
Abstract
Hypoxic postconditioning (HPC) was reported to stabilize nuclear β-catenin by inhibiting lysine (K)-specific demethylase 2 A (KDM2A) in hippocampal CA1 against transient global cerebral ischemia (tGCI). Herein we investigate how HPC inhibits the K48-linked poly-ubiquitination (K48-Ub)-related degradation of nuclear β-catenin in CA1 after tGCI. We confirmed that SCFKDM2A complex targets nuclear β-catenin for degradation via ubiquitin proteasome pathway in vitro. HPC reduced SCFKDM2A complex and the K48-Ub of β-catenin, and increased ubiquitin-specific peptidase 22 (USP22) in nucleus after tGCI. Furthermore, KDM2A knockdown decreased the K48-Ub of nuclear β-catenin and nuclear β-catenin-SCFKDM2A complex interaction after tGCI. Moreover, β-catenin knockdown suppressed nuclear survivin expression and attenuated neuroprotection induced by HPC. In contrast, the overexpression of USP22 promoted nuclear β-catenin deubiquitination and enhanced the neuroprotective effects offered by HPC. Taken together, this study supports that HPC downregulated the K48-Ub of nuclear β-catenin through suppressing SCFKDM2A and increasing USP22, thereby inducing cerebral ischemic tolerance.
Similar content being viewed by others


Introduction
Acute ischemic stroke is a major health hazard that causes physical disability, cognitive impairment and even death, affecting millions of people worldwide each year1. However, the available treatment strategies for stroke are limited. Therefore, it is of great significance to search for potential brain protectors or initiate endogenous neuroprotective mechanisms against acute cerebral ischemia. Accumulating studies have shown that β-catenin, a key component of the canonical Wingless/Int (Wnt) /β-catenin signaling pathway, plays an essential role in ischemic neuronal survival2,3. Our previous study revealed that hypoxic postconditioning (HPC) with 8% O2 activates the Wnt/β-catenin pathway in the hippocampal CA1 subregion, thereby restoring the level of nuclear β-catenin and alleviating neuronal damage after transient global cerebral ischemia (tGCI) in Wistar rats4. Recently, we demonstrated that HPC increases the methylation of nuclear β-catenin and promotes the stabilization of β-catenin in CA1 through downregulating tGCI-induced expression of lysine (K)-specific demethylase 2 A (KDM2A), which catalyzes the demethylation of nuclear β-catenin5. However, the underlying mechanism by which the demethylation of nuclear β-catenin catalyzed by KDM2A affects the stability of nuclear β-catenin in HPC-mediated ischemic tolerance remains unclear.
Previous research has shown that the stability of nuclear β-catenin is regulated through the ubiquitin-proteasome system (UPS)6. In a series of enzymatic reactions involved in ubiquitination, ubiquitin ligase (E3) is not only a key enzyme in determining substrate specificity, but also capable of generating various types of ubiquitin chains through seven internal lysine (K) residues on ubiquitin7. It is recognized that the K48-linked poly-ubiquitination (K48-Ub) serves as the most prominent proteolytic marker for target proteins in UPS8. Importantly, the SKP1-CUL1-F-box (SCF) E3 ligase complex, composed of S-phase kinase-associated protein 1 (SKP1), cullin 1 (CUL1) and one F-box protein, promotes the K48-Ub of nuclear β-catenin for subsequent degradation via the ubiquitin proteasome pathway9,10. Notably, KDM2A acts as a kind of F-box protein in the SCF E3 ligase (SCFKDM2A) complex to guarantee substrate recognition11,12. Its demethylase activity is required to mediate nuclear β-catenin degradation via UPS13. We previously demonstrated that tGCI promoted the degradation of nuclear β-catenin by the proteasome in the CA1 region, which was reversed by HPC, whereas adenovirus-mediated KDM2A overexpression abolished the effect of HPC on nuclear β-catenin degradation5. Thus, it is intriguing to explore whether KDM2A acts as an E3 ligase targeting nuclear β-catenin for ubiquitylation-related degradation in a SCFKDM2A-dependent manner during the neuroprotection induced by HPC against tGCI.
In addition to E3 ligase, ubiquitination is under negative regulation of deubiquitinating enzymes (DUBs), which hydrolyzes the peptide bonds between ubiquitin and substrates to remove ubiquitin chains and reverses ubiquitination14. So far, a few ubiquitin-specific peptidases (USPs), which belong to the family with the most members of DUB, have been identified to deubiquitinate β-catenin15,16. Among them, USP22 has been reported strongly expressed in brain of mice, which contains a nuclear localization signal (NLS) allowing itself to shuttle between cytoplasm and nucleoplasm17,18. Also, USP22 increases nuclear β-catenin expression in several types of cancers19,20,21, indicating the involvement of USP22 in regulating the stability of nuclear β-catenin. There are evidences that the downregulation of USP22 is closely linked to poor prognosis in cerebral, intestinal and myocardial ischemia-reperfusion injury22,23,24. Besides, USP22 expression was upregulated under hypoxic condition25. Hence, it is worth investigating whether HPC induces USP22 expression in CA1 to catalyze nuclear β-catenin deubiquitination, thereby promoting nuclear β-catenin stabilization and mediating neuroprotection against tGCI.
Herein we aim to explore the mechanisms by which HPC regulates β-catenin ubiquitination to stabilize nuclear β-catenin in CA1 after tGCI. Specifically, HPC suppresses the formation of SCFKDM2A E3 ligase complex after tGCI to reduce nuclear β-catenin ubiquitination. On the other hand, HPC increases USP22 expression to induce the deubiquitination of nuclear β-catenin, thereby inhibiting the degradation of nuclear β-catenin to enhance nuclear β-catenin stability and finally promoting neuronal survival after tGCI.
Results
SCFKDM2A E3 ligase complex targets nuclear β-catenin for ubiquitylation and degradation in vitro
To reconfirm the subcellular expression pattern of β-catenin of rats after tGCI in our previous studies4, the in vitro experiments were conducted on primary rat hippocampal neurons. In accordance with the previous study in hippocampal CA14, a progressive decline of nuclear β-catenin was observed compared with the control group, whereas no differences were found in total and cytoplasmic β-catenin at 2-24 h after oxygen-glucose deprivation/reoxygenation (OGD/R) (Fig. 1A). Next, the proteasome inhibitor MG132 was used to investigate whether OGD/R promotes nuclear β-catenin degradation through the ubiquitin-proteasome pathway. As expected, the OGD/R-induced downregulation of nuclear β-catenin was completely blocked by MG132 (Fig. 1B). Consistently, OGD/R treatment significantly increased the K48-Ub of nuclear β-catenin in primary hippocampal neurons (Fig. 1C).

A Western blot assay showing the expressions of total, nuclear and cytoplasmic β-catenin at 2, 6, 12, 24 h after OGD/R in in primary hippocampal neurons, respectively. GAPDH is used as loading control to normalize the relative expression of whole cellular/cytoplasmic β-catenin and Lamin B1 is used as loading control to normalize the relative expression of nuclear β-catenin. Data are expressed as percentage of value of the control group. *p < 0.05 versus control group (p = 0.232 for total β-catenin, n = 3-5, Kruskal-Wallis Test; p < 0.001 for nuclear β-catenin, n = 6, One-Way ANOVA Test, p = 0.048 at OGD/R 2 h, p = 0.001 at OGD/R 6 h, p = 0.011 at OGD/R 12 h, p = 0.019 at OGD/R 24 h, Tamhane post-test; p = 0.345 for cytoplasmic β-catenin, n = 4, One-Way ANOVA Test). B Effects of MG132 on nuclear and cytoplasmic β-catenin proteins in primary hippocampal neurons at 24 h after OGD/R using western blot analysis. GAPDH and Lamin B1 are used as loading control to normalize the relative expression of cytoplasmic and nuclear β-catenin, respectively. Data are expressed as percentage of value of the control group with DMSO. *p < 0.05 versus the control (ctrl) group with DMSO (p = 0.04 for nuclear β-catenin, p = 0.977 for cytoplasmic β-catenin, unpaired t-test), #p < 0.05 versus the same group with DMSO (p < 0.001for nuclear β-catenin in the ctrl group or OGD/R group, p = 0.211 for cytoplasmic β-catenin in the ctrl group, p = 0.03 for cytoplasmic β-catenin at the OGD/R group, unpaired t-test, n = 3 in each group). C Immunoprecipitation assay showing the K48-Ub of nuclear β-catenin. β-catenin was immunoprecipitated (IP) using anti-β-catenin antibody. IgG antibody was used as a negative control. K48-Ub was detected by western blot using anti-ubiquitin (linkage-specific K48) antibody. Data are expressed as percentage of value of the control group with MG132. *p < 0.05 versus the control group with MG132 (p = 0.018, n = 3-4, unpaired t-test). D Immunofluorescence assay showing that the effects of Flag-KDM2A (a-d, e-h) or Flag-KDM2A-∆F-box (i-l, m-p) plasmids treatment on the fluorescence intensity of β-catenin with or without MG132 treatment. Representative photomicrographs with fluorescent staining of β-catenin (white), Flag (red) and DAPI (blue) in SH-SY5Y cells. White arrows indicate the positive cells expressed exogenous KDM2A protein. Scale bar: 10 μm. E Western blot assay showing the expression of Flag, KDM2A and β-catenin in nuclear lysates of HEK-293T and SH-SY5Y cells which were transfected with the indicated plasmids and then treated with 20 μM MG132 or DMSO for 6 h. The histogram presents the quantitative analyses of nuclear β-catenin levels. PCNA is used as a loading control to normalize the relative expression of nuclear β-catenin. Data are expressed as percentage of value of control group with DMSO. *p < 0.05 versus Flag-Con group with DMSO (p = 0.026 for HEK-293T, p = 0.026 in the Flag-KDM2A group with DMSO, p = 0.207 in the Flag-KDM2A-∆F-box group with DMSO; p = 0.008 for SH-SY5Y, p = 0.01 in the Flag-KDM2A group with DMSO, p = 0.05 at the Flag-KDM2A-∆F-box group with DMSO, One-Way ANOVA Test followed by Bonferroni post-test), #p < 0.05 versus the same group with DMSO (for HEK293T, p = 0.003 in the Flag-Con group, p = 0.005 in the Flag-KDM2A group, p < 0.001 in the Flag-KDM2A-∆F-box group; for SH-SY5Y, p = 0.036 in the Flag-Con group, p = 0.010 in the Flag-KDM2A group, p = 0.002 in the Flag-KDM2A-∆F-box group, unpaired t-test, n = 8 in each group). F Effects of Flag-KDM2A or Flag-KDM2A-∆F-box plasmids treatment on the ubiquitination of Myc-β-catenin were assessed by in vivo ubiquitination assay. HA-Ub and Myc-β-catenin were co-transfected into HEK-293T cells with KDM2A constructs or empty vector control. G Western blots and Immunoprecipitation blots showing the expression of SKP1, CUL1 and Flag and the assembly of SCFKDM2A ligase complex in nuclear lysates of HEK-293T cells which were transfected with the indicated plasmids. HNs, hippocampal neurons; Flag, flag-tagged protein. The nuclear fraction purity was assessed by absence of β-tubulin staining in Figure E–G. Each bar represents the mean ± SD (error bars).
In view of the regulation of KDM2A in nuclear β-catenin stability in CA1 after tGCI and the ubiquitin ligase activity of KDM2A F-box domain5, we investigated whether KDM2A functions as a potential ubiquitin ligase of nuclear β-catenin both in SH-SY5Y and HEK-293T cells. The immunofluorescent assay showed that the cells with Flag-KDM2A overexpression reduced the fluorescent intensity of nuclear β-catenin compared with adjacent cells without ectopic KDM2A, which was reversed by MG132. Moreover, compared to WT-KDM2A, Flag-KDM2A-∆F-box failed to affect nuclear β-catenin. Additionally, there was an enhanced signal of nuclear β-catenin when MG132 and Flag-KDM2A-∆F-box overexpression were combined in SH-SY5Y cells (Fig. 1D). Flag-KDM2A, but not the Flag-KDM2A-∆F-box, reduced nuclear β-catenin expression both in HEK-293T cells and SH-SY5Y cells. These results were further confirmed by western blot (Fig. 1E). To further determine whether the effect of KDM2A on β-catenin stability depends on its E3 ligase activity, we assessed the ubiquitination level of β-catenin in HEK-293T cells over-expressing WT-KDM2A or the F-box-deficient KDM2A mutant. As shown in Fig. 1F, WT KDM2A rather than KDM2A-∆F-box mutant significantly increased the ubiquitination of ectopic β-catenin in HEK-293T cells. To clarify the role of F-box domain of KDM2A in the assembly of SCFKDM2A E3 ligase complex, we examined the interaction among endogenous SKP1, CUL1 and ectopic KDM2A by co-immunoprecipitation assay. The results revealed the interactions between exogenous FLAG-KDM2A and endogenous SKP1, and between exogenous FLAG-KDM2A and endogenous CUL1 in HEK-293T cells with MG132 treatment. Unlike wild-type KDM2A, KDM2A-∆F-box mutant does not interact with the endogenous SKP1 and CUL1 (Fig. 1G)
HPC reduces the K48-Ub of nuclear β-catenin and alleviates neuronal damage by inhibiting the SCFKDM2A E3 ligase complex in CA1 after tGCI
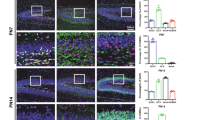
To determine whether SKP1 and CUL1 together with KDM2A participate in HPC-mediated ischemic tolerance in rats, firstly we detected the expressions of SKP1 and CUL1 in CA1 after tGCI with or without hypoxia. As shown in Fig. 2A, SKP1-positive cells were concentrated in the pyramidal cell layer with the typical neuron-like morphology of sham-operated brain. The number of SKP1-positive neuron-like cells increased at 26 h after tGCI, while HPC inhibited this increase of neuron-like cells at the same time points of reperfusion after tGCI (Fig. 2B). Also, western blot analysis showed that HPC obviously attenuated the tGCI-induced upregulation of SKP1 in the early stage of reperfusion (Fig. 2C). However, the morphology of SKP1-positive cells markedly changed in the late stage of reperfusion. At 168 h of reperfusion after tGCI, SKP1 was expressed mainly in glia-like cells with polymorphic somata and processes. Notably, HPC reversed the increase of SKP1-positive glia-like cells and the reduction of SKP1-positive neuron-like cells induced by tGCI in the late stage of reperfusion (Fig. 2A, B). As shown in Fig. 2D, almost all SKP1-positive cells in Sham rats were neuronal nucleus (NeuN) -positive (Fig. 2D, a-h). Notably, at 168 h after tGCI, most of SKP1-positive cells were colocalized with glial fibrillary acidic protein (GFAP) (Fig. 2D, i-p), while mainly with NeuN in the HPC group (Fig. 2D, q-x).

A Immunohistochemistry of SKP1 in the hippocampus after tGCI with or without hypoxia. Representative images show the sham-operated group (a, b), 26 h after reperfusion of tGCI groups (c, d) and HPC groups (e, f), 168 h after reperfusion of tGCI groups (g, h) and HPC groups (i, j), respectively. Scale bars: 250 μm (a, c, e, g, i) and 25 μm (b, d, f, h, j). B Quantitative analyses of SKP1-positive neuron-like and glia-like cells in CA1 (n ≥ 4 in each group). *p < 0.05 versus Sham animals (for SKP1-positive neuron-like cells, p < 0.001 in the tGCI and HPC groups, One-Way ANOVA Test, p < 0.001 at tGCI 26 h, p = 0.307 at tGCI 50 h, p < 0.001 at tGCI 168 h, p = 0.001 at HPC 26 h, p = 0.175 at HPC 50 h, p < 0.001 at HPC 168 h, Bonferroni post-test; for SKP1-positive glia-like cells, p < 0.001 in the tGCI and HPC groups, One-Way ANOVA Test, p = 1.000 at tGCI 26 h, p = 1.000 at tGCI 50 h, p < 0.001 at tGCI 168 h, Bonferroni post-test, p = 0.997 at HPC 26 h, p = 0.732 at HPC 50 h, p < 0.001 at HPC 168 h, Tamhane post-test), #p < 0.05 versus tGCI group at the same time point (for SKP1-positive neuron-like cells, p = 0.001 at 26 h, p = 0.028 at 50 h, p = 0.003 at 168 h; for SKP1-positive glia-like cells, p = 0.833 at 26 h, p = 0.011 at 50 h, p < 0.001 at 168 h; n = 4-6, unpaired t-test). C Western blot analysis of SKP1 in CA1. The histogram presents the quantitative analyses of SKP1 levels. GAPDH is used as loading control to normalize the relative expression of whole cellular SKP1. Data are expressed as percentage of value of Sham animals. *p < 0.05 versus Sham animals (p = 0.003 in the Hypoxia groups, p < 0.001 in the tGCI groups, One-Way ANOVA Test, p = 0.591 at Hypoxia 0 h, p = 0.042 at Hypoxia 24 h, p = 0.090 at tGCI 0 h, p = 0.005 at tGCI 4 h, p = 0.011 at tGCI 26 h, p = 0.309 at tGCI 50 h, p = 0.227 at HPC 26 h, p = 0.474 at HPC 50 h, Tamhane post-test), #p < 0.05 versus tGCI group at the same time point (p = 0.046 at 26 h, p = 0.030 at 50 h, n = 6-8, unpaired t-test). D Representative photomicrographs with fluorescent staining of SKP1 (green), NeuN/GFAP (red) and DAPI (blue) in CA1. Scale bars: 75 μm. Each bar represents the mean ± SD (error bars).
As shown in Fig. 3A, CUL1 immunopositive cells were seen to be distributed predominantly in the pyramidal cell layer in CA1 with a round and clear appearance. In line with SKP1, the number of CUL-positive cells increased at 26 h, while decreased markedly at 168 h after tGCI. However, when compared with tGCI groups at corresponding time points of reperfusion, the expression of CUL1 reduced in the HPC groups (Fig. 3B). The above results were further confirmed by western blot analysis (Fig. 3C). Immunofluorescent assay demonstrated that CUL1 in CA1 of Sham animals were colocalized with NeuN, only a few were colocalized with ionized calcium-binding adaptor molecule-1 (Iba-1), but not with GFAP (Fig. 3D). We also confirmed that the trend of SKP1 expression in the cytoplasmic fraction was similar to that of whole cell lysate in CA1, whereas CUL1 was less expressed in cytoplasm. There were no significant differences in cytoplasmic CUL1 between tGCI and HPC groups (Fig. 3E). Subsequently we measured SKP1 and CUL1 levels in the nucleus. In line with the expressions of total SKP1 and CUL1, the expressions of nuclear SKP1 and CUL1 were significantly increased after tGCI compared with the Sham group. However, HPC markedly reduced the tGCI-induced elevation of nuclear SKP1 and CUL1 in CA1 (Fig. 3F). Co-immunoprecipitation assay showed that the interactions of KDM2A-SKP1 and KDM2A-CUL1 were increased within the nucleus in CA1 after tGCI, which was inhibited by HPC (Fig. 3G).

A Immunohistochemistry of CUL1 in the hippocampus after tGCI with or without hypoxia. Representative images show the sham-operated group (a, b), 26 h after reperfusion of tGCI groups (c, d) and HPC groups (e, f), respectively. Scale bars: 250 μm (a, c, e) and 25 μm (b, d, f). B Quantitative analysis of CUL1-positive cells in CA1. *p < 0.05 versus Sham animals (p = 0.391 in the Hypoxia groups, p < 0.001 in the tGCI groups, p = 1.000 at tGCI 0 h, p = 0.220 at tGCI 4 h, p = 0.014 at tGCI 26 h, p = 0.062 at tGCI 50 h, p < 0.001 at tGCI 168 h, One-Way ANOVA Test followed by Bonferroni post-test; p = 0.003 in the HPC groups, p = 1.000 at HPC 26 h, p = 1.000 at HPC 50 h, p = 0.035 at HPC 168 h, Kruskal Wallis Test, #p < 0.05 versus tGCI group at the same time point (p = 0.010 at 26 h, p = 0.145 at 50 h, p = 0.969 at 168 h, n = 4-6, unpaired t-test). C Western blot analysis of CUL1 in CA1. The histogram presents the quantitative analyses of CUL1 levels. GAPDH is used as loading control to normalize the relative expression of whole cellular CUL1. Data are expressed as percentage of value of Sham animals. *p < 0.05 versus Sham animals (p = 0.205 in the Hypoxia groups, p < 0.001 in the tGCI groups, p = 0.002 in the HPC groups, One-Way ANOVA Test, p = 0.732 at tGCI 0 h, p = 0.428 at tGCI 4 h, p = 0.044 at tGCI 26 h, p = 0.897 at tGCI 50 h, p = 0.434 for HPC 26 h, p = 0.011 for HPC 50 h, Tamhane post-test), #p < 0.05 versus tGCI group at the same time point (p = 0.001 at 26 h, p = 0.001 at 50 h, n = 4, unpaired t-test). D Representative photomicrographs with fluorescent staining of CUL1 (green), NeuN/GFAP/Iba-1 (red) and DAPI (blue) in CA1. Scale bars: 75 μm. E, F Representative immunoblots and quantitative data show SKP1 and CUL1 in cytoplasmic and nuclear fractions, respectively. GAPDH and Lamin B1 are used as loading controls to normalize the relative expressions of cytoplasmic and nuclear proteins of interest, respectively. Data are expressed as percentage of value of sham-operated animals. *p < 0.05 versus Sham animals (for cytoplasmic SKP1, n = 4, p = 0.056 for Hypoxia, p = 0.004 for tGCI, p = 0.443 for HPC, One-Way ANOVA Test, p = 1.000 at tGCI 0 h, p = 0.523 at tGCI 4 h, p = 0.042 at tGCI 26 h, p = 1.000 at tGCI 50 h; for cytoplasmic CUL1, n = 6, p = 0.616 for Hypoxia, p = 0.538 for tGCI, p = 0.376 for HPC, One-Way ANOVA Test; for nuclear SKP1, n = 4, p = 0.024 in the Hypoxia groups, p = 0.019 at Hypoxia 0 h, p = 0.517 at Hypoxia 24 h, Kruskal Wallis Test; p = 0.094 at tGCI 0 h, p = 0.249 at tGCI 4 h, p = 0.011 at tGCI 26 h, p = 0.070 at tGCI 50 h, unpaired t-test; for nuclear CUL1, n = 4, p = 0.005 in the Hypoxia groups, p = 0.048 at Hypoxia 0 h, p = 1.000 at Hypoxia 24 h, One-Way ANOVA Test followed by Tamhane post-test, p = 0.041 at tGCI 0 h, p = 0.355 at tGCI 4 h, p = 0.003 at tGCI 26 h, p < 0.001 at tGCI 50 h, unpaired t-test). #p < 0.05 versus tGCI group at the same time point (for cytoplasmic SKP1, p = 0.016 at 26 h, p = 0.152 at 50 h; for cytoplasmic CUL1, p = 0.933 at 26 h, p = 0.485 at 50 h; for nuclear SKP1, p = 0.016 at 26 h, p = 0.048 at 50 h; for nuclear CUL1, p = 0.006 at 26 h, p = 0.001 at 50 h; n = 4, unpaired t-test). G Immunoprecipitation assay showing the level of SCFKDM2A E3 ligase complex in CA1 of tGCI and HPC groups. KDM2A was immunoprecipitated (IP) using anti-KDM2A antibody. IgG antibody was used as a negative control. SKP1 and CUL1 was detected by western blot using anti-SKP1 and anti-CUL1 antibodies. Data are expressed as percentage of value of sham-operated animals. *p < 0.05 versus Sham animals (p = 0.034 for SKP1/KDM2A, p = 0.036 at tGCI 26 h, p = 0.201 at tGCI 50 h; p = 0.001 for CUL1/KDM2A, p = 0.001 at tGCI 26 h, p = 0.328 at tGCI 50 h, One-Way ANOVA Test followed by Bonferroni post-test), #p < 0.05 versus tGCI group at the same time point (n = 7 in each group for SKP1/KDM2A, p = 0.103 at 26 h, p = 0.041 at 50 h; n=6 in each group for CUL1/KDM2A, p = 0.009 at 26 h, p = 0.207 at 50 h, unpaired t-test). Each bar represents the mean ± SD (error bars).
Further, we observed an obvious increase of K48-Ub of nuclear β-catenin in tGCI group at 26 h and 50 h after reperfusion via co-immunoprecipitation assay. Conversely, in HPC group, the increased K48-Ub of nuclear β-catenin was reversed (Fig. 4A). To investigate the role of KDM2A in mediating the recognition of substrate β-catenin by SCFKDM2A E3 ligase complex and subsequently catalyzing the K48-Ub of nuclear β-catenin in CA1 after tGCI, KDM2A small-interfering RNA (siKDM2A) was then utilized. The validity in inducing KDM2A silence, the maintenance for nuclear β-catenin stabilization after tGCI and the safety for CA1 neurons of siKDM2A administration had been previously confirmed5. As shown in Fig. 4B, nuclear KDM2A was analyzed after siKDM2A administration and the result was consistent with our previous studies5. There was no significant difference on nuclear CUL1 with or without siKDM2A administration. Interestingly, siKDM2A administration inhibited nuclear SKP1 in tGCI rats when compared to NC group (Fig. 4B). As expected, the increase of K48-Ub level of nuclear β-catenin and the interactions among KDM2A, CUL1, SKP1 and nuclear β-catenin in CA1 induced by tGCI were counteracted by siKDM2A administration (Fig. 4C). To clarify the relation between KDM2A-mediated demethylation and K48-Ub of nuclear β-catenin, daminozide, a potent small-molecule inhibitor of demethylase activity of KDM2A, was utilized. Daminozide at 20 μM and 40 μM up-regulated total lysine methylation of Sham rats when compared to vehicle, and the effect at 40 μM on total lysine methylation was more significant (Supplementary Fig. 1). However, no effects on total KDM2A level were observed after daminozide administration. Thus, 40 μM of daminozide was applied for subsequent studies. Further experiments confirmed that daminozide treatment reversed the tGCI-induced reductions of nuclear β-catenin and methylated β-catenin, as well as the elevation of the K48-Ub of nuclear β-catenin in CA1 at 26 h of reperfusion, whereas there were no significant differences in CA1 of Sham rats between daminozide and vehicle treatments (Fig. 4D, E).

A Immunoprecipitation assay showing the K48-Ub of nuclear β-catenin in CA1 of tGCI and HPC groups. Data are expressed as percentage of value of sham-operated animals. *p < 0.05 versus Sham animals (p = 0.006 in the tGCI group, p = 0.698 in the HPC group, One-Way ANOVA Test, p = 0.041 at tGCI 26 h, p = 0.050 at tGCI 50 h, Tamhane post-test), #p < 0.05 versus tGCI group at the same time point (p = 0.007 at 26 h, unpaired t-test, p = 0.015 at 50 h, Mann-Whitney test, n = 6 in each group). B Representative images of western blot showing the expressions of KDM2A, CUL1 and SKP1 in nuclear fraction after tGCI with or without siKDM2A administration. Lamin B1 is used as loading controls to normalize the relative expressions of nuclear proteins of interest. Data are expressed as percentage of Sham animals value. *p < 0.05 versus Sham group with NC (p < 0.001 for KDM2A, n = 4; p = 0.045 for CUL1, n = 8; p = 0.015 for SKP1, n = 6, unpaired t-test), &p < 0.05 versus the same group with NC (For KDM2A, p < 0.001 in the Sham and tGCI group with siKDM2A; For CUL1, p = 0.363 in the Sham group with siKDM2A, p = 0.218 at the tGCI group with siKDM2A; For SKP1, p = 0.097 in the Sham group with siKDM2A, p = 0.001 in the tGCI group with siKDM2A; unpaired t-test). C Immunoprecipitation assays showing the effects of siKDM2A administration on the K48-Ub of nuclear β-catenin and the interaction between β-catenin and three subunits of SCFKDM2A within nucleus of Sham and tGCI rats. Data are expressed as percentage of Sham animals value. *p < 0.05 versus Sham group with NC (p = 0.046 for K48-Ub/β-catenin, n = 7; p < 0.001 for KDM2A/β-catenin, n = 5; p = 0.028 for CUL1/β-catenin, n = 5; p < 0.001 for SKP1/β-catenin, n = 6; unpaired t-test, &p < .05 versus the same group with NC (for K48-Ub/β-catenin, p = 0.537 in the Sham group with siKDM2A, p = 0.006 in the tGCI group with siKDM2A; for KDM2A/β-catenin, p = 0.617 in the Sham group with siKDM2A, p = 0.001 in the tGCI group with siKDM2A; for CUL1/β-catenin, p = 0.233 in the Sham group with siKDM2A, p = 0.006 in the tGCI group with siKDM2A; for SKP1/β-catenin, p = 0.706 in the Sham group with siKDM2A, p = 0.022 in the tGCI group with siKDM2A; unpaired t-test). D Representative images of western blot showing the expression of nuclear β-catenin in CA1 of Sham and tGCI groups with or without daminozide administration. Lamin B1 is used as loading controls to normalize the relative expressions of nuclear proteins of interest. Data are expressed as percentage of Sham animals value. *p < 0.05 versus Sham group with vehicle (p = 0.004, unpaired t-test), &p < 0.05 versus the same group with vehicle (p = 0.118 in the Sham group with daminozide, p = 0.045 in the tGCI group with daminozide, unpaired t-test, n = 4 in each group). The nuclear fraction purity was assessed by absence of GAPDH staining in Figure B and D. E Immunoprecipitation assays showing the level of nuclear K48-ubiquitinated β-catenin and me-β-catenin in CA1 of Sham and tGCI groups with or without daminozide administration. Methylation was detected by western blot using antimethyl-lysine antibody. *p < 0.05 versus Sham group with vehicle (p = 0.019 for K48-Ub/β-catenin, n = 4; p = 0.022 for Methyl-K/β-catenin, n = 7, unpaired t-test), &р < 0.05 versus the same group with vehicle, injection intracerebroventricularly with ddH2O (for K48-Ub/β-catenin, p = 0.294 in the Sham group with daminozide, p = 0.023 in the tGCI group with daminozide; for Methyl-K/β-catenin, p = 0.252 in the Sham group with daminozide, p = 0.045 in the tGCI group with daminozide, unpaired t-test). Each bar represents the mean ± SD (error bars).
To further support the involvement of β-catenin in HPC-mediated cerebral ischemia tolerance, the adeno-associated viruses (AAV) containing Ctnnb1 small interfering RNA (AAVi-Ctnnb1) to inhibit the expression of β-catenin was injected into bilateral CA1 region. First, the effective transfection of AAVi-CON or AAVi-Ctnnb1 in CA1 was confirmed by fluorescent images (Fig. 5A). The expression of β-catenin significantly decreased after AAVi-Ctnnb1 administration at the dosage of 7.92 × 109 v.g or 15.84 × 109 v.g in Sham rats, which was more effective at the higher dosage (Fig. 5B). Thus, the dosage of 15.84 × 109 v.g was selected in the following experiments. As expected, AAVi-Ctnnb1 administration dramatically aggravated neuronal damage in CA1 with HPC, whereas AAVi-Ctnnb1 or AAVi-CON did not alter the numbers of surviving and NeuN-positive cells in Sham rats (Fig. 5C–E). Besides, compared with the AAVi-CON group, AAVi-Ctnnb1 significantly reduced nuclear β-catenin and survivin in CA1 of Sham and HPC rats (Fig. 5F, G).

A Phase contrast and fluorescent images from coronal sections of CA1 following injection of AAVi-CON (a, b) or AAVi-Ctnnb1 (c, d) in Sham animals. Black arrowhead shows the needle trace. Scale bar: 75 μm. B Representative immunoblots of β-catenin expression in CA1 after AAVi-Ctnnb1 administration at the dosage of 3.96 × 109 v.g, 7.92 × 109 v.g or 15.84 × 109 v.g in Sham rats. Data are expressed as percentage of value of Sham animals with AAVi-CON. &p < 0.05 vs. Sham group with AAVi-CON (p = 0.001, n = 3, One-Way ANOVA Test, p = 0.871 at 3.96× 109 v.g, p = 0.015 at 7.92× 109 v.g, p = 0.002 at 15.84× 109 v.g, Bonferroni post-test). C Representative microphotographs of cresyl violet staining and NeuN immunostaining in CA1 from rats administered bilaterally with either AAVi-CON or AAVi-Ctnnb1 at 7 d after tGCI with hypoxia or without hypoxia. Boxes indicate that the magnified regions displayed in the right panel. Scale bar: a, c, e, g, i, k, m, o, q, s, u w: 250 μm; b, d, f, h, j, l, n, p, r, t, v, w: 25 μm. D, E Quantitative analyses of surviving and NeuN-positive cells in CA1. *p < 0.05 vs. Sham animals (p < 0.001 for surviving cells, p < 0.001 for NeuN-positive cells, n = 3, unpaired t-test), &p < 0.05 vs. the same group with AAVi-CON (for surviving cells, p = 0.438 in the Sham group with AAVi-Ctnnb1, p = 0.040 in the HPC group with AAVi-Ctnnb1; for NeuN-positive cells, p = 0.714 in the Sham group with AAVi-Ctnnb1, p = 0.046 in the tGCI group with AAVi-Ctnnb1, unpaired t-test). F, G Western blot analyses of nuclear β-catenin and survivin in CA1. The histogram presents the quantitative analyses of nuclear β-catenin and survivin levels. Data are expressed as percentage of value of Sham animals. The nuclear fraction purity was assessed by absence of GAPDH staining in Figure F and G. *p < 0.05 vs. Sham group (p = 0.023 for β-catenin, n = 3; p = 0.013 for survivin, n = 4, unpaired t-test), &p < 0.05 vs. the same group with AAVi-CON (for β-catenin, p = 0.047 at the Sham group with AAVi-Ctnnb1, p = 0.041 in the HPC group with AAVi-Ctnnb1; for survivin, p = 0.016 in the Sham group with AAVi-Ctnnb1, p = 0.007 in the HPC group with AAVi-Ctnnb1, unpaired t-test). Each bar represents the mean ± SD (error bars).
HPC promotes nuclear β-catenin deubiquitination and neuronal survival through increasing nuclear USP22 in CA1 after tGCI
With immunohistochemical assay we observed that USP22-poitive cells mainly existed in the pyramidal cell layer with a neuron-like appearance in Sham and ischemic rats at 26 h of reperfusion after tGCI with or without hypoxia. However, at 168 h after reperfusion, the majority of USP22-positive cells exhibited elongated and irregular nuclei with polymorphic processes (Fig. 6A). Compared with sham-operated rats, the number of USP22-positive cells presented a sustained decrease during 26-168 h after reperfusion of tGCI. Conversely, HPC reversed tGCI-induced reductions of USP22-positive cells to a certain extent (Fig. 6B). Double-fluorescent labeling immunohistochemistry confirmed the predominant neuronal location of USP22 in Sham rats, as evidenced by the colocalization of USP22 with NeuN, but not GFAP and Iba-1. Nevertheless, at 168 h of reperfusion after tGCI, the fluorescence of USP22 was reduced. They were mainly co-labelled with GFAP, only a few with NeuN, but did not with Iba-1. Conversely, with HPC treatment, the fluorescence of USP22 was enhanced. They were mainly located in hippocampal microglia, as shown by the co-localization with Iba-1. Parts of them located in neurons, as shown by the co-localization with NeuN. Few of USP22-positive cells were astrocytes, as shown by the co-localization with GFAP (Fig. 6C). Also, the reduction of USP22 in CA1 at 26 and 50 h of reperfusion after tGCI was confirmed by western blot. Inconsistently, no statistical differences were observed in the level of total USP22 between tGCI and HPC groups (Fig. 6D). In line with total USP22, tGCI significantly down-regulated cytoplasmic and nuclear USP22 in CA1. However, HPC remarkably inhibited the reduction of nuclear USP22 induced by tGCI, but had no effect on cytoplasmic USP22 (Fig. 6E, F).

A Immunohistochemistry of USP22 in the hippocampus after tGCI with or without hypoxia. Representative images show the sham-operated group (a, b), 26 h after reperfusion of tGCI groups (c, d) and HPC groups (e, f), 168 h after reperfusion of tGCI groups (g, h) and HPC groups (i, j), respectively. Scale bars: 250 μm (a, c, e, g, i) and 25 μm (b, d, f, h, j). B Quantitative analysis of USP22-positive cells in CA1. *p < 0.05 versus Sham animals (p = 0.345 in the hypoxia group, One-Way ANOVA Test; p < 0.001 in the tGCI group, One-Way ANOVA Test, p = 0.017 at tGCI 0 h, p < 0.001 at tGCI 4 h, p = 0.024 at tGCI 26 h, p = 0.001 at tGCI 50 h, p < 0.001 at tGCI 168 h, Tamhane post-test; p < 0.001 in the HPC group, One-Way ANOVA Test, p = 0.006 at HPC 26 h, p = 0.001 at HPC 50 h, p < 0.001 at HPC 168 h, Bonferroni post-test, n = 4-8), #p < 0.05 versus tGCI group at the same time point (p = 0.006 at 26 h, p = 0.023 at 50 h, p = 0.001 at 168 h, unpaired t-test). C Representative photomicrographs with fluorescent staining of USP22 (red), NeuN/GFAP/Iba-1 (green), and DAPI (blue) in CA1 after tGCI with or without hypoxia. Scale bars: 75 μm. D Western blot analysis of USP22 in CA1. GAPDH is used as loading control to normalize the relative expression of whole cellular USP22. Data are expressed as percentage of value of Sham animals. The histogram presents the quantitative analyses of USP22 levels. *p < 0.05 versus Sham animals (p = 0.536 in the hypoxia group, One-Way ANOVA Test; p = 0.019 in the tGCI group, One-Way ANOVA Test, p = 0.999 at tGCI 0 h, p = 0.982 at tGCI 4 h, p = 0.031 at tGCI 26 h, p = 0.045 at tGCI 50 h, Tamhane post-test; p < 0.015 in the HPC group, One-Way ANOVA Test, p = 0.136 at HPC 26 h, p = 0.014 at HPC 50 h, Bonferroni post-test, n = 3-5), #p < 0.05 versus tGCI group at the same time point (p = 0.103 at 26 h, p = 0.984 at 50 h, unpaired t-test). E, F Representative immunoblots and quantitative data show USP22 in cytoplasmic and nuclear fractions, separately. GAPDH and Lamin B1 are used as loading controls to normalize the relative expressions of cytoplasmic and nuclear USP22, respectively. Data are expressed as percentage of value of Sham animals (n = 4 in each group for cytoplasmic USP22, n = 6 for nuclear USP22). *p < 0.05 versus Sham animals (for cytoplasmic USP22, p = 0.016 at hypoxia 24 h, Kruskal-Wallis Test; p < 0.001 in the tGCI group, One-Way ANOVA Test, p = 0.502 at tGCI 0 h, p = 0.999 at tGCI 4 h, p = 0.018 at tGCI 26 h, p = 0.001 at tGCI 50 h, Tamhane post-test; p = 0.001 in the HPC group, One-Way ANOVA Test, p = 0.030 at HPC 26 h, p = 0.024 at HPC 50 h, Tamhane post-test, n = 4; for nuclear USP22, p = 0.121 in the hypoxia group, One-Way ANOVA Test; p < 0.001 in the tGCI group, One-Way ANOVA Test, p = 0.734 at tGCI 0 h, p = 1.000 at tGCI 4 h, p = 0.023 at tGCI 26 h, p = 0.024 at tGCI 50 h, Tamhane post-test; p = 0.466 in the HPC group, One-Way ANOVA Test, n = 6), #p < 0.05 versus tGCI group at the same time point (for cytoplasmic USP22, p = 0.894 at 26 h, p = 0.334 at 50 h, for nuclear USP22, p = 0.003 at 26 h, p = 0.025 at 50 h, unpaired t-test). G Immunoprecipitation blots showing the interaction between nuclear USP22 and nuclear β-catenin in the CA1 of tGCI and HPC groups. USP22 was immunoprecipitated (IP) using anti-USP22 antibody. IgG antibody was used as a negative control. β-catenin was detected by western blot. Data are expressed as percentage of Sham animals value. *p < 0.05 versus Sham animals (p = 0.005 in the tGCI group, p = 0.121 in the HPC group, n = 4, One-Way ANOVA Test, p = 1.000 at 26 h, p = 0.025 at 50 h, Bonferroni post-test), #p < 0.05 versus tGCI group at the same time point (p = 0.711 at 26 h, p = 0.006 at 50 h, unpaired t-test). Each bar represents the mean ± SD (error bars).
Next, we examined the interaction between USP22 and β-catenin within the nucleus after tGCI with or without hypoxia by co-immunoprecipitation assay. The results showed that the interaction between USP22 and β-catenin in the nucleus was significantly weakened at 50 h after tGCI. In contrast, HPC completely reversed this reduction induced by tGCI (Fig. 6G). Next, USP22-carried AAV vector (AAV-USP22) or AAV-Con was administrated into the bilateral CA1 (Fig. 7A). Notably, AAV-Con only carries a single mCherry without USP22. As shown in Fig. 7B, in Sham group with AAV-Con administration, the red fluorescence (mCherry) represents the effectiveness of AAV transfection, whereas in Sham group with AAV-USP22, mCherry represents the exogenous expression of USP22, which located dominantly in the nucleus of the CA1 pyramidal cells. Western blot analysis showed that USP22 expression was augmented significantly after AAV-USP22 administration in Sham rats (Fig. 7C). Similar change was observed in total β-catenin level (Fig. 7D). AAV-USP22 or AAV-Con had no impact on the neuronal number of Sham rats. Obviously, AAV-USP22 treatment markedly ameliorated neuronal damage in CA1 after tGCI, which was demonstrated by an increase in the number of surviving and NeuN-positive cells as well as a decrease in Fluoro-Jade B (F-JB)-positive cells. Additionally, an additive neuroprotective effect was observed in HPC group with AAV-USP22 administration (Fig. 7E-H).

A Experimental design/timeline of AAV vectors injection bilaterally into dorsal CA1 pyramidal layer. Posttranslational modifications of β-catenin, learning and memory, as well as cellular damage in CA1 were measured in Sham, tGCI and HPC rats injected with the AAV-Con/USP22. B Phase contrast and fluorescent images from coronal sections of CA1 following injection of AAV-Con or AAV-USP22 in Sham animals. White arrows show the needle trace. Scale bar: 100 μm. C, D Representative immunoblots of USP22 and β-catenin expressions in CA1. GAPDH is used as loading control to normalize the relative expression of whole cellular USP22 and β-catenin. Data are expressed as percentage of value of Sham animals. *p < 0.05 versus Sham animals and &p < 0.05 versus Sham group with AAV-Con (p = 0.048 for USP22, p = 0.004 for β-catenin, n = 3, unpaired t-test). E Representative microphotographs of cresyl violet staining, NeuN immunostaining, and F-JB staining in the hippocampus at 7 days after tGCI with AAV-Con/USP22. Scale bar: 250 μm (a, e, i, m, q, u), 25 μm (b-c, f-g, j-k, n-o, r-s, v-w) and 75 μm (d, h, l, p, t, x). F–H Quantitative analyses of surviving, NeuN-positive cells and F-JB-positive cells in CA1. *p < 0.05 versus Sham group with AAV-Con (for surviving cells, p < 0.001 in the tGCI group with AAV-Con, p < 0.001 in the HPC group with AAV-Con; for NeuN-positive cells, p < 0.001 in the tGCI group with AAV-Con, p < 0.001 in the HPC group with AAV-Con; For F-JB-positive cells, p < 0.001 in the tGCI group with AAV-Con, p = 0.001 in the HPC group with AAV-Con, n = 5–8, unpaired t-test), #p < 0.05 versus tGCI group with AAV-Con (p = 0.015 for surviving cells, Mann-Whitney test; p = 0.041 for NeuN-positive cells, p = 0.042 for F-JB-positive cells, unpaired t-test), &р<0.05 versus the same group with AAV-Con (for surviving cells, p = 0.521 in the Sham group, p = 0.002 in the tGCI group, p = 0.023 in the HPC group, unpaired t-test; for NeuN-positive cells, p = 0.095 in the Sham group, p = 0.013 in the tGCI group, p < 0.001 in the HPC group, unpaired t-test; For F-JB-positive cells, p = 0.005 in the tGCI group, unpaired t-test, p = 0.016 in the HPC group, Mann-Whitney test). MWM, Morris water maze; PTMs, posttranslational modifications. Each bar represents the mean ± SD (error bars).
Further, Morris water maze (MWM) test was performed to measure learning and memory functions (Fig. 8A). The swimming path tracings during the training period (learning) and probe trial (memory) in each group were shown as Fig. 8B. The escape latency and path length of rats in six groups decreased in a time-dependent manner over 5 d hidden-platform training sessions. For the rats injected with AAV-Con, the path length and escape latency in the tGCI group were significantly prolonged compared with Sham group. Inversely, HPC improved the spatial learning ability of rats at 4rd to 6th day after tGCI (Fig. 8C, D). In the probe phase, the percentage of time that rats spent in the target quadrant after removing the platform was recorded to assess memory function. As shown in Fig. 8E, HPC partially reversed the reduction induced by tGCI in target quadrant occupancy. These results were consistent with our previous study in the absence of AAV intervention5. Compared with AAV-Con administration, USP22 overexpression significantly enhanced the spatial learning ability and long-term memory of tGCI rats, as presented by the shorter time in the swimming path length and escape latency as well as the longer time in the target quadrant occupancy (Fig. 8B-E). Hence, the spatial learning and memory functions of rats after tGCI were effectually improved by AAV-USP22 administration.

A Schematic diagram of the experimental design of the Morris water maze (MWM) test after tGCI with or without hypoxia. B Representative swimming path tracings in each group during the training period (learning) and probe trial (memory). C, D The escape latency and path length were recorded among AAV-Con/USP22 treated rats in Sham, tGCI and HPC group. With Mauchly’s test of sphericity, p = 0.001 for the escape latency, p = 0.009 for the path length. *p < 0.05 versus the Sham group with AAV-Con (For the escape latency, in the tGCI+AAV-Con groups, p = 0.016 at day1, p < 0.001 at day2, p = 0.001 at day3, p < 0.001 at day4 and day5, in the HPC + AAV-Con groups, p = 0.648 at day1, p = 1.000 at day2-5; For the path length, in the tGCI+AAV-Con groups, p = 0.008 at day1, p = 0.001 at day2, p < 0.001 at day3 and day 4, p = 0.004 at day5, in the HPC + AAV-Con groups, p = 0.264 at day1, p = 1.000 at day2-5, MANOVA Test followed by Bonferroni post-test), #p < 0.05 represents HPC groups versus tGCI groups with AAV-Con (For the escape latency, p = 1.000 at day1, p = 0.020 at day2, p = 0.009 at day3, p = 0.001 at day4, p = 0.002 at day5; For the path length, p = 1.000 at day1, p = 0.056 at day2, p = 0.004 at day3, p = 0.006 at day4, p = 0.029 at day5, MANOVA Test followed by Bonferroni post-test), and &р<0.05 versus the same group with AAV-Con (For the escape latency, p = 1.000 at day1-5 in the Sham and the HPC groups, p = 0.416 at day1, p = 0.057 at day2, p = 0.086 at day3, p = 0.004 at day4, p = 0.014 at day5 in the tGCI groups; For the path length, p = 1.000 at day1-5 in the Sham and HPC groups, p = 1.000 at day1, p = 0.180 at day2, p = 0.032 at day3, p = 0.007 at day4, p = 0.067 at day5 in the tGCI groups; n = 6-8, MANOVA Test followed by Bonferroni post-test). E The percentage of time spent in the target quadrant was recorded at day 7 after tGCI when the total time of probe trial was 30 s. *p < 0.05 versus Sham group with AAV-Con (p < 0.001 in the tGCI group with AAV-Con, p = 0.041 in the HPC group with AAV-Con, unpaired t-test), #p < 0.05 versus tGCI group with AAV-Con (p = 0.016, unpaired t-test) and &р<0.05 versus the same group with AAV-Con (p = 0.264 in the Sham group, p < 0.001 in the tGCI group, p=0.015 in the HPC group, n = 7-10, unpaired t-test). F, G Representative immunoblots and quantitative data show USP22, β-catenin and survivin in cytoplasmic and nuclear fractions, separately. Lamin B1 and GAPDH are used as loading controls to normalize the relative expressions of cytoplasmic and nuclear proteins of interest, respectively. Data are expressed as percentage of value of sham-operated animals. *p < 0.05 versus Sham group with AAV-Con (for cytoplasmic USP22, p < 0.001 in the tGCI group with AAV-Con, p < 0.001 in the HPC group with AAV-Con, n = 5; for cytoplasmic β-catenin, p = 0.179 in the tGCI group with AAV-Con, p = 0.546 in the HPC group with AAV-Con, n = 4; for nuclear USP22, p = 0.001 in the tGCI group with AAV-Con, p = 0.397 in the HPC group with AAV-Con, n = 8-15; for nuclear β-catenin, p = 0.017 in the tGCI group with AAV-Con, p = 0.941 in the HPC group with AAV-Con, n = 8–14; for nuclear survivin, p = 0.003 in the tGCI group with AAV-Con, p = 0.545 in the HPC group with AAV-Con, n = 8, unpaired t-test), #p < 0.05 versus tGCI group with AAV-Con (p = 0.474 for cytoplasmic USP22, p = 0.175 for cytoplasmic β-catenin, p = 0.026 for nuclear USP22, p = 0.008 for nuclear β-catenin, p = 0.001 for nuclear survivin, unpaired t-test), &р<0.05 versus the same group with AAV-Con (For cytoplasmic USP22, p = 0.089 in the Sham group, p = 0.269 in the tGCI group, p = 0.169 in the HPC group; For cytoplasmic β-catenin, p = 0.148 in the Sham group, p = 0.862 in the tGCI group, p = 0.923 in the HPC group; For nuclear USP22, p = 0.030 in the Sham group, p = 0.030 in the tGCI group, p = 0.005 in the HPC group; For nuclear β-catenin, p = 0.038 in the Sham group, p = 0.040 in the tGCI group, p = 0.018 in the HPC group; For nuclear survivin, p = 0.309 in the Sham group, p = 0.021 in the tGCI group, p = 0.313 in the HPC group, unpaired t-test). H Immunoprecipitation assay showing the level of nuclear K48-ubiquitinated β-catenin and me-β-catenin in CA1 of Sham, tGCI and HPC groups with AAV-Con/USP22. IgG antibody was used as a negative control. Data are expressed as percentage of value of sham-operated animals. Each bar represents the mean ± SD. *p < 0.05 versus Sham group with AAV-Con (for K48-Ub/β-catenin, p = 0.002 in the tGCI group with AAV-Con, p = 0.904 at the HPC group with AAV-Con; for methyl-K/β-catenin, p = 0.073 in the tGCI group with AAV-Con, p = 0.698 in the HPC group with AAV-Con, unpaired t-test), #p < 0.05 versus tGCI group with AAV-Con (p = 0.002 for K48-Ub/β-catenin, p = 0.040 for methyl-K/β-catenin, unpaired t-test), &р<0.05 versus the same group with AAV-Con (for K48-Ub/β-catenin, p=0.454 in the Sham group, p = 0.041 in the tGCI group, p = 0.442 in the HPC group; for methyl-K/β-catenin, p = 0.703 in the Sham group, p=0.012 in the tGCI grou, p = 0.430 in the HPC group, n = 4-6, unpaired t-test). MWM,Morris water maze; S, second; cm, centimeter; d, day. Each bar represents the mean ± SD (error bars).
Considering that mCherry representing exogenous USP22 mainly localized in the nucleus, we further analyzed cytoplasmic and nuclear USP22 expressions after AAV-USP22 administration and the effects of USP22 overexpression on β-catenin and downstream survivin. As shown in Fig. 8F, there was a slight increase in cytoplasmic USP22 of CA1 after treatment with AAV-USP22, but the difference was not statistically significant. Additionally, AAV-USP22 did not alter cytoplasmic β-catenin expression. Consistent to our previous study, no survivin expression could be observed in cytoplasmic fraction of CA14. As expected, compared to AAV-Con group, AAV-USP22 significantly upregulated the nuclear USP22 and β-catenin in CA1 of Sham, tGCI and HPC groups. Moreover, AAV-USP22 reversed the tGCI-induced reduction of survivin in CA1 (Fig. 8G). Furthermore, the elevation of K48-Ub of β-catenin and the reduction of methylated β-catenin within nucleus in CA1 after tGCI were counteracted with AAV-USP22 treatment) (Fig. 8H).
Discussion
We previously reported that HPC reverses tGCI-induced upregulation of KDM2A, which inhibits nuclear β-catenin demethylation and promotes nuclear β-catenin stabilization, thus reducing ischemic neuronal death in CA1. In this study, we further demonstrated that HPC inhibited nuclear β-catenin degradation via the negative regulation on its ubiquitylation in two ways. Concretely, HPC suppressed the formation of SCFKDM2A E3 ligase complex within the neuronal nucleus in CA1 after tGCI to inhibit ubiquitylation of demethylated β-catenin. On the other hand, HPC increased the expression of USP22 within the neuronal nucleus in CA1 after ischemia to promote nuclear β-catenin deubiquitination. As a result, the co-effects of HPC on SCFKDM2A and USP22 significantly hindered nuclear β-catenin degradation through the ubiquitin-proteasome pathway, leading to nuclear β-catenin stabilization and cerebral ischemic tolerance (Fig. 9).

The expression of SCFKDM2A complex composed of SKP1, CUL1 and KDM2A increases within neuronal nucleus in CA1 after tGCI. Next, SCFKDM2A complex targets methylated β-catenin for degradation. On one hand, SCFKDM2A E3 ligase complex demethylates β-catenin through KDM2A demethylase activity. On the other hand, SCFKDM2A complex catalyzes the K48-Ub of β-catenin after demethylation, thereby promoting β-catenin degradation through the proteasome pathway. In addition, tGCI decreases the expression of deubiquitinating enzyme USP22 in neurons, which further promotes the ubiquitination and degradation of nuclear β-catenin. Subsequently, the binding of nuclear β-catenin to the T-cell factor/lymphoid enhancing factor (TCF/LEF) is reduced, which in turn, inhibits the expression of downstream target gene survivin, ultimately leading to cellular death in CA1. In contrast, HPC inhibits the assembly of SCFKDM2A and enhaces the expression of USP22 within neuronal nucleus in CA1 after tGCI, which reduces nuclear β-catenin demethylation and ubiquitination, thereby stabilizing nuclear β-catenin. Eventually, the interaction between β-catenin and the TCF/LEF is enhanced, which upregulates the expression of survivin and protects CA1 neurons against tGCI.
As a core factor of the canonical Wnt signaling pathway, β-catenin is widely expressed in various cells of the central nervous system and plays a neuroprotective role against cerebral ischemia26. For example, β-catenin is able to inhibit apoptosis, alleviates oxidative stress, hinders excessive autophagy in neurons and promotes the differentiation of neural progenitor cells toward mature neurons, thereby attenuating ischemia-induced brain damage27,28,29,30. Astrogial β-catenin facilitate reprogramming of reactive astrocytes into neurons, inhibit astrogliosis and repair damaged blood-brain barrier31,32,33. In this study, the administration of AAVi-Ctnnb1 significantly reduced nuclear survivin and abolished HPC-mediated neuroprotection against tGCI. These results demonstrated that β-catenin plays an essential role in the neuroprotection offered by HPC after tGCI through upregulating the expression of survivin. Growing evidences showed distinct expression patterns of subcellular β-catenin across different cerebral ischemic models and regions of interest34,35,36. In line with our previous results in vivo4, β-catenin expression was decreased in the nucleus instead of the cytoplasm after OGD/R in primary hippocampal neurons. Moreover, MG132 treatment significantly reversed the downregulation of nuclear β-catenin induced by OGD/R, suggesting that OGD/R mainly promoted nuclear β-catenin degradation via the proteasome pathway. As K48-Ub is a key signal marker mediating β-catenin degradation by the proteasome in the canonical Wnt signaling pathway6, we further investigated whether the UPS is involved in nuclear β-catenin degradation induced by OGD/R. As expected, the K48-Ub of nuclear β-catenin was increased. Our studies in vitro and in vivo are consistent with the notion that ubiquitin-protein conjugates were increased obviously under pathological stress such as cerebral ischemia37,38,39. On the contrary, HPC attenuated tGCI-induced elevation of the K48-Ub of nuclear β-catenin in CA1. Although hypoxia is an undisputed cellular stressor that regulates the activity and level of β-catenin40, the mechanism by which HPC regulates the K48-Ub of nuclear β-catenin after tGCI remains unexplored.
As aforementioned, KDM2A is a potential E3 ubiquitin ligase11. Our previous study showed that KDM2A overexpression promoted nuclear β-catenin degradation by the proteasome and eliminated the neuroprotection induced by HPC after tGCI5. The current study illustrates that KDM2A forms the SCFKDM2A E3 ligase complex through its F-box domain and mediates the ubiquitination and following proteasome degradation of nuclear β-catenin. Notably, the increase of SCFKDM2A complex induced by tGCI was reversed by HPC. Moreover, KDM2A silencing significantly inhibited the K48-Ub increase of nuclear β-catenin mediated by tGCI, accompanied by the decreased interaction among nuclear β-catenin and three subunits of the SCFKDM2A complex in CA1. Although KDM2A has been reported to regulate the ubiquitination of nuclear β-catenin13, few studies focused on the function of SCFKDM2A complex as a whole. Our study revealed that KDM2A regulates the K48-Ub and degradation of nuclear β-catenin in an SCF-dependent manner after cerebral ischemia.
It was documented that the UPS was activated in response to cerebral ischemia, thereby disrupting protein homeostasis and leading to neuronal death41.The generation and accumulation of a large number of ubiquitin-protein conjugates in neurons lead to the overload and functional decompensation of proteasome, which would induce neurotoxic effect and aggravate cerebral ischemic injury38. As two invariant subunits of SCF complex, SKP1 and CUL1 can bind to a variety of F-box proteins in addition to KDM2A11,42. In this study, we confirmed that tGCI significantly induced the expressions of both SKP1 and CUL1 in CA1, which were offset by the administration of HPC. Notably, the cells that expressed SKP1 and CUL1 in CA1 were predominantly neurons in the early stage after tGCI. These results suggest that tGCI promotes the generation of SCF complexes by increasing neuronal SKP1 and CUL1 in CA1, thus leading to the ubiquitination of excess substrate proteins and the decompensation of the proteasome. Conversely, HPC inhibits the formation of SCF complexes through downregulating SKP1 and CUL1, thereby protecting the proteasome from overload and mediating the neuroprotection. In addition to the UPS, astrocytes were activated after cerebral ischemia43. At 168 h of reperfusion after tGCI, the predominant expression of SKP1 in astrocytes was observed, which was reversed by HPC. In view of the fact that SKP1 is an important part of UPS, we speculate that the activation of UPS might be involved in the activation of astrocytes after cerebral ischemia. While HPC inhibits astrocytic activation through hindering the formation of SCF E3 ligase complexes. Additionally, silencing KDM2A inhibited nuclear SKP1 in tGCI rats, but there was no any effect on the expression of CUL1, suggesting that KDM2A positively regulates SKP1 expression without affecting CUL1 in cerebral ischemia. After a complete literature search, we did not find relevant studies on the positive regulation of SKP1 by KDM2A. It is known that the demethylation of H3K36me2/3 mediated by KDM2A plays a role in inhibiting the transcription and expression of target gene44. Equally important, KDM2A could negatively regulate the expression of specific substrates through the E3 ubiquitin ligase activity8. For this reason, SKP1 might be not a direct target of KDM2A and KDM2A indirectly up-regulates SKP1 expression through other unknown intermediate mediators. Further studies are needed to elucidate the role and regulatory mechanism of SKP1 in HPC-mediated cerebral ischemic tolerance.
USP22, a deubiquitination enzyme containing an NLS and positively regulating β-catenin19,45, has been reported to stabilize the substrate protein through deubiquitinating enzyme activity23. So far, the expression and exact role of USP22 in cerebral ischemia remains controversial22,46. Xie et al. revealed that the upregulation of USP22 alleviates inflammation and hinders apoptosis caused by cerebral ischemia-reperfusion in vivo and in vitro22. Another study showed that USP22 was upregulated in OGD/R-induced pheochromocytoma-12 cells and in mouse brain tissues after middle cerebral artery occlusion/reperfusion. Conversely, USP22 knockdown exerts neuroprotective effects in cerebral ischemia/reperfusion injury46. Our results exhibited that the overexpression of USP22 markedly ameliorated neuronal apoptosis in CA1 and improved the neurological outcome of ischemic rats. HPC enhanced the neuroprotective effects of USP22. The discrepancies in the expression and function of USP22 after ischemic stimuli may be related to different cerebral ischemia models and the duration of ischemic stimuli. The implication for the increase of USP22 in microglia in HPC group is not fully understood. Di et al. reported that USP22 could promote nuclear factor-kappa-B (NF-κB)/oligomerization domain-like receptors protein (NLRP) 3 inflammasome degradation and inhibit NLRP3 inflammasom activation in mouse peritoneal macrophages and human THP-1cells47. Previously, we also identified that HPC alleviated microglia activation through inhibiting NLRP3 inflammasome activation after tGCI48. Therefore, we speculate that the increase of USP22 in microglia in the HPC group at late phase of tGCI may contribute to the inhibition of NLRP3 inflammasome activation, thus suppressing the microglia activation. Further investigations are necessary to understand this interesting phenomenon.
To date, the role of USP22 subcellular localization remains controversial. It is known that the localization and function of proteins are closely related to its domains. USP22 contains an ubiquitin hydrolase domain at the carboxy-terminal, a zinc finger motif domain at the amino-terminal, and two different sequences of NLS45. USP22 catalyzes the deubiquitylation of histone H2A and H2B in the nucleus, thus participating in gene regulation and cell-cycle progression49. It has been demonstrated that cytoplasmic USP22 promotes nuclear translocation of key transcription factors50. Considering that USP22 does not possess any nuclear export signals45, it is reasonable to speculate that USP22 mainly plays a role in the nucleus, and the final destination of cytoplasmic USP22 also be cellular nucleus during mediating nuclear translocation of the substrate proteins. Despite that USP22 was detectable in both nucleus and cytoplasm of CA1 cells, cytoplasmic USP22 exhibited no significant change in our ischemic model, whereas HPC restored the tGCI-induced downexpression of nuclear USP22. These results suggested that nuclear USP22 rather than cytoplasmic, plays a major role in HPC-mediated cerebral ischemic tolerance.
Herein, we also noted that the expression of USP22 was line with the trend of β-catenin in nucleus4. Previous researches indicated that USP22 is a positive regulator of nuclear β-catenin in several types of cancers19,20,21. In line with those findings, HPC reduces the K48-Ub of nuclear β-catenin and maintains the stability of nuclear β-catenin by increasing nuclear USP22 in CA1 after tGCI, thereby promoting the survival of neurons. Besides, survivin has been identified as a potential target protein of USP22. It was reported that USP22 silence by small interfering RNA reduced the expression of survivin in oral squamous cell carcinoma51. Lin et al. confirmed that USP22 could regulate survivin via deubiquitination, thereby promoting the proliferation of renal cell carcinoma52. Consistently, our results uncovered that USP22 overexpression significantly increased nuclear survivin in CA1 after tGCI. Hence, the expression of survivin might be regulated by β-catenin transcriptionally and USP22 post-translationally in HPC-mediated cerebral ischemic tolerance.
Actually, the intercellular function of β-catenin is regulated by the posttranslational modifications (PTMs) on its lysine residues, including ubiquitination, methylation, acetylation, and glycation. A characteristic feature of PTMs at lysine residue is the competitive relationship between different types of modification53. It was reported that β-catenin methylation catalyzed by enhancer of zeste homolog 2 (EZH2) prevents β-catenin from being ubiquitinated and ultimately stabilizes β-catenin in liver cancer cells54. Lei et al. found that methyltransferases EZH2 regulates β-catenin stability via recruiting USP7, a well-studied deubiquitinating enzyme, to mediate neuronal gene expression in neural progenitor cells55. It follows that methylation regulates nuclear β-catenin turnover by interfering with β-catenin ubiquitination. To better understand the regulation of nuclear β-catenin stability in the neuroprotection induced by HPC after tGCI, we further studied the nature of cross-talk between KDM2A-mediated demethylation and ubiquitination of nuclear β-catenin. Treatment with daminozide, a selective demethylase inhibitor of KDM2A, significantly increased methylated β-catenin and inhibited the K48-Ub of nuclear β-catenin in CA1 after tGCI, thus promoting the accumulation of nuclear β-catenin. These findings indicates that the demethylation of β-catenin catalyzed by KDM2A is a requisite for KDM2A-mediating the K48-Ub of β-catenin in nucleus of CA1 after tGCI. Another characteristic feature of PTMs at lysine residue is reversibility, such as methylation/demethylation and ubiquitination/deubiquitination53. Our data revealed that nuclear β-catenin deubiquitination induced by USP22 overexpression significantly increased the methylation of nuclear β-catenin after tGCI. Therefore, USP22 may couple with KDM2A to mediate the crosstalk between ubiquitination/deubiquitination and methylation/demethylation of nuclear β-catenin in HPC-offered neuroprotection against cerebral ischemia.
Finally, the limitations of our study should be mentioned. Although we demonstrated that β-catenin was involved in HPC-mediated neuroprotection against tGCI as a substrate of KDM2A for ubiquitination, we could not completely rule out whether KDM2A modulated other downstream factors besides β-catenin in such situation. Therefore, β-catenin might not be alone as the direct mediator of neuroprotection against tGCI after removal of KDM2A. In addition, considering that the ubiquitination of β-catenin could be reduced by the inhibition of demethylase activity of KDM2A and the methylation of β-catenin increased by USP22, we speculate that there may be a competition between the methylation and ubiquitination modification of β-catenin. However, no direct results were available to confirm whether KDM2A and USP22 regulate methylation and ubiquitination on the same lysine residue of β-catenin. Future studies will be needed to focus on the high-throughput analyses of protein methylation and ubiquitination and the identification of specific substrate subsets of KDM2A and USP22.
In conclusion, our findings uncovered the regulatory mechanisms of nuclear β-catenin ubiquitylation in the neuroprotection mediated by HPC against tGCI and the relation between ubiquitylation and methylation in HPC-induced nuclear β-catenin stabilization. Briefly, HPC inhibits the K48-Ub of nuclear β-catenin in CA1 through suppressing the assembly of the SCFKDM2A complex and upregulating nuclear USP22 after tGCI. Further, the upregulation of nuclear USP22 induced by HPC promotes the methylation of nuclear β-catenin after USP22-mediated deubiquitination, which enhances the stability of nuclear β-catenin in CA1 and protects ischemic neuron against cerebral ischemia. Elucidation of the molecular mechanism underlying nuclear β-catenin PTMs mediated by SCFKDM2A complexes and USP22 may provide novel therapeutic targets for ischemic stroke.
Methods
Culture of primary hippocampal neurons and cell lines
For isolation and culture of primary hippocampal neurons, the hippocampi was isolated from embryos at 18 day (E18) of gestational Wistar rats provided by Southern Medical University, Guangzhou, China. Embryonic hippocampal tissues were gently minced into pieces using a sterile scalpel and incubated with 0.25% trypsine (Gibco, Cat# 25200-072) at 37 °C for 15 min. The plating medium prepared from Dulbecco’s Modified Eagle’s Medium (DMEM) (Gibco, New York, USA, Cat# C11995500BT), 10% F12 (Gibco, New York, USA, Cat# 11765-054), 10% newborn calf serum (Gibco, Cat# 16000-044) and 50 μg/mL of penicillin and streptomycin (Gibco, Cat# 15140-122) was used to stop the enzymatic reaction. Following 5 min of centrifugation at 1000 rpm, supernatant was discarded. The precipitate was mixed with 1 mL plating medium mentioned above. After standing for 1 min, the upper cell suspension was collected. Then, the steps of suspension and collection were repeated twice. For the immunoblotting, the cells were seeded onto Poly-D-Lysine (Pricella, Wuhan, China, Cat# PB180523) coated 6-well plates (Corning) at a density of 1×106 cells/well. For fluorescence assays, cells were seeded on PolyD-Lysine coated 24-well plates (Corning) at a density of 2×105 cells/well. After 4 h, the plating medium was replaced with neurobasal feeding medium, which was composed of neurobasal medium (Gibco, Cat# 21103-049), B-27 (Gibco, Cat# 17504-044) and 0.5% L-glutamine (Gibco, Cat# 25030081) and 50 μg/mL of penicillin and streptomycin (Gibco, Cat# 15140-122). The hippocampal cells were cultured in a 37 °C normoxic incubator with 5% CO2 and 95% air. To reduce the fibroblasts and the glial cells, cytarabine (Sigma-Aldrich, USA, Cat# C1768) was dissolved in DMSO to form a 5 mM stock solution which was diluted at 1:1000 with neurobasal feeding medium prior to supplementation at 5 µM for 24 h incubation. The neurons were fed every 2 days by removing half of the old media and replacing it with the same volume of fresh neurobasal feeding medium. After 7 days of culture, primary hippocampal neurons were subjected to OGD/R to mimic cerebral ischemia-reperfusion injury.
For cell cultures, human neuroblastoma SH-SY5Y cells and human embryonic kidney HEK-293T cell lines were obtained from the American Type Culture Collection (ATCC). Then, these cells were fed with Dulbecco’s Modified Eagle’s Medium (Gibco, New York, USA, Cat# 8121336) supplemented with 10% newborn calf serum (Gibco, Cat# 16000- 044), 50 μg/mL of penicillin and streptomycin (Gibco, Cat# 15070063) and cultured in a 37°C incubator with 5% CO2.
OGD/R model construction and drug treatment
For the OGD/R procedure, hippocampal neurons were gently washed with phosphate-buffered saline (PBS, pH7.4) (Gibco, Cat# 10010-023) for 3 times and then were cultured with glucose-free and serum-free DMEM (Gibco, Cat# RPMI-1640) medium in a sealed chamber (Billups-rothenberg, USA) pre-flushed with a mixture of 95% N2 and 5% CO2 for 5 min at a flow rate of 30 L/min. The anaerobic chamber was placed in an incubator for 90 min at 37 °C in 5% CO2. Subsequently, the cultures were replaced with the regular neurobasal feeding medium and returned to a normoxic incubator for reoxygenation. The samples were collected at certain indicated time for following experiments. Normal controls were obtained from the neurons that were kept in medium containing glucose and under normoxic condition throughout the experiment. To inhibit proteasome, the cultures were treated with 20 μM MG132 (Sigma-Aldrich, USA, Cat# C2211) for 6 h before cell collection.
Plasmids construction and transfection
The plasmids carrying the sequence of rat KDM2A, β-catenin and the corresponding negative control sequence were constructted by Genechem (Shanghai, China). As previously described5, the fusion gene containing rat KDM2A (GenBank accession number NM_001108515) and N-terminal NLS sequence was cloned into the CMV-MCS-3FLAG-SV40-EGFP (GV314) vector. The KDM2A-∆F-box mutant deleted for the amino acids 888-935 was generated and subcloned into GV314 vector containing a N-terminal Flag tag and a NLS. Besides, full-length Rat β-catenin (GenBank accession number NM_053357) was cloned into the CMV-MCS-Myc-SV40-mcherry (GV230) vector, which containing a NLS.
Cells were implanted in a 6-well plate and were transfected with different plasmids after reaching 80% confluence. For each well transfection, 3 ug plasmids and 9 ug transfection reagents were dissolved in 125 uL Opti-MEM (Gibco, Cat# 31985-070) for 5 min, mixed for 20 min and then added to cell medium. Notably, SH-SY5Y cells transfection was mediated by Lipofectamine 2000 transfection reagent (Invitrogen, Cat# 11668019) according to the manufacturer’s instruction and the medium was DMEM (Gibco, New York, USA, Cat# 8121336) during transfection. After 6 h of transfection, medium was replaced with fresh complete medium prepared with 40% DMEM, 10% newborn calf serum, 50 μg/mL of penicillin and streptomycin. For HEK-293T cells transfection, polyethylenimine (MilliporeSigma, Cat# 408727) was used as transfection reagent and cells was in incubated with fresh complete medium during transfection. The corresponding empty plasmid was used as a supplement to ensure that cells from different pores were transfected the same number of plasmids in the same experiment.
Immunocytochemistry
The cultured cells were fixed with 4% cold paraformaldehyde (PFA) prepared in PBS for 10 min, and then permeabilized with 0.2% triton X-100 for 15 min and blocked with 5% normal serum for 1 h. Cells were incubated overnight at 4 °C with mouse anti-Flag (1:200; MilliporeSigma, Cat# F4049, RRID: AB_439701) and rabbit anti-β-catenin (diluted 1:200). After that, the cells were incubated with the following secondary antibodies: goat anti-rabbit IgG H&L (Alexa FluorTM 4647) (1:200; Cat# A21244, Invitrogen) and Cy3-conjugated goat anti-mouse IgG antibody (1:200; Cat# AP124C; MilliporeSigma). Next, slides were mounted with mounting medium containing 4’, 6-diamidino-2-phenylindole (DAPI). Fluorescent images were captured and analyzed with a confocal laser microscope (SP8, Leica Microsystems).
In vivo ubiquitination assay
For in vivo ubiquitylation assay, at 24-48 h after transfection, HEK-293T cells were treated with MG132 for 6 h to avoid polyubiquinated β-catenin degradation and thus increase the sensitivity of detection. HEK-293T cells were collected and separated using a NE-PER nuclear cytoplasmic extraction reagent kit (Pierce, Rockford, IL, USA) according to the manufacturer’s protocol. For immunoprecipitation, 20% of nuclear lysates was mixed directly with 2x Laemmli sample buffer (Bio-RAD, Cat# 1610737) and used as input on western blot. The rest were incubated with a mouse monoclonal antibody against Myc (1:50; Abcam, Cat# ab32) for immunoprecipitation. The ubiquitylation levels of exogenetic β-catenin were determined by western blot with rabbit anti-HA (1:2000; Abcam, Cat# ab9110) and mouse anti-Myc (1:1000) antibody.
Animals
Adult male Wistar rats (weighing 220–280 g, 7–8 wk old) were obtained from Southern Medical University, Guangzhou, China. Before the experiment, these rats were kept in a specific-pathogen-free and temperature-controlled (21–23°C) environment with a relative humidity of 50-60%, a light/dark cycle of 12-h/12-h, as well as allowed free access to water and food for at least 1 week. All animal procedures were conducted in strict accordance with the Animal Research: Reporting In Vivo Experiments (ARRIVE) guidelines. Experiments involving animals were approved by the Animals Care and Use Committee of Guangzhou Medical University (Guangzhou, China). All relevant ethical regulations for animal use were complied. Every effort had been made to reduce the amount, suffering and distress of the animals on the basis of ensuring the needs of the experiments.
In total, 497 rats were used. Four rats in the tGCI and 3 in the HPC groups died during the tGCI procedure. Three rats in the tGCI and 3 in the HPC groups died after the tGCI procedure. In addition, 4 rats died during HPC process and 4 died during anesthesia. Eighteen rats that presented with convulsion and dyspnea after ischemia were also excluded.
Experimental models
An improved four-vessel occlusion method was used to establish tGCI model. Briefly, animal anesthesia was induced with 3-4% isoflurane in a box flowing with air at 2 L/min and maintained with 1.5-2.5% isoflurane in air at 800 mL/min, delivered through a nasal mask (SurgiVet, Waukesha, WI, United States) during the whole operation. After cauterizing bilateral vertebral arteries, a teflon/silastic occluding device was assembled loosely around the two carotid arteries to separate the common carotid arteries without blocking blood flow. At 24 h after operation, global cerebral ischemia was induced by blocking the blood flow of both common carotid arteries in awake rats for 10 min. Rats that had mydriasis and lost righting reflex within 1 min were enrolled for the following experiments. The sham-operated (Sham) rats received the same surgical procedures without the interruption of common carotid arteries for 10 min. All operations are performed under aseptic conditions by mature technicians.
Then, rats were treated with the HPC process by exposure to flowing mixed gas (8% O2 + 92% N2) in a sealed chamber of 9000 cm3 for 120 min at 24 h after 10-min ischemia. For hypoxia treated groups, rats were exposed to 120-min hypoxia at 24 h after sham-operation without tGCI.
Western blot
Cultured cells were collected at 2, 6, 12 and 24 h after OGD/R or 24-48 h after transfection by immersing in cold PBS for 3 times and then centrifuging at 3000 rpm at 4 °C for 3 min. To obtain tissue samples, rats were sacrificed at 0, 4, 26, and 50 h after reperfusion with or without hypoxia, respectively. Bilateral hippocampal CA1 subregions of each group were immediately dissected under the stereomicroscope. Total, cytosolic, and nuclear proteins were extracted as previously described4,56. The nuclear and cytosolic proteins of cell and tissue samples were extracted with NE-PER nuclear cytoplasmic extraction reagent kit (Pierce, Rockford, IL, USA). According to the manufacturer’s protocol, cytoplasmic protein lysate (composed of cytoplasmic protein lysate CERI, protein phosphatase inhibitor, and protease inhibitor phenylmethylsulfonyl fluorid (PMSF) at a ratio of 100:1:1was added to cell or tissue samples. Next, samples were homogenized on ice and left for 10 min. After swirling for 5 s, cytoplasmic protein lysate CERII was added and continued vortexing for 5 s, and then left for 1 min on ice. The homogenate was centrifuged at 16000 g/min in a 4 °C centrifuge for 5 min, and the supernatant was collected carefully as cytoplasmic fraction. Afterwards, the pellets were re-suspended with nucleolysis solution (which was composed of nucleolysis solution NER, protein phosphatase inhibitor and protease inhibitor PMSF at a ratio of 100:1:1) on the ice for 50 min, during which oscillation was carried out for 15 s every 10 min. After centrifuging for 10 min at 16000 g/min at 4 °C, the supernatant was saved as nuclear fraction. The purity of each fraction was guaranteed by detecting the expression of corresponding reporters, including Lamin B1, glyceraldehyde 3-phosphate dehydrogenase (GAPDH), β-tubulin, and proliferating cell nuclear antigen (PCNA). Western blot was performed as described previously57 with the primary antibodies, including rabbit anti-β-catenin (1:4000; Abcam, Cambridge, MA, USA, Cat# ab32572, RRID: AB_725966), rabbit anti-K48 Ub (1:4000; Abcam, Cambridge, MA, USA, Cat# ab14060, RRID: AB_2783797), rabbit anti-Flag (1:2000; Abcam, Cat# ab205606), KDM2A (1:2000; Abcam, Cat# ab191387), rabbit anti-SKP1 (1:2000; Abcam, Cambridge, MA, USA, Cat# ab76502, RRID: AB_1524396), rabbit anti-CUL1 (1:6000; Abcam, Cambridge, MA, USA, Cat# ab75817, RRID: AB_1310103), rabbit anti-USP22 (1:4000; Abcam, Cambridge, MA, USA, Cat# ab195289, RRID: AB_2801585), rabbit anti-survivin (1:2000; Abcam, Cat# ab76424, RRID: AB_1524459), rabbit anti-methylated lysine (1:2000; Abcam, Cat# ab76118, RRID: AB_1523945), rabbit anti-Lamin B1 (1:2000; Proteintech Group, Cat# 12987-1-AP, RRID: AB_2136290), mouse anti-GADPH (1:10000; Proteintech Group, Cat# 60004-I-Ig, RRID: AB_2107436), rabbit anti-PCNA (1:2000; Abcam, Cat# ab29, RRID: AB_303394) and mouse anti-β-tubulin (1:10,000; Proteintech Group, Wuhan, China, Cat# 10094-1-AP, RRID: AB_2210695). Optical densities analysis for the quantification of the bands was performed with ImageJ (NIH, Bethesda, MD, United States) and then calibrated with Lamin B1, GAPDH or PCNA as well as normalized to those in negative control (NC) cells or Sham rats.
Immunoprecipitation
To determine the K48-Ub of nuclear β-catenin, the interaction among KDM2A, SKP1 and CUL1, as well as the binding of β-catenin to three subunits of the SCFKDM2A complex in vitro and in vivo, an amount of 0.2 mg of nuclear proteins from each group was incubated with indicated antibodies, including a rat anti-β-catenin (diluted 1:75) or a rat anti-Flag (diluted 1:50) or a rat anti-KDM2A (diluted 1:50) overnight at 4 °C. Next, the protein/antibody complexes were added to protein A/G agarose beads (MilliporeSigma, Cat# IP05) with agitation for 4 h at 4 °C. The immuno-complexes were eluted with cold PBS containing 1% Tween by centrifugation at 10000 rpm at 4°C for 3 min, and then replaced carefully PBS. After repeating centrifugation steps described-above 5 times, bound proteins were collected and eluted via boiling in 15 uL of 2x Laemmli sample buffer (Bio-RAD, Cat# 1610737) prior to sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE). The eluted proteins were analyzed by western blot with indicated antibodies. Densitometric analysis for the quantification of the relative precipitated protein bands were calibrated with the bands of β-catenin or KDM2A and normalized to those in NC cells or Sham rats.
Immunohistochemistry
Rats were perfused intracardially with normal saline (NS), followed by a cold PBS mixture of 4% PFA and 1% glutaraldehyde at 0, 4, 26, 50, and 168 h after reperfusion of tGCI with or without hypoxia, respectively. Next, brain tissues were postfixed with 10%, 20% and 30% sucrose with the above fixation as the solvent. Brains specimens were sliced into 30-μm-thick coronal sections with cryotome (Leica, Wetzlar, Hessen, Germany) at -22 °C. The slices containing dorsal hippocampus (between anterior-posterior (AP) 4.8 and 5.8 mm, interaural or AP-3.3 to 3.4 mm, bregma) were collected for immunohistochemistry.
Single-label immunohistochemistry was undertaken with the avidin-biotin complex (ABC) peroxidase method as described previously58. In short, brain slices were incubated with 3% hydrogen peroxide for 30 min at room temperature, followed by 5% normal serum for 1 h. Meanwhile, primary antibodies including rabbit anti-SKP1 (1:200), rabbit anti-CUL1 (1:2000), rabbit anti-USP22 (1:100) and mouse against neuronal nucleus (NeuN) (1:600; MilliporeSigma, Cat# MAB377, RRID: AB_2298772) were prepared with PBS containing 0.2% Triton and 5% BSA. Then, primary antibody incubation was performed in chromatography freezer (Cat# GYCX-890) overnight and the secondary antibody was incubated with biotinylated IgG at room temperature for 2 h. After incubation of primary and secondary antibodies, the sections were washed with PBS for 6 times. Subsequently, the sections were incubated with the ABC peroxidase for 30 min at room temperature. Eventually, DAB substrate kit (Abcam, Cat# ab64238) was applied to visualization of the peroxidase reaction according to the instructions. The light microscope (Leica Microsystems, Wetzlar, Hessen, Germany) was adopted for observation and capture. For semi-quantitative assessment, immunopositive cells were counted within 3 non-repeated random fields (0.037mm2/field×3 = 0.111mm2 in total) in CA1 and data were quantified bilaterally in 4 sections from each rat with double-blind method.
Double-fluorescent labeling immunohistochemistry was utilized to observe cell types and the subcellular location of SKP1 and CUL1 in CA1. As described previously59, NeuN, glial fibrillary acidic protein (GFAP) and ionized calcium binding adaptor molecule-1 (Iba-1) were used to confirm neuron, astrocytes and microglia, respectively. Antibodies used in this part of experiments included rat anti-SKP1 (1:100) and rat anti-CUL1 (1:200), rabbit anti-USP22 (1:50; Cat# HPA044980, Sigma), mouse anti-NeuN (1:1000), mouse anti-GFAP (1:3000; MilliporeSigma, Cat# MAB360, RRID: AB_2109815), mouse anti-Iba-1 (1:500, Millipore, Cat# MABN92), goat anti-rabbit IgG H&L (Alexa Fluor® 488) (1:100; Cat# ab150077, Abcam) and Cy3-conjugated goat anti-mouse IgG antibody (1:100; Cat# AP124C; MilliporeSigma).
KDM2A RNAi knockdown
To inhibit KDM2A expression, three sequences of KDM2A small-interfering RNAs (siKDM2A) were designed by BlueLife Biotechnology (Guangzhou, Guangdong, China), as described previously5. The best-performing siKDM2A, whose sequence was GCCAACGAATTGAGCTCAA, was purchased from RIB Bio (Guangzhou, Guangdong, China) and dissolved with normal RNase free water for following experiments. Then, hemagglutinating virus of Japan HVJ-E vector kit (CosmoBio, Tokyo, Japan, Cat# 808920) were utilized for effective delivery of siRNA to CA1 region by membrane-fusion activity of fusion protein. On the basis of manufacturer’ protocols, the HVJ-E siKDM2A complex or NC vector was a blend of HVJ-E and equivalent siKDM2A solution (10 μL; 2 μg/μL) or equivalent sterilized RNase free water. Twenty-four hours before tGCI, the above complexes at 5-μL were delivered to the bilateral CA1 region (3.5 mm posterior to bregma, both 2.3 mm lateral to bregma, and 2.5 mm below the dura) of rats by stereotaxic injection using a 25-μL Hamilton syringe with a 34-gauge needle and micro-injection pump (No. D-306370, Harvard Apparatus) at a flow rate of 0.5 μL/min. Rats were sacrificed at 50 h after injection of vectors in Sham or 26 h after reperfusion in tGCI group.
Pharmacologic interventions
To inhibit KDM2A demethylase activity, daminozide (Cat# HY-13643, MedChemExpress) or vehicle (isometric normal saline) was intracerebrally injected into the right ventricle (1.0 mm posterior, 1.5 mm lateral, and 3.6 mm dorsal with respect to bregma) at 48 h before tGCI (at 48 h before ischemia in tGCI rats and at 24 h after sham-operation in Sham rats). The stereotaxic injection was performed via a 25 μL Hamilton syringe at a speed of 1 μL/min.
Adeno-associated virus construction and administration
Plasmids containing the sequence of rat USP22 (GenBank accession number NM_001191644) were designed by Genechem (Shanghai, China). Briefly, USP22 sequence was inserted into KpnI and BamHI sites of CMV bGlobin-MCS-mCherry-3FLAG-WPRE-hGH polyA (GV389) adeno-associated virus (AAV) vector. The titers were approximately 1.13×1013 v.g./mL. As mentioned above, a 2-μL volume containing 1×1010 v.g./mL particles of scrambled AAV vector (AAV-control, AAV-Con)/AAV-USP22 was respectively injected into bilateral hippocampal CA1 region using a 25-μL Hamilton syringe with a 34-gauge needle and micro-injection pump at a speed of 0.5 μL/min.
Three small-interfering RNA sequences targeted rat β-catenin (Ctnnb1, GenBank accession number NM_053357.2) and a negative control vector were designed by Genechem (Shanghai, China). The sequences for Ctnnb1-RNAi 1, 2, 3 were ACCGGGCAGTTTGATGCCGCTCATCCTTCAAG AGAGGATGAGCGGCATCAAACTGCTTTT; ACCGGGCTGCATAATCTCCTGCTACATTCAAG AGATGTAGCAGGAGATTATGCAGCTTTT and ACCGGGCTTACGGCAATCAGGAAAGCTT CAAGAGAGCTTTCCTGATTGCCGTAAGCTTTT, respectively. Briefly, RNAi was inserted into the U6-MCS-CAG-EGFP (GV478) AAV8 vector according to the manufacture’s instruction. The shuttle vector and viral packaging system were cotransfected into AAV-293 cells, which derived from HEK293 cells, to produce recombinant AAV particles. The best-performing Ctnnb1-RNAi sequence was Ctnnb1-RNAi 3. Therefore, Ctnnb1 RNAi 3 was used for subsequent experiments. The titers were ~1.98 × 1012 v.g/mL. To select the best inhibitory dose, 1 or 2 or 4-ul AAV was injected into rat hippocampal CA1, respectively. Subsequently, two 4-μL volume containing 15.84 ×109 v.g of particles was bilaterally injected into CA1 region.
After the AAV treatment, rats were allowed to recover for up to 14 days to ensure the overexpression of target gene before being used for the following experiments.
Assessment of cellular damage
Nissl, NeuN and F-JB staining were conducted to evaluate the effects of AAV on neuronal damage in the CA1 region at 168 h after reperfusion of tGCI with or without hypoxia. Details of cresyl violet and F-JB powders as well as specific procedures are available from our previous study60. NeuN staining was performed with the single-label immunohistochemistry described above using anti-NeuN primary antibody. Then, Nissl-, NeuN- and F-JB-stained images were captured with an inverted fluorescent microscope (Leica Microsystems, Wetzlar, Hessen, Germany). Immunoreactive cells in CA1 were analyzed as above-mentioned.
Morris water maze test
Morris water maze (MWM) test was performed as previously described5. In brief, the maze consists of a circular black pool with a diameter of 210 cm and a height of 60 cm (Taimeng Technology Co., Ltd, Chengdu, China) which was pre-filled with clear water (23 ± 1 °C, 20 cm in depth). The pool was divided into four equal-sized quadrants (Q1, Q2, Q3 and Q4), and a transparent platform (escape platform) with a diameter of 10 cm was placed 2.0 cm below the water surface of Q1. The MWM test includes two periods of the training period and probe trial. During the training period, the platform remained in the same location. For 5 consecutive days from the 2nd day after tGCI, rats were placed in the water pool facing toward the wall and the fixed starting point of every quadrant was used in a different order each day. All rats completed the training to find the platform within 120 s and then stayed on the platform for 15 s. When failed to find the hidden platform, rats were guided to there, an escape latency of 120 s was recorded. Rats were subjected to 4 sequential trains per day with a 10-min interval between each session. The path length and the escape latency for each trial were recorded. At 7nd day after tGCI, a 30-sec probe trial was conducted without the platform. Rats were put into a maze at a fixed starting point farthest from the platform position during training. The time and percentage in the target quadrant occupancy were recorded. To minimize variability in the performance of rats, the experiment place should be keep quiet, the location of various experimental equipment is fixed, and all the experiments are conducted at the same time each day. Data from each trial was recorded using a video tracking system (SMART, Polyvalent video-tracking system, 35B73-C6C, PANLAB, Spain).
Statistics and Reproducibility
Statistical analyses were performed with Statistical Package for Social Sciences Software for Windows, version 25.0 (SPSS, Inc., Chicago, IL, USA). The group size “n” simply refers to biologically independent cell/animal experiments rather than technical replications. The evaluation of sample size was based on our and others published studies and the results from our preliminary experiments in this study. All data represent at least 3 independent experiments. For comparison between two groups conforming to the normal distribution, the unpaired two-tailed Student’s t-test was applied. Multiple comparisons were conducted using one-way ANOVA when the data between Sham and tGCI groups were normally distributed. Analyses were followed by a Bonferroni or Tamhane’s T2 post hoc test according the results of homogeneity of variance test. When the data did not conform to normal distribution, nonparametric tests were used, including Mann-Whitney’s U test for the comparison between two groups and Kruskal-Wallis’ for multiple comparisons. As the data from path length, escape latency, and swimming speed are repeatedly measured and time-dependent variables, these data were treated with Mauchly’s test of sphericity and Multivariate Analysis of Variance (MANOVA). Data graphing was performed with GraphPad Prism version 6.0. All variables were expressed as mean ± standard deviation (SD) and represented by scatter plots. Values of p < 0.05 were considered statistically significant. The exact p value was also described in the figure legends.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
The underlying source data for this study can be found in the Supplementary Data file and DRYAD repository at https://doi.org/10.5061/dryad.q573n5tt0. The uncropped and unedited blot images are available in the Supplementary Figs. The numerical source data for the graphs are available in the file Supplementary Data 1. All other data are available from the corresponding author on reasonable request.
Code availability
The analyzing code for the integration data set is available from DRYAD repository at https://doi.org/10.5061/dryad.q573n5tt0.
References
Martin, S. S. et al. 2024 Heart Disease and Stroke Statistics: A Report of US and Global Data From the American Heart Association. Circulation 149, e347–e913 (2024).
He, X. et al. Role of Wnt/beta-catenin in the tolerance to focal cerebral ischemia induced by electroacupuncture pretreatment. Neurochem Int 97, 124–132 (2016).
Zhang, B., Zheng, G., Han, Y., Chen, X. & Jiang, Q. Ilexonin A Promotes Neuronal Proliferation and Regeneration via Activation of the Canonical Wnt Signaling Pathway after Cerebral Ischemia Reperfusion in Rats. Evid. Based Complement Altern. Med. 2016, 9753189 (2016).
Zhan, L. et al. Hypoxic postconditioning activates the Wnt/beta-catenin pathway and protects against transient global cerebral ischemia through Dkk1 Inhibition and GSK-3beta inactivation. FASEB J. 33, 9291–9307 (2019).
Zuo, Y. et al. Stabilization of nuclear beta-catenin by inhibiting KDM2A mediates cerebral ischemic tolerance. FASEB J. 37, e22796 (2023).
Paul, D. et al. F-box protein FBXO16 functions as a tumor suppressor by attenuating nuclear beta-catenin function. J. Pathol. 248, 266–279 (2019).
Kud, J. et al. Functional Characterization of RING-Type E3 Ubiquitin Ligases In Vitro and In Planta. J. Vis. Exp. 5, 154 (2019).
Lim, K., Song, M. & Baek, K. Decision for cell fate: deubiquitinating enzymes in cell cycle checkpoint. Cell Mol. Life Sci. 73, 1439–1455 (2016).
Xue, J. et al. (2015) Tumour suppressor TRIM33 targets nuclear beta-catenin degradation. Nat. Commun. 6, 6156 (2015).
Chitalia, V. et al. c-Cbl, a ubiquitin E3 ligase that targets active beta-catenin: a novel layer of Wnt signaling regulation. J. Biol. Chem. 288, 23505–23517 (2013).
Gorelik, M. et al. A Structure-Based Strategy for Engineering Selective Ubiquitin Variant Inhibitors of Skp1-Cul1-F-Box Ubiquitin Ligases. Structure 26, 1226–1236 (2018).
Liu, X. et al. KDM2A Targets PFKFB3 for Ubiquitylation to Inhibit the Proliferation and Angiogenesis of Multiple Myeloma Cells. Front Oncol. 11, 653788 (2021).
Lu, L. et al. Kdm2a/b Lysine Demethylases Regulate Canonical Wnt Signaling by Modulating the Stability of Nuclear beta-Catenin. Dev. Cell 33, 660–674 (2015).
Kowalski, J. R. & Juo, P. The role of deubiquitinating enzymes in synaptic function and nervous system diseases. Neural Plast. 2012, 892749 (2012).
Yang, B. et al. Deubiquitinase USP9X deubiquitinates beta-catenin and promotes high grade glioma cell growth. Oncotarget 7, 79515–79525 (2016).
Ma, P. et al. The ubiquitin ligase RNF220 enhances canonical Wnt signaling through USP7-mediated deubiquitination of beta-catenin. Mol. Cell Biol. 34, 4355–4366 (2014).
Lee, H. J. et al. The expression patterns of deubiquitinating enzymes, USP22 and Usp22. Gene Expr. Patterns 6, 277–284 (2006).
Melo-Cardenas, J., Zhang, Y., Zhang, D. & Fang, D. Ubiquitin-specific peptidase 22 functions and its involvement in disease. Oncotarget 7, 44848–44856 (2016).
Ning, Z. et al. USP22 promotes the G1/S phase transition by upregulating FoxM1 expression via beta-catenin nuclear localization and is associated with poor prognosis in stage II pancreatic ductal adenocarcinoma. Int J. Oncol. 45, 1594–1608 (2014).
Jiang, S. et al. MiR-30-5p suppresses cell chemoresistance and stemness in colorectal cancer through USP22/Wnt/beta-catenin signaling axis. J. Cell Mol. Med. 23, 630–640 (2019).
Xin, R., Li, W., Hu, Z., Wu, Z. & Sun, W. MiR-329-3p inhibits hepatocellular carcinoma cell proliferation and migration through USP22-Wnt/beta-Catenin pathway. Eur. Rev. Med. Pharm. Sci. 24, 9932–9939 (2020).
Xie, X., Cao, Y., Dai, L. & Zhou, D. Bone marrow mesenchymal stem cell-derived exosomal lncRNA KLF3-AS1 stabilizes Sirt1 protein to improve cerebral ischemia/reperfusion injury via miR-206/USP22 axis. Mol. Med. 29, 3 (2023).
Ma, S. et al. USP22 Protects Against Myocardial Ischemia-Reperfusion Injury via the SIRT1-p53/SLC7A11-Dependent Inhibition of Ferroptosis-Induced Cardiomyocyte Death. Front Physiol. 11, 551318 (2020).
Ji, A. et al. Ubiquitin-specific protease 22 enhances intestinal cell proliferation and tissue regeneration after intestinal ischemia reperfusion injury. World J. Gastroenterol. 25, 824–836 (2019).
Qiu, G. et al. Hypoxia-induced USP22-BMI1 axis promotes the stemness and malignancy of glioma stem cells via regulation of HIF-1alpha. Life Sci. 247, 117438 (2020).
Mo, Z. et al. Activation of Wnt/Beta-Catenin Signaling Pathway as a Promising Therapeutic Candidate for Cerebral Ischemia/Reperfusion Injury. Front Pharm. 13, 914537 (2022).
Pan, W. & Xu, Z. Triptolide mediates Wnt/beta-catenin signalling pathway to reduce cerebral ischemia-reperfusion injury in rats. Folia Neuropathol. 58, 324–333 (2020).
Mu, Q. et al. NPD1 inhibits excessive autophagy by targeting RNF146 and wnt/beta-catenin pathway in cerebral ischemia-reperfusion injury. J. Recept Signal Transduct. Res. 40, 456–463 (2020).
Hu, Z. et al. Momordica charantia polysaccharides modulate the differentiation of neural stem cells via SIRT1/Beta-catenin axis in cerebral ischemia/reperfusion. Stem Cell Res. Ther. 11, 485 (2020).
Kaga, S., Zhan, L., Altaf, E. & Maulik, N. Glycogen synthase kinase-3beta/beta-catenin promotes angiogenic and anti-apoptotic signaling through the induction of VEGF, Bcl-2 and survivin expression in rat ischemic preconditioned myocardium. J. Mol. Cell Cardiol. 40, 138–147 (2006).
Fan, H. et al. IRES-mediated Wnt2 translation in apoptotic neurons triggers astrocyte dedifferentiation. NPJ Regen. Med. 7, 42 (2022).
Yuan, Y. et al. p-hydroxy benzaldehyde revitalizes the microenvironment of peri-infarct cortex in rats after cerebral ischemia-reperfusion. Phytomedicine 105, 154379 (2022).
Garcia-Martin, G., Alcover-Sanchez, B., Wandosell, F. & Cubelos, B. Pathways Involved in Remyelination after Cerebral Ischemia. Curr. Neuropharmacol. 20, 751–765 (2022).
Cappuccio, I. et al. Induction of Dickkopf-1, a negative modulator of the Wnt pathway, is required for the development of ischemic neuronal death. J. Neurosci. 25, 2647–2657 (2005).
Zhang, Q., Wang, R., Khan, M., Mahesh, V. & Brann, D. W. Role of Dickkopf-1, an antagonist of the Wnt/beta-catenin signaling pathway, in estrogen-induced neuroprotection and attenuation of tau phosphorylation. J. Neurosci. 28, 8430–8441 (2008).
Zhao, B. et al. JLX001 attenuates blood-brain barrier dysfunction in MCAO/R rats via activating the Wnt/beta-catenin signaling pathway. Life Sci. 260, 118221 (2020).
Hayashi, T., Takada, K. & Matsuda, M. Changes in ubiquitin and ubiquitin-protein conjugates in the CA1 neurons after transient sublethal ischemia. Mol. Chem. Neuropathol. 15, 75–82 (1991).
Hochrainer, K. Protein Modifications with Ubiquitin as Response to Cerebral Ischemia-Reperfusion Injury. Transl. Stroke Res. 9, 157–173 (2018).
Hu, B. R. Protein aggregation after focal brain ischemia and reperfusion. J. Cereb. Blood Flow. Metab. 21, 865–875 (2001).
Liu, L. et al. Activation of beta-catenin by hypoxia in hepatocellular carcinoma contributes to enhanced metastatic potential and poor prognosis. Clin. Cancer Res. 16, 2740–2750 (2010).
Caldeira, M. V., Salazar, I. L., Curcio, M., Canzoniero, L. M. & Duarte, C. B. Role of the ubiquitin-proteasome system in brain ischemia: friend or foe? Prog. Neurobiol. 112, 50–69 (2014).
Zhou, W., Wei, W. & Sun, Y. Genetically engineered mouse models for functional studies of SKP1-CUL1-F-box-protein (SCF) E3 ubiquitin ligases. Cell Res. 23, 599–619 (2013).
Shen, X. et al. Activation and Role of Astrocytes in Ischemic Stroke. Front Cell Neurosci. 15, 755955 (2021).
Vacik, T., Ladinovic, D. & Raska, I. KDM2A/B lysine demethylases and their alternative isoforms in development and disease. Nucleus 9, 431–441 (2018).
Xiong, J., Wang, Y., Gong, Z., Liu, J. & Li, W. Identification of a functional nuclear localization signal within the human USP22 protein. Biochem Biophys. Res. Commun. 449, 14–18 (2014).
Li, Y. et al. USP22 knockdown protects against cerebral ischemia/reperfusion injury via destabilizing PTEN protein and activating the mTOR/TFEB pathway. Naunyn Schmiedebergs Arch. Pharm. 396, 3163–3175 (2023).
Di, Q. et al. USP22 suppresses the NLRP3 inflammasome by degrading NLRP3 via ATG5-dependent autophagy. Autophagy 19, 873–885 (2023).
Lu, X. et al. Hypoxic Preconditioning Attenuates Neuroinflammation via Inhibiting NF-kappaB/NLRP3 Axis Mediated by p-MLKL after Transient Global Cerebral Ischemia. Mol. Neurobiol. 61, 1080–1099 (2024).
Stanek, T. J. et al. The SAGA complex regulates early steps in transcription via its deubiquitylase module subunit USP22. EMBO J. 40, e102509 (2021).
Cai, Z. et al. USP22 promotes IRF3 nuclear translocation and antiviral responses by deubiquitinating the importin protein KPNA2. J. Exp. Med. 217, e20191174 (2020).
Liu, T. et al. Expression of USP22 and the chromosomal passenger complex is an indicator of malignant progression in oral squamous cell carcinoma. Oncol. Lett. 17, 2040–2046 (2019).
Lin, Y. et al. USP22 promotes proliferation in renal cell carcinoma by stabilizing survivin. Oncol. Lett. 20, 246 (2020).
Balmik, A. A. & Chinnathambi, S. Methylation as a key regulator of Tau aggregation and neuronal health in Alzheimer’s disease. Cell Commun. Signal 19, 51 (2021).
Zhu, P. et al. lnc-beta-Catm elicits EZH2-dependent beta-catenin stabilization and sustains liver CSC self-renewal. Nat. Struct. Mol. Biol. 23, 631–639 (2016).
Lei, A. et al. EZH2 Regulates Protein Stability via Recruiting USP7 to Mediate Neuronal Gene Expression in Cancer Cells. Front Genet 10, 422 (2019).
Zhan, L. et al. Activation of Akt/FoxO and inactivation of MEK/ERK pathways contribute to induction of neuroprotection against transient global cerebral ischemia by delayed hypoxic postconditioning in adult rats. Neuropharmacology 63, 873–882 (2012).
Zhan, L. et al. Activation of Akt/FoxO signaling pathway contributes to induction of neuroprotection against transient global cerebral ischemia by hypoxic pre-conditioning in adult rats. J. Neurochem 114, 897–908 (2010).
Wen, H. et al. Hypoxic postconditioning promotes mitophagy against transient global cerebral ischemia via PINK1/Parkin-induced mitochondrial ubiquitination in adult rats. Cell Death Dis. 12, 630 (2021).
Zhan, L., Lu, X., Xu, W., Sun, W. & Xu, E. Inhibition of MLKL-dependent necroptosis via downregulating interleukin-1R1 contributes to neuroprotection of hypoxic preconditioning in transient global cerebral ischemic rats. J. Neuroinflammation 18, 97 (2021).
Wen, H. et al. Hypoxic postconditioning restores mitophagy against transient global cerebral ischemia via Parkin-induced posttranslational modification of TBK1. Neurobiol. Dis. 179, 106043 (2023).
Acknowledgements
This work was supported by the National Natural Science Foundation of China (Grant 82071281, 82471309) and Basic and Applied Basic Research Foundation of Guangdong Province (Grant 2021A1515011269). The authors express their gratitude to Dr. Shanshan Duan for the experimental assistance during the article revising stage. They also thank the reviewers for their useful comments on an earlier version of this article.
Author information
Authors and Affiliations
Contributions
En Xu, Yunyan Zuo, and Lixuan Zhan designed the research. Yunyan Zuo collected and analyzed the data. Yunyan Zuo, Jiahui Xue and Meiyan Chen performed all the experiments with assistance from Weiwen Sun. The manuscript was written by En Xu, Yunyan Zuo and Haixia Wen. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Communications Biology thanks the anonymous reviewers for their contribution to the peer review of this work. Primary Handling Editor: Christina Karlsson Rosenthal. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Zuo, Y., Xue, J., Wen, H. et al. Inhibition of SCFKDM2A/USP22-dependent nuclear β-catenin ubiquitylation mediates cerebral ischemic tolerance. Commun Biol 8, 214 (2025). https://doi.org/10.1038/s42003-025-07644-5
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s42003-025-07644-5
