MLP, we use message passing with TransformerConv49. When processing graph data, we utilize a multi-head attention mechanism to enhance interaction between nodes, while also considering edge feature information during this process. We denote the hidden feature vectors of node i and edge j − > i in layer l as \({h}_{i}^{\left(l\right)}\) and eij, respectively. Specifically, we calculate multi-head attention for each edge from j to i as follows:
Where \( < q,k > =exp(\frac{{q}^{T}k}{\sqrt{d}})\) represents the exponential scale dot-product function, and d denotes the hidden size of each head. We transform the \({h}_{i}^{\left(l\right)}\) and \({h}_{j}^{\left(l\right)}\) into query vector \({q}_{c,i}^{\left(l\right)}\) and key vector \({k}_{c,j}^{\left(l\right)}\) respectively by Wq, Wk. The edge features eij will be encoded and incorporated into the key vector as supplementary information for each layer.
To update node i, the message passing in layer l is:
Where ∣∣ denotes the concatenation operation for C-head attention. \({{\mathcal{N}}}(i)\) denotes the neighbors of node i.
Before generating the final output, we employ self-attention pooling19 to automatically emphasize important residues. Our studies have two types of tasks: regression and three-class classification. Therefore, the multi-layer perceptron (MLP) of the output dimensions is set to one and three, respectively. In the classification task, the three outputs represent the probability values of each class, and we select the class with the highest probability as the result.
Implementations and analyses
We performed five-fold cross-validation on the training data. The training sets were randomly divided into five folds, and each time the model was trained on four folds and tested on the remaining fold. This process is repeated five times and the average validation performance is used to optimise the hyperparameters of the network. Specifically, GeoPoc consists of 2 GNN layers, 256 hidden units, and 4 attention heads. During training, we use the early stopping technique based on the validation performance for a maximum of 50 epochs, where the batch size is set to 12. In the testing phase, all five trained models from the cross-validation are used for prediction, and the average is taken as the final prediction result of GeoPoc. In the testing phase, predictions are made using all five trained models in cross-validation, and the average is taken as the final prediction result of GeoPoc.
We conduct species-level validation to predict optimal temperatures and evaluate the performance of our model when applied to new species. Under the premise that proteins from the species in the training dataset are excluded from the test set, we randomly split the proteins for the training and test set in a ratio of 8:2. Consequently, the test set contained 19,817 proteins from 626 species, while the training set contained 79,272 proteins from 3,148 species. To evaluate the performance of GeoPoc in predicting the species optimal temperature, we calculate the species optimal temperature by taking the average of the predicted optimal temperatures for all proteins within the species. Moreover, we excluded species from the test data set that had fewer than three proteins, which are sensitive to deviations in the prediction of its protein.
For qualitative analysis, we calculate the importance of residues in the sequence towards predictions of the GeoPoc using the GNNExplainer method50. Amino acid importance for each model in the ensemble is summed and normalized across the protein sequence using the L2-norm. To compute residue conservation scores for protein families, we utilize the tool provided by51 with scaled Shannon entropy. We choose protein families using MMseqs2 with a minimum sequence identity of 0.2 and then query in InterPro52.
Evaluation metrics
We use the Pearson correlation coefficient (PCC), mean absolute error (MAE), Accuracy, area under the receiver operating characteristic curve (AUC), the area under the precision-recall curve (AUPR), F1-score (F1), and Matthews correlation coefficient (MCC) to evaluate the prediction performance:
Where x and y are the two variables being compared, and xi and yi are the individual data points in the samples. TP (True Positive) refers to the number of correct positive predictions made by a classifier, while TN (True Negative) refers to the number of correct negative predictions. FP (False Positive) represents the number of incorrect positive predictions, and FN (False Negative) represents the number of incorrect negative predictions. In the evaluation of the three-class predictors for pH and salt concentration, we used macro-averaging for the AUC, F1, and AUPR metrics to account for the performance across all classes equally. For MCC, micro-averaging was applied, treating all samples equally regardless of class, to provide a global measure of performance.
PCC is sensitive to linear relationships between variables, making it an ideal metric for evaluating the performance of regression models that aim to capture such relationships. The other metrics are used for evaluating the performance of classification models.
Statistics and Reproducibility
Data for n ≥3 independent experiments were expressed as mean absolute error (MAE). The statistical significances in this work were analyzed via a two-sample independent t-test from the SciPy (version 1.10.1). In this research, we made use of datasets that are available at https://github.com/ZhummCrow/GeoPoc/tree/main/datasets. The protein structures are downloaded from the AF2 database which is accessible to the public. It’s important to note that our experiments were not randomized.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
This study constructed a new dataset by linking the Uniprot (https://www.uniprot.org/), BacDive (https://bacdive.dsmz.de/), and HoloDom (http://halodom.bio.auth.gr/?view=all_data) databases. The protein optimal condition data sets are available at https://github.com/ZhummCrow/GeoPoc/tree/main/datasets. The source data behind the graphs in the paper can be found in Supplementary Data 1.
Code availability
All source codes and trained models used in our experiments have been deposited at https://github.com/ZhummCrow/GeoPoc.
References
Stetter, K. O. Extremophiles and their adaptation to hot environments. FEBS Lett. 452, 22–25 (1999).
Dumorné, K., Córdova, D. C., Astorga-Eló, M. & Renganathan, P. Extremozymes: a potential source for industrial applications J. Microbiol. Biothechnol. 27, 649–659 (2017).
Cowan, D. A., Ramond, J.-B., Makhalanyane, T. P. & De Maayer, P. Metagenomics of extreme environments. Curr. Opin. Microbiol. 25, 97–102 (2015).
Fujiwara, S. Extremophiles: Developments of their special functions and potential resources. J. Biosci. Bioeng. 94, 518–525 (2002).
Brininger, C., Spradlin, S., Cobani, L. & Evilia, C. The more adaptive to change, the more likely you are to survive: protein adaptation in extremophiles. In Seminars In Cell & Developmental Biology (ed. Mao, Y.) 158–169 (Elsevier, 2018).
Sahoo, K., Sahoo, R. K., Gaur, M. & Subudhi, E. Cellulolytic thermophilic microorganisms in white biotechnology: a review. Folia Microbiol. 65, 25–43 (2020).
Zhou, X.-X., Wang, Y.-B., Pan, Y.-J. & Li, W.-F. Differences in amino acids composition and coupling patterns between mesophilic and thermophilic proteins. Amino Acids 34, 25–33 (2008).
Zhang, G. & Fang, B. Logitboost classifier for discriminating thermophilic and mesophilic proteins. J. Biotechnol. 127, 417–424 (2007).
Meng, C., Ju, Y. & Shi, H. Tmppred: A support vector machine-based thermophilic protein identifier. Anal. Biochem. 645, 114625 (2022).
Charoenkwan, P. et al. Sapphire: A stacking-based ensemble learning framework for accurate prediction of thermophilic proteins. Comput. Biol. Med. 146, 105704 (2022).
Lin, H. & Chen, W. Prediction of thermophilic proteins using feature selection technique. J. Microbiol. Methods 84, 67–70 (2011).
Ahmed, Z. et al. ithermo: a sequence-based model for identifying thermophilic proteins using a multi-feature fusion strategy. Front. Microbiol. 13, 790063 (2022).
Zhao, J., Yan, W. & Yang, Y. Deeptp: a deep learning model for thermophilic protein prediction. Int. J. Mol. Sci. 24, 2217 (2023).
Suplatov, D. et al. Computational design of a ph stable enzyme: understanding molecular mechanism of penicillin acylase’s adaptation to alkaline conditions. PLoS One 9, e100643 (2014).
Hu, S. et al. Hpclas: A data-driven approach for identifying halophilic proteins based on catboost. bioRxiv https://doi.org/10.1101/2023.11.30.569348 (2023).
Ma, F. et al. Sequence homolog-based molecular engineering for shifting the enzymatic ph optimum. Synth. Syst. Biotechnol. 1, 195–206 (2016).
Gado, J. E. et al. Deep learning prediction of enzyme optimum pH. bioRxiv https://doi.org/10.1101/2023.06.22.544776 (2023).
Joo, W.-A. & Kim, C.-W. Proteomics of halophilic archaea. J. Chromatogr. B 815, 237–250 (2005).
Yuan, Q., Xie, J., Xie, J., Zhao, H. & Yang, Y. Fast and accurate protein function prediction from sequence through pretrained language model and homology-based label diffusion. Brief. Bioinforma. 24, bbad117 (2023).
Song, Y. et al. Fast and accurate protein intrinsic disorder prediction by using a pretrained language model. Brief. Bioinforma. 24, bbad173 (2023).
Yuan, Q., Chen, S., Wang, Y., Zhao, H. & Yang, Y. Alignment-free metal ion-binding site prediction from protein sequence through pretrained language model and multi-task learning. Brief. Bioinforma. 23, bbac444 (2022).
Panja, A. S., Maiti, S. & Bandyopadhyay, B. Protein stability governed by its structural plasticity is inferred by physicochemical factors and salt bridges. Sci. Rep. 10, 1822 (2020).
Jumper, J. et al. Highly accurate protein structure prediction with alphafold. Nature 596, 583–589 (2021).
Yuan, Q. et al. Alphafold2-aware protein–dna binding site prediction using graph transformer. Brief. Bioinforma. 23, bbab564 (2022).
Song, Y., Yuan, Q., Zhao, H. & Yang, Y. Accurately identifying nucleic-acid-binding sites through geometric graph learning on language model predicted structures. Brief. Bioinforma. 24, bbad360 (2023).
Reimer, L. C. et al. Bac dive in 2022: the knowledge base for standardized bacterial and archaeal data. Nucleic Acids Res. 50, D741–D746 (2022).
Loukas, A., Kappas, I. & Abatzopoulos, T. J. Halodom: a new database of halophiles across all life domains. J. Biol. Res. Thessalon. 25, 1–8 (2018).
Uniprot: the universal protein knowledgebase in 2023. Nucleic Acids Res. 51, D523–D531 (2023).
Charoenkwan, P., Chotpatiwetchkul, W., Lee, V. S., Nantasenamat, C. & Shoombuatong, W. A novel sequence-based predictor for identifying and characterizing thermophilic proteins using estimated propensity scores of dipeptides. Sci. Rep. 11, 23782 (2021).
Taylor, T. J. & Vaisman, I. I. Discrimination of thermophilic and mesophilic proteins. BMC Struct. Biol. 10, 1–10 (2010).
Matarredona, L., Camacho, M., Zafrilla, B., Bonete, M.-J. & Esclapez, J. The role of stress proteins in haloarchaea and their adaptive response to environmental shifts. Biomolecules 10, 1390 (2020).
Petukhov, M., Kil, Y., Kuramitsu, S. & Lanzov, V. Insights into thermal resistance of proteins from the intrinsic stability of their α-helices. Proteins Struct. Funct. Bioinforma. 29, 309–320 (1997).
Blaber, M., Zhang, X.-j & Matthews, B. W. Structural basis of amino acid α helix propensity. Science 260, 1637–1640 (1993).
Nakariyakul, S., Liu, Z.-P. & Chen, L. Detecting thermophilic proteins through selecting amino acid and dipeptide composition features. Amino Acids 42, 1947–1953 (2012).
Baldwin, R. L. Energetics of protein folding. J. Mol. Biol. 371, 283–301 (2007).
Zhang, G. & Fang, B. Application of amino acid distribution along the sequence for discriminating mesophilic and thermophilic proteins. Process Biochem. 41, 1792–1798 (2006).
Szilágyi, A. & Závodszky, P. Structural differences between mesophilic, moderately thermophilic and extremely thermophilic protein subunits: results of a comprehensive survey. Structure 8, 493–504 (2000).
Hu, M. et al. Exploring evolution-aware &-free protein language models as protein function predictors. Adv. Neural Inf. Process. Syst. 35, 38873–38884 (2022).
Li, G., Rabe, K. S., Nielsen, J. & Engqvist, M. K. Machine learning applied to predicting microorganism growth temperatures and enzyme catalytic optima. ACS Synth. Biol. 8, 1411–1420 (2019).
Steinegger, M. & Söding, J. Mmseqs2 enables sensitive protein sequence searching for the analysis of massive data sets. Nat. Biotechnol. 35, 1026–1028 (2017).
Schäfer, G., Purschke, W. & Schmidt, C. L. On the origin of respiration: electron transport proteins from archaea to man. FEMS Microbiol. Rev. 18, 173–188 (1996).
Burns, D. G. et al. Natronomonas moolapensis sp. nov., non-alkaliphilic isolates recovered from a solar saltern crystallizer pond, and emended description of the genus natronomonas. Int. J. Syst. Evolut. Microbiol. 60, 1173–1176 (2010).
Pikuta, E. V., Hoover, R. B. & Tang, J. Microbial extremophiles at the limits of life. Crit. Rev. Microbiol. 33, 183–209 (2007).
Baker-Austin, C. & Dopson, M. Life in acid: ph homeostasis in acidophiles. Trends Microbiol. 15, 165–171 (2007).
Krulwich, T. A. Alkaliphiles:‘basic’molecular problems of ph tolerance and bioenergetics. Mol. Microbiol. 15, 403–410 (1995).
Preiss, L., Hicks, D. B., Suzuki, S., Meier, T. & Krulwich, T. A. Alkaliphilic bacteria with impact on industrial applications, concepts of early life forms, and bioenergetics of atp synthesis. Front. Bioeng. Biotechnol. 3, 75 (2015).
Takai, K., Nakagawa, S., Sako, Y. & Horikoshi, K. Balnearium lithotrophicum gen. nov., sp. nov., a novel thermophilic, strictly anaerobic, hydrogen-oxidizing chemolithoautotroph isolated from a black smoker chimney in the suiyo seamount hydrothermal system. Int. J. Syst. Evolut. Microbiol. 53, 1947–1954 (2003).
Yuan, Q., Tian, C. & Yang, Y. Genome-scale annotation of protein binding sites via language model and geometric deep learning. bioRxiv https://doi.org/10.1101/2023.11.02.565344 (2023).
Shi, Y. et al. Masked label prediction: Unified message passing model for semi-supervised classification. arXiv https://doi.org/10.48550/arXiv.2009.03509 (2020).
Ying, Z., Bourgeois, D., You, J., Zitnik, M. & Leskovec, J. Gnnexplainer: Generating explanations for graph neural networks. Adv. Neural Inform. Process. Syst. https://doi.org/10.48550/arXiv.1903.03894 (2019).
Capra, J. A. & Singh, M. Predicting functionally important residues from sequence conservation. Bioinformatics 23, 1875–1882 (2007).
Paysan-Lafosse, T. et al. Interpro in 2022. Nucleic Acids Res. 51, D418–D427 (2023).
Acknowledgements
This study has been supported by the National Natural Science Foundation of China (T2394502).
Author information
Authors and Affiliations
Contributions
Y.Y. and Q.Y. designed research. M.Z. and Y.S. performed research. All the authors analyzed data. M.Z., Y.S., and Q.Y. wrote the paper. Y.Y. reviewed and edited the manuscript.
Corresponding author
Ethics declarations
Competing interests
Y.Y. is an Editorial Board Member for Communications Biology, but was not involved in the editorial review of, nor the decision to publish this article. All the other authors declare no competing interests.
Peer review
Peer review information
Communications Biology thanks Yandong Huang, Xiao Wang and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. Primary Handling Editors: Aylin Bircan and Joao Manuel de Sousa Valente.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Zhu, M., Song, Y., Yuan, Q. et al. Accurately predicting optimal conditions for microorganism proteins through geometric graph learning and language model. Commun Biol 7, 1709 (2024). https://doi.org/10.1038/s42003-024-07436-3
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s42003-024-07436-3
This article is cited by
-
Macromolecular crystallography for mammalian body temperature in support of molecular biophysics methods
Biophysical Reviews (2025)
