
Abstract
The multi-petal “Hutou” jasmine (Jasminum sambac var. Trifoliatum) is highly valued for bonsai cultivation and landscape design, however, the aroma profile and mechanisms underlying floral scent formation remain elusive. In this study, we generate a nearly complete telomere-to-telomere (T2T) genome assembly of “Hutou” jasmine (487.45 Mb with contig N50 of 38.93 Mb). Metabolomic profiling unveils that 16 significantly differential volatiles (SDVs) may play a crucial role in the formation of flower aroma. Among them, five scented SDVs, particularly α-farnesene and pentanoic acid 1-ethenyl-1,5-dimethyl-4-hexenyl ester, contribute to the characteristic aroma profile of “Hutou” jasmine flowers. Weighted gene co-expression network analysis (WGCNA) identifies HTWRKY41, HTWRKY53, and HTHSP90 as the hub genes potentially regulating the production of these 16 metabolites. The expression of selected genes and duplication events drive the increased relative content of major sesquiterpenoids in terpenoid biosynthetic pathway. Four structural genes (BEAT3, BSMT1, BPBT2, and BPBT3) are potentially implicated in the emission of downstream key volatile esters (benzyl acetate, methyl benzoate, and benzyl benzoate) in the phenylpropanoids synthesis. Our integrated dataset of genomics, transcriptomics, and metabolomics present here provides a theoretical basis for the practical utilization of fragrance and genetic improvement in horticultural applications of “Hutou” jasmine.
Similar content being viewed by others

Introduction
Jasmine [Jasminum sambac (L.) Aiton, Oleaceae], commonly grown in tropical and subtropical regions, is a perennial evergreen shrub highly prized for its exquisite white flowers and distinctive fragrance1. The flowers emit a sweet and potent smell when blooming and have various applications in the extraction of essential oils, the perfume industry, and aromatherapy2,3. Originating from India and the Arabian Peninsula, jasmine cultivation boasts a rich history spanning over 2000 years4. In China, the jasmine flowers are utilized in the manufacture of jasmine-scented tea, where the fresh blossoms naturally infuse their floral aroma into base tea5,6. Cultivated jasmine varieties exhibit single-petal (SP), double-petal (DP), and multi-petal (MP) phenotypes, which mainly differ from each other in flower yield, floral scent, and stress resistance7. SP and DP are the predominant commercial germplasm. SP flowers are renowned for their fresh and elegant aroma, while DP plants are valued for their rather high-yield flowers and resistance to abiotic stresses, such as low temperatures and water scarcity8. The MP jasmine is typically characterized by its multi-whorled leaves, dense clusters of tight flower buds, long flowering duration, and weak disease resistance. Due to the poor pollen viability (0.13–0.19%) of jasmine flowers, cutting propagation is the most convenient method in jasmine floriculture9, which has led to the accumulation of deleterious mutations in jasmine genomes. The cultivar “Hutou” (J. sambac var. Grand Duke of Tuscany, MP) has long been regarded as an unstable or mutable genotype, however, a definitive genetic basis for this has not yet been established10. In the realm of fragrant plants in Oleaceae family, “Hutou” jasmine flower has large white petals (over 50 petals) and mellow refreshing fragrance, embodying unique horticultural and aesthetic values.
Floral scents comprise the full range of volatile organic compounds (VOCs) emitted by flowering plants to attract pollinators and provide defense against microbial pathogens11,12. The variations and abundances of these VOCs in fragrances are among the most crucial factors for assessing the ornamental appeal of scented plants. Based on their origin, synthesis, and function, flower fragrances are mainly categorized as terpenoids, phenylpropanoids/benzenoids, fatty acid-derived compounds and amino acids13. Among these, terpenes play a pivotal role in diverse biological processes and are crucial for the formation of floral aromas14,15. Various types of terpenoids compounds are synthesized through the mevalonate (MVA) pathway in the cytoplasm and the plastid-based 2-C-methyl-D-erythritol 4-phosphate (MEP) pathway, which are responsible for the synthesis of sesquiterpenes and monoterpenes, respectively16. Phenylpropanoids/benzenoids represent the second most abundant class of fragrance components and are generated from the aromatic amino acid phenylalanine17. In jasmine, flower buds are scentless, but mature flowers emit fragrance at night throughout the flowering period18, reflecting dynamic changes in VOC emission as flowering progressed. The inherent diversity in aroma composition is influenced by factors such as the time of day for harvesting, weather conditions, and the specific cultivar in question. Thanks to the advances in omics technologies and trends in floral market preferences, the fragrance profiles of several exemplary jasmine cultivars have been elucidated in recent years. The floral scent of post-harvest DP jasmine flowers was analyzed, revealing that linalool, α-farnesene, and d-nerolidol were the most abundant aromatic compounds18. Subsequently, 158 volatiles were determined in the flowers of SP and DP jasmine, indicating that the high content of α-farnesene and low content of other key volatiles may likely contribute to shaping their distinct aroma profiles and differences19. Meanwhile, certain candidate genes associated with fragrance biosynthetic pathways have been pinpointed in jasmine. For instance, several critical protein-coding genes involved in the biosynthesis of α-farnesene, such as 3-hydroxy-3-methylglutaryl coenzyme A reductase (HMGR) and terpene synthase (TPS), have been isolated and subjected to a thorough analysis in DP jasmine20. Previous studies have vastly expanded our understanding of floral scent in jasmine. Despite the considerable economic and ornamental value of the “Hutou” jasmine, the genetic and breeding efforts, as well as studies on fragrance composition, have lagged behind those of other jasmine varieties. The currently available MP genome presents numerous gaps and assembly errors21, necessitating further refinement. A high-quality reference genome assembly will emerge as a compelling tool for elucidating the VOCs profile and molecular mechanisms behind floral aroma synthesis in MP jasmine varieties.
In the present study, we assembled and annotated the “Hutou” multi-petal jasmine genome using a combination of PacBio High fidelity (HiFi) sequencing and high-throughput Chromosome Conformation Capture (Hi-C) technology. The comprehensive analysis of widely-targeted volatilomics coupled with time-course transcriptome analysis provided a thorough understanding of the floral fragrance profile and potential pivotal regulatory genes involved in floral scent production. This work will fill the research gap regarding the floral scent and fragrance-related genes involved in MP jasmine flowering, providing a blueprint for applications in perfumery, cosmetics, and trait improvement.
Results
Genome assembly and annotation
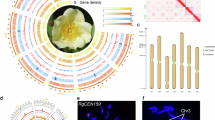
The karyotype analysis and fluorescence in situ hybridization (FISH) mapping indicated that “Hutou” jasmine cell has a diploid genome with 13 pairs of chromosomes (2n = 26) (Supplementary Fig. 1). The 17-mer profile based on Illumina short reads predicted the genome size to be ~492.88 Mb, with a heterozygosity rate of ~1.19% and a repeat proportion of ~58.70% (Supplementary Fig. 2). We generated the “Hutou” jasmine genome by integrating PacBio HiFi sequencing data with Hi-C data. A total of 21.86 Gb (~44×) HiFi data and 53.13 Gb (109×) Hi-C data were obtained (Supplementary Table 1 and Supplementary Fig. 3). The initial de novo assembly using Hifiasm22 produced contig-level sequences of 514.72 Mb comprising 480 contigs with N50 of 26.90 Mb (Supplementary Table 2). The purged assembly achieved an improved contig N50 of 28.29 Mb after removing haplotigs and contig overlaps (Supplementary Table 2 and Supplementary Fig. 4). Then, the Hi-C data were utilized to cluster, order, and orient the sequences using the ALLHiC23 pipeline, and 483.45 Mb (99.18%) were anchored into 13 super-scaffolds representing all chromosomes. Strong interactive signals were observed along the diagonals of the Hi-C heatmap, while significantly lower enrichment was detected off the diagonal (Fig. 1a). The final genome assembly spanned 487.45 Mb, comprising 26 contigs with an N50 of 38.93 Mb and a GC content of 34.56%. The lengths of the pseudochromosomes varied from 29.13 Mb to 45.82 Mb (Fig. 1b, Supplementary Tables 2 and 3). No other microbial contamination was observed in this assembly, as reveal by the GC-depth distribution (Supplementary Fig. 5).

a High-throughput chromosomal conformation capture (Hi-C) contact map among 13 chromosomes visualized by HapHiC (v 1.0.6) with a 200-kb resolution (with units in Mb). Hi-C scale was indicated in Knight-Ruiz (KR)-normalized counts, with a darker red color indicating higher contact probability. b Circos plot of gene features at 500-kb intervals across 13 chromosomes of “Hutou” jasmine. The track from A to F is as follows: chromosomes (in Mb), gene density, LTR/Gypsy density, LTR/Copia density, GC content and color ribbons representing the genome-wide syntenic blocks. c The BUSCO completeness of the “Hutou” genome in this study and seven published genomes with the default gene predictor MetaEuk against embryophyta_odb10 dataset. JSU-FSP, J. sambac var. unifoliatum cultivar Fuzhou Single-Petal55; FZCLSP, Fuzhou Changle Single-Petal118; DP, Double-Petal119; YZDP, Yangzhou Double-Petal45; FZDP, Fuzhou Double-Petal118; JSDP, Jiangsu Double-Petal120; MP, Multi-Petal21. d The violin plot of longest terminal repeat (LTR) index (LAI) among eight jasmine assemblies. The horizontal axis represents different jasmine varieties, while the number in each box indicates the LAI value.
Multiple metrics were employed to evaluate the quality of assembly, including mapping rates, Busco scores, LAI values, and QV assessments. The mapping rates for HiFi reads, DNA-seq, and RNA-seq from flowers and leaves were 99.68%, 98.65%, and at least 88.35%, respectively (Supplementary Table 4). The BUSCO evaluation revealed that the “Hutou” assembly contained 98.50% complete core orthologous genes with a relatively low proportion of duplicates (2.6%), ranking it among the top three jasmine genomes (Fig. 1c). Our assembly showed improvements in genome completeness compared with the recently published MP jasmine genome21. The longest terminal repeat (LTR) index (LAI) was 21.81, meeting the assembly gold quality standard (LAI > 20)24 (Fig. 1d). Notably, the genomic assembly achieved a remarkable quality value (QV) score of 63.40 (Supplementary Table 5). We also observed that 11 out of the 13 chromosomes possessed telomeric repeats on both ends (Supplementary Fig. 6), indicating a near T2T jasmine assembly and representing the highest level of continuity and completeness of jasmine monoploid genome.
A total of 23,968 protein-coding genes were predicted with 95.20% BUSCO completeness (Supplementary Table 6), and 98.16% (23,527/23,968) of these genes showed hits to at least one protein-related database (Supplementary Table 7 and Supplementary Fig. 7). Totally, 303.77 Mb (62.32%) repetitive sequences were identified, of which long terminal repeat retrotransposons (LTR-RTs), mainly characterized by Gypsy and Copia, were the most abundant (23.92%) (Supplementary Table 8). Additionally, 653 tRNAs, 5,822 rRNAs, 145 miRNAs, 2,085 snRNAs, and one sRNA were annotated (Supplementary Table 9).
Flowering Metabolic Profiling Unveiling Aroma Composition Variations
To investigate the composition and variations of aroma in “Hutou” jasmine flowers, headspace solid-phase microextraction (HS-SPME) combined with gas chromatography-mass spectrometry (GC-MS) was utilized to detect the volatiles at five different flowering stages. The total ion current (TIC) diagram of the quality-controlled samples displayed the reproducibility of the data (Supplementary Fig. 8). The principal component analysis (PCA) revealed that the biological replicates for each period tended to cluster together, indicating significant variations in volatile compounds across the five blooming stages (Fig. 2a). All samples were grouped into five clusters on the heatmap, with F3 showing distinct volatile levels compared to other flowering stages (Fig. 2b). The total volatile compounds exhibited an increase from F1 (2,853,961.80 μg/kg) to F3 (3,841,905.25 μg/kg), followed by a sharp decline towards F5 (387,463.21 μg/kg), which recorded the lowest volatile content (Fig. 2c and Supplementary Data 1). A total of 912 volatiles were identified across all flower samples (Supplementary Data 2), with the four most abundant classes being terpenoids (198), esters (170), heterocyclic compounds (125), and hydrocarbons (75) (Fig. 2d and Supplementary Table 10). It is worth noting that terpenoids and esters comprised the majority (40.35%) of the identified volatile compounds.

a The principal component analysis (PCA) model among “Hutou” jasmine flower samples by HS-SPME-GC/MS at five flowering stages. b Hierarchical cluster analysis (HCA) of the identified metabolites in different flower samples. Each sample is represented by a column, and metabolite information is represented by a row. The relative contents are indicated by the different shade colors in the Z-score (i.e., red and green), with red representing high contents and green indicating low contents. c Variation trend of total volatiles content. Data are represented as means±standard deviations (SD) d Classes and proportions of a total of 912 metabolites detected in “Hutou” jasmine flowers. e Venn diagram shows the numbers of the overlapping and unique significant differential metabolites (SDVs) among the four comparisons. f, g KEGG enrichment analysis of SDVs in F1 vs. F2 and F1 vs. F3 comparison groups. The size and color of the bubbles represent the count of metabolites involved in a certain pathway and the P-value of the rich factor, respectively. The red fonts indicate metabolic pathways associated with fragrance synthesis (P < 0.05).
The types and amounts of volatile compounds emitted by blossoms varied significantly at different floral phases. In order to explore the variations in floral fragrance compounds across the flowering process, a total of 821 SDVs (VIP ≥ 1.0, Fold change≥1.2 or ≤0.67) were identified in comparisons of F1 vs. F2, F1 vs. F3, F1 vs. F4, and F1 vs. F5, with 285 SDVs common to all comparisons (Fig. 2e). Considering the dramatic rise in aroma intensity from the F1 to the F2 and F3 stages in “Hutou” flowering (Fig. 2c), all 673 SDVs from the F1 vs. F2 and F1 vs. F3 comparison sets were examined in the subsequent analysis (Fig. 2e). These SDVs were divided into 16 groups, including terpenoids, esters, heterocyclic compounds, among others (Supplementary Data 3, 4). Additionally, KEGG enrichment analysis revealed that these SDVs were markedly enriched in the “Terpenoid backbone biosynthesis” and “Biosynthesis of secondary metabolites” pathways (Fig. 2 f, g).
To better understand the influence of key components contributing to the characteristic floral aroma of “Hutou” jasmine, we focused on the SDVs in the aforementioned two groups with a relative content percentage exceeding 1% at the F2 or F3 period. This led to the identification of sixteen predominant volatiles in jasmine flowers, including six terpenoids, five esters, two heterocyclic compounds, one acid, one aromatic, and one alcohol (Fig. 3a and Supplementary Data 5). As the flowering process unfolded, the accumulation patterns of these compounds initially increased and then declined. Among six terpenoids, 1-methyl-4-(1,2,2-trimethylcyclopentyl) cyclohexa-1,3-diene and 2-isopropyl-5-methyl-9-methylene bicyclo[4.4.0]dec-1-ene had significantly higher content than other volatiles, yet they were scentless (Fig. 3b). This observation prompted a more thorough examination of the specific constituents that are pivotal in defining jasmine’s characteristic fragrance. Of particular note, the content of scented volatile α-farnesene, associated with the floral balsamic aroma, increased from the F1 to F3 stages and subsequently decreased (Fig. 3b). Likewise, only scented volatiles were examined in the other groups. Among the five esters studied, three scented volatiles were identified, including pentanoic acid 1-ethenyl-1,5-dimethyl-4-hexenyl ester, (Z)-3-hexen-1-ol benzoate, and geranyl isobutyrate, which exhibited sweet, fruity, floral, aromatic, and leafy odor qualities (Fig. 3c). Their content generally increased from the F1 to F3 stage and reached their peaks at F3. Additionally, benzyl alcohol, with a mild and pleasant odor, had the highest concentration at F2 compared with other stages (Fig. 3d). Compared to the F1 stage, the increased content of these five SDVs during the F2 and F3 stages likely contribute to the aromatic profile of the “Hutou” jasmine flowers through their various odor characteristics.

a The heatmap of 16 predominant SDVs (relative content percentage>1%) created by TBtools with “ZeroToOne” option. The circle size and color represent content magnitude. b Quantification of six terpenoids identified across different flowering stages. c Quantification of five esters identified across different flowering stages. d Quantification of five other high-abundance compounds. Structural formulas of five scented SDVs are displayed on the right. Data are represented as means ± standard deviations (SD). All datasets for the 16 predominant volatiles are available in Supplementary Data 5.
Flowering transcriptomic dynamics
To elucidate the transcriptional dynamics and regulatory mechanisms of genes involved in aroma synthesis, RNA samples from five flowering periods were used for transcriptomic profiling (Fig. 4a). A total of 118.25 Gb of clean data composed of 788.41 million reads was produced by Illumina sequencing platform NovaSeq (Supplementary Data 6). The number of clean reads per library ranged from 41.74 to 64.82 million, with Q30 values of at least 91%. After mapping the RNA-seq reads against the “Hutou” jasmine reference genome, over 90.89% of reads from each sample aligned uniquely to the genome (87.77–90.40%), while the remaining reads had multiple matches (2.39–3.22%) (Supplementary Table 11).

a Five flowering stages of “Hutou” jasmine. F1: flower at the closed bud stage; F2: flower starts opening; F3: flower fully opening; F4: flower starts senescence; F5: withered flower stage. Bar = 1 cm. b The correlation analysis using Pearson’s Correlation Coefficient (PCC) between each pair of samples. c The principal component analysis (PCA) of five groups of flowering stages. PC1 and PC2 represent the first and second principal components, respectively. d Statistics of up-regulated and down-regulated differentially expressed genes (DEGs) in different comparison sets. e The Venn diagram depicting DEGs across four different comparisons.
Correlation and PCA analysis suggested that the samples in each group had good reproducibility and significant differences among various flowering stages (Fig. 4b, c). The gene expression analysis revealed an increase in the number of highly expressed genes (10 < FPKM < 100) at F3 (Supplementary Table 12). Differential gene expression analysis was conducted across ten comparison groups spanning five flowering periods (Fig. 4d). The comparisons from F1 vs. F2 to F1 vs. F5 revealed a significant increase in the number of DEGs, with upregulated genes rising from 904 to 4,638, and downregulated genes increasing from 549 to 4,143. This suggested the levels of numerous transcripts altered significantly throughout the blossom phase. Additionally, 633 DEGs were shared by the comparisons between F1 vs. F2, F1 vs. F3, F1 vs. F4, and F1 vs. F5 (Fig. 4e), with 350, 370, 424, and 1,830 specific DEGs identified for each comparison, respectively. We primarily focused on the comparisons from F1 vs. F2 to F1 vs. F5 in the following analysis.
To explore the biological processes and molecular components involved in scent production in “Hutou” jasmine flowers, Gene Ontology (GO) enrichment analysis was utilized to categorize the functions of DEGs. The top 25 GO terms of DEGs were distributed in three major GO categories: biological process, molecular function, and cellular component (Supplementary Fig. 9). Among the four comparisons, biological process had the most significantly enriched GO terms, followed by molecular function. In all groups, the term “oxidoreductase activity” was consistently enriched, implying that flowering is a highly active metabolic process. Increased metabolic activity may raise the temperature within the flower tissue25. DEGs from the F1 vs. F2 and F1 vs. F3 comparisons were primarily enriched in the pathways of the “benzene-containing compound metabolic process”, “secondary metabolic process”, “apoplast”, “phenol-containing compound metabolic process”, and “monooxygenase activity”. In the F1 vs. F4 and F1 vs. F5 comparisons, DEGs were mainly involved in pathways such as “UDP-glycosyltransferase activity”, “hydrolase activity, hydrolyzing glycosyl compounds”, “active transmembrane transporter activity”, and “isoprenoid metabolic process”, which contributed to the synthesis, modification, and transport of secondary metabolites during the later time of blossom. Besides, several plant hormone response pathways were enriched, including the “salicylic acid metabolic process”, “salicylic acid-mediated signaling pathway”, and “jasmonic acid mediated signaling pathway”.
Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway analysis was used to identify metabolic pathways for these DEGs. In total, 57 significantly enriched pathways were obtained (Supplementary Figs. 10, 11). Among them, 24, 29, 30, and 37 KEGG pathways were markedly enriched in the four comparison sets, indicating a shift in metabolic activity as the flowering progressed. Six pathways, “Sesquiterpenoid and triterpenoid biosynthesis”, “Phenylpropanoid biosynthesis”, “Cyanoamino acid metabolism”, “alpha-Linolenic acid metabolism”, “ABC transporters”, and “Glycerolipid metabolism”, were shared among all four comparison groups. Moreover, except for the F1 vs. F2 group, the “MAPK signaling pathway-plant” was enriched in the remaining three comparisons.
Temporal expression of genes involved in terpene biosynthesis
Terpenes play a crucial role in the formation of floral fragrances26. (MVA pathway: HMGRs, 3-hydroxy-3-methylglutaryl coenzyme A reductase genes; SQSs, squalene synthase genes; AACTs, acetoacetyl-CoA thiolase genes; HMGSs, 3-hydroxy-3-methylglutaryl coenzyme A synthase genes; MVK, mevalonic acid kinase gene; PMK, phosphomevalonate kinase gene; MVD, mevalonate diphosphate decarboxylase gene; IDI, isopentenyl diphosphate isomerase gene; IPK, isoprene kinase gene; FPPS, farnesyl diphosphate synthase gene) In the MVA pathway, twenty genes potentially involved in sesquiterpenoids biosynthesis were identified, including 5 HMGRs, 5 SQSs, 2 AACTs, 2 HMGSs and one each of MVK, PMK, MVD, IDI, IPK, and FPPS (Supplementary Data 7). These genes were expressed across various tissues and flowering periods (Fig. 5a), with seven genes (AACT1, HMGS2, HMGR3, MVD, IDI, FPPS, and SQS1) exhibiting relatively high expression levels throughout the bloom. Homologous genes showed differential expression across all tissues and phases, with certain homologs showing higher expression levels than their counterparts. HMGR, the first rate-limiting enzyme of the MVA pathway27, possessed five homologous copies, with their gene expression levels peaking at F2 or F3. For instance, HMGR1 had the highest expression level at F3, while the expression levels of HMGR3 and HMGR4 peaked at F2. This pattern was consistent with the trend in the accumulation of major sesquiterpenes content (Fig. 5a, b). In contrast, HMGR2 and HMGR5 had negligible expression. FPPS, responsible for the synthesis of farnesyl diphosphate (FPP), was detected to have the highest expression level during the F2 period. IDI, modulating the ratios of isopentenyl pyrophosphate (IPP) to dimethylallyl diphosphate (DMAPP), also exhibited peak expression levels at F2.

a Expression profiles of genes encoding enzymes (in red) involved in terpenoids synthesis across different flowering stages (F1-F5) and tissues (leaf, root, and stem). F1, flower at the closed bud stage; F2, flower starts opening; F3, flower fully opening; F4, flower starts senescence; F5, withered flower stage. Homologous genes are termed from top to bottom in Arabic numerical order. Enzymatic steps are represented by arrows, and multiple enzymatic steps are represented by dotted lines. The expression level ranging from low to high is depicted by bule to red. b Total relative content of sesquiterpenes and monoterpenes in datasets of 16 predominant volatiles across different flowering stages. Each value is displayed as means ± standard deviations (SD). c Circos plot showing genes in the terpene biosynthesis pathway. Genes with tandem clusters are marked in blue. Gene pairs with segmental duplications are indicated by red lines. acetyl-CoA acetyl coenzyme A, acetoacetyl-CoA acetoacetyl coenzyme A, HMG-CoA 3-hydroxy-3-methylglutaryl coenzyme A, MVA mevalonic acid, MVAP mevalonic acid-5-phosphate, MVAPP mevalonic acid-5-diphosphate, DMAPP dimethylallyl diphosphate, IP isopentenyl pyrophosphate, IPP isopentenyl pyrophosphate, FPP farnesyl diphosphate, GAP glyceraldehyde-3-phosphate, DXP 1-deoxy-D-xylulose 5phosphate, MEP 2-C-methylerythritol 4-phosphate, CDP-ME2P 2-phospho-4-(cytidine 5’-diphospho)-2-C-methyl-D-erythritol, ME-CPP 2-C-phospho-4-(cytidine-5’-diphospho)-2C-methyl-d-erythritol, HMBDP 1-hydroxy-2-methyl-2-(E)-butenyl 4-diphosphate, GPP geranyl pyrophosphate, GGPP geranylgeranyl diphosphate, AACT acetoacetyl-CoA thiolase, HMGS HMG-CoA synthase, HMGR HMG-CoA reductase, MVK MVA kinase, PMK phosphomevalonate kinase, MVD mevalonate diphosphate decarboxylase, IPK isoprene kinase, IDI isopentenyl diphosphate isomerase, FPPS FPP synthase, SQS squalene synthase, TPS terpenoid synthase, DXS DXP synthase, DXR DXP reducto-isomerase, MCT, 2-C-methyl-D-erythritol-4-(cytidine-5-diphospho) transferase, CMK CDP-ME2P kinase, MDS ME-CPP synthase, HDS HMBDP synthase, HDR HMBDP reductase, GPPS GPP synthase, GGPPS GGPP synthase, SPS sucrose-phosphate synthase.
For the (MEP pathway: GGPPSs, geranylgeranyl diphosphate synthase genes; DXSs, 1-deoxy-D-xylulose 5phosphate synthase genes; HDRs, 1-hydroxy-2-methyl-2-(E)-butenyl 4-diphosphate reductase genes; GPPSs, geranyl pyrophosphate genes; DXR, 1-deoxy-D-xylulose 5phosphate reducto-isomerase gene; MCT, 2-C-methyl-D-erythritol-4-(cytidine-5-diphospho) transferase gene; CMK, 2-phospho-4-(cytidine 5’-diphospho)-2-C-methyl-D-erythritol kinase gene; MDS, 2-C-phospho-4-(cytidine-5’-diphospho)-2C-methyl-d-erythritol synthase gene; HDS, 1-hydroxy-2-methyl-2-(E)-butenyl 4-diphosphate synthase gene; SPS, sucrose-phosphate synthase gene) MEP pathway, twenty-one genes related to monoterpene biosynthesis were determined, including 7 GGPPSs, 4 DXSs, 2 HDRs, 2 GPPSs, as well as one each of DXR, MCT, CMK, MDS, HDS, and SPS (Supplementary Data 8). Out of the 21 members, 18 genes were expressed across various tissues and flowering periods, while the remaining three (GGPPS2, GGPPS4, and GGPPS6) were expressed at almost undetectable levels. The enzyme DXS, which initiates isoprenoid biosynthesis28, was identified in four copies and further classified into three distinct classes (class I, II, and III) (Supplementary Fig. 12). These homologous exhibited differential expression patterns, similar to those observed in other plant species29. HTDXS3 (class I) serves housekeeping functions and provides building blocks for essential isoprenoid synthesis30. HTDXS4 was categorized under class III, whereas HTDXS1 and HTDXS2 belonged to class II, each playing distinct roles in plant secondary metabolism31. Notably, HTDXS3 stood out with the highest expression level, showing an increase from F1 to F2 followed by a decrease from F2 to F5. Similarly, DXR displayed a comparable tend.
Taken together, the expression levels of genes associated with the MVA pathway were relatively higher than those in the MEP pathway, aligning with the significantly higher abundance of major sesquiterpenes compared to monoterpenes (Fig. 5a, b and Supplementary Data 5). Furthermore, seven segmental duplication events related to the MVA and MEP pathways were identified on 7 out of the 13 chromosomes (Fig. 5c). The MVA pathway comprised 7 genes derived from segmental duplications (HMGS1-HMGS2, HMGR3-HMGR4, SQS1-SQS4, SQS4-SQS5), while the MEP pathway included 6 genes (DXS1-DXS2, HDR1-HDR2, GGPPS1-GGPPS2). Furthermore, SQS1-SQS2-SQS3 and GGPPS3-GGPPS4 were recognized as tandem duplications.
Terpene synthases (TPSs) are key gatekeepers in the MVA and MEP pathways, responsible for the final catalytic reaction to produce terpenoid compounds32. A total of 53 TPS homologous genes were identified (Supplementary Data 9). Phylogenetic analysis of TPS protein sequences from “Hutou” jasmine, Arabidopsis thaliana, Osmanthus fragrans, and Fraxinus excelsior revealed that “Hutou” jasmine has more TPS genes than A. thaliana (32), but fewer than O. fragrans (62) and F. excelsior (65) (Supplementary Fig. 13). The phylogenetic tree divided these TPSs into five subgroups: TPS-a, TPS-b, TPS-c, TPS-e/f, and TPS-g. The majority (75.47%) of “Hutou” jasmine TPSs were classified into the TPS-a (28 genes) and TPS-b (12 genes) subfamilies, which are primarily responsible for the biosynthesis of sesquiterpenes and monoterpenes, respectively. Remarkably, HTTPS3, HTTPS4, HTTPS14, and HTTPS39 were upregulated at the F2 or F3 flowering stages, coinciding with the emission profile of terpene compounds (Fig. 5 b and Supplementary Fig. 14).
Comprehensive Analysis of Genes Involved in Phenylpropanoids/Benzenoids Production
Phenylpropanoids/benzenoids constitute the second most ubiquitous group of floral VOCs, which are derived from L-phenylalanine (L-Phe) via the shikimate pathway. In total, 42 genes were involved in (phenylpropanoid pathway: PALs, phenylalanine ammonia-lyase genes; PAASs, phenylacetaldehyde synthase genes; BALDHs, benzaldehyde dehydrogenase genes; CCoAOMTs, Caffeoyl-CoA O-Methyltransferase genes; CNLs, cinnamoyl-CoA ligase genes; KATs, 3-ketoacyl-CoA-thiolase genes; BSMTs, benzoic acid/salicylic acid carboxyl methyltransferase genes; BPBTs, benzoyl-CoA:benzylalcohol/2-phenylethanol benzoyltransferase genes; BEATs, acetyl-CoA:benzyl alcohol acetyltransferase genes; CHDs, cinnamoyl-CoA dehydrogenase genes) phenylpropanoid pathway in “Hutou” jasmine flowers (Fig. 6a and Supplementary Data 10). Both segmental and tandem duplication events have significantly contributed to the expansion of genes in the phenylpropanoids pathway (Fig. 6b), thereby influencing the aroma metabolism. Specifically, 12 genes were derived from segmental duplications (PAL1-PAL4, PAAS1-PAAS3, BALDH1-BALDH2, CCoAOMT1-CCoAOMT2, CNL1-CNL2, and KAT1-KAT2), and 6 arose through tandem duplications (PAL2-PAL3, BSMT1-BSMT2, and BPBT1-BPBT2) (Fig. 6b). The initial committed step in phenylpropanoids/benzenoids biosynthesis is the conversion of L-Phe to trans-cinnamic acid (CA) by PAL33. Among the four identified PAL genes, PAL3 was exclusively highest expressed over five flowering stages and peaked at F2 (Fig. 6a), indicating its potential key role in promoting the phenylpropanoids/benzenoids biosynthesis in “Hutou” jasmine flower. On the other hand, PAL4 had a peak expression at F5, implying its active involvement during the late flowering stage.

a Expression profiles of genes encoding enzymes (in red) possibly associated with the phenylpropanoid/benzenoid biosynthesis in “Hutou” jasmine flowers. The gradient color for each gene represents the gene expression levels in five flowering stages (F1-F5) and different tissues (leaf, root, and stem). F1, flower at the closed bud stage; F2, flower starts opening; F3, flower fully opening; F4, flower starts senescence; F5, withered flower stage. Orthologous genes are numbered sequentially in descending order using Arabic numerals. Enzymatic reactions are represented with arrows, and multiple enzymatic steps are displayed by dashed lines. The eleven phenylpropanoids compounds detected in “Hutou” jasmine flowers are circled in red boxes while non-detected metabolites are boxed with blue dotted lines. b Circos plot showing genes in the phenylpropanoids/benzenoids biosynthesis pathway. Genes with tandem clusters are marked in blue. Gene pairs with segmental duplications are indicated by red lines. c Histogram depicting four phenylpropanoids identified in “Hutou” jasmine flowers at different flowering stages. Data are represented as means ± standard deviations (SD), and “*” indicates significant difference (P-value ≤ 0.05) compared to F1. PEP phosphoenolpyruvate, E4P erythose-4-phosphate, DAHP 3-deoxy-D-arabinoheptulosonate 7-phosphate, PAL phenylalanine ammonia-lyase, C4H cinnamate-4-hydroxylase, 4CL 4-hydroxycinnamoyl CoA ligase, CCoAOMT Caffeoyl-CoA O-Methyltransferase, CFAT coniferyl alcohol acyltransferase, IGS isoeugenol synthase, EGS eugenol synthase, PAAS phenylacetaldehyde synthase, CNL cinnamoyl-CoA ligase, CHD cinnamoyl-CoA dehydrogenase, KAT 3-ketoacyl-CoA-thiolase, BPBT benzoyl-CoA:benzylalcohol/2-phenylethanol benzoyltransferase, BALDH benzaldehyde dehydrogenase, BSMT benzoic acid/salicylic acid carboxyl methyltransferase, BEAT acetyl-CoA:benzyl alcohol acetyltransferase, SAMT salicylic acid carboxyl methyltransferase.
A total of eleven volatiles from the phenylpropanoids/benzenoids metabolic pathway were detected in “Hutou” jasmine flowers, including aldehydes (phenylacetaldehyde and benzaldehyde), alcohols (phenethyl alcohol and benzyl alcohol), esters (benzyl acetate, benzyl benzoate, methyl salicylate, and methyl benzoate), phenols (isoeugenol and eugenol), and salicylic acid (Fig. 6c and Supplementary Fig. 15). Among these, benzyl acetate, a sweetly-scented ester derived from benzyl alcohol and acetic acid, was consistently detected throughout the entire flowering period in “Hutou” jasmine (Fig. 6c and Supplementary Data 2). This compound serves as a principal component of the phenylpropanoids in both SP and DP jasmine varieties, where it significantly contributes to their floral aromas19. It is produced through an acetyl-CoA-dependent reaction catalyzed by enzyme BEAT, and three BEAT gene copies were discovered in the “Hutou” genome. BEAT3 exhibited high expression level at F2, correlating strongly with the peak emission of benzyl acetate (Fig. 6a, c). BEAT2 was primarily expressed in roots and stems, while BEAT1 was not expressed at any floral stages or in other tissues. Methyl benzoate, a major phenylpropanoid compound in “Hutou” jasmine flowers (Supplementary Data 2), showed a similar emission pattern to its upstream gene BSMT1, with its levels peaking at F2 (Fig. 6a, c). Methyl salicylate, also present in flowers (Supplementary Data 2), exhibited higher concentrations from F1 to F3, with its levels significantly greater than those of methyl benzoate (Fig. 6c). Benzyl benzoate, the dominant phenylpropanoid compound found in flowers (Supplementary Data 2), is synthesized from benzoyl-CoA as the acyl donor via the β-oxidative pathway catalyzed by BPBT enzyme34. Three BPBT genes were identified and two of them (BPBT1 and BPBT2) were highly expressed at the F2 stage. Nine genes involved in β-oxidative pathway were identified in jasmine genome, including two CNLs, four CHDs and three KATs (Supplementary Data 10). Key structural genes such as CNL1, CHD2, CHD4, KAT2, and KAT3 were notably upregulated during F2, consistent with the emission of the downstream product benzyl benzoate throughout the flowering process (Fig. 6a, c). Notably, phenylethyl benzoate was absent in jasmine plants, contrasting with the findings in Petunia hybrida35.
Transcription Factors Positively Correlated with Terpene Biosynthesis
To investigate the expression pattern of DEGs throughout the entire flowering stage, K-means clustering analysis was performed, and all DEGs in four comparison groups were distinctly divided into 10 subclasses (Fig. 7a). The expression profiles of the DEGs exhibited various changes in blossoming time. Subclass 2, consisting of 252 genes, showed a significant upward trend from F2 to F4. However, 2603 genes of subclass 7 and subclass 10 displayed a significantly consistent decline throughout the entirety of the flowering phases. The expression levels of DEGs in subclass 3, 4, 5, 6, 8, and 9 showed a significant increase compared to the F1 stage. Notably, in subclass 4, 6, and 9, gene expression was significantly higher in F2 and F3, respectively, mirroring the changing trends of the primary SDVs emitted during the flowering process (Figs. 3a, 7a). KEGG enrichment analysis of the genes in subclass 4, 6, and 9 indicated that they were primarily involved in the following pathways: “MAPK signaling pathway-plant”, “Plant hormone signal transduction”, “Sesquiterpenoid and triterpenoid biosynthesis”, and “Phenylalanine, tyrosine and tryptophan biosynthesis” (Fig. 7b–d). The orchestrated production of floral volatile compounds is not solely determined by the enzymatic functions encoded by corresponding biosynthetic genes, but also critically requires the involvement of pertinent transcription factors36. A multitude of transcription factors (TFs) families, including AP2/ERF (136), MYB (123), bHLH (116), C2H2 (103), NAC (92), WRKY (70), MADS (68), and bZIP (64), were identified in “Hutou” jasmine genome (Supplementary Fig. 16). In subclass 4, 6, and 9, the most abundantly differentially expressed TFs among the 195 DEGs were AP2/ERF (21), followed by MYB (18), bHLH (15), and WRKY (15) (Fig. 7e). These TFs, activated during the F2 or F3 stages, may influence the process of flowering and govern the floral scent emission.

a K-means cluster analysis of DEGs and their expression patterns. b–d KEGG enrichment analysis of DEGs in subclass 4, 6, and 9, respectively. The pathways related to aroma production are highlighted in red color. e Differentially expressed transcriptome factors (TFs) in subclass 4, 6 and 9. f Correlation analysis of terpene biosynthesis-related genes and SDVs in subclass 4, 6 and 9 (Pearson correlation coefficient>0.9 or <-0.9 and P-value < 0.05). The blue ellipses and green rectangles represent the index of terpenoid SDVs and biosynthesis-related genes, respectively. Solid edges represent positive correlations, while dashed edges represent negative correlations. g Correlation analysis of terpene biosynthesis-related genes and differentially expressed TF genes in subclass 4, 6 and 9 (Pearson correlation coefficient>0.9 or <−0.9 and P-value < 0.05). Octagons represent structural genes, and spheres of different colors represent different TF families. The networks were drawn by Cytoscape (v 3.10.2).
In order to further explore the relationship between terpene volatile compounds and DEGs in subclass 4, 6, and 9, a correlation network analysis of terpene SDVs and differentially expressed terpene biosynthesis-related genes was performed (Pearson Correlation Coefficient>0.9 or <−0.9, P-value < 0.05) (Supplementary Data 11). A total of 72 pairs of SDVs exhibited significant correlation, of which 51 pairs were positively correlated, and 21 pairs were negatively correlated (Fig. 7f). Among these SDVs, (Z)-3,7-dimethylocta-1,3,6,-triene (KMW0235) was linked to the largest number of DEGs in the terpene synthesis. The pivotal the MEP pathway gene HTDXS2, was associated with five fragrance terpenes: α-phellandrene (KMW0223), limonene (KMW0226), α-terpinene (KMW0223), myrcene (KMW0199), and geranyl acetate (KMW0530). Most TPS genes (10/12) in the network showed higher expression in the F2 or F3 stages, except for HTTPS9 and HTTPS12 (Supplementary Fig. 14), indicating their key function in modulating terpene metabolism. Basically, the network exhibited strong correlations among 20 terpenoids SDVs and 31 DEGs related to the terpene synthesis (Fig. 7f), suggesting the intricate regulatory interactions between gene expression and downstream terpene accumulation. Furthermore, a regulatory network of differentially expressed TFs genes was established, with 142 TFs regulating target genes associated with terpene biosynthesis (Fig. 7g and Supplementary Data 12). Among them, the ERF, MYB, WRKY, and bHLH families were the most abundant among the positively correlated TFs, highlighting their pivotal role in regulating the terpenoids production at different blooming stages.
WGCNA modules associated with floral volatiles emission
To explore the genetic regulatory network underlying aroma production during the blooming period of “Hutou” jasmine, co-expressed gene sets were identified through Weighted Gene Co-expression Network Analysis (WGCNA). A total of 15,628 genes were organized into eleven color-coded modules after filtering out non-expression (FPKM < 1) genes (Fig. 8a). The correlation heatmap between modules and traits was created, using the relative contents of sixteen SDVs as phenotypic data (Fig. 8b). The findings indicated a strong positive relationship between the magenta/purple modules and distinct volatile compounds. Expression heatmaps and consensus profiles revealed a progressive upregulation of co-expressed genes within the magenta module from F1 to F3 stages, followed by a decline (Fig. 8c). In the purple module, gene expression peaked at F2 before gradually declining (Supplementary Fig. 17). These trends were consistent with the accumulation pattern of sixteen floral SDVs at various flowering stages (Fig. 3a).

a Hierarchical cluster dendrogram. Each branch represents a module, and each leaf corresponds to an individual gene. Different colors represent different modules. b Correlation heatmap between the 16 floral SDVs and 11 gene modules. The x-axis presents each volatile index (trait), while the y-axis presents each module. The colored cells represent the correlation coefficient between the module and SDV content, with darker shades of red reflecting a stronger positive correlation. c Expression heatmaps and consensus profiles of co-expressed genes in the MEmagenta module. The upper section displays a clustered heatmap of genes within the module (red and green represent high and low expression, respectively), while the lower section represents the expression pattern of the module eigengene across different samples. d Hub gene network (top 150 genes) in the MEmagenta module visualized by Cytoscape (v 3.10.2). The red solid points indicate the key hub genes. e Phylogenetic tree of HTWRKY41, HTWRKY53 and AtWRKY family (Arabidopsis thaliana) proteins. f Multiple alignment of HTWRKY41 and HTWRKY53 proteins with AtWRKY41 and AtWRKY53 was performed using DNAMAN (v 10). The sequence in red represents the conserved heptapeptide WRKYGQK domain, and the sequence in blue represents the C2-HC (C-X7-C-X23-H-X-C)-type zinc finger protein domain. Black, pink, and blue colors indicate 100%, 75%, and 50% sequence consistency, respectively. g Subcellular localization of the HTWRKY53 protein in tobacco protoplast. GFP: green fluorescent protein; mKATE: nuclear marker; Cy5: chloroplast; Bright: bright field; Merged: combined mKATE and GFP and bright field; Bars, 10 μm. h Transcriptional capacity of hub transcription factor HTWRKY53 in tobacco leaves. Data are presented as mean ± standard deviations (SD) from three biological replicates. BD-62SK vector and BD-62SK-VP16 are used as negative and positive controls, respectively. Significant differences compared to the empty BD-62SK vector are marked with asterisks above the bars (**P-value < 0.01).
In the magenta module, HTWRKY41 (HT24808), HTWRKY53 (HT00049), and HTHSP90 (HT02416) were recognized as the key hub genes with highly connected nodes in the network (Fig. 8d). Six heat shock proteins (HSP) genes, including four HSP20 (HT01061, HT01063, HT13281, and HT17322), one HSP70 (HT25660), and one HSP90 (HT02416) were also found in magenta module (Supplementary Data 13). Most of them (5/6) had high expression levels and peaked at F3 stage (Supplementary Fig. 18). Meanwhile, three hub genes, TTL3 (HT19096), GRAS (HT30595), and R1A10 (HT29050), were identified in the purple module (Supplementary Fig. 19). These six hub genes were associated with “plant-pathogen interaction”, “protein processing in endoplasmic reticulum” and “plant hormone signal transduction” pathways (Supplementary Table 13). Notably, in the magenta module, the expression trends of HTWRKY41 and HTWRKY53 were similar to the flower scent emission during the flowering period (Supplementary Fig. 20). The phylogenetic analysis showed that they were clustered in the group III WRKY family, along with AtWRKY41 and AtWRKY53 (Fig. 8e). The multiple protein sequence alignment revealed that both HTWRKY41 and HTWRKY53 contained a highly conserved heptapeptide WRKYGQK domain and a C2-HC (C-X7-C-X23-H-X-C)-type zinc finger protein domain37 (Fig. 8f). Given the central role of HTWRKY53 in the gene network and higher expression level throughout the flowering (Fig. 8d and Supplementary Fig. 20), it was selected for further functional characterization. The HTWRKY53-GFP fusion vector and the GFP empty vector were independently introduced into tobacco (Nicotiana benthamiana) protoplasts, respectively. The HTWRKY53-GFP signal predominantly localized in the nucleus, whereas the GFP empty vector displayed signals in both the cytosol and nucleus (Fig. 8g). This confirms that HTWRKY53 is localized in the nuclear, aligning with its function as a transcription factor. The dual-luciferase assay results showed that the LUC/REN ratio of BD-62SK-HTWRKY53 was markedly reduced compared to the BD-62SK (negative control) and BD-62SK-VP16 (positive control) (Fig. 8h). This indicated that HTWRKY53 was a potential nucleus-localized transcriptional repressor.
Quantitative analysis of differentially expressed aroma-related genes
Based on transcriptomic and metabolomic data, six structural genes related to floral volatile compounds were selected to perform qRT-PCR analysis. Overall, the relative expression trend of candidate genes measured with quantitative real-Time PCR (qRT-PCR) was similar to the RNA-Seq results across various flowering periods (Fig. 9). Of the genes detected, five were highly expressed at F2 or F3, with the exception of HTDXR. Hence, the transcriptome data could accurately reflect the expression of terpene and phenylpropanoid biosynthesis-related genes in “Hutou” jasmine.

The relative expression levels of six scent-related genes were calculated using the 2-ΔΔCt method; JsActin2 served as an internal reference gene. Error bars indicate the standard deviation of three independent replicates.
Discussion
Metabolomic insights into “Hutou” jasmine fragrance
Jasmine, celebrated for its fragrant blossoms, has been extensively studied for diverse aroma compounds, particularly in SP and DP jasmine varieties38. The “Hutou” jasmine (MP) is appreciated in ornamental floriculture for its distinctive white overlapping petals and captivating fragrance. However, the floral composition and underlying mechanisms of aroma production in this variety, remain largely unknown. In this study, we presented a near-complete T2T genome of “Hutou” jasmine, providing an essential resource for floral scent research and applications in fragrance-based industries. The HS-SPME coupled with GC-MS was utilized to collect and measure VOCs from five flowering stages, unveiling a total of 912 volatile metabolites significantly more than previously reported in SP and DP varieties (Supplementary Data 2). Based on their chemical structures, these compounds can be categorized into 16 groups, which collectively contribute to the intricate metabolism of flower scent (Fig. 2d). Terpenoids and esters were the two major floral scent classes (40.35%) detected in “Hutou” jasmine flowers.
Aromatic substances are swiftly synthesized and emitted during the night-time blossoming of jasmine flowers, with the fragrance lasting for approximately one day39. In order to further investigate the compounds that shape the aroma profile of “Hutou” jasmine flowers, a screening of SDVs with a relative abundance percentage greater than 1% was performed in F1 vs. F2 and F1 vs. F3 comparisons (Supplementary Data 5). A total of sixteen SDVs were identified, and their concentrations increased as the flowering progressed, peaking at the F2 or F3 stages (Fig. 3a). Among these, eleven volatiles were scentless, even though two terpenoids were present at significantly high concentrations (Fig. 3b). These non-odorous volatiles did not directly contribute to the floral aroma, but may influence other physiological processes, such as plant-insect interactions. Among the scented volatiles, α-farnesene, known for its sweet and fruity odor, is a major volatile constituent of jasmine flowers39 (Fig. 3b). Three esters including pentanoic acid 1-ethenyl-1,5-dimethyl-4-hexenyl ester, (Z)-3-hexen-1-ol benzoate and geranyl isobutyrate contributed sweet, fruity, aromatic, and leafy notes to the overall scent (Fig. 3c). Also, benzyl alcohol added a mild and pleasant fragrance (Fig. 3d). These five scented compounds likely play a crucial role in shaping the distinct aroma characteristics of the “Hutou” jasmine flowers, offering potential for the development of diverse aromatic products across the essential oil, fragrance, and cosmetics sectors. Importantly, our metabolomics profiling further revealed that “Hutou” jasmine flowers contain several floral aroma compounds that are key contributors to the fragrance profile of jasmine tea, such as α-farnesene, methyl salicylate, and benzyl acetate (Figs. 3b and 6c)20,40. For example, benzyl acetate, which gives off a fruity and jasmine-like aroma, is especially notable for its role in imparting fragrance to jasmine tea41. The presence of these compounds opens new perspectives for material selection in the jasmine-scented tea industry.
Mechanisms Underlying Increased Sesquiterpene Biosynthesis
To unravel the molecular mechanisms underlying the release of aromatic compounds in “Hutou” jasmine flower, it is necessary to identify and analyze the biosynthesis-related genes that regulate the production of flower scent volatiles. Based on the genome annotation and transcriptome data, critical genes involved in terpene synthesis were determined in “Hutou” jasmine genome. The concentration of terpenoid compounds in plants is regulated not only by the expression pattern of structural genes, but also by the rate-limiting reactions over which they catalyze42,43. In this study, the gene expression levels in the MVA pathway were comparatively higher than those in the MEP pathway, which aligned with the elevated content of major sesquiterpenes in the floral scent compounds (Fig. 5a, b). Five HMGR homologous genes were observed in the “Hutou” jasmine genome, and four of these copies were highly expressed at F2 or F3 stages (Fig. 5a). The peak emission of sesquiterpenes occurred at F3, whereas the highest expression levels of HMGR3 and HMGR4 were found at F2 (Fig. 5a, b). This indicated that the scent-related genes were initially expressed, and then the subsequent metabolites were produced in response to the physiological requirements of plant, in line with earlier research33,44. Similar to the HMGRs, the expression profiles of AACT1, HMGS2, MVD, IDI, and FPPS reached their peaks at the F2 stage (Fig. 5a), providing additional evidence that elevated expression of genes preceded the increased biosynthesis of downstream compounds. Consistent with the recent studies20,45, the expression profiles of HMGSs, HMGRs and FPPS during five flowering stages closely mirrored the emission pattern of α-farnesene (Figs. 3a and 5a), emphasizing their crucial involvement in the biosynthesis of this key floral component. In the MEP pathway, DXS3 gene exhibited the highest expression among the four homologs (Fig. 5a), suggesting its potential utility in genetic engineering to augment specific monoterpenoids. The preponderance of TPS-a subfamily (Supplementary Fig. 13), which specializes in the production and diversification of sesquiterpenes46, likely facilitates the synthesis and accumulation of sesquiterpenes in “Hutou” jasmine flowers. The peak expression levels of HTTPS3, HTTPS4, HTTPS14, and HTTPS39 were prominently observed during the F2 and F3 flowering stages, displaying a robust correlation with the emission profiles of floral terpene compounds (Fig. 5b and Supplementary Fig. 14). Additional research is required to explore how these TPSs contribute to the terpenoid fragrance of “Hutou” jasmine.
Frequent segmental and tandem duplication events have resulted in increased gene copy numbers, thereby exerting the dosage effect47. Notably, the rate-limiting enzyme genes HMGS (HMGS1-HMGS2) and HMGR (HMGR3-HMGR4) in the MVA pathway both originated from these segmental duplication events (Fig. 5c). This gene amplification, along with elevated expression of specific genes such as HMGS2, HMGR3 and FPPS in the MVA pathway has the potential to enhance sesquiterpene synthesis, leading to higher levels of major sesquiterpenes compared to monoterpenes (Fig. 5b).
Dynamic gene regulation of phenylpropanoids/benzenoids biosynthesis
A comprehensive set of 42 candidate genes involved in phenylpropanoids/benzenoids biosynthesis was discerned, including the rate-limiting gene PALs and terminal enzyme genes such as BEATs, BSMTs, and BPBTs (Supplementary Data 10). The conversion of L-phenylalanine to trans-cinnamic is catalyzed by PAL, which facilitates the biosynthesis of aromatic compounds in plants48. In “Hutou” genome, four PALs were detected, with PAL3 and PAL4 showing elevated expression levels during the F2 and F3 periods, likely regulating downstream compound production. (Fig. 6a). The synthesis of benzyl acetate is facilitated by the enzyme BEAT, which is closely related to the emission profile of volatile benzenoids. Recent studies have shown that the ectopic expression of JsBEAT1 and JsBEAT2 led to enhanced biosynthesis of benzyl benzoate and benzyl acetate49. Furthermore, through the overexpression of BEAT genes from Clarkia breweri and the provision of alcoholic substrates, lisianthus plants have been demonstrated to produce higher levels of benzyl acetate, suggesting potential applications for the development of novel fragrances50. Our study uncovered three copies of the BEAT gene in the “Hutou” jasmine genome and particularly, the expression pattern of BEAT3 paralleled the emission of benzyl acetate (Fig. 6a, c). Methyl salicylate and methyl benzoate, compounds synthesized through enzymatic reactions catalyzed by benzenoid carboxyl methyltransferases, function as plant volatiles that are instrumental in their potential participation in pollinator attraction and pathogen response51. Our study confirmed their presence in “Hutou” jasmine flowers, in concert with the observations reported in SP and DP jasmines19 (Fig. 6c). The higher content of methyl salicylate may play a crucial role in response to pathogen attack during the growth of “Hutou” jasmine plant. This observation also indicates significant diversity in the emission of these methyl esters, consistent with previous studies52,53, underscoring the need for further research on benzenoid carboxyl methyltransferases. Benzyl benzoate, synthesized by BPBT, was also identified in the floral compounds of “Hutou” jasmine flowers (Fig. 6c and Supplementary Data 2). In the present study, three BPBTs were discovered, with BPBT2 and BPBT3 showing high expression at the F2 stage, which coincided with the release pattern of benzyl benzoate (Fig. 6a, c). Generally, the integrated metabolomic and transcriptomic analysis revealed that the change trends of benzyl benzoate, methyl benzoate, and benzyl acetate closely followed the expression profiles of their corresponding structural genes during various blooming times (Fig. 6a, c). This suggests that the expression of upstream genes may contribute to the accumulation of final products54. Segmental duplication and tandem duplication events in the “Hutou” jasmine genome have likely played a critical role in the expansion of fragrance-related genes, optimizing their function in the production of a variety of aromatic compounds3,55.
Hormonal regulation and transcription factors in foral scent synthesis
Time-series transcriptome analyses revealed that the “alpha-Linolenic acid metabolism” and “MAPK signaling pathway-plant” pathways were consistently enriched throughout the entire flowering period (Supplementary Fig. 9). Jasmonates (JAs) have been demonstrated to act as signaling molecules that integrate biotic and abiotic stresses with endogenous responses, thereby enhancing plant persistence and survival56. The biosynthesis of JAs is initiated by alpha-linolenic acid as a precursor57. Given that the “Hutou” jasmine is susceptible to disease infection and flower malformation, we hypothesized that the plant may utilize alpha-linolenic acid to synthesize JAs, there enhancing its stress resistance during the blooming. The MAPK signaling cascade, a critical pathway for signal transduction, amplifies signals via phosphorylation reactions in response to external stimuli, transmitting them to downstream receptors to adapt to changes in the external environment58,59. This pathway can promote the transition of plants from vegetative growth to reproductive growth and plays an important role in regulating the development of floral organs60,61,62. In summary, these findings illustrate that floral scent production is closely linked to plant hormone synthesis and MAPK signaling.
The synthesis of floral scent is complex and tightly regulated, where the orchestration of gene transcription by TFs serves as a fundamental component within this process63. In this study, the correlation analysis of terpene biosynthesis-related genes and TFs unveiled that the members of the ERF, MYB, WRKY, and bHLH transcription factor families may positively regulate the biosynthesis of terpene metabolites (PCC > 0.9) (Fig. 7g). These TFs have been recognized as key regulators in controlling the expression of structural genes involved in the biosynthesis of aroma compounds64,65,66,67. For example, it has been demonstrated that ERF61 binds to the CCD4 promoter and stimulates CCD4 expression, thereby regulating β-ionone synthesis in sweet osmanthus64. In Catharanthus roseus, CrWRKY1 was identified as a positive regulator of the terpenoid indole alkaloid biosynthesis65. Overexpression of CpMYC2 and CpbHLH13 in Chimonanthus praecox significantly increased floral volatile linalool and β-caryophyllene, respectively66. Furthermore, the MYB-related TF JsLHY, can bind to the promoters of six fragrance-related structural genes and enhance their expression directly in jasmine flowers67. In this study, WGCNA analysis identified HTWRKY41, HTWRKY53 and HTHSP90 as hub genes in the magenta module (Fig. 8d). The expression pattern of HTWRKY41 and HTWRKY53 closely mirror the floral scent emission during the flowering process (Supplementary Fig. 20). Subcellular localization and dual-luciferase assay further confirmed that HTWRKY53 was a potential nuclear-localized transcription factor with transcriptional repression activity (Fig. 8g, h). The temporal variation in HTWRKY53 expression levels may indicate its dynamic regulatory role in floral scent synthesis throughout the flowering process, ensuring the controlled release of scents. Additional experiments may further accurately identify the specific target genes regulated by HTWRKY53 and elucidate their relationship to floral scent synthesis.
Temperature is crucial in regulating the blooming and fragrance emission of jasmine flowers68. Typically, a high abundance of jasmine flower volatiles is produced from July through September, when daytime temperatures frequently exceed 30 °C69. However, strong heat stress may have adverse effects on the growth and flowering regulation of jasmine. Previous research has demonstrated that the elevated expression of HSP genes may play an important role in the heat resistance of post-harvest jasmine flowers and other plant species55,70. In magenta module, five HSP family members, including HT02416, HT25660, HT17322, HT01063, and HT01061, showed high expression levels during the flowering period (Supplementary Fig. 18). These HSPs protein likely function as molecular chaperones, preventing protein misfolding and aggregation71,72.
Conclusion
In summary, we have constructed a nearly complete T2T multi-petal jasmine genome. The metabolome analyses uncovered that 16 primary SDVs showed significant variation as flowering progressed, with five scented compounds, particularly α-farnesene and pentanoic acid 1-ethenyl-1,5-dimethyl-4-hexenyl ester, likely responsible for the mellow and elegant aroma of “Hutou” jasmine flowers. As revealed by WGCNA, HTWRKY41, HTWRKY53 and HTHSP90 in the magenta module are positively correlated with these 16 primary volatiles. The time-course transcriptomic analysis highlighted that the floral scent synthesis is orchestrated not only by plant hormones but also by essential transcription factors, such as ERF, MYB, WRKY, and bHLH. It is noteworthy that the expression patterns of structural genes involved in the biosynthesis of terpenoid and phenylpropanoid compounds play a pivotal role in modulating the relative content of subsequent aromatic compounds. The duplication events appeared to be the primary driving force for the generation and functional diversification of these fragrance-related genes. This study represents the first comprehensive analysis of floral fragrance chemistry and its intricate molecular mechanisms in “Hutou” jasmine (MP) flowers. Our findings lay a solid foundation for the practical utilization of fragrance and the genetic improvement in horticulture for this ornamental variety.
Materials and Methods
Plant materials
The multi-petal cultivar “Hutou” (Jasminum sambac var. Trifoliatum) was conserved at the Botanical Garden of Minrong Tea Industry Co. Ltd, Fuzhou, China, under the specimen voucher number GDHTML3. Three-month-old rooted cuttings of this specimen were kindly provided by the chief scientific officer of Minrong Tea Industry and transplanted into plastic flowerpots with a top diameter of 27.5 cm and a depth of 31 cm in a greenhouse of Fujian Normal University (26°01'36.5” N, 119°12'33.5” E), Fuzhou, Fujian Province, China. The soil moisture and plants were checked daily and watered as needed. Yong leaves of “Hutou” jasmine were collected to extract DNA for Illumina, PacBio, and Hi-C sequencing. Samples from each of the various blooming stages of the jasmine flower (designated as F1-F5, Fig. 4a), as well as from tissues of roots, mature stems, young and mature leaves were collected for RNA extraction and RNA sequencing (RNA-seq). All the samples were immediately frozen in liquid nitrogen for at least 15 min, and then stored at -80 °C in the lab freezer prior to RNA isolation. Three biological replicates were collected for each tissue.
DNA extraction, library construction and sequencing
The stored leaf samples were ground with liquid nitrogen, and high-quality genomic DNA was extracted using the Dneasy Plant Mini Kit (Qiagen, USA) following the manufacturer’s instructions. The BluePinppin system was employed for 20-kb size DNA fragment selection. The final libraries were sequenced on the PacBio Sequel II platform (Pacific Biosciences, USA) in Circular Consensus Sequencing (CCS) mode. Young leaves tissues taken from the same individual were used for Hi-C library construction. The DNA was digested by the restriction enzyme HindIII, and fragments were sheared into ~350 bp, and subsequently sequenced using the Illumina NovaSeq platform (Illumina, USA) in paired-end (PE) 150 bp mode. For genomic DNA resequencing, a paired-end library with an insert size of 300~350 bp was constructed and sequenced on the Illumina NovaSeq platform.
Genome size estimation and assembly
The chromosome number of the “Hutou” jasmine genome was determined using root tip tissues of cultured “Hutou” jasmine plants, following a previously established method73. The root tip cells were digested with an enzyme mixture containing cellulase and pectolyase, and subsequently macerated with a dissecting needle. The resulting cell suspension was examined under an optical microscope to obtain suitable chromosome preparations. For fluorescence in situ hybridization (FISH), the 5S and 45S rDNA were individually labeled with Dig-dUTP and Biotin-dUTP to mark nuclear locations10,74. After hybridization, the slides were examined and photographed under a fluorescence microscope.
The raw short reads generated from the Illumina platform were filtered by fastp (v 0.19.3)75 to eliminate low-quality reads and adapters. The clean PacBio HiFi reads were obtained after being filtered and corrected using pbccs (v4.02) (https://github.com/PacificBiosciences/ccs) (Supplementary Table 1). The k-mer profile (17-mers) of quality-controlled Illumina short reads was measured using Jellyfish (v2.2.10)76, and the frequency distribution was plotted by genomescope (v2.0)77. The PacBio HiFi long reads of “Hutou” genome were processed to Hifiasm (v0.16.1)22 with the default parameters to generate the contig-level jasmine assembly. Redundant sequences in the diploid draft genome were removed using a whole-genome alignment strategy and read-depth analysis with the purge_dups tool (v1.25)78. For chromosome-level assembly, the raw Hi-C data were primarily filtered using Hi-C-Pro (v2.11.4)79, and then the clean reads were aligned to the purged “Hutou” genome using BWA (v0.7.17)80. The ALLHiC (v0.9.13)23 pipeline was used to cluster, order and orientate the sequences based on the HindIII restriction sites. The Juicebox (v1.11.08)81 was utilized to manually rearrange misplaced contigs by identifying abrupt long-range contacts, and the contact map was visualized by HapHiC (v1.0.6)82. The telomeric repeats were identified by the Telomere Identification toolkit (TIDK v0.2.63) (available at https://github.com/tolkit/telomeric-identifier). The Circos83 (v0.69.8) was used to visualize the chromosome landscape of gene density, GC content, and distribution of repetitive elements.
Genome assembly evaluation and annotation
The PacBio HiFi reads, DNA short reads, and RNA-seq reads were mapped to the genome assembly to assess assembly accuracy using minimap284 (v2.22), BWA, and HISAT285 (v2.1.0), respectively. The completeness of the final “Hutou” genome and seven published jasmine genomes was assessed using Benchmarking Universal Single-Copy Orthologs (BUSCO v5.3.0)86 with the default gene predictor MetaEuk against embryophyta_odb10 dataset. The Long Terminal Repeats (LTR)24 sequences were firstly detected by LTR_FINDER_parallel87 and LTRharvest88 with default parameters, and then results were merged to calculate the scores with LTR_retriever89. The Merqury90 (v1.3) software was used to assess the consensus quality value (QV) (K = 20). The RepeatModeler91 package (v1.0.8) was initially employed for de novo repeat prediction. Next, the de novo repeat library were merged together to annotate the repetitive sequences using RepeatMasker92 (v4.07), and TEclass93 (v2.1.3) was further used to classify the unknown TEs. In addition, the tandem repeats were further identified by the Tandem Repeat Finder (TRF)94 package (v0.9).
The GETA (v2.4.12) pipeline (https://github.com/chenlianfu/geta) was employed to integrate annotations from ab initio, transcriptome-based, and homolog-based gene predictions. The protein sequences from Arabidopsis thaliana, Oryza sativa, Vitis vinifera, Solanum lycopersicum, Medicago sativa, Olea europaea, and Camellia sinensis were downloaded from Phytozome database95 and then merged into a single homologous protein file as the query sequences. The final gene annotation results were generated after being filtered by the GetaFilter96 (https://github.com/Datapotumas/GetaFilter). The gene annotation completeness of the final predicted protein-coding genes was evaluated by BUSCO with 1617 genes from embryophyta_odb10 orthologous database. All protein-coding genes were functionally annotated through sequence similarity searches using BLASTP, with an E-value threshold of 1e-5 against multiple public protein databases: NCBI non-redundant (Nr) protein database, Gene Ontology (GO) terms, Kyoto Encyclopedia of Genes and Genomes (KEGG), InterProScan, Swiss-Prot, Clusters of Orthologous Groups (COGs) and EggNOG. The protein domains were annotated against InterPro database (v4.8) and Pfam database. The transfer RNA gene (tRNAs), ribosomal RNA genes (rRNAs), small nuclear RNA genes (snRNAs), and microRNAs (miRNAs) were predicted by tRNAscan-SE97 (v2.0.9), RNAmmer98 (v1.2), and INFERNAL99 (v1.1.2), respectively.
RNA extraction and library construction
Total RNAs were isolated from different samples with three biological replicates using the RNAprep Pure Plant Plus Kit (Tiangen Biotech, Beijing, China) and purified using RNeasy MiniElute Cleanup Kit (Qiagen, Hilden, Germany) following the manufacturer’s protocols. The purity, quantity, and quality of total RNAs were assessed on a NanoPhotometer spectrophotometer (IMPLEN, CA, USA) and an Agilent 5400 Bioanalyzer (Agilent, CA, USA), respectively. After quantity and quality determination, high-quality RNA samples (RIN ≥ 8.6, Supplementary Table 14) were further used for Illumina RNA sequencing library construction. A total of 24 cDNA libraries (including samples from flowers, stems, roots, and leaves) were constructed using NEBNext UltraTM RNA Library Prep Kit for Illumina, and subsequently sequenced on the Illumina NovaSeq platform (Illumina Inc., CA, USA) in paired-end (PE) 150 nt mode by the Novogene Company (Beijing, China).
RNA-seq data mapping and transcriptomic analyses
Prior to downstream transcriptome analysis, raw Illumina data were initially subjected to quality checks to obtain clean reads. The empty reads, reads with low-quality bases (Phred quality score (Q) < 20), and Illumina adapters were filtered out through fastp (v0.19.3). Quality reports for the raw and clean RNA-seq data are shown in Supplementary Data 6. Approximately 788.41 million clean PE reads, totaling 118.25 Gb transcriptome data were generated for five flowering stages.
The quality-controlled PE 150-bp reads were aligned to the “Hutou” reference genome using HISAT2 (v2.1.0) with parameters: --new-summary -p -x -1 -2 -S. We mapped the reads against the reference genome for each biological replicate independently. The output SAM (Sequence Alignments/Map) files were converted into BAM (Binary Alignment/Map) files, sorted by Samtools (v1.9)100, and subsequently fed into FeatureCounts (v1.6.2)101 for transcript quantification. The FPKM (Fragments Per Kilobase of transcript per Million fragments mapped) was calculated and the differentially expressed genes (DEGs) were identified using the DESeq2 (v1.22.1)102 (|log2FC | >1, padj<0.05, and FDR < 0.05). Venn diagram was generated by the VennDiagram R package to visualize the overlap of genes with differential transcript abundance among comparisons. Sample correlation analysis and principal component analysis (PCA) were performed by corrplot (v0.84) and base (v3.5.1) packages in the R software. Gene ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG) enrichment analyses (P < 0.05) were conducted by clusterProfiler (v4.6.1) R package103.
Analysis of volatile compounds by a widely-targeted metabolomics method
The headspace solid-phase microextraction (HS-SPME) technique was applied to extract floral compounds from five flowering stages, and each assay was conducted in triplicate. The samples were ground in liquid nitrogen, and 500 mg of the powder was immediately placed into a 20 mL head-space vial (Agilent, Palo Alto, CA, USA), containing a 10 μL of internal standard solution (50 μg/mL, 3-Hexanone) and a saturated NaCl solution. The HS-SPME procedure was carried out under the following conditions: each vial was shaken at 60 °C for 5 min and then a 120 μm divinylbenzene/carboxen/polydimethylsilioxan fiber was utilized for volatile enrichment for 15 min at 60 °C. The samples were resolved at 250 °C for 5 min and then separated and identified using gas chromatography-mass spectrometry (GC-MS) (Agilent, 8890-7000D, USA). The GC-MS analysis was completed under the following conditions104: Chromatographic analysis was carried out using an Agilent DB-5MS capillary column (30 m × 0.25 mm × 0.25 μm, Folsom, CA, USA) in splitless mode. High-purity helium (purity>99.999%) was used as the carrier gas at a constant flow rate of 1.2 mL/min. The injector temperature was maintained at 250 °C with a solvent delay of 3.5 minutes. The oven temperature program was as follows: the initial temperature was set at 40 °C and held for 3.5 minutes, then ramped up at 10 °C/min to 100 °C, followed by an increase at 7 °C/min to 180 °C, and finally increased at 25 °C/min to 280 °C, where it was held for 5 minutes. Mass spectra were recorded in electron impact (EI) ionization mode at 70 eV. The ion source temperature was set to 230 °C, and the interface temperature was maintained at 280 °C. Data acquisition was performed in selected ion monitoring (SIM) mode. The reproducibility of the analysis process was monitored by using a quality control (QC) sample (prepared by mixing sample extracts) every 10 test samples. Overlapping analysis of the total ion chromatograms (TIC) of the mixed samples was used to assess the instrumental stability for GC-MS analysis. The volatile substances were recognized by comparing the mass spectra with the MWGC database (http://www.metware.cn/) and linear retention index. Then, the relative content (μg/kg) of specific volatile elements was calculated using 3-Hexanone as an internal standard according to the following formula: Ci = (Ai /Ais * mis)/mi. Where Ci represents the relative content of the target compound (μg/kg); Ai and Ais are the peak areas of the compound and internal standard, respectively; mi and mis are the masses of the volatile compound and internal standard (μg), respectively18. The sum of the volatile compounds detected was considered to be the total volatile content. The relative content percentage was determined through the ratio of the relative content to the total content. The structural formulas of VOCs were available on the Guidechem website (https://www.guidechem.com/). For odor characteristics, preferential access can be obtained through the Guidechem website initially, followed by consultation of the PubChem database (https://pubchem.ncbi.nlm.nih.gov/) if needed. After unit variance scaling (UV), hierarchical cluster analysis (HCA) was performed using the R package ComplexHeatmap (v2.8.0). The orthogonal projections to latent structure-discriminant analysis (OPLS-DA) were employed to screen the significantly differential volatiles (SDVs) between groups applying the MetaboAnalystR (v1.0.1) package in R, with the criteria of variable importance in projection (VIP) score≥1.0 and Fold change≥1.2 or ≤0.67.
Analysis of protein-coding genes and transcription factors related to floral aroma synthesis
Based on the homologous protein sequences of flower fragrance-related genes downloaded from the National Center for Biotechnology Information (NCBI) database, the protein-coding genes responsible for the terpene and phenylpropanoids biosynthetic pathway in “Hutou” jasmine were determined through a local blast search and KEGG annotation against the jasmine genome using filtered parameters (E-value < 10-5, max_target_seqs 5, identity>40%, and coverage>30%). The synteny and collinearity analysis of these genes were performed using MCScanX105. For terpene synthase genes (TPSs), the hidden Markov model (HMM) of the TPS domains (PF01397 and PF03936) was downloaded from the Pfam database (http://pfam.xfam.org/) to predict TPS genes using HMMER (v3.1)106 with an E-value threshold of <1e-5. The Arabidopsis thaliana TPS proteins (https://www.arabidopsis.org/) were utilized as query sequences by BLASTP (2.15.0 + )107 searches (E-value < 1e-5, identity>40%, and coverage>30%). Then, the candidate TPSs identified from both approaches were combined and further searched against the NCBI-CDD database (https://www.ncbi.nlm.nih.gov/cdd/) and SMART to verify the presence of the TPS domain. Multiple sequence alignments between the TPS protein sequences of “Hutou” jasmine, Arabidopsis thaliana, Osmanthus fragrans, and Fraxinus excelsior were performed using Muscle (v3.8.1)108, and the phylogenetic tree was constructed using RAxML (v8.2.12)109 by maximum likelihood method. Sequences of DXS proteins of closely related species were obtained from the NCBI database. Finally, the phylogenetic tree was visualized using iTOL110 (https://itol.embl.de/). Plant transcription factors were predicted using the iTAK pipeline111 and classified based on the comprehensive database. The heat map was created by TBtools (v1.113)112 with the “Log2 Scale” option, based on the gene expression levels in floral and other tissues.
Transcript-metabolite network analysis
The K-means clustering method was performed using the FPKM values of DEGs across groups of samples by the base package (v3.5.1) in R software after standardization and centralization. To explore the potential correlation between terpenoid volatiles and DEGs, as well as between DEGs and differentially expressed TFs within subclass 4, 6, and 9, the Pearson Correlation Coefficient (PCC) was utilized by base and Hmisc (v 4.4.0) packages, with correlation coefficients >0.9 or <-0.9 and P-value < 0.05 as the selection criteria. The resulting transcript-metabolite network was visualized using Cytoscape (v3.10.2).
Weighted gene co-expression network analysis
Weighted gene co-expression network analysis (WGCNA) of the transcriptome data was performed using the R package WGCNA (v4.02)113 to identify co-expressed genes. Genes with FPKM > 1 were chosen for constructing the co-expression network. The minimum module size and merge cut height were set to 50 and 0.25, respectively, while other parameters remained at their default settings. Each module was characterized by the module eigengene (ME), and the content data of 16 primary SDVs were utilized as phenotypic traits. The association between ME and floral volatiles was analyzed based on PCC analysis. The candidate hub genes in the magenta and purple modules were identified using the CytoHubba function embedded in Cytoscape. The top 150 associated genes were selected based on their weight values to construct hub gene subnetworks. The visualization of gene co-expression networks was constructed using Cytoscape (v3.10.2). The AtWRKY protein sequences were obtained from TAIR (http://www.arabidopsis.org/) and the phylogenetic tree was constructed using RAxML. The multiple alignments of WRKY protein sequences were conducted using DNAMAN (v10). The heat maps of hub genes in the magenta module were generated by TBtools with the “Log2 Scale” option.
Validation of RNA-seq by quantitative real-time PCR
To verify the reliability of RNA-seq data, total RNAs were extracted from samples collected at five flowering periods (F1-F5) and then cDNAs were synthesized using the GoScriptTM Reverse Transcription Mix, Oligo (dT) (Promega, USA) for quantitative real-Time PCR (qRT-PCR). JsActin2114 was used as an internal control gene and the primer sequences used for qRT-PCR analysis were designed using Primer Premier (v5.0) (Supplementary Table 15). The qRT-PCR reaction was completed in three replicates using ChamQ SYBR qPCR Master Mix (Vazyme, Nanjing, China) on a CFX Connect device (Bio-Rad, USA). The PCR conditions were as follows: 95 °C for 2 min, 39 cycles of 95 °C for 15 s, and 60 °C for 1 min. The relative expression level was calculated according to the 2-ΔΔCt method115.
Analysis of HTWRKY53 subcellular localization
Total RNAs were extracted from various flowering samples using RNAprep Pure Plant Plus Kit (Tiangen Biotech, Beijing, China), following the manufacturer’s protocol. The full-length coding sequence of HTWRKY53, excluding the termination codon, was amplified using gene-specific primers (Supplementary Table 15). The purified PCR product was then fused with the upstream GFP (green fluorescent protein) gene to construct the pBWA(V)H2STMV Ω-HTWRKY53-GFP fusion vector, with the 35S::GFP empty vector as a control. NLS (nuclear localization signal)-mKATE served as the nuclear localization marker. The HTWRKY53-GFP and NLS-mKATE vectors were co-transformed into the tobacco protoplasts, which were then gently mixed with 110 μL PEG4000 solution. After incubation in darkness at 25 °C for 18–24 h, the green fluorescence was visualized by confocal laser scanning microscopy (Nikon C2-ER), and images were captured.
Dual-luciferase reported experiment
The coding sequence of HTWRKY53 was inserted into pGreenII 62-SK vector, which contains GAL4 DNA-binding sites, to function as an effector. The reporter construct was generated using the pGreenII 0800-LUC reporter vector, which included firefly luciferase (LUC), five GAL DNA-binding sites and 35S::renilla luciferase (REN) as internal control116. The empty pGreenII 62SK vector and pGreenII 62SK-VP16 were used as negative and positive controls, respectively. Both the effector and reporter plasmids were transferred into the Agrobacterium tumefaciens GV3101 strain and subsequently co-infiltrated into tobacco leaves117. Samples were collected 48 hours after transient expression. The transcriptional ability of HTWRKY53 was assessed by measuring the LUC/REN ratio using a Dual Luciferase Reporter Gene Assay Kit (Vazyme, Nanjing, China) on a Full-Featured Microplate Reader (PerkinElmer, USA). Three biological replicates were performed for each pair.
Statistics and reproducibility
For genomic sequencing (DNA), transcriptome sequencing (RNA-seq), and metabolite profiling, three biological replicates were used for each analysis. DEGs were identified using the DESeq2 with criteria of |log2FC | >1, padj<0.05, and FDR < 0.05. Sample correlation and PCA were performed using the corrplot and base R packages. GO and KEGG enrichment analyses (P < 0.05) were conducted by clusterProfiler. To ensure the reproducibility of the GC-MS analysis, a quality control sample was included after every 10 test samples. The instrumental stability was assessed by overlapping analysis of the TIC of the mixed samples. OPLS-DA was applied to screen the SDVs between groups, using a VIP score≥1.0 and a fold change ≥1.2 or ≤0.67 as the selection criteria. The relative content of terpenes and phenylpropanoids/benzenoids is presented as means ± SD, with statistical significance determined by t-tests. For transcript-metabolite network analysis, correlation coefficients >0.9 or <-0.9 and P-value < 0.05 were used as selection thresholds. qRT-PCR reactions were performed in triplicate, and the dual-luciferase reporter assay was conducted with three biological replicates, with statistical significance evaluated by t-tests.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
The Illumina RNA-sequencing raw data of “Hutou” jasmine (J. sambac var. Trifoliatum, MP) transcriptome across different tissues are available in the Genome Sequence Archive (GSA) (https://ngdc.cncb.ac.cn/gsa/), part of the National Genomics Data Center (NGDC), under the accession number CRA012747. The “Hutou” jasmine genome are publicly available in the NGDC under project number PRJCA019962. The source data behind the graphs are available in the Supplementary Data. All other data are available from the corresponding authors on reasonable request.
Code availability
The GetaFilter used in this study is available at GitHub (https://github.com/Datapotumas/GetaFilter).
References
Bera, P. et al. Inter-specific variation in headspace scent volatiles composition of four commercially cultivated jasmine flowers. Nat. Prod. Res. 29, 1328–1335 (2015).
Hongratanaworakit, T. Stimulating Effect of Aromatherapy Massage with Jasmine Oil. Nat. Prod. Commun. 5, 157–162 (2010).
Shang, J. Z. et al. The chromosome-level wintersweet (Chimonanthus praecox) genome provides insights into floral scent biosynthesis and flowering in winter. Genome Biol. 21, 28 (2020).
Deng, Y. M. et al. Morphological anatomy, sporogenesis and gametogenesis in flowering process of jasmine (Jasminum sambac. Aiton). Sci. Hortic. 198, 257–266 (2016).
Shen, J. X. et al. Differential contribution of jasmine floral volatiles to the aroma of scented green tea. J. Food Qual., 10, https://doi.org/10.1155/2017/5849501 (2017).
Ito, Y. et al. Identification of potent odorants in Chinese jasmine green tea scented with flowers of Jasminum sambac. J. Agric. Food Chem. 50, 4878–4884 (2002).
Wang, P. J. et al. Genomes of single- and double-petal jasmines (Jasminum sambac) provide insights into their divergence time and structural variations. Plant Biotechnol. J. 20, 1232–1234 (2022).
Deng, Y. M. et al. Identification of pre-fertilization reproductive barriers and the underlying cytological mechanism in crosses among three petal-types of Jasminum sambac and their relevance to phylogenetic relationships. Plos One 12, 19 (2017).
Deng, Y. et al. Ultrastructural abnormalities in pollen and anther wall development may lead to low pollen viability in jasmine (Jasminum sambac (L.) Aiton, Oleaceae). S. Afr. J. Bot. 114, 69–77 (2018).
Fang, J. et al. Cytoplasmic genomes of Jasminum sambac reveal divergent sub-mitogenomic conformations and a large nuclear chloroplast-derived insertion. BMC Plant Biol. 24, 861 (2024).
Schiestl, F. P. The evolution of floral scent and insect chemical communication. Ecol. Lett. 13, 643–656 (2010).
Junker, R. R. et al. Composition of epiphytic bacterial communities differs on petals and leaves. Plant Biol. 13, 918–924 (2011).
Mostafa, S. et al. Floral scents and fruit aromas: functions, compositions, biosynthesis, and regulation. Front. Plant Sci. 13, 23 (2022).
de Groot, A. C. et al. Essential Oils, Part VI: Sandalwood Oil, Ylang-Ylang Oil, and Jasmine Absolute. Dermatitis 28, 14–21 (2017).
Baldwin, I. T. Plant volatiles. Curr. Biol. 20, R392–R397 (2010).
Lichtenthaler, H. K. The 1-deoxy-D-xylulose-5-phosphate pathway of isoprenoid biosynthesis in plants. Annu. Rev. Plant Physiol. Plant Mol. Biol. 50, 47–65 (1999).
Qian, Y. C. et al. Completion of the cytosolic post-chorismate phenylalanine biosynthetic pathway in plants. Nat. Commun. 10, 15 (2019).
Zhou, C. et al. Integrated volatile metabolome, multi-flux full-length sequencing, and transcriptome analyses provide insights into the aroma formation of postharvest jasmine (Jasminum sambac) during flowering. Postharvest Biol. Technol. 183, https://doi.org/10.1016/j.postharvbio.2021.111726 (2022).
Wang, P. et al. High-resolution transcriptome and volatile assays provide insights into flower development and aroma formation in single- and double-petal jasmines (Jasminum sambac). Ind, Crops Prod. 189, https://doi.org/10.1016/j.indcrop.2022.115846 (2022).
Yu, Y. et al. Volatiles Emitted at Different Flowering Stages of Jasminum sambac and Expression of Genes Related to α-Farnesene Biosynthesis. Molecules 22, 13 (2017).
Xu, S. X. et al. A high-quality genome assembly of Jasminum sambac provides insight into floral trait formation and Oleaceae genome evolution. Mol. Ecol. Resour. 22, 724–739 (2022).
Cheng, H. Y. et al. Haplotype-resolved de novo assembly using phased assembly graphs with hifiasm. Nat. Methods 18, 170 (2021).
Zhang, X. T. et al. Assembly of allele-aware, chromosomal-scale autopolyploid genomes based on Hi-C data. Nat. Plants 5, 833–845 (2019).
Ou, S. J. et al. Assessing genome assembly quality using the LTR Assembly Index (LAI). Nucleic Acids Res. 46, 11 (2018).
Li, P. L. et al. CmDOF18 positively regulates salinity tolerance in Chrysanthemum morifolium by activating the oxidoreductase system. BMC Plant Biol. 24, 12 (2024).
Muhlemann, J. K. et al. Floral volatiles: from biosynthesis to function. Plant Cell Environ. 37, 1936–1949 (2014).
Liu, Y. et al. HMGR and CHS gene cloning, characterizations and tissue-specific expressions in Polygala tenuifolia Willd. Plos One 19, 36 (2024).
Saladié, M. et al. The 2-C-methylerythritol 4-phosphate pathway in melon is regulated by specialized isoforms for the first and last steps. J. Exp. Bot. 65, 5077–5092 (2014).
Phillips, M. A. et al. The plastidial MEP pathway: unified nomenclature and resources. Trends Plant Sci. 13, 619–623 (2008).
Rodríguez-Concepción, M. et al. Breaking new ground in the regulation of the early steps of plant isoprenoid biosynthesis. Curr. Opin. Plant Biol. 25, 17–22 (2015).
de Luna-Valdez, L. et al. Reassessing the evolution of the 1-deoxy-D-xylulose 5-phosphate synthase family suggests a possible novel function for the DXS class 3 proteins. Plant Sci. 310, 13 (2021).
Xu, J. et al. Identification and expression analysis of TPS family gene in Cannabis sativa L. Heliyon 10, 16 (2024).
Bera, P. et al. Enzymatic production and emission of floral scent volatiles in Jasminum sambac. Plant Sci. 256, 25–38 (2017).
Dexter, R. J. et al. Tissue-specific PhBPBT expression is differentially regulated in response to endogenous ethylene. J. Exp. Bot. 59, 609–618 (2008).
Boatright, J. et al. Understanding in Vivo Benzenoid Metabolism in Petunia Petal Tissue. Plant Physiol. 135, 1993–2011 (2004).
Chuang, Y. C. et al. Diurnal regulation of the floral scent emission by light and circadian rhythm in the Phalaenopsis orchids. Botan. Stud. 58, 9 (2017).
Javed, T. et al. WRKY transcription factors in plant defense. Trends Genet 39, 787–801 (2023).
Younis, A. et al. Supercritical carbon dioxide extraction and gas chromatography analysis of jasminum sambac essential oil. Pak. J. Bot. 43, 163–168 (2011).
Edris, A. E. et al. Isolation and characterization of the volatile aroma compounds from the concrete headspace and the absolute of Jasminum sambac (L.) Ait. (Oleaceae) flowers grown in Egypt. Eur. Food Res. Technol. 226, 621–626 (2008).
Hong, Y. P. et al. Aroma formation in single- and double-petal jasmines (Jasminum sambac) during flowering via volatile metabolome and transcriptome. Sci. Hortic. 333, 9 (2024).
Issa, M. Y. et al. Volatiles distribution in jasmine flowers taxa grown in Egypt and its commercial products as analyzed via solid-phase microextraction (SPME) coupled to chemometrics. Ind. Crops Prod. 144, 10 (2020).
Henry, L. K. et al. Contribution of isopentenyl phosphate to plant terpenoid metabolism. Nat. Plants 4, 721–729 (2018).
Vranová, E. et al. in Annual Review of Plant Biology, Vol. 64 Annual Review of Plant Biology (ed S. S. Merchant) 665–700 (2013).
Kondo, M. et al. Floral scent diversity is differently expressed in emitted and endogenous components in Petunia axillaris lines. Ann. Bot. 98, 1253–1259 (2006).
Chen, G. et al. The Jasmine (Jasminum sambac) genome provides insight into the biosynthesis of flower fragrances and Jasmonates. Genomics Proteom. Bioinform. 21, 127–149 (2023).
Chen, F. et al. The family of terpene synthases in plants: a mid-size family of genes for specialized metabolism that is highly diversified throughout the kingdom. Plant J. 66, 212–229 (2011).
Shang, J. Z. et al. Evolution of the biosynthetic pathways of terpene scent compounds in roses. Curr. Biol. 34, 23 (2024).
Feng, L. et al. Mechanistic, mutational, and structural evaluation of a Taxus Phenylalanine Aminomutase. Biochemistry 50, 2919–2930 (2011).
Wang, Y. T. et al. Characterization of Two BAHD Acetyltransferases Highly Expressed in the Flowers of Jasminum sambac (L.) Aiton. Plants-Basel 11, 14 (2022).
Aranovich, D. et al. Post-harvest enhancement of aroma in transgenic lisianthus (Eustoma grandiflorum) using the Clarkia breweri benzyl alcohol acetyltransferase (BEAT) gene. Postharvest Biol. Technol. 43, 255–260 (2007).
Effmert, U. et al. Floral benzenoid carboxyl methyltransferases: From in vitro to in planta function. Phytochemistry 66, 1211–1230 (2005).
Pott, M. B. et al. Evening specific oscillations of scent emission, SAMT enzyme activity, and SAMT mRNA in flowers of Stephanotis floribunda. J. Plant Physiol. 159, 925–934 (2002).
Jürgens, A. et al. Flower scent composition in Dianthus and Saponaria species (Caryophyllaceae) and its relevance for pollination biology and taxonomy. Biochem. Syst. Ecol. 31, 345–357 (2003).
Battilana, J. et al. Functional effect of grapevine 1-deoxy-D-xylulose 5-phosphate synthase substitution K284N on Muscat flavour formation. J. Exp. Bot. 62, 5497–5508 (2011).
Zhou, C. Z. et al. The chromosome-scale genome assembly of Jasminum sambac var. unifoliatum provides insights into the formation of floral fragrance. Hortic. Plant J. 9, 1131–1148 (2023).
Wasternack, C. et al. Jasmonates in flower and seed development. Biochimie 95, 79–85 (2013).
Floková, K. et al. A previously undescribed jasmonate compound in flowering Arabidopsis thaliana - The identification of cis-(+)-OPDA-Ile. Phytochemistry 122, 230–237 (2016).
Ichimura, K. et al. Mitogen-activated protein kinase cascades in plants: a new nomenclature. Trends Plant Sci. 7, 301–308 (2002).
Yin, Z. J. et al. Molecular characterization, expression and interaction of MAPK, MAPKK and MAPKKK genes in upland cotton. Genomics 113, 1071–1086 (2021).
Li, X. D. et al. Calcium-dependent protein kinase CPK32 mediates calcium signaling in regulating Arabidopsis flowering time. Natl. Sci. Rev. 9, 10 (2022).
Meng, X. Z. et al. A MAPK cascade downstream of ERECTA receptor-like protein kinase regulates Arabidopsis inflorescence architecture by promoting localized cell proliferation. Plant Cell 24, 4948–4960 (2012).
Tang, Q. et al. The mitogen-activated protein kinase phosphatase PHS1 regulates flowering in Arabidopsis thaliana. Planta 243, 909–923 (2016).
Samad, A. F. A. et al. MicroRNA and transcription factor: key players in plant regulatory network. Front. Plant Sci. 8, 18 (2017).
Han, Y. J. et al. Mechanism of floral scent production in Osmanthus fragrans and the production and regulation of its key floral constituents, β-ionone and linalool. Horticulture Res. 6, 12 (2019).
Suttipanta, N. et al. The Transcription Factor CrWRKY1 positively regulates the Terpenoid Indole alkaloid biosynthesis in Catharanthus roseus. Plant Physiol. 157, 2081–2093 (2011).
Aslam, M. Z. et al. Molecular cloning and functional characterization of CpMYC2 and CpBHLH13Transcription factors from Wintersweet (Chimonanthus praecox L.). Plants-Basel 9, 21 (2020).
Zhou, C. Z. et al. Multiomics analysis reveals the involvement of JsLHY in controlling aroma production in jasmine flowers. J. Agric. Food Chem. 71, 17455–17468 (2023).
Deng, Y. M. et al. Differential responses of double petal and multi petal jasmine to shading: I. Photosynthetic characteristics and chloroplast ultrastructure. Plant Physiol. Biochem. 55, 93–102 (2012).
Zhou, H. C. et al. Large scale preparation, stress analysis, and storage of headspace volatile condensates from Jasminum sambac flowers. Food Chem. 286, 170–178 (2019).
Woolf, A. B. et al. Postharvest responses to high fruit temperatures in the field. Postharvest Biol. Technol. 21, 7–20 (2000).
Waters, E. R. et al. Plant small heat shock proteins - evolutionary and functional diversity. N. Phytol. 227, 24–37 (2020).
Rosenzweig, R. et al. The Hsp70 chaperone network. Nat. Rev. Mol. Cell Biol. 20, 665–680 (2019).
Zhang, X. T. et al. Genomes of the Banyan tree and pollinator wasp provide insights into Fig-Wasp coevolution. Cell 183, 875 (2020).
Huang, Y. J. et al. The formation and evolution of centromeric satellite repeats in Saccharum species. Plant J. 106, 616–629 (2021).
Chen, S. F. et al. fastp: an ultra-fast all-in-one FASTQ preprocessor. Bioinformatics 34, 884–890 (2018).
Marcais, G. et al. A fast, lock-free approach for efficient parallel counting of occurrences of k-mers. Bioinformatics 27, 764–770 (2011).
Ranallo-Benavidez, T. R. et al. GenomeScope 2.0 and Smudgeplot for reference-free profiling of polyploid genomes. Nat. Commun. 11, 10 (2020).
Guan, D. F. et al. Identifying and removing haplotypic duplication in primary genome assemblies. Bioinformatics 36, 2896–2898 (2020).
Servant, N. et al. HiC-Pro: an optimized and flexible pipeline for Hi-C data processing. Genome Biol. 16, 11 (2015).
Li, H. et al. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 25, 1754–1760 (2009).
Durand, N. C. et al. Juicer provides a one-click system for analyzing loop-resolution Hi-C experiments. Cell Syst. 3, 95–98 (2016).
Zeng, X. F. et al. Chromosome-level scaffolding of haplotype-resolved assemblies using Hi-C data without reference genomes. Nat. Plants 10, 31 (2024).
Krzywinski, M. et al. Circos: An information aesthetic for comparative genomics. Genome Res. 19, 1639–1645 (2009).
Li, H. Minimap2: pairwise alignment for nucleotide sequences. Bioinformatics 34, 3094–3100 (2018).
Kim, D. et al. HISAT: a fast spliced aligner with low memory requirements. Nat. Methods 12, 357–U121 (2015).
Simao, F. A. et al. BUSCO: assessing genome assembly and annotation completeness with single-copy orthologs. Bioinformatics 31, 3210–3212 (2015).
Xu, Z. et al. LTR_FINDER: an efficient tool for the prediction of full-length LTR retrotransposons. Nucleic Acids Res 35, W265–W268 (2007).
Ellinghaus, D. et al. LTRharvest, an efficient and flexible software for de novo detection of LTR retrotransposons. BMC Bioinforma. 9, 14 (2008).
Ou, S. J. et al. LTR_retriever: A highly accurate and sensitive program for identification of long terminal repeat Retrotransposons. Plant Physiol. 176, 1410–1422 (2018).
Rhie, A. et al. Merqury: reference-free quality, completeness, and phasing assessment for genome assemblies. Genome Biol. 21, 27 (2020).
Flynn, J. M. et al. RepeatModeler2 for automated genomic discovery of transposable element families. Proc. Natl. Acad. Sci. USA 117, 9451–9457 (2020).
Tarailo-Graovac, M. et al. Using RepeatMasker to identify repetitive elements in genomic sequences. In Current protocols in bioinformatics Ch. 4, https://doi.org/10.1002/0471250953.bi0410s25 (2009).
Abrusán, G. et al. TEclass-a tool for automated classification of unknown eukaryotic transposable elements. Bioinformatics 25, 1329–1330 (2009).
Benson, G. Tandem repeats finder: a program to analyze DNA sequences. Nucleic Acids Res. 27, 573–580 (1999).
Goodstein, D. M. et al. Phytozome: a comparative platform for green plant genomics. Nucleic Acids Res. 40, D1178–D1186 (2012).
Bao, Y. et al. A chromosomal-scale genome assembly of modern cultivated hybrid sugarcane provides insights into origination and evolution. Nat. Commun. 15, 3041 (2024).
Chan, P. P. et al. tRNAscan-SE 2.0: improved detection and functional classification of transfer RNA genes. Nucleic Acids Res 49, 9077–9096 (2021).
Lagesen, K. et al. RNAmmer:: consistent and rapid annotation of ribosomal RNA genes. Nucleic Acids Res. 35, 3100–3108 (2007).
Nawrocki, E. P. et al. Infernal 1.1: 100-fold faster RNA homology searches. Bioinformatics 29, 2933–2935 (2013).
Li, H. et al. The sequence alignment/map format and SAMtools. Bioinformatics 25, 2078–2079 (2009).
Liao, Y. et al. featureCounts: an efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics 30, 923–930 (2014).
Love, M. I. et al. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 15, 38 (2014).
Yu, G. C. et al. clusterProfiler: an R package for comparing biological themes among gene clusters. OMICS-J. Integr. Biol. 16, 284–287 (2012).
Zhang, W. et al. HS-SPME and GC/MS volatile component analysis of Yinghong No. 9 dark tea during the pile fermentation process. Food Chem. 357, https://doi.org/10.1016/j.foodchem.2021.129654 (2021).
Wang, Y. P. et al. MCScanX: a toolkit for detection and evolutionary analysis of gene synteny and collinearity. Nucleic Acids Res. 40, 14 (2012).
Wheeler, T. J. et al. nhmmer: DNA homology search with profile HMMs. Bioinformatics 29, 2487–2489 (2013).
Altschul, S. F. et al. Basic local alignment search tool. J. Mol. Biol. 215, 403–410 (1990).
Edgar, R. C. MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 32, 1792–1797 (2004).
Stamatakis, A. RAxML version 8: a tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 30, 1312–1313 (2014).
Letunic, I. et al. Interactive Tree Of Life (iTOL) v5: an online tool for phylogenetic tree display and annotation. Nucleic Acids Res. 49, W293–W296 (2021).
Zheng, Y. et al. ITAK: A program for genome-wide prediction and classification of plant transcription factors, transcriptional regulators, and protein kinases. Mol. Plant 9, 1667–1670 (2016).
Chen, C. J. et al. TBtools: An integrative toolkit developed for interactive analyses of big biological data. Mol. Plant 13, 1194–1202 (2020).
Langfelder, P. et al. WGCNA: an R package for weighted correlation network analysis. BMC Bioinforma. 9, 13 (2008).
Wang, P. P. et al. Cloning and functional assessments of floral-expressed SWEET transporter genes from Jasminum sambac. Int. J. Mol. Sci. 20, 16 (2019).
Livak, K. J. et al. Analysis of relative gene expression data using real-time quantitative PCR and the 2-ΔΔCT method. Methods 25, 402–408 (2001).
Hellens, R. P. et al. Transient expression vectors for functional genomics, quantification of promoter activity and RNA silencing in plants. Plant Methods 1, 14 (2005).
Yang, X. H. et al. DgMYB2 improves cold resistance in chrysanthemum by directly targeting DgGPX1. Hortic. Res. 9, 10 (2022).
Wang, P. et al. Genomes of single - and double-petal jasmines (Jasminum sambac) provide insights into their divergence time and structural variations. Plant Biotechnol. J. 20, 1232–1234 (2022).
Xu, M. et al. A novel genome sequence of Jasminum sambac helps uncover the molecular mechanism underlying the accumulation of jasmonates. J. Exp. Bot. 74, 1275–1290 (2023).
Qi, X. Y. et al. The chromosome-level genome of double-petal phenotype jasmine provides insights into the biosynthesis of floral scent. Hortic. Plant J. 10, 259–272 (2024).
Acknowledgements
This work was supported by the Fujian Provincial Natural Science Foundation of China (Grant number 2023J01508) and the China Scholarship Council (Grant number 201908350014). All authors greatly appreciate helpful suggestions and comments on the manuscript from the editor and anonymous reviewers.
Author information
Authors and Affiliations
Contributions
J.F. conceived this project and coordinated the research activities. J.F. and Q.C. designed the experiments. J.F. and Q.C. collected the samples and prepared the transcriptomics and metabolomics datasets. L.Z., Q.C., and J.F. carried out the experiments and processed the genome assembly and annotation, as well as the transcriptomics and metabolomics data. Q.C., L.Z., J.F., S.L., J.W., Y.Z., K.Z., and H.Y. performed the following bioinformatic analyses. J.Z. and R.H. contributed to the study design and conceptualization. L.Z., J.F. and Q.C. wrote the draft manuscript. J.F., L.Z., and all other authors have read, revised, and approved the final version of the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Communications Biology thanks Monica Borghi and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. Primary Handling Editors: Heejin Yoo, David Favero, and Aylin Bircan.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Fang, J., Zhou, L., Chen, Q. et al. Integrated multi-omics analysis unravels the floral scent characteristics and regulation in “Hutou” multi-petal jasmine. Commun Biol 8, 256 (2025). https://doi.org/10.1038/s42003-025-07685-w
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s42003-025-07685-w
