
Abstract
Conjugative IncX plasmids are vital for spreading clinically significant antibiotic resistance genes. We identified key factors governing the conjugative process of IncX plasmids, the plasmid encoded activator PrfaH and inhibitor H-NS. Deletion of prfaH completely abolishes conjugative transfer, and the PrfaH binding site is an ops-like sequence located downstream of the prfaH promoter. We solved the crystal structure of PrfaH and identified the residues that likely mediate interactions with its target. The IncX3 plasmid-encoded H-NS inhibits conjugation by directly repressing PrfaH expression, while simultaneously enhancing host fitness. This tradeoff between plasmid conjugation and fitness is indispensable for plasmid persistence in nutrient-deprived environments. The presence of PrfaH paralogs in various antibiotic resistance plasmids suggests its fundamental role in regulating plasmid transfer. Our study not only elucidates the regulatory mechanisms behind the horizontal transfer of IncX plasmids but also highlights PrfaH as a potential target for strategies aimed at combating antimicrobial resistance.
Similar content being viewed by others

Introduction
Antimicrobial resistance (AMR) in bacterial pathogens is a growing crisis that constitutes a severe threat to human health, with ~4.95 million deaths linked to bacterial AMR each year1. The rapid development of multidrug-resistant (MDR) Gram-negative bacteria, especially Gram-negative bacteria resistant to last-resort antibiotics that have been classified as critical priority pathogens by the World Health Organization (WHO), is a major AMR challenge2,3,4,5. The key factor that contributes to the rapid increase and global dissemination of MDR Gram-negative bacteria is the horizontal transfer of resistance genes mediated by mobile genetic elements, such as plasmids6. Plasmid backbones usually contain a variety of core gene groups with different functions, including toxin–antitoxin (TA) systems, partitioning systems, and conjugation systems7. Conjugative transfer of plasmids from one strain to another plays a crucial role in the dissemination of resistance genes and is mediated by the type IV secretion system (T4SS)8. Since conjugation may impose a fitness cost on the host bacteria, its efficiency is tightly regulated to ensure that the process does not impose a significant burden on the strain, thus maintaining optimal bacterial growth9,10. However, research on the regulation of plasmid conjugation has long been neglected, especially in the current context of the widespread dissemination of antibiotic resistance.
To date, the regulatory mechanisms controlling transfer gene expression have been well studied in F-like plasmids in Gram-negative bacteria11,12, which may provide a paradigm for understanding the transfer mechanisms of other types of conjugative plasmids. The transfer of F-like plasmids is tightly controlled by plasmid-encoded and host factors. The F-like tra operon (traY-X) is activated by TraJ, while the expression of traJ is directly inhibited by the plasmid-encoded FinOP system and the host-encoded global factors including H-NS and Hfq13,14,15,16,17. Furthermore, the CpxAR two-component system indirectly regulates the expression of the tra operon by promoting the degradation of TraJ18. These findings suggest that natural plasmids exhibit intricate cross-talk mechanisms in the regulation of conjugation with their respective host bacteria19.
In recent years, IncX plasmids have garnered widespread attention from the public health community due to their ability to carry antimicrobial resistance genes that confer resistance to last-resort antibiotics, including carbapenem, colistin, and tigecycline, and to transmit them horizontally between bacteria20,21,22,23,24,25,26,27,28. IncX plasmids are commonly isolated from members of the Enterobactericeae, particularly Escherichia coli, Salmonella, and Klebsiella pneumoniae29. These plasmids were initially grouped on the basis that they confer sensitivity to the filamentous bacteriophage X-230, and can be divided into nine subgroups (IncX1~IncX9) based on the differences in the taxC gene and pir gene, and their sizes range from 30 to 50 kb31. A core set of backbone genes, including pir-bis-par-hns-topB-pilX(virB)-actX-taxCA, is shared among IncX plasmids32. The pir and bis genes are involved in replication29. The pilX(virB) gene cluster encodes a T4SS that mediates plasmid conjugation8,33. As in many conjugative plasmids that have been studied, the IncX conjugative pilus is naturally repressed34, implying that the expression of genes involved in conjugation is tightly regulated. However, the regulation of conjugation in IncX plasmids has not been studied in detail.
Previously, we have identified and characterized the activator of conjugative transfer of mcr-1-bearing IncX4 plasmids, named PixR, which activates IncX4 plasmid transfer by directly binding the promoter of the transfer operon and is essential for plasmid persistence27. In the present work, we focused on the blaNDM-bearing IncX3 plasmids and tet(X4)-bearing IncX1 plasmids, as they have spread in clinical settings and pose a growing threat to public health. We characterized the regulatory mechanism of IncX3 and IncX1 plasmids and found that the NusG family protein PrfaH is essential for activation of IncX3 and IncX1 virB gene expression. We also determined the structure of IncX3 plasmid PrfaH by X-ray crystallography and identified its binding target. Bioinformatics analysis indicates that PrfaH paralogs are present in many antibiotic-resistant plasmids. The plasmid-encoded H-NS was shown to repress the expression of PrfaH and is crucial for the persistence of IncX3 plasmids in nutrient-deprived environments. These findings significantly enhance the understanding of the mechanisms behind IncX plasmid transfer and persistence, providing potential targets for controlling their spread.
Results
Identification of regulators for IncX3 plasmid conjugative transfer
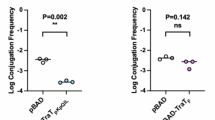
The backbone of IncX3 plasmids is highly conserved. We analyzed the genetic structure of the prototype IncX3 plasmid pHNYX644. The genes of pHNYX644 could be divided into five functional groups: replication, partitioning, maintenance, conjugation, and antibiotic resistance (Fig. 1a). To define the key regulators required for conjugation, we initially focused on genes encoding the conserved hypothetical proteins in the conjugation and nearby regions (Fig. 1a). To investigate the roles of these genes in conjugation, we constructed deletion mutants for each and then tested conjugative transfer rate of each mutant plasmid in comparison to the wild-type plasmid (Fig. 1b). Deletion of rs00145, which encodes a putative histone-like nucleoid-structuring protein (H-NS), led to ~320-fold increase in transfer rate. Deletion of rs00245, which encodes a putative N-terminal domain of transcription termination/antitermination protein RfaH, completely abolished conjugative transfer. To avoid confusion with homologs of the two genes that are present in the E. coli chromosome, rs00245 and rs00145 were renamed prfaH and phns, for plasmid-encoded RfaH and H-NS, respectively. Deletion of the other hypothetical genes had little (less than 12-fold of WT) or no impact on transfer efficiency. In the case of pHNYX644\(\Delta\)rs00145(phns) or pHNYX644\(\Delta\)rs00245(prfaH), enhanced or abolished transfer frequencies were restored to their wild-type transfer rates when complemented by plasmids containing phns or prfaH under the control of their native promoters (Fig. 1c). These results suggest that RS00245 has a positive effect on IncX3 conjugation, while RS00145 exerts a negative effect on conjugation. Complementation with the chromosomal RfaH could not restore conjugation ability, while complementation with chromosomal H-NS repressed conjugation to the wild-type level (Fig. 1c).

a Genetic map of the pHNYX644 plasmid. This plasmid belongs to the IncX3 incompatibility group and contains carbapenemase gene blaNDM-5 and bleomycin-resistant gene ble-MBL. Colors are coded by function as follows: blue, conjugation; cyan, prfaH (a gene that activates conjugative transfer); yellow, phns (a gene that inhibits conjugative transfer); red, replication; orange, antibiotic resistance; gray, transposase; white, unknown; partitioning and maintenance, pink. b Conjugation rates of pHNYX644∆rs00140, pHNYX644∆00145(phns), pHNYX644∆rs00175, pHNYX644∆rs00245(prfaH), pHNYX644∆rs00240, pHNYX644∆rs00260, pHNYX644∆rs00270, pHNYX644∆rs00275, pHNYX644∆rs00285, pHNYX644∆rs00290, and pHNYX644∆rs00300. Individual values were obtained from n = 3 independent conjugation assays and represented by black dots; bars represent the mean. Statistical analyses were performed using t-test with Welch’s correction. The p-values are shown when making comparisons with the wild-type (WT). The asterisk denotes that the transfer rate fell beneath the detectable threshold (~1\(\times\)10−18). c The effect of chromosomal homologs RfaH and H-NS on the IncX3 plasmid conjugation. WT represents the wild-type pHNYX644 plasmid; ∆prfaH represents the pHNYX644∆prfaH plasmid;∆phns represents the pHNYX644∆phns plasmid. Complementation assays were conducted by expressing prfaH, chromosomal rfaH, phns, and chromosomal hns with their corresponding native promoters on pHSG575 plasmids, and the resulting plasmids were named P-prfaH, P-chr-rfaH, P-phns, and P-chr-hns, respectively. Conjugation assays were performed with n = 3 biological replicates. Statistical comparisons of ∆phns with ∆phns/P-phns and ∆phns/P-chr-hns were performed using one-way ANOVA with Dunnett’s T3 correction. Statistical comparison of ∆prfaH/P-prfaH with WT was performed using t-test with Welch’s correction. d RT-qPCR analysis of the effect of PrfaH on the expression of virB genes. WT (wild type): BW25113/pHNYX644, ∆prfah: BW25113/pHNYX644∆prfah, ∆prfah/pHSG575-prfaH: BW25113/pHNYX644∆prfah + pHSG575-prfaH. RT-qPCR experiments were performed with n = 3 biological replicates. The statistical comparison of differences between the two groups were performed using t-test with Welch’s correction. e PrfaH enhances the expression of virB operon but not as a promoter activator. The promoter region of virB1 or prfaH was fused with a promoterless lacZ gene in pHGR01 plasmid. The +1 represents the first nucleotide of the open reading frame of prfaH or virB1. Dashed arrows without color annotation indicate gene deletions. IGR represents the intergenic region between rs000240 and virB1. β-Galactosidase activity for each construct was detected in the absence or presence of PrfaH. f The effect of the potential termination signal ahead of virB1 on the function of PrfaH. IGRm1 represents IGR lacking m1 region(gray); IGRm2 represents IGR lacking m2 region(purple); IGRm3 represents IGR harboring GG-AA mutations (brown font); IGRm4 represents deletion of the IGR fragments. All \({{\rm{\beta }}}\)-Galactosidase assays were performed with n = 3 biological replicates. The t-test with Welch’s correction was used for comparison of differences between the two groups.
PrfaH is a master activator of the T4SS in IncX3 plasmids
To test whether PrfaH has an impact on the expression of genes for the IncX3 T4SS, the mRNA levels of virB genes in the \(\Delta\)prfaH mutant were measured by reverse transcription-quantitative polymerase chain reaction (RT-qPCR) relative to wild-type strain (BW25113/pHNYX644) (Fig. 1d). Deletion of prfaH significantly reduced the expression of virB genes in the pHNYX644 plasmid. Complementation of the ∆prfaH mutation by expressing prfaH under its native promoter not only restored but even exceeded the expression of virB genes compared to the wild-type strain, confirming that prfaH regulates IncX3 plasmid conjugation by activating the expression of virB genes of IncX3 plasmids (Fig. 1d).
Next, we tested whether prfaH could activate the expression of virB genes from the promoter PvirB1. The PvirB1 was fused with a promoterless lacZ gene and β-galactosidase activity was monitored upon expression of prfaH. The PvirB1 activity was not affected by prfaH (Fig. 1e). Since prfaH is located just upstream of virB clusters, reverse transcription-polymerase chain reaction (RT-PCR) was used to determine whether prfaH and virB1 are co-transcribed. The total RNA isolated from BW25113/pHNYX644 and specific primers spanning prfaH and virB1 were used for RT-PCR analysis. As shown in Supplementary Fig. 1, the DNA band covering the prfaH and virB1 were detected when the cDNA was used as a template, as in the case of the positive control when the genomic DNA was served as a template. No band was detected when the total RNA was used as a template. These results suggest that prfaH and virB1 were co-transcribed within a singular operon.
Then, we tested whether prfaH could enhance the expression of virB genes from PprfaH. The promoter region of prfaH was fused with lacZ to detect the β-galactosidase activity (Fig. 1e). Expression of prfaH had no impact on PprfaH activity (Fig. 1e), indicating that PrfaH does not enhance operon expression by increasing promoter initiation efficiency. Since PrfaH is predicted to exhibit structural similarity to RfaH, which promotes the transcription of long operons by inhibiting Rho-dependent termination, the elevation of virB gene expression by prfaH is likely achieved through its transcription antitermination activity.
To validate this hypothesis, the fragment containing the promoter of prfaH extending to virB1 but with deletion of prfaH coding region was cloned and ligated with the open reading frame of lacZ (Fig. 1e). The β-galactosidase activity was detected upon expression of prfaH. As shown in Fig. 1e, overexpression of prfaH in BW25113/PprfaH-virB1-lacZ results in a significant increase in β-galactosidase activity compared to the negative control. Next, we analyzed the intergenic region (IGR) between rs000240 and virB1 using the RNAfold web server, and found a predicted stem-loop (SL) structure serving as a potential termination signal upstream of the virB1 start codon (Fig. 1f). To examine if this putative termination signal plays a role in PrfaH-mediated regulation of virB1, we introduced several deletions or mutations to disrupt the SL structure (Fig. 1f). Disruption or point mutations in the stem-loop region (IGRm1, IGRm2, and IGRm3) resulted in a significant increase in β-galactosidase activity compared to the wild-type strain, while complete deletion of the stem-loop (IGRm4) led to a slight decrease in β-galactosidase activity compared to the wild-type strain (Fig. 1f). Simultaneously, the expression of prfaH no longer affected enzyme activity in these mutants. These data suggest that PrfaH might boost virB1 expression by assisting RNA polymerase to overcome this potential termination signal.
Identification of the PrfaH binding site in the IncX3 plasmid backbone
The in-silico analysis revealed that PrfaH contains the N-terminal domain of RfaH, which is recruited by the operon polarity suppressor (ops) signal sequence in the non-template DNA strand. This led us to hypothesize that prfaH promotes the expression of virB genes via the recruitment of PrfaH by an ops-like signal. Initially, we examined whether the IGR upstream of the virB1 functions as an ops signal. To test this, we constructed pHNYX644\(\Delta\)prfaH mutants lacking IGR, or with IGR replaced by a Shine-Dalgarno (SD) sequence, and named pHNYX644\(\Delta\)IGR or pHNYX644\(\Delta\)IGR-SD, respectively (Fig. 2a). If this IGR is the target of PrfaH, reintroducing prfaH to the BW25113/pHNYX644\(\Delta\)prfaH would likely be ineffective in restoring the plasmid conjugation ability. However, deletion or substitution of the IGR did not abolish the activation of conjugation by ectopic expression of prfaH (Fig. 2a), indicating that IGR is not the target of PrfaH.

a The deletion of the intergenic region (IGR) between rs000240 and virB1 does not affect plasmid conjugation. The diagram shows mutations in IGR. ∆IGR refers to the pHNYX644∆IGR plasmid. A synthetic SD sequence (5’-AAAGAGGAGAAA-3’) was fused upstream of virB1, resulting in the pHNYX644∆IGR-SD plasmid. b The 5’UTR of PrfaH is indispensable for the function of PrfaH. The diagram shows mutations in the upstream region of prfaH. ∆prfaH refers to the pHNYX644∆prfaH plasmid. ∆prfaH-PJ23119 represents a construct where the prfaH promoter was replaced with the PJ23119 promoter in the pHNYX644∆prfaH plasmid, with a synthetic SD sequence fused upstream of prfaH. ∆prfaH-PJ23119-UTR indicates that the synthetic SD sequence was replaced by the 5’ UTR of prfaH. Dashed arrows without color annotations indicate gene deletions. c Identification of the target of PrfaH in 5’UTR of PrfaH. The pHNYX644∆prfaH plasmids with mutations in the 5’UTR of prfaH (W1 to W12), and the mutated bases are indicated in red font. ∆prfaH represents the pHNYX644∆prfaH plasmid. The conjugation rate of pHNYX644∆prfaH and its derivatives with mutations in the 5’UTR of PrfaH were measured in the presence or absence of PrfaH. The conjugation rates of all prfaH-complemented mutants were compared with the conjugation rate of ∆prfaH complemented with prfaH using t-test with Welch’s correction. Conjugation assays were conducted with n = 3 biological replicates, and statistical analysis was performed using t-test with Welch’s correction.
Since prfaH is located upstream of virB1 and co-transcribed with it, we attempted to identify the ops-like signal within the promoter region of prfaH (PprfaH). To do this, we initially replaced the promoter region of prfaH (350 bp upstream of prfaH ATG) with the constitutive PJ23119 promoter in pHNYX644\(\Delta\)prfaH using the λRed recombination system, and the resulting plasmid was named pHNYX644\(\Delta\)prfaH-PJ23119 (Fig. 2b). After replacing the PprfaH with PJ23119 promoter, complementation with prfaH did not restore the conjugative transfer of pHNYX644\(\Delta\)prfaH-PJ23119, indicating that the target of PrfaH was located in the promoter region of prfaH (PprfaH).
To further define the binding site of PrfaH within the promoter region of prfaH(PprfaH) and test whether PrfaH could restore the conjugation, the 62 bp upstream of prfaH start codon (−62 to −1, relative to the prfaH start codon) was reintroduced downstream of the PJ23119 promoter (the resulting plasmid mutant was named pHNYX644\(\Delta\)prfaH-PJ23119-UTR) (Fig. 2b). Fortuitously, we found that complementation of this mutant with prfaH restored and even exceeded wild-type level transfer rate of pHNYX644\(\Delta\)prfaH with ectopic expression of prfaH, indicating that the target of PrfaH was located within the 62 bp upstream of the start codon of prfaH (Fig. 2b). To precisely define the binding site of PrfaH within this 62 bp region, we introduced a series of mutations in increments of three nucleotides per step to this region in pHNYX644\(\Delta\)prfaH (Fig. 2c). The resulting mutants were sequentially named W1 to W12. Reintroduction of prfaH did not or poorly restore conjugation ability to wild-type levels in W3 through W7 mutants, but it did in other mutants (Fig. 2c). The result suggests that the −42 to −25 region is indispensable for the activation of conjugative transfer by PrfaH.
To demonstrate the interaction between PrfaH and a non-template strand of the −42 to −25 region, the synthesized non-template DNA fragment corresponding to −48 to −13 region (Fig. 2c), and its mutants (ss-W1–ss-W12) was used for EMSA assay. We expressed and purified full-length PrfaH, but found that it is prone to degradation (Supplementary Fig. 2a). The AlphaFold2-predicted structure of PrfaH contains a flexible structure at the C terminus, potentially contributing to its proneness to degradation (Supplementary Fig. 2b). Hence, we expressed a truncated PrfaH (∆14-PrfaH) lacking the C-terminal 14 amino acids under its native promoter and detected its impact on plasmid conjugation. As shown in Supplementary Fig. 3a, ∆14-PrfaH activated plasmid conjugation with efficiency comparable to that of the full-length PrfaH. The truncated N-terminal 6\(\times\)His-tagged ∆14-PrfaH was expressed from a T7 promoter and purified by Ni-NTA affinity chromatography. The ∆14-PrfaH protein was obtained through PreScission protease cleavage and Heparin ion purification (Supplementary Fig. 3b and c). Then, these single-strand DNA (ss-WT, ss-W1–ss-W12) were incubated with increasing concentrations of ∆14-PrfaH, respectively. The mobility of ssDNA including ss-WT, ss-W1, ss-W2, ss-W9, ss-W10, ss-W11, and W12 were retarded by the addition of 25 μg/mL (~1.4 \(\mu\)M) PrfaH (Fig. 3a). While the ss-W3 to ss-W8 were not or slightly retarded by ∆14-PrfaH in the same concentration (Fig. 3a). These data demonstrate that ∆14-PrfaH exactly binds to the non-template strand of the −42 to –25 region upstream of prfaH (Figs. 3a and 2c).

a EMSA analysis of the interaction between the 5’ UTR of prfaH and ∆14-PrfaH protein. In the EMSA experiment, each ssDNA (~0.4 μM) was incubated with different concentrations (0, 0.5, and 1.3 μM) of ∆14-PrfaH protein, respectively. b Precise analysis of the key nucleotides affecting PrfaH function. The core target sequence (the −42 to −25 region, relative to prfaH ATG) is marked as +1 to +18, and the mutated bases are indicated in red font. WT represents pHNYX644 plasmid. T1A represents a point mutation at T1A in the 5’ UTR of prfaH on the pHNYX644 plasmid. Similarly, C2A, G3A, …, G18A represent point mutations at C2A, G3A, …, G18A, respectively, in the 5’ UTR of prfaH on the pHNYX644 plasmid. The conjugation assays were carried out with n = 3 biological replicates. The asterisk denotes that the transfer rate fell beneath the detectable threshold(~1 × 10−18). The conjugation rate of each mutant was compared with that of the WT using t-test with Welch’s correction. The DNA secondary structure of the core sequence was predicted by RNAfold web server (http://rna.tbi.univie.ac.at/cgi-bin/RNAWebSuite/RNAfold.cgi).
Furthermore, a series of single-base mutations were introduced to this region to identify the key nucleotides involved in the regulation of PrfaH on conjugation (Fig. 3b). We found that C2A, G4A, G5A, T7A, A9T, C12A, and G15A substitutions completely abolish plasmid transfer ability. The G3A, C11A, G13A, and A14T substitutions led to a 20–30-fold decrease in conjugation rate relative to the wild-type. The non-template DNA strand of this region is predicted to form a stem-loop structure. The stem of this SL is formed by C2:G13, G3:C12, and G4:C11 base pairs, and G5, G6, T7, C8, A9, and T10 are located in the loop region (Fig. 3b). These data implies that −42 to −25 region (relative to prfaH ATG) is the target of PrfaH (Figs. 3b and 2c).
Analysis of the crystal structure of PrfaH
To investigate the structural basis of PrfaH function, we crystalized the ∆14-PrfaH protein and determined its structure by X-ray crystallography. The protein was crystallized at 4 °C in the space group P43 (a = 57.695 Å, b = 57.695 Å, c = 116.465 Å), with two PrfaH molecules per asymmetric unit. The crystals diffracted to 2\(\mathring{\rm A}\) resolution (Table 1). The structure of ∆14-PrfaH was solved by molecular replacement using AlphaFold predicted structure as the search model. We observed two conformations of ∆14-PrfaH, namely the Mg2+ bound and non-bound states (Supplementary Fig. 4). However, the dimer interface between the two ∆14-PrfaH molecules is not evident through Pymol analysis. Size-exclusion chromatography analysis revealed that ∆14-PrfaH is monomeric in solution (Supplementary Fig. 3b). Overall, ∆14-PrfaH consists of two domains (N-terminal and C-terminal domains) connected by a flexible linker (amino acids H104-P117) (Fig. 4a, b). The core of the N-terminal domain (M1-S103) is composed of four antiparallel β-folds, with an α-helix on each side of the core and two consecutive β-folds connected by a loop (R38-K41). The C-terminal domain (H118-D155) contains two longer antiparallel α-helices. A magnesium ion binding site is present at C-terminal residues S144-T146 (Fig. 4a, b). In the Mg2+ bound state, the loop connecting the N-terminal β-folds flips relative to the Mg2+-free state (Supplementary Fig. 4).

a Amino acid sequence and secondary structure of ∆14-PrfaH. The α-helices represented as red cylinders and β-strands illustrated as yellow arrows. The amino acids forming the N-terminal domain are highlighted in green, those forming the C-terminal domain are shown in blue, and the amino acids constituting the linker region are represented in gray. b The cartoon illustration and electrostatic potential map of the surface of the ∆14-PrfaH crystal structure. The N-terminal domain is mainly colored green and the C-terminal domain is colored blue. The linker is colored gray. The electrostatic potential map of the ∆14-PrfaH crystal structure is shown in the same orientation as the cartoon illustration. c Analysis of the effect of α1 and α2-α3-β6 on the function of PrfaH. The single amino acid mutation was separately introduced to α1 and α2-α3-β6 by substitution with Ala or Gly. The function of PrfaH mutants was tested via mating assay using pHNYX644∆prfaH. WT represents the conjugation rate of pHNYX644∆prfaH complemented with pHSG575-prfaH(wild type). The others represent the conjugation rates of pHNYX644∆prfaH complemented with pHSG575 expressing PrfaH with a single amino acid mutation at different positions within the α1, α2-α3-β6 regions, respectively. The key residues are shown in the stick representation. Conjugation assays were conducted with n = 3 biological replicates, and statistical analysis was performed using t-test with Welch’s correction. The p-values are shown when making comparisons with the wild-type (WT).
The structure of PrfaH is remarkably similar to that of E. coli chromosomal RfaH (Supplementary Fig. 5). Since the crucial amino acid residues involved in the interaction with the ops element are situated in the α1 and α2-α3-β6 of RfaH, we constructed a series of mutations in the corresponding region of PrfaH using low copy number plasmid pHSG575. A series of single-amino acid mutations were introduced in α1 and α2-α3-β6 of PrfaH. Then, these PrfaH mutants were reintroduced into BW25113/pHNYX644\(\Delta\)prfaH to assess their impacts on plasmid conjugation efficiency. As shown in Fig. 4c, three amino acid residues T11, R12, and F17 in α1, are essential for the function of PrfaH. Introduction of T11A completely abolished the plasmid conjugation, while the R12A and F17A led to an 80–130-fold decrease in conjugation rate compared with the wild-type. For α2-α3-β6, the introduction of T72A renders the plasmid unable to conjugate, and Y76A resulted in a 60-fold decrease in conjugation rate. These data indicate that α1and α2-α3-β6 plays a crucial role in the function of PrfaH.
The PrfaH paralogs are widely distributed in antibiotic resistance plasmids and exhibit extensive variation
We next analyzed the PrfaH paralogs from a collection of completely sequenced plasmid datasets sourced from GenBank in January 17, 2024 and found that 3212 plasmids contain annotated nusG family genes (Fig. 5a). PrfaH paralogs are present in various plasmids including IncX plasmids (31%), IncI-complex plasmids (45%), IncFII plasmids (10%), and other untyped plasmids (14%) (Fig. 5a). The positioning of prfaH and its paralogs adjacent to the type IV secretion system coding region is consistent with their function (Fig. 5b). Sequence alignment showed that the amino acids sequences of PrfaH paralogs encoded by different groups of plasmids vary extensively (Supplementary Fig. 6). The region ranging from R49 to E92 (β5-α2-α3-β6) was relatively conserved within PrfaH paralogs. The highly conserved residues were mainly located in β5, while most residues within α2-α3-β6 were less conserved (Supplementary Fig. 6). The phylogenetic analysis was conducted on the distinct representatives of PrfaH and its homologs derived from diverse groups of plasmids and revealed that PrfaH genes exhibit great diversity among different plasmid groups (Supplementary Fig. 7). The clustering of many PrfaH paralogs alongside plasmid groups, particularly IncX and IncI plasmids, suggests that these prfaH genes evolved with plasmids (Supplementary Fig. 7). Since T72A and Y76 in α2-α3-β6 of PrfaH are indispensable for its function, we analyzed the key residues at the corresponding positions in PrfaH paralogs derived from different groups of plasmids and observed that residues at these positions are highly diverse (Supplementary Fig. 7). Given that PrfaH paralogs are typically located upstream of the conjugation region, we propose that the function of PrfaH paralogs may be highly specialized in regulating plasmid conjugation.

a The distribution of PrfaH paralogs carried by different plasmid types. We totally collected 3214 plasmids carrying the NusG family protein from Genbank. These plasmids were classified using PlasmidFinder. The plasmid types were indicated by different colors. NA represents the untyped plasmids. b The locus of genes encoding PrfaH paralogs on different plasmid types. The genes encoding PrfaH paralogs were colored by red. The T4SS-associated genes were colored by blue. Green represents the genes coding type IV pili. The hns genes on IncX plasmids were colored by orange. The genes with unknown functions were not colored.
pHNYX644 encoded H-NS inhibits conjugation by repression of prfaH
Since pHNYX644-encoded H-NS can significantly inhibit plasmid conjugation and PrfaH is the core regulatory factor activating conjugation, we constructed a pHNYX644 plasmid mutant lacking both prfaH and phns to explore the mechanism of phns-mediated conjugation inhibition. As shown in Fig. 6a, deletion of both prfaH and phns abolished conjugation, which is consistent with that of \(\Delta\)prfaH, suggesting that phns inhibit conjugation by directly or indirectly regulating PrfaH. Then, we test whether phns could repress the activity of PprfaH. The promoter region of prfaH was cloned and fused with promoterless lacZ and β-galactosidase activity was detected upon expression of phns. As shown in Fig. 6b, the β-galactosidase activity decreased significantly upon expression of phns, indicating that pHNYX644-encoded H-NS could repress the promoter activity of prfaH.

a Deletion of prfaH abolishes the conjugation of pHNYX644∆phns. WT represents the pHNYX644 plasmid. ∆phns∆prfaH represents the pHNYX644 plasmid with deletion of phns and prfaH. ∆phns∆prfaH was complemented by pHSG575-prfaH, pHSG575-phns or pHSG575-prfaH-phns, respectively. ∆phns∆prfaH complemented by pHSG575 was used as the negative control. The statistical analysis was performed using the t-test with Welch’s correction. The p-values are shown when making comparisons with the wild-type (WT). b Plasmid-encoded H-NS inhibits the promoter activity of prfaH. The \({{\rm{\beta }}}\)-Galactosidase assays were carried out with three biological replicates. The statistical analysis was performed using t-test with Welch’s correction. c The effect of plasmid-encoded H-NS and PrfaH on the conjugation of IncX plasmids. The IncX1b(pHN21SC1631T-3) and its mutants are presented in the first group, and the IncX3(pHNYX644) and its mutants are presented in the second group. In the first group, WT represents wild-type IncX1b plasmid; ∆phns represents IncX1b with deletion of phns; ∆phns/P-phns(X1b) represents complementation of ∆phns with pHSG575-phns(IncX1b); ∆phns/P-phns(X3) represents complementation of ∆phns with pHSG575-phns(IncX3); ∆prfaH represents pHN21SC1631T-3 with deletion of prfaH; ∆prfaH/P-prfaH(X1b) represents complementation of ∆prfaH with pHSG575-prfaH(IncX1b); ∆phns/P-prfaH(X3) represents complementation of ∆prfaH with pHSG575-prfaH(IncX3). In the second group, WT represents wild-type IncX3 plasmid; ∆phns represents IncX3 with deletion of phns; ∆phns/P-phns(X1b) represents complementation of ∆phns with pHSG575-phns(IncX1b); ∆prfaH represents pHNYX644 with deletion of prfaH; ∆prfaH/P-prfaH(X1b) represents complementation of ∆prfaH with pHSG575-prfaH(IncX1b). The mating assays were performed to detect the transfer rate of these plasmids. Individual values were obtained from n = 3 independent experiments and represented by dots. The asterisk denotes that the transfer rate fell beneath the detectable threshold (~1\(\times\)10−18). Statistical analyses were performed using t-test with Welch’s correction. The p-values are shown when making comparisons with the wild-type (WT).
PrfaH and H-NS mediate mutual regulation of conjugative transfer between IncX plasmids
To investigate if the regulatory mechanisms of prfaH and phns in the control of conjugative transfer can be extended to other IncX plasmids, we compared the IncX3 plasmid pHNYX644 with other IncX plasmids (Supplementary Fig. 8). The conjugative transfer region of plasmid pHNYX644 exhibits a sequence similarity of >56% with the conjugative transfer regions of IncX1a, IncX1b, IncX2, IncX5, IncX8, and IncX9 type plasmids. However, the sequence similarity with the conjugative transfer regions of IncX4, IncX6, and IncX7 type plasmids is relatively low (15–46%) (Supplementary Fig. 8).
Plasmids of the IncX1a, IncX1b, IncX2, IncX5, IncX6, IncX8, and IncX9 types harbor genes that encode homologous PrfaH and H-NS proteins. The PrfaH homologs encoded by IncX1b, IncX2, IncX5, and IncX8 plasmids are ~60% similar to that of IncX3, while those encoded by IncX1a, IncX6, and IncX9 plasmids are more distantly related (~20% similarity) (Supplementary Table 1). The H-NS encoded by IncX3 plasmids shows relatively high sequence similarity (60%-80%) with the H-NS proteins encoded by all other IncX plasmids (Supplementary Table 2). These data suggest that there is a strong likelihood of mutual regulatory interactions among IncX plasmids in terms of conjugative transfer. To test this hypothesis, we focused on IncX1b plasmids, as they are widely distributed and known to carry various antibiotic resistance genes, including the tigecycline resistance gene tet(X4).
Firstly, we investigated the effect of IncX1b (pHN21SC1631T-3) plasmid-encoded PrfaH and H-NS on plasmid transfer (Fig. 6c). As expected, the deletion of prfaH (IncX1b) completely abolished the conjugation ability of pHN21SC1631T-3 plasmids, and deletion of phns led to ~150-fold increase in transfer rate (Fig. 6c). Complementation of the ∆phns and ∆prfaH in IncX1 plasmids by ectopic expression of the corresponding genes, phns(X1b) and prfaH(X1b), under control of their native promoters from the plasmids P-phns(X1b) and P-prfaH(X1b), restored the wild-type conjugation rate (Fig. 6c). Since the IncX1b plasmid-encoded PrfaH and H-NS share 64% and 77% identity with those of IncX3 (pHNYX644), we complemented the ∆phns and ∆prfaH mutations in IncX1b plasmids by expressing the IncX3-encoded PrfaH and H-NS, prfaH(X3) and phns(X3), respectively. Unexpectedly, complementation with phns(X3) did not inhibit the conjugation rate of IncX1b ∆phns but rather increased it. In contrast, complementation with prfaH(X3) significantly elevated the conjugation rate of IncX1b ∆prfaH, surpassing the wild-type level. Similarly, we also performed complementation of PrfaH and H-NS encoded by IncX1b into ∆prfaH and ∆phns in IncX3 plasmids. We found that complementation of phns(X1) significantly inhibited conjugation of IncX3∆phns plasmids, while complementation of prfaH(X1b) restored the conjugation ability of IncX3∆prfaH plasmid to the wild-type level (Fig. 6c). Next, we constructed various combinations of coexisting plasmids through conjugation: BW25113/IncX3+IncX1b, BW25113/IncX3+IncX1b\(\Delta\)prfaH, BW25113/IncX1b+IncX3\(\Delta\)prfaH, and determined the transfer frequencies of each plasmid (Supplementary Fig. 9). It was observed that both IncX1b∆prfaH and IncX3∆prfaH could restore the ability for conjugative transfer when co-existing with an intact IncX3 or IncX1b plasmid (Supplementary Fig. 9).
The antagonistic regulation of plasmid transfer by PrfaH and H-NS promotes the persistence of IncX3 plasmids
Since plasmid conjugation is an energy-intensive process, it often incurs a fitness cost to the host bacteria9. To investigate whether PrfaH and H-NS impact the fitness of E. coli harboring an IncX3 plasmid, we performed bacterial competition experiments in vitro. The pHNYX644 variant lacking phns was associated with a significant decrease in the fitness of host bacteria relative to BW25113/pHNYX644 (Fig. 7a). In contrast, deletion of prfaH endows the host bacteria with a fitness advantage relative to BW25113/pHNYX644 (Fig. 7a). It is noteworthy that deletion of prfaH can completely alleviate the cost imposed by pHNYX644∆phns (Fig. 7a). These results suggest that the expression of prfaH imposes a cost on the host, while phns rescues the host fitness by repressing prfaH.

a Relative fitness of BW25113 harboring pHNYX644∆phns::kan, pHNYX644∆prfaH::kan, pHNYX644∆prfaH∆phns::kan, and pHNYX644∆prfaH∆phns::kan-pHSG575-prfaH. These strains were competed with the reference strain BW25113/pHNYX644 in vitro separately. All competition assays were carried out with three biological replicates, and the relative fitness of each strain was detected at 24, 48, 72, and 96 h. b Co-cultures with BW25113 and BW25113/pHNYX644 in LB broth. c Co-cultures with BW25113 and BW25113/pHNYX644 in M9 medium. d Co-cultures with BW25113 and BW25113/pHNYX644∆phns::kan in LB broth. e Co-cultures with BW25113 and BW25113/pHNYX644∆phns::kan in M9 medium. f Co-cultures with BW25113, BW25113/pHNYX644, and BW25113/pHNYX644∆phns::kan in LB broth. g Co-cultures with BW25113, BW25113/pHNYX644, and BW25113/pHNYX644∆phns::kan in M9 medium. Plasmid-containing strains were mixed with a 10-fold excess of plasmids-free BW25113 at the beginning of the invasion assay. Bars represent SD of n = 3 biological replicates.
Since natural environments often present poor nutrient conditions, we further investigated the effect of H-NS on the persistence of an IncX3 plasmid in a bacterial population under two nutritional conditions (LB or M9 medium). We examined the persistence of pHNYX644 and pHNYX644∆phns by assessing their capacity to invade plasmid-free populations, individually or in competitive co-cultures (Fig. 7b–g). The proportion of cells carrying pHNYX644 remained constant when co-cultured with the plasmid-free bacterial population under either LB broth or M9 medium, even though it does not dominate within the population (Fig. 7b, c). However, the cells harboring plasmids lacking phns exhibited a different scenario when comparing with that of wild-type. The plasmid pHNYX644∆phns invaded and was present in most cells after 5 days in LB broth, whereas it failed to invade the plasmid-free cells in M9 medium and slightly decreased after 6 days (Fig. 7d, e). In the competitive co-cultures, the dynamics of cells harboring pHNYX644∆phns or pHNYX644 were similar to those of the individual co-cultures (Fig. 7f, g). To unravel why the dynamics of pHNYX644∆phns vary under different nutritional conditions, we investigated the conjugation rate of the pHNYX644∆phns plasmid in LB or M9 medium. We observed that the conjugation rate of this plasmid is comparable in M9 and LB media (Supplementary Fig. 10). However, the cost imposed by the plasmid is higher in M9 compared to LB (Fig. 7a). These data imply that the plasmid’s conjugation capability plays a dominant role in its persistence under nutrient-rich conditions, whereas under nutrient scarcity, host fitness appears to take precedence in determining plasmid persistence.
Discussion
IncX plasmids are crucial vectors for antibiotic resistance gene dissemination in Gram-negative bacteria31. Although IncX plasmids share a relatively conservative backbone structure, the regulatory mechanisms involved in their conjugative transfer remain unclear. Previously, we identified an activator of conjugative transfer, PixR, encoded by IncX4 plasmids, which directly activates the promoter of the transfer operon27. Here, we identified different transfer activators, PrfaH, in IncX3 and IncX1 plasmids. The mechanism by which PrfaH regulates the conjugative transfer gene cluster differs from that of PixR. Our data show that PrfaH does not activate the promoter activity of the transfer gene cluster, but rather enhances the expression of the gene cluster as a transcription anti-termination factor (Fig. 8a). This study experimentally establishes that IncX3 PrfaH functions as a transcription anti-termination protein. Furthermore, we solved the crystal structure of PrfaH, and found that it closely resembles the structure of RfaH encoded by the E. coli chromosome, despite notable disparities in their amino acid sequences. E. coli RfaH is known to promote the expression of operons encoding hemolysin, the F pilus of F-like plasmids, lipopolysaccharide core, and exopolysaccharide35,36. It is a transcription antiterminator and its function depends critically on an ops sequence (~12 nt conserved sequences 5’-GGCGGTAGCGTG-3’) in the promoter-proximal 5’ untranslated region of the operon37,38,39. The α1(K10, R16, and H20) and α2 (T68, T72, and R73) located in the N-terminal domain of RfaH make direct contacts with the hairpin loop of ops DNA, which is formed by G4-A739. Here, we investigated the amino acid residues in α1 and α2-β6 that are crucial for the function of PrfaH. Substitution of T11 and T72 located with Ala completely nullified PrfaH activity, while mutations in R12, F17, or Y76 resulted in a 60–130-fold decrease in conjugation rate. The differences in key amino acid residues between RfaH and PrfaH likely reflect their distinct target sequences. These data suggest that although RfaH and PrfaH share a high structural similarity, the key amino acid residues responsible for their interactions with ops differ and their functions are highly specialized.

a Model of conjugation regulation of IncX3 plasmids. The expression of PrfaH is directly repressed by H-NS (colored by orange). The prfaH and virB genes are co-transcribed. The PrfaH activates the expression of virB cluster via its transcriptional antitermination activity. RNA polymerase (RNAP) (colored by gray) paused at the ops site (colored by red) upstream of the ATG codon of prfaH. Then, PrfaH was recruited by this ops signal and binds with RNAP. The RNAP-PrfaH complex can further recruit ribosomes. Through the mechanism of transcription and translation coupling, this complex enhances the expression of virB cluster by resisting the potential termination signals. Deletion of prfaH completely abolished the conjugation of the IncX3 plasmid. b The PrfaH-H-NS regulon’s role in the persistence of IncX3 plasmids under LB or M9 medium conditions. The plasmid-free strain represents the E. coli BW25113 without plasmids. The phns+ prfaH+ strain represents the E. coli BW25113 with the wild-type IncX3 plasmid. The phns− prfaH+ strain represents the E. coli BW25113 with the phns-deficient IncX3 plasmid. The phns− prfaH- strain represents the E. coli BW25113 harboring the IncX3 plasmid with deletion both of phns and prfaH. The fitness and conjugation ability of each strain were shown in the diagram. PrfaH is the master activator for the conjugation of IncX3 plasmids. However, overactivation of the conjugative transfer system usually impairs the fitness of the host strain. The IncX3 plasmids encoded H-NS maintain the host fitness by directly repressing the expression of prfaH. So, the phns+ prfaH+ strain could persist stably in the environment (nutrition-rich or poor condition) for the long term. The phns-deficient plasmids possess the highest transfer ability but incur a high cost on their host. Their persistence is highly dependent on the growth environment. Under nutrition-rich conditions (e.g., LB broth), these plasmids could quickly invade large plasmid-free populations. However, under conditions of nutritional deprivation (e.g., M9 medium), the contribution of high-frequency conjugative transfer to plasmid persistence is counteracted by the high fitness cost.
PrfaH homologs are also found in plasmids outside of the IncX group. Since these PrfaH homologs are often located within or in close proximity to their cognate transfer operons, they are likely to activate the transfer of the plasmids that encode them. For example, the IncI1 plasmid R64 encoded TraB, the homologs of PrfaH, is required for the conjugative transfer of this plasmid40. However, the amino acid sequence of PrfaH homologs varies extensively. This is reminiscent of another activator of plasmid transfer, TraJ, which is encoded by F-like plasmids12. TraJ activates the expression of the F-like transfer (tra) operon by directly binding to the PY promoter41,42. The amino acid sequences of TraJ homologs exhibit extensive variation, which is associated with the plasmid-specific activation of tra12. PrfaH and TraJ, serving as activation factors for plasmid transfer, are highly specialized in their function. They specifically activate the horizontal transfer of their respective plasmids, which should prevent potentially destabilizing crosstalk when unrelated plasmids reside in the same bacterial host.
The conjugation system is often strictly repressed by plasmid or host factors, presumably to prevent the host bacteria from incurring a cost due to overexpression of transfer genes9,17. In this study, we found that the IncX plasmid-encoded H-NS significantly inhibits conjugation, reducing the transfer rate by ~100-fold, via directly suppressing the expression of the activator PrfaH. We hypothesize that inhibiting conjugation ensures the fitness of the host bacteria within microbial communities. The successful dissemination of plasmids is likely the outcome of a delicate balance between the fitness of various hosts and the ability of the plasmid to transfer between them. Here, we explored the role of the H-NS-PrfaH regulon in the persistence of IncX3 plasmids via plasmid invasion experiments. Our data indicate that derepression of conjugation assists plasmids in infecting more plasmid-free cells in nutrient-rich environments (e.g., LB broth). However, in nutrient-poor environments (e.g., M9 medium), which more closely resemble natural living conditions, high-frequency transfer does not guarantee that plasmid-bearing strains become dominant in bacterial population (Fig. 8b). High conjugation frequency can indeed increase the chances of plasmids entering more bacteria and contributes to their widespread, but the fitness of plasmid-bearing host bacteria is the key factor determining the long-term presence of plasmids in natural environments, which is consistent with previous reports43.
Studying the mechanism of conjugative transfer contributes to the development of conjugation inhibitors for controlling the spread of antibiotic resistance genes. However, the extent to which inhibitors can effectively control transmission by suppressing conjugation is unclear. Partially inhibiting conjugation seems to have limited efficacy in controlling the spread of antibiotic resistance plasmids, as plasmids still have opportunities to enter the most suitable bacterial hosts, persisting in the environment along with them. Based on the findings of this study, we advocate for research into the development of conjugation abolishment agents, which may be more effective. We consider PrfaH as one of the most suitable candidates, as deletion of prfaH results in the complete cessation of plasmid conjugation. The highly specific activity of PrfaH described here also suggests that it might be possible to design highly specific inhibitors for the transfer of IncX plasmids associated with antibiotic resistance.
In conclusion, we present the intrinsic master regulators of IncX3 and IncX1 plasmid conjugation. We have identified the conjugative transfer activator PrfaH and elucidated its structure and function. Notably, PrfaH paralogs are found in various types of plasmids, implying that the activation of pilus gene cluster expression by PrfaH is likely to be a common and crucial regulatory mechanism in conjugative transfer. Furthermore, we discovered that plasmid-encoded H-NS can directly suppress the expression of PrfaH, and found that this H-NS is indispensable for the long-term persistence of IncX3 plasmids. Our findings significantly contribute to understanding the mechanisms governing IncX plasmid transfer and persistence, partly explaining their widespread dissemination and offering potential targets for controlling their spread.
Methods
Bacterial strains and growth conditions
The strains used in this study are described in Supplementary Table 3. All strains were routinely cultured at 37 °C in LB broth (180 rpm) and on LB agar plates at 37 °C, and appropriate antibiotics were added as needed: ampicillin (100 μg/mL), kanamycin (30 μg/mL), chloramphenicol (30 μg/mL), and tetracycline (20 μg/mL). All strains were stored in 25% glycerol broth at −80 °C.
Construction of plasmids and strains
The plasmids used in this study are listed in Supplementary Table 3 and primers used are presented in Supplementary Table 4. Deletion mutants of pHNYX644 were constructed using the λ Red recombination system44. For non-overlapping genes, we constructed full ORF deletion mutants, whereas for overlapping genes, we created partial ORF deletion mutants to prevent interference with the expression of adjacent genes. Deletion of rs00140, phns, rs00175, prfah, rs00240, rs00260, rs00270, rs00275, rs00285, rs00290, and rs00300 genes were constructed using primers knock-rs00140-F/knock-rs00140-R, knock-phns-F/knock-phns-R, knock-rs00175-F/knock-rs00175-R, knock-prfah-F/knock-prfah-R, knock-rs00240-F/knock-rs00240-R, knock- rs00260-F/knock-rs00260-R, knock-rs00270-F/knock-rs00270-R, knock- rs00275-F/knock-rs00275-R, knock-rs00285-F/knock-rs00285-R, knock-rs00290-F/knock-rs00290-R, knock-rs00300-F/knock- rs00300-R, respectively. Deletion of IGR was constructed using primers IGR-up-F/IGR-up-R, IGR-kan-F/IGR-kan-R, IGR-down-F/IGR-down-R. The primers IGR-kan-F/IGR-kan-R were used to amplify the kanamycin resistance gene. The primers IGR-up-F/IGR-up-R were used to amplify the region (~600 bp) that are homologous to prfaH. The primers IGR-down-F/IGR-down-R were used to amplify the region covering rs00240 to virB1 but lacking IGR. The primer IGR-down-SD-F was used for constructing pHNYX644\(\Delta\)IGR-SD. All products were cloned into pHSG575 using the Seamless Cloning Kit (Genesand Biotech Co., Ltd, China) to generate a template for amplifying the fragments for homologous recombination. The other mutations in the promoter region of prfaH, including pHNYX644-PJ23119, pHNYX644-PJ23119-UTR, pHNYX644-W1-W12, and pHNYX644-T1A to T16A were constructed using the same strategy, and the corresponding primers are listed in Supplementary Table 4.
The pHN21SC1631T-3 with deletion of phns (X1) or prfaH (X1) were constructed using primers knock-phnsX1-F/knock-phnsX1-R and knock-prfaHX1-F/knock-prfaHX1-R, respectively. The kanamycin resistance genes in all mutants were eliminated by FLP expression plasmid pCP20. The putative promoter region of prfaH (350 bp upstream of the prfaH start codon) was cloned using primers promoter-prfaH-F/promoter-prfaH-R, and cloned into the upstream of a promoterless lacZ gene of pHGR01 plasmid to produce PprfaH-lacZ. The putative promoter region of virB1 (230 bp upstream of virB1 start codon) was cloned using primers promoter-virB1-F/promoter-virB1-R, and cloned into the pHGR01 plasmid to generate PvirB1-lacZ. The fragment ranging from prfaH promoter to virB1 but lack of prfaH was cloned from pHNYX644\(\Delta\)prfaH using primers promoter-prfaH-F/promoter-virB1-R, and cloned into the pHGR01 plasmid to generate PprfaH-virB1-lacZ. The IGRm1, IGRm2, IGRm3 and IGRm4 mutations were created based on PprfaH-virB1-lacZ using primers IGRm1-F/IGRm1-R, IGRm2-F/IGRm2-R, IGRm3-F/IGRm3-R, and IGRm4-F/IGRm4-R, respectively. The fragments of prfaH and phns with their native promoter were amplified from pHNYX644 using primers pro-prfaH-F/pro-prfaH-R and pro-phns-F/pro-phns-R, respectively, and cloned into pHSG575 to generate pHSG575-prfaH and pHSG575-phns. The prfaH mutants (prfaH-T9A to prfaH-F80G) were obtained by SOE PCR, and cloned into pHSG575. All constructs were confirmed by PCR and sequencing.
Bacterial conjugation assays
The Simonsen method45 was used to estimate the plasmid transfer rate (γ). The donor (D) strain used was E. coli BW25113/pHNYX644 or E. coli BW25113/ pHN21SC1631T-3 and their derivatives, while the recipient strain (R) was kanamycin-resistant E. coli BW25113::kan46. The numbers of donor (D) cells harboring pHNYX644 or its derivatives were estimated on LB plates containing ampicillin (100 μg/mL), while the donor (D) harboring pHN21SC1631T-3 or its derivatives were selected on LB plates containing tetracycline (20 μg/mL). Recipient (R) numbers were estimated on LB plates containing kanamycin (30 μg/mL). Transconjugants (T) were selected on LB plates containing kanamycin (30 μg/mL) and ampicillin (100 μg/mL)/ tetracycline (20 μg/mL). At the beginning of the experiment, the donor and recipient strains were individually cultured in 5 mL of LB broth overnight at 37 °C. Overnight cultures were adjusted to an OD600 = 1, and 3 µL of each culture was added to 3 mL of LB broth. At the start of mating, 100 µL of the mixture was collected to measure the initial cell density (N0). The cultures were incubated at 37 °C with shaking for 12 h. During the exponential phase, optical densities were measured at two time points, tb and ta, to calculate the growth rate using the formula:
where \(\psi\) is the growth rate and ODb and ODa are the optical densities at times tb and ta, respectively. This model assumes that donors (D), recipients (R), and transconjugants (T) grow at the same rate, but differences in growth dynamics could introduce bias, with the minimum detectable conjugation rate being approximately 1 × 10−18.
After 12 h, appropriate dilutions of the mating cultures were plated on selective LB agar plates to determine the total cell density (N), as well as the counts for donors (D), recipients (R), and transconjugants (T). Plates containing kanamycin, ampicillin, or tetracycline, as well as plates with combinations of these antibiotics, were used. The number of donors (D) was calculated by subtracting the count on the plate with both antibiotics from the ampicillin or tetracycline-only plate count. Similarly, the number of recipients (R) was calculated by subtracting the count on the plate with both antibiotics from the kanamycin-only plate count. The number of transconjugants (T) was determined from the plates containing both antibiotics. The total cell density (N) was the sum of donors (D), recipients (R), and transconjugants (T). The transfer rate (\({\gamma }\)) was calculated as45.
RNA extraction and RT-qPCR
According to the product manual, the same numbers of bacterial cells at the logarithmic growth stage were harvested for use with the E.Z.N. A. Bacterial RNA Kit (OMEGA BIO-TEK, USA) to extract total bacterial RNA. Then, 1% agarose gel electrophoresis was used to determine the integrity of the total RNA, and the NanoDrop 2000c to measure RNA concentration. cDNA synthesis was performed on RNA samples with a 23S/16S rRNA ratio of approximately 2.0 and an A260/A280 ratio of 1.8–2.2. According to the product manual, the HiScript® III All-in-one RT SuperMix Perfect for qPCR (Nuowei Zan Biotechnology Company, China) was used to reverse transcribe with approximately 1 µg of total RNA. The TB Green Premix Ex Taq II reagent (TaKaRa, Japan) and the LightCycle® 96 real-time PCR detection system (Roche, Switzerland) were used for detection. The reaction mixture contained 10 µL TBGreenExTaqII, 1 µL cDNA (diluted 1:10), 0.5 µL of each primer (10 µM), and 8 µL double-distilled water. Amplification conditions are as follows: 95 °C for 5 min, 40 cycles of 95 °C for 30 s, 60 °C for 30 s, and 72 °C for 20 s, followed by a melting reaction from 65 to 95 °C. The relative gene expression level was calibrated using the 2−ΔΔCt method47.
\({{\mathbf{\beta }}}\)-galactosidase assay
The cells carrying the lacZ fusion vector were cultured overnight at 37 °C in LB broth containing kanamycin. After overnight culture, the cells were diluted 1:10 into fresh medium and shaken at 37 °C (180 rpm) until the bacteria reached the logarithmic phase. The cells were collected and resuspended in lysis buffer (50 mM Tris–HCl pH 7.3, 1 mM DTT, 5% glycerol, and 1 mM EDTA). 1 mL of the cell suspension was mixed with 0.1 mL chloroform and 0.05 mL of 0.1% SDS, vortexed for 30 s, and centrifuged at 4 °C and 12,000×g for 5 min. The supernatant was used to measure protein concentration using the Bradford protein detection kit (TaKaRa, Japan). About 50 μL of the supernatant was mixed with 450 μL of lysis buffer. Subsequently, 0.1 mL of o-nitrophenyl-β-d-galactopyranoside (4 mg/L) was added immediately and the mixture was incubated at room temperature. When the color of the mixture turned yellow, 250 μL of 1 M Na2CO3 was added to terminate the reaction. The optical density (OD) at 420 nm was measured using a Multiskan spectrum microplate spectrophotometer (Thermo Labsystems, Franklin, MA, USA).
Protein expression, crystallization, and crystal structure determination
BL21 Gold (DE3) harboring pET28b-6\(\times\)Histag-PrfaH (1–155) or pET28b-6\(\times\)Histag-PrfaH(full length) was grown to OD600 = 0.5 at 37 °C, and induced by 0.2 mM IPTG for 16 h. The His-tag-PrfaH (1–155) (renamed as His-tag-∆14-PrfaH) or PrfaH(full-length) was purified by Ni-NTA affinity chromatography. Then, the purified protein was digested by the PreScission enzyme and the His-tag was removed by Ni-NTA column.
The purified ∆14-PrfaH was used for crystallization. Crytals were grown at 4 °C by the hanging drop vapor diffusion method. For the hanging drop, 1 μL of a 60 mg/mL protein solution in 50 mM Tris pH7.0, 500 mM NaCl, 0.1 mM PMSF, 5% glycerol, 1 mM TCEP, and 5 mM MgCl2 was mixed with 1 μL reservoir solution (0.1 M Sodium citrate tribasic dihydrate pH4.8, 5 mM MgCl2, and 18% Polyethylene glycol 20,000). Crystals were collected in a reservoir solution with 20% (V/V) glycerol, and flash-frozen in liquid nitrogen. Diffraction data for crystals were collected using X-ray at a wavelength of 0.97915 Å with beamline SSRF_BL10U2 at the Shanghai Synchrotron Radiation Facility (SSRF), at a temperature of 100 K. The collected data were analyzed by XDS48 and AimLess49. The AlphaFold model used for molecular replacement was generated using ColabFold version 1.3.050,51. The model was run with the following parameters: one query, template usage enabled, and Amber refinement applied. The multiple sequence alignment (MSA) was generated using MMseqs2, utilizing both the UniRef and Environmental databases. The model type selected was AlphaFold2-ptm, with five model predictions and three recycling steps. Models were ranked based on the pLDDT score, and the host URL used was https://api.colabfold.com. The AlphaFold predicted prfaH structure was used as a search model to perform molecular replacement using Phaser52. The model was built in Coot53 and optimized by Phenix54. The Ramachandran statistics for the final refined model were: 97.32% of residues in the favored regions, 2.68% in the allowed regions, and 0.00% in the outlier regions. Structural visualizations were performed by PyMOL (www.pymol.org).
Electrophoretic mobility shift assay (EMSA)
The purified ∆14-PrfaH was used for EMSA. Single-stranded DNAs (ss-WT, ss-W1–ss-W12) were synthesized by Tsingke Biotech Co., Ltd. The sequences of these ssDNAs are provided in Supplementary Table 4. The EMSA experiments were performed using the Electrophoretic Mobility-Shift Assay (EMSA) kit according to the manufacturer’s instructions (Thermo Fisher Scientific, USA). The binding assay was carried out on the ice in 10 μl reaction mixtures based on binding reaction buffer (150 mM KCl, 0.1 mM EDTA, 0.1 mM dithiothreitol, 10 mM Tris, pH 7.4). An appropriate amount of ssDNA (~50 ng,~4 pmol) was mixed with different concentrations of ∆14-PrfaH protein and incubated for 1 h at 25 °C. Subsequently, the mixture was subjected to 6% non-denaturing PAGE (40 min at 220 V). The gels were stained with SYBR® Green and imaged using a Bio-Rad Molecular Imager Chemi Doc XRS+ Imaging System.
Competition experiments and plasmids invasion assays
Competition experiments were conducted to assess the relative fitness of strains carrying pHNYX644 or its derivatives. Overnight cultures of the two strains were individually diluted 1:1000 in LB broth or M9 medium and mixed in a 1:1 ratio. The mixture was incubated at 37 °C with shaking at 180 rpm for 24 h, after which the population was diluted 1:100 into fresh LB broth. This process was repeated for a total duration of 4 days. Every 24 h, samples from the competitive mixtures were properly diluted and plated onto LB agar plates, both with and without antibiotics (kanamycin or ampicillin). The relative fitness (RF) was calculated using the following formula: RF = (log10 S2dt−log10 S2d0)/ (log10 S1dt−log10 S1d0), where RF represents the relative fitness of strain S2 compared to strain S1, and S1dt and S1d0 are the cell densities (CFU/mL) of S1 at the end and start of the competition, respectively, while S2dt and S2d0 are the corresponding values for S2. Each competition experiment was performed with three biological replicates.
The plasmid invasion test was performed according to the method described in previous studies43. All plasmid invasion experiments were carried out using three biological replicates. The overnight cultures of BW25113 were diluted 1:10 into 2 mL LB broth medium or M9 basal medium containing 0.4% glucose, and then mixed and cultured with BW25113/pHNYX644, BW25113/pHNYX644Δphns::kan, or all three strains.
Every 24 h, the cultures were diluted 1:100 into fresh medium and cultured for 10 days. Dilution plate counts were performed every 24 h. At each time point, the diluted mixed cultures were dropped onto LB plates without antibiotics, with kanamycin or ampicillin. For the mixed system of BW25113 and BW25113/pHNYX644: BW25113 colony count = colony count on the antibiotic-free plate−colony count on the ampicillin plate; BW25113/pHNYX644 colony count = colony count on the ampicillin plate; For the mixed system of BW25113 and BW25113/pHNYX644Δphns::kan: BW25113 colony count = colony count on the antibiotic-free plate−colony count on the kanamycin plate; BW25113/pHNYX644Δphns::kan colony count = colony count on the kanamycin plate; For the mixed system of all three strains: BW25113 colony count = colony count on the antibiotic-free plate−colony count on the ampicillin plate; BW25113/pHNYX644 colony count = colony count on the ampicillin plate−colony count on the kanamycin plate; BW25113/pHNYX644Δphns::kan colony count = colony count on the kanamycin plate.
Statistics and reproducibility
Statistical analyses were performed using GraphPad Prism 10.1.1. Graphical data are shown as the mean ± standard deviation from at least three independent biological replicates. Comparisons were made using one-way ANOVA with Dunnett’s T3 correction or two-tailed Student’s t-test with Welch’s correction. A 95% confidence interval was applied. Results were considered significant when the p-value was <0.05.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
All data supporting the findings of this study are included in the article and supplementary information files. The structure of PrfaH has been deposited in the Protein Data Bank (PDB ID 9IK0). GenBank accession number NZ_MK033578 for pHNYX644 and BioProject number PRJNA893238 (GCA_025909815.1) for pHN21SC1631T-3. The accession numbers for the remaining plasmids shown in Figure 5b are as follows: pKp189_4 (CP114757),pLAO96 (NZ_OP242301), BK31567 (JX193302), pTJ30-KPC (MZ853143), DE2 (CP132355), pB16EC1113-2 (CP088727), JT1-188 (CP099539), pNBBC01-3 (CP099731), p26019-4 (CP119052), pRES21 (CP034710), OXWO6B1 (LWLR01000003), pAR24_C (CP134760), p21254 (MN419432), pIP72 (MN612051). Source data for the graphs can be found in a single Excel file titled ‘Supplementary Data 1’ and any additional information can be provided by the corresponding author upon request. Uncropped and unedited gel images are available as Supplementary Fig. 11 in “Supplementary Information”.
References
Collaborators, A. R. Global burden of bacterial antimicrobial resistance in 2019: a systematic analysis. Lancet 399, 629–655 (2022).
De Oliveira, D. M. P. et al. Antimicrobial resistance in ESKAPE pathogens. Clin. Microbiol. Rev. 33, e00181–19 (2020).
Holmes, A. H. et al. Understanding the mechanisms and drivers of antimicrobial resistance. Lancet 387, 176–187 (2016).
Huemer, M., Mairpady Shambat, S., Brugger, S. D. & Zinkernagel, A. S. Antibiotic resistance and persistence-Implications for human health and treatment perspectives. EMBO Rep. 21, e51034 (2020).
Tacconelli, E. et al. Discovery, research, and development of new antibiotics: the WHO priority list of antibiotic-resistant bacteria and tuberculosis. Lancet Infect. Dis. 18, 318–327 (2018).
Castañeda-Barba, S., Top, E. M. & Stalder, T. Plasmids, a molecular cornerstone of antimicrobial resistance in the One Health era. Nat. Rev. Microbiol. 22, 18–32 (2024).
Wein, T. & Dagan, T. Plasmid evolution. Curr. Biol. 30, R1158–R1163 (2020).
Ilangovan, A., Connery, S. & Waksman, G. Structural biology of the Gram-negative bacterial conjugation systems. Trends Microbiol. 23, 301–310 (2015).
Bethke, J. H. et al. Vertical and horizontal gene transfer tradeoffs direct plasmid fitness. Mol. Syst. Biol. 19, e11300 (2023).
San Millan A., MacLean R. C. Fitness costs of plasmids: a limit to plasmid transmission. Microbiol. Spectr. 5, https://doi.org/10.1128/microbiolspec.MTBP-0016-2017(2017).
Wong, J. J., Lu, J. & Glover, J. N. Relaxosome function and conjugation regulation in F-like plasmids—a structural biology perspective. Mol. Microbiol. 85, 602–617 (2012).
Koraimann, G. Spread and persistence of virulence and antibiotic resistance genes: a ride on the F plasmid conjugation module. EcoSal Plus 8, https://doi.org/10.1128/ecosalplus.ESP-0003-2018 (2018).
Will, W. R. & Frost, L. S. Hfq is a regulator of F-plasmid TraJ and TraM synthesis in Escherichia coli. J. Bacteriol. 188, 124–131 (2006).
Gubbins, M. J., Arthur, D. C., Ghetu, A. F., Glover, J. N. & Frost, L. S. Characterizing the structural features of RNA/RNA interactions of the F-plasmid FinOP fertility inhibition system. J. Biol. Chem. 278, 27663–27671 (2003).
Arthur, D. C. et al. FinO is an RNA chaperone that facilitates sense-antisense RNA interactions. EMBO J. 22, 6346–6355 (2003).
Will, W. R. & Frost, L. S. Characterization of the opposing roles of H-NS and TraJ in transcriptional regulation of the F-plasmid tra operon. J. Bacteriol. 188, 507–514 (2006).
Koraimann, G., Teferle, K., Markolin, G., Woger, W. & Högenauer, G. The FinOP repressor system of plasmid R1: analysis of the antisense RNA control of traJ expression and conjugative DNA transfer. Mol. Microbiol. 21, 811–821 (1996).
Lau-Wong, I. C., Locke, T., Ellison, M. J., Raivio, T. L. & Frost, L. S. Activation of the Cpx regulon destabilizes the F plasmid transfer activator, TraJ, via the HslVU protease in Escherichia coli. Mol. Microbiol. 67, 516–527 (2008).
Vial, L. & Hommais, F. Plasmid-chromosome cross-talks. Environ. Microbiol. 22, 540–556 (2020).
Liu, J. H. et al. Plasmid-mediated colistin-resistance genes: mcr. Trends Microbiol. 32, 365–378 (2024).
Wu, W. et al. NDM metallo-β-lactamases and their bacterial producers in health care settings. Clin. Microbiol. Rev. 32, e00115–e00118 (2019).
Cui, C. Y. et al. Comprehensive analysis of plasmid-mediated tet(X4)-positive Escherichia coli isolates from clinical settings revealed a high correlation with animals and environments-derived strains. Sci. Total Environ. 806, 150687 (2022).
He, T. et al. Emergence of plasmid-mediated high-level tigecycline resistance genes in animals and humans. Nat. Microbiol. 4, 1450–1456 (2019).
Sun, J. et al. Plasmid-encoded tet(X) genes that confer high-level tigecycline resistance in Escherichia coli. Nat. Microbiol. 4, 1457–1464 (2019).
Walsh, T. R., Weeks, J., Livermore, D. M. & Toleman, M. A. Dissemination of NDM-1 positive bacteria in the New Delhi environment and its implications for human health: an environmental point prevalence study. Lancet Infect. Dis. 11, 355–362 (2011).
Pitout, J. D. D., Peirano, G., Kock, M. M., Strydom, K. A. & Matsumura, Y. The global ascendency of OXA-48-type carbapenemases. Clin. Microbiol. Rev. 33, e00102–e00119 (2019).
Yi, L. et al. PixR, a Novel activator of conjugative transfer of IncX4 resistance plasmids, mitigates the fitness cost of mcr-1 carriage in Escherichia coli. mBio 13, e0320921 (2022).
Wu, R. et al. Fitness Advantage of mcr-1-Bearing IncI2 and IncX4 plasmids in vitro. Front. Microbiol. 9, 331 (2018).
Johnson, T. J. et al. Expansion of the IncX plasmid family for improved identification and typing of novel plasmids in drug-resistant Enterobacteriaceae. Plasmid 68, 43–50 (2012).
Coetzee, J. N, Bradley, D. E, du Toit, L & Hedges, R. W Bacteriophage X-2: a filamentous phage lysing IncX-plasmid-harbouring bacterial strains. J. Gen. Microbiol. 134, 2535–2541 (1988).
Rozwandowicz, M. et al. Plasmids carrying antimicrobial resistance genes in Enterobacteriaceae. J. Antimicrob. Chemother. 73, 1121–1137 (2018).
Norman, A., Hansen, L. H., She, Q. & Sørensen, S. J. Nucleotide sequence of pOLA52: a conjugative IncX1 plasmid from Escherichia coli which enables biofilm formation and multidrug efflux. Plasmid 60, 59–74 (2008).
Costa, T. R. D. et al. Type IV secretion systems: advances in structure, function, and activation. Mol. Microbiol. 115, 436–452 (2021).
Bradley, D. E. Determination of pili by conjugative bacterial drug resistance plasmids of incompatibility groups B, C, H, J, K, M, V, and X. J. Bacteriol. 141, 828–837 (1980).
Bailey, M. J., Hughes, C. & Koronakis, V. RfaH and the ops element, components of a novel system controlling bacterial transcription elongation. Mol. Microbiol. 26, 845–851 (1997).
Wang, B., Gumerov, V. M., Andrianova, E. P., Zhulin, I. B. & Artsimovitch, I. Origins and molecular evolution of the NusG paralog RfaH. mBio 11, e02717–e02720 (2020).
Santangelo, T. J. & Roberts, J. W. RfaH, a bacterial transcription antiterminator. Mol. Cell 9, 698–700 (2002).
Zuber, P. K., Schweimer, K., Rösch, P., Artsimovitch, I. & Knauer, S. H. Reversible fold-switching controls the functional cycle of the antitermination factor RfaH. Nat. Commun. 10, 702 (2019).
Zuber, P. K. et al. The universally-conserved transcription factor RfaH is recruited to a hairpin structure of the non-template DNA strand. Elife 7, e36349 (2018).
Kim, S. R., Funayama, N. & Komano, T. Nucleotide sequence and characterization of the traABCD region of IncI1 plasmid R64. J. Bacteriol. 175, 5035–5042 (1993).
Ziegelin, G., Fürste, J. P. & Lanka, E. TraJ protein of plasmid RP4 binds to a 19-base pair invert sequence repetition within the transfer origin. J. Biol. Chem. 264, 11989–11994 (1989).
Lu, J., Wu, R., Adkins, J. N., Joachimiak, A. & Glover, J. N. Crystal structures of the F and pSLT plasmid TraJ N-terminal regions reveal similar homodimeric PAS folds with functional interchangeability. Biochemistry 53, 5810–5819 (2014).
Haft, R. J., Mittler, J. E. & Traxler, B. Competition favours reduced cost of plasmids to host bacteria. ISME J. 3, 761–769 (2009).
Datsenko, K. A. & Wanner, B. L. One-step inactivation of chromosomal genes in Escherichia coli K-12 using PCR products. Proc Natl Acad Sci USA 97, 6640–6645 (2000).
Simonsen, L., Gordon, D. M., Stewart, F. M. & Levin, B. R. Estimating the rate of plasmid transfer: an end-point method. Microbiology 136, 2319–2325 (1990).
Yang, J. et al. The evolution of infectious transmission promotes the persistence of mcr-1 plasmids. mBio 14, e0044223 (2023).
Livak, K. J. & Schmittgen, T. D. Analysis of relative gene expression data using real-time quantitative PCR and the 2−ΔΔCT method. Methods 25, 402–408 (2001).
Kabsch, W. Integration, scaling, space-group assignment and post-refinement. Acta Crystallogr. Sect. D 66, 133–144 (2010).
Evans, P. R. & Murshudov, G. N. How good are my data and what is the resolution? Acta Crystallogr. D Biol. Crystallogr. 69, 1204–1214 (2013).
Mirdita, M. et al. ColabFold: making protein folding accessible to all. Nat. Methods 19, 679–682 (2022).
Kim, G. et al. Easy and accurate protein structure prediction using ColabFold. Nat. Protoc. https://doi.org/10.1038/s41596-024-01060-5 (2024).
McCoy, A. J. Solving structures of protein complexes by molecular replacement with Phaser. Acta Crystallogr. D Biol. Crystallogr. 63, 32–41 (2007).
Emsley, P., Lohkamp, B., Scott, W. G. & Cowtan, K. Features and development of Coot. Acta Crystallogr. Sect. D 66, 486–501 (2010).
Afonine, P. V. et al. Towards automated crystallographic structure refinement with phenix.refine. Acta Crystallogr. D Biol. Crystallogr. 68, 352–367 (2012).
Acknowledgements
This work was funded by the National Natural Science Foundation of China (No. 32141002) and the National Key Research and Development Program of China (No. 2022YFC2303900). We are grateful to Vincent Burrus from Université de Sherbrooke and Beile Gao from the South China Sea Institute of Oceanology for their helpful comments on this paper. We would like to thank ReadCrystal Technology Co., Ltd for providing the protein crystallization technology platform.
Author information
Authors and Affiliations
Contributions
Conceptualization: J.H.L. and J.Y.; Investigation: J.Y., Y.Y.L., J.J.Y., and X.H.C.; Writing: J.H.L. and J.Y.; Review and editing: C.Z.W., L.C.L., R.A.M., X.L.Z., Z.Y.H., and M.Z.D.; Supervision: J.H.L. and J.Y.; Funding acquisition: J.H.L.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Communications Biology thanks Joanna Timmins and the other, anonymous, reviewers for their contribution to the peer review of this work. Primary Handling Editors: Wendy Mok and Tobias Goris.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Yang, J., Lu, Y., Yu, J. et al. Comprehensive analysis of Enterobacteriaceae IncX plasmids reveals robust conjugation regulators PrfaH, H-NS, and conjugation-fitness tradeoff. Commun Biol 8, 363 (2025). https://doi.org/10.1038/s42003-025-07782-w
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s42003-025-07782-w
