
Abstract
To address the pressing need to improve breast cancer outcomes, we identify 9 plasma proteins with significant associations to breast cancer, namely ULK3, CSK, ASIP, TLR1 in breast cancer, ADH5, SARS2, ULK3, UBE2N in Luminal A subtype, PEX14 in Luminal B subtype. Tumor immune cell infiltration analysis and mutation phenotypes in mice further demonstrate a complex pattern of interaction between these genes and immune responses. Compared to normal tissues, tumor tissues exhibit reduced expression of ULK3 and CSK. Notably, elevated ULK3 expression in both breast cancer and the Luminal A subtype is significantly associated with prolonged recurrence-free survival. Overexpression of CSK and ULK3 is confirmed to significantly inhibit the proliferation and migratory ability of MCF-7 cells. Additionally, three drug candidates—TG100801, Hydrochlorothiazide, and Imatinib—show promise in targeting these proteins, contributing valuable insights for prioritizing drug development in realm of breast cancer.
Similar content being viewed by others


Introduction
Breast cancer (BC) is a malignant neoplasm that originates from cells within the breast tissue. It is the most prevalent cancer in women and also constitutes the primary cause of cancer-related mortality in women globally. BC encompasses four distinct molecular subtypes: human epidermal growth factor receptor 2 positive (HER2), luminal A, luminal B and triple-negative breast cancer (TNBC). Significantly, the clinical attributes and biological behaviors of these distinct subtypes exhibit substantial variations. The HER2 and TNBC subtypes manifest heightened invasiveness, increased metastatic potential, susceptibility to relapse, and an unfavorable prognosis. It is noteworthy that approximately 46% of TNBC patients present with distant metastasis at the point of diagnosis1.
In 2020, the International Agency for Research on Cancer (IARC, https://www.iarc.who.int/) released that there were approximately 2.3 million new cases of BC worldwide, accounting for 24.5% of all malignancy in women. In addition, the worldwide fatality count attributed to breast cancer in 2020 reached approximately 685,000, constituting 15.5% of female cancer-related deaths. The widespread occurrence of BC has resulted in substantial physical and economic burdens on individuals and nations globally. Therefore, there exists a compelling necessity to explore targeted drugs of BC.
Plasma proteins encompass both actively secreted and passively leaked proteins originating from diverse organs and cells. They play pivotal roles in numerous biological processes and are commonly dysregulated or upregulated in a myriad of diseases, including cardiovascular disease and inflammatory bowel disease2,3. These proteins function as biomarkers, facilitating the diagnosis of diverse diseases. Consequently, they hold paramount significance in clinical practice and drug development over the past decade. Circulating proteins, being readily targetable using small molecules or biologics (such as monoclonal antibodies), emerge as compelling candidates for potential drug targets.
Mendelian randomization (MR) analysis was employed to evaluate potential causal relationships between plasma proteins and BC. MR stands as an invaluable tool for inferring causal relationship between exposure and outcome, gaining widespread usage in numerous prominent studies in recent years4,5. Its fundamental principle relies on the random allocation of genes during the mitosis stage, enabling the inference of the impact of biological factors on disease through their effects on phenotypes6,7. This approach offers the advantages of the randomized controlled trial, overcoming potential confounding and reverse causality, while also eliminating the disadvantage for the randomized controlled trial, thus saving financial and human resources. Protein quantitative trait locus (pQTLs) represent genetic variants related with plasma proteins at the Genome-wide Association Study (GWAS) level2,8,9, facilitating the ability to distinguish the relationship between plasma proteins and BC10. We performed a systematic exploration into the potential causality between plasma proteins and breast cancer, including its four subtypes. Finally, plasma proteins involved in prognosis were prioritized as potential therapeutic targets for drug repurposing or the development of new agents.
Colocalization analysis, a statistical method employed in genetics and genomics, aims to discern whether there exists a common genetic foundation for the observed associations of distinct traits. It assists in ascertaining whether a specific genetic loci influences both traits or if the observed associations are, in fact, independent11. It has been applied to enhance resolution and validate the robustness of protein instrumental variables across a diverse spectrum of diseases, including hematological disorders and neurological diseases12,13.
Previous studies have used MR to explore the relationship between plasma proteins and breast cancer, laying important groundwork but leaving gaps in understanding14. Mälarstig et al. measured plasma protein levels using Olink PEA Explore in a cohort of 598 patients with a follow-up of two years. While informative, the study was limited by its small sample size and short duration14. Similarly, Papier et al. assessed 1463 plasma proteins across 19 cancer types in the UK Biobank, though their findings on breast cancer were inconclusive15. Smith-Byrne et al. further explored potential causal links between plasma proteins and breast cancer using two-sample Mendelian randomization (TSMR) and colocalization analysis16. However, these studies have not yet conducted deeper validation and exploration behind the causal relationship.
In this study, we employed an integrated approach that connected genetic variations with breast cancer, plasma proteins, biological pathways, and drug discovery. The analysis identified 62 circulating proteins, including ULK3, TLR1, CSK, ASIP, ADH5, SARS2, UBE2N, and PEX14, which were strongly supported by colocalization analysis. Through experimental studies and validation in clinical cohorts, CSK and ULK3 were confirmed as critical targets closely linked to Luminal A breast cancer. Additionally, three drug candidates—TG100801, Hydrochlorothiazide, and Imatinib—demonstrated potential for targeting these proteins in realm of breast cancer treatment.
Results
Proteome-wide MR analysis
The deCode database contains a total of 4907 plasma protein data. Among them, 1815 proteins successfully met the four established criteria and were slated for inclusion in the analysis (Supplementary Data 1). The cohort included 133,384 breast cancer patients and 113,789 controls. Among the patients, 45,253 were classified as effective luminal A-like cases, while 8602 were diagnosed with triple-negative breast cancer. We conducted TSMR and Summary-data-based Mendelian randomization (SMR) analyses involving 1815 plasma proteins and their association with breast cancer and its subtypes. Upon excluding associations that failed the Heterogeneity in dependent instrument (HEIDI) test and implementing multiple testing correction, we identified a final set of 62 circulating proteins. Among these, 33 were associated with overall breast cancer, 3 with Luminal B, and 24 with Luminal A subtypes, 2 with triple-negative breast cancer. (Tabel 1, Supplementary Data 2-11) The comprehensive results of the MR analysis were visually depicted in Fig. 1a and Supplementary Figs. 1-2. It can be seen that the circulating proteins of various subtypes are not consistent, indicating heterogeneity among different subtypes of breast cancer. The position of 62 genetically predicted circulating protein genes was shown in Fig. 1b. More detailed characteristic of 62 proteins were shown in Table 1. Based on the MR power analysis, our study had sufficient statistical power to detect associations between plasma proteins and breast cancer (Supplementary Data 12)

a The identified gene proteins after colocalization analysis and mendelian analysis. * & # stand different probes in the Decode database: *13094_75 #8427_118 PH4 posterior probability of H4. The diagram depicts concentric circles, with each circle’s distinct coloration corresponding to specific groups. The innermost circle denotes different groups, while the middle circle signifies the colocalization result, and the outermost circle represents the TSMR result. Color gradients, ranging from red to blue, symbolize β values spanning from −0.05 to 0.5. In this gradient, red signifies protective factors, whereas blue indicates risk factors. b Manhattan plots for identified 62 circulating proteins in mendelian analysis stage. Results are plotted by gene start position. Each point signifies an individual association test between a gene and breast cancer, arranged by genomic position on the x-axis and association strength on the y-axis, represented as the −log10(P) of a z-score test.
Colocalization analysis results
To ascertain the robustness of the previously identified 62 circulating proteins, we performed colocalization analysis. As a result, we identified 9 circulating proteins with high support (posterior probability of H4, PH4 > 0.8, Tier 1) and 13 with medium support (0.5 ≤ PH4 ≤ 0.8, Tier 2), 40 with limited support (PH4 < 0.5, Tier 3) as shown in the Fig. 1a.
We have identified four robust support proteins in breast cancer: Unc-51 Like Kinase 3 (ULK3), Toll Like Receptor 1 (TLR1), C-terminal Src kinase (CSK), Agouti Signaling Protein (ASIP). Notably, ULK3 (Odds ratio [OR] 0.680, 95% confidence interval [CI] 0.586–0.790) and CSK (OR 0.639, 95%CI 0.527–0.775) exhibited protective effects, whereas the ASIP (OR 1.097, 95%CI 1.049–1.148) and TLR1 (OR 1.375, 95%CI 1.179–1.604) were associated with an increased risk of breast cancer. In the context of luminal A breast cancer, four plasma proteins exhibited high support: Alcohol dehydrogenase 5 (ADH5), Seryl-tRNA Synthetase 2 (SARS2), Ubiquitin-conjugating enzyme E2 N (UBE2N), ULK3. ULK3 (OR 0.597, 95%CI 0.489–0.727) and UBE2N (OR 0.499, 95%CI 0.356–0.698) were identified as protective factors, while ADH5 (OR 1.200, 95%CI 1.079–1.335) and SARS2 (OR 1.323, 95%CI 1.142–1.532) were deemed risk factors. Additionally, one high-support protein in luminal B breast cancer, namely peroxisomal biogenesis factor 14 (PEX14), was identified as a risk factor (OR 3.268, 95%CI 1.834–5.822).
In the domain of medium support proteins, nine instances of breast cancer were identified, namely collagen type VI alpha 3 chain (COL6A3, OR 1.200, 95%CI 1.086–1.326), ADH5 (OR 1.128, 95%CI 1.053–1.210), SARS2 (OR 1.209, 95%CI 1.08–1.353), R-spondin 3 (RSPO3, OR 0.895, 95%CI 0.84–0.953), R-spondin 3 (RSPO3, OR 0.905, 95%CI 0.854–0.958), Switching B Cell Complex Subunit SWAP70 (SWAP70, OR 1.074, 95%CI 1.03–1.12), Snurportin 1 (SNUPN, OR 0.802, 95%CI 0.732–0.878), Layilin(LAYN, OR 1.650, 95%CI 1.312–2.076), Recombinant Karyopherin Alpha 2 (OR 0.634, 95%CI 0.489–0.822). Coincidentally, two proteins share the designation RSPO3; however, they employ distinct gene probes, with serial numbers 13094_75 and 8427_118, respectively, in the deCODE database. Meanwhile, Luminal A breast cancer exhibited three proteins (SNUPN, LAYN, CSK) with medium support, (Supplementary Data 13) and Luminal B type presented a single case (Ubiquitin-associated and SH3 domain-containing protein B, UBASH3B). (Supplementary Data 14) Identified potential targets for the HER-2 enriched subtype and triple-negative breast cancer in MR analysis were all classified as tier 3 targets in the colocalization stage. Additional details can be found in Table 1. (Supplementary Data 15 and 16)
Protein-protein interaction network analysis results
Proteins CSK, ULK3, LAYN, ASIP, SWAP70, ADH5, SARS2, and SNUPN have been identified as playing noteworthy roles in breast cancer. To elaborate, CSK, ULK3, LAYN, SWAP70, ADH5, and SNUPN exhibited particular importance in Luminal A breast cancer. Within TNBC, the proteins Interleukin 33, killer cell lectin like receptor B1, and interleukin 1 receptor like 1 emerged as core nodes, underscoring their pivotal role in this particular subtype of breast cancer. Similarly, in Luminal B breast cancer, proteins Peroxisomal biogenesis factor 13, PEX14, Cadherin 17, UBASH3B, and ELAV like RNA binding protein 3 served as core nodes, indicating their significance within this subtype. Further details can be seen in the Supplementary Figs. 3-6 and Supplementary Data 17. The depicted nodes in the Supplementary Figs. 3-6 represented proteins, while the variously colored lines between the nodes signified distinct types of interactions, including physical (protein-protein) interactions, co-expression, colocalization, genetic interactions, enrichment pathways, and predictions from websites.
Pathway enrichment results
In addition, we also conducted pathway enrichment analysis to further explore the pathways involved in breast cancer and its subtypes. Due to limitations in the number of genes, we conducted pathway analysis on breast cancer and Luminal A breast cancer.
In the context of breast cancer, Gene Ontology (GO) analysis revealed significant enrichment in the following biological processes (BP): nuclear transport (p = 0.002), nucleocytoplasmic transport (p = 0.002), and blood coagulation (p = 0.005). In the cellular components (CC) category, the endoplasmic reticulum lumen (p = 0.001) was notably enriched, with 4 genes involved. For molecular functions (MF), serine-type endopeptidase inhibitor activity was significantly represented (p = 0.0005). Disease Ontology (DO) analysis identified several diseases with substantial enrichment, including nephritis (p = 0.026), glomerulonephritis (p = 0.014), lipid metabolism disorders (p = 0.007), and cervical squamous cell carcinoma (p = 0.017). Furthermore, Kyoto Encyclopedia of Genes and Genomes (KEGG) analysis highlighted enriched pathways such as fatty acid degradation (p = 0.004), drug metabolism via cytochrome P450 (p = 0.011), metabolism of xenobiotics by cytochrome P450 (p = 0.013), nucleocytoplasmic transport (p = 0.023), and the HIF-1 signaling pathway (p = 0.024). (Fig. 2 and Supplementary Data 18)

a KEGG pathway enrichment analysis of identified proteins associated with breast cancer. b DO disease categorization analysis of identified proteins associated with breast cancer. c GO biological process analysis of identified proteins associated with breast cancer. BP biological processes, CC cellular components, MF molecular functions.
For luminal A breast cancer, the GO analysis identified enriched biological processes, including negative regulation of phagocytosis, disaccharide metabolism, and skeletal muscle cell proliferation (all p < 0.05). The CC analysis revealed significant enrichment of the collagen-containing extracellular matrix (p = 0.0001). Notable molecular functions included phosphatidylserine binding (p = 0.003) and nucleocytoplasmic carrier activity (p = 0.0007). KEGG analysis underscored key pathways such as nucleocytoplasmic transport (p = 0.015), highlighting their potential significance in this research. (Supplementary Fig. 7 and Supplementary Data 19)
Mouse knock-out model results
Eight plasma proteins were identified as highly relevant through colocalization analysis: ULK3, TLR1, CSK, ASIP, ADH5, SARS2, UBE2N, and PEX14. Except for ASIP and SARS2, which were not available in the database, mutations related to the remaining six proteins were documented.
Mutations in the ULK3 gene in mice were associated with diverse phenotypes affecting the endocrine/exocrine, reproductive, hematopoietic, and immune systems, underscoring the gene’s critical role in these areas. Similarly, TLR1 was implicated in the hematopoietic and immune systems; CSK influenced the endocrine/exocrine, hematopoietic, and immune systems, as well as aging and mortality; ADH5 was involved in hematopoiesis and immunity, in addition to aging and mortality; UBE2N played a significant role in the endocrine/exocrine, hematopoietic, reproductive and immune systems, alongside aging and mortality; and PEX14 was important in the system related to aging and mortality. Collectively, five of the six proteins engaged the hematopoietic and immune systems. Further exploration of the mutation phenotypes primarily revealed involvement in immunoglobulin levels, cytokines, immune cells, and immune organs (thymus, spleen, lymph nodes), with specific details available in Table 2 and Supplementary Data 20.
Tumor immune cell infiltration
In murine knockout models, we identified specific plasma proteins associated with the immune system that may play a significant role in the pathogenesis of breast cancer. Consequently, we further validated the relationship between the expression of specific genes and immune cell infiltration in a breast cancer cohort. The results indicated a complex interaction pattern between gene expression and immune responses in breast cancer. Specifically, ULK3 expression was positively correlated with CD4 + T cell infiltration (r = 0.13, p = 7.08E−05) but negatively correlated with CD8 + T cell (r = −0.10, p = 1.41E−03) and macrophage infiltration (r = −0.10, p = 1.12E−03). TLR1 exhibited a positive correlation with all six studied immune cells (r ranging from 0.42 to 0.72, p < 0.05). CSK showed positive correlations with B cells, CD8 + T cells, CD4 + T cells, neutrophils, and dendritic cells (r values were 0.34, 0.10, 0.41, 0.33, and 0.37, respectively). In hormone receptor-positive breast cancer, ADH5 was positively correlated with CD8 + T cells, CD4 + T cells, macrophages, neutrophils, and dendritic cells, with r values of 0.28, 0.15, 0.21, 0.20, and 0.14, respectively. Similarly, UBE2N displayed positive correlations with B cells, CD8 + T cells, macrophages, neutrophils, and dendritic cells (all P < 0.05). Further details are available in Supplementary Data 21.
Survival analysis results
Survival analyses were performed on a platform integrating data from Gene Expression Omnibus (GEO), European Genome-Phenome Archive (EGA), The Cancer Genome Atlas (TCGA), and Molecular Taxonomy of Breast Cancer International Consortium (Metabric) databases. Recurrence free survival (RFS) and overall survival (OS) were considered as the primary outcome measures, with a follow-up period spanning 150 to 300 months. As shown in Figs. 3–4 and Supplementary Figs. 8-9, heightened expressions of ULK3 (Hazard Ratio (HR) 0.67, 95% CI 0.58–0.78, P = 1.7e−07), SNUPN (HR 0.82, 95% CI 0.74–0.91, P < 0.001), and ASIP (HR 0.82, 95% CI 0.74–0.91, P < 0.001) were correlated with a statistically significant extension of RFS. Additionally, breast cancer patients with high RSPO3 expression (HR 0.7, 95% CI 0.54–0.92, P = 0.010) and ASIP expression (HR 0.76, 95% CI 0.63–0.92, P = 0.004) had longer OS compared to those with low expression, with statistically significant differences.

Overall and recurrence free survival analysis of identified plasma proteins with high support in breast cancer.

Overall and recurrence free survival analysis of identified plasma proteins with high support in Luminal breast cancer.
In Luminal A breast cancer, elevated expression levels of ULK3 (HR 0.65, 95% CI 0.51–0.83, P < 0.001) and SNUPN (HR 0.77, 95% CI 0.66–0.91, P = 0.002) were significantly linked to prolonged RFS. Meanwhile, there was no notable disparity in RFS between high and low expression groups of CSK and ADH5 proteins. Similarly, no significant distinctions in OS were discerned between high and low expression groups for CSK, ULK3 and ADH5 proteins. (Figs. 3–4 and Supplementary Fig. 10)
In Luminal B breast cancer, there were no statistically significant differences in RFS and OS between high and low expression groups for PEX14 proteins. (Figs. 3–4 and Supplementary Fig. 11)
Druggable protein
As one protein concurrently manifested in breast cancer and its subtypes, we identified a total of eight high-support proteins actually. We searched for the 8 proteins obtained above in the mentioned database: 3 proteins were found in the database, namely ULK3, CSK, ADH5, as shown in Fig. 5. No information was available for the remaining high-support proteins (ASIP, TLR1, PEX14, SARS2, UBE2N).

Discovered drug targeting identified plasma proteins with robust support in breast cancer.
CSK functions as a protective factor in breast cancer by inhibiting SRC. TG100801, an SRC inhibitor, emerges as a potential drug target for breast cancer and is currently in the investigation stage. Hydrochlorothiazide was reported to be associated with CSK, yet the specific direction of this relationship remains unknown, warranting further exploration in breast cancer17. ULK3 serves as a protective factor in breast cancer. Previous literature suggested a connection between Imatinib and the ULK3 protein, but the specific nature of this relationship was unclear and required additional investigation18. Hydrochlorothiazide and Imatinib are both in approved stage. On the contrary, ADH5 is identified as a risk factor for breast cancer. Inhibitors of ADH5, namely Nitrefazole, N6022, and Cavosostat, have been recognized previously. While Nitrefazole has obtained approval, the latter two await approval.
Immunohistochemistry
We collected tumor and adjacent non-tumor tissues from 25 patients with Luminal A subtype breast cancer. All cases were pathologically classified as invasive ductal carcinoma, with a mean patient age of 40 years and clinical stages ranging from I to III. Detailed information is provided in Supplementary Data 22. Among these high-confidence proteins, only three—ULK3, CSK, and ADH5—were identified in the database with corresponding drugs. Consequently, we performed further validation on these proteins. This study conducted an immunohistochemical analysis of three proteins—CSK, ULK3, and ADH5—in both tumor and adjacent normal tissues. Qualitative comparisons and quantitative assessments of the immunohistochemistry images revealed that the expression levels of CSK and ULK3 were significantly lower in tumor tissues compared to adjacent normal tissues, with markedly reduced staining intensity (P < 0.001, 0.005 respectively). In contrast, ADH5 expression was significantly downregulated in tumor tissues relative to normal tissues (P = 0.006). All relevant immunohistochemical results and their quantitative analyses were presented in the Fig. 6 and Supplementary Data 23.

a Representative images showing CSK, ULK3, and ADH5 expression in Luminal A breast cancer tissues and adjacent normal tissues. Positive staining is indicated by brown coloration. b Quantitative analysis of protein expression in tumor and adjacent tissues in Luminal A breast cancer. Quantification of CSK, ULK3, and ADH5 expression levels in cancer tissues compared to normal tissues. AOD average optical density, T tumor, N normal. *P < 0.05, **P < 0.01, ***P < 0.001. Data are presented as mean ± standard deviation. (N = 25 biologically independent samples).
Cell proliferation and migration results
To elucidate the functional roles of potential drug targets in breast cancer, MCF-7 cells were transfected with lentiviral vectors carrying the CSK and ULK3 genes. Real-time quantitative polymerase chain reaction (qPCR) analysis confirmed a significant upregulation of CSK and ULK3 expression in transfected MCF-7 cells (P < 0.001) (Fig. 7a, b and Supplementary Data 24). Cell Counting Kit-8 (CCK-8) assay results demonstrated that overexpression of CSK and ULK3 significantly inhibited the proliferation of MCF-7 cells (P < 0.001) (Fig. 7c and Supplementary Data 25).

a, b CSK and ULK3 were significantly overexpressed in MCF-7 cells 24 hours after lentiviral transfection. c Cell proliferation was assessed using the CCK-8 assay at 24, 48, 72, and 96 hours in MCF-7 cells overexpressing CSK and ULK3b (N = 5 per group). d–g Cell migration was evaluated using a wound healing assay in MCF-7 cells overexpressing CSK and ULK3 at 24 hours. *P < 0.05, **P < 0.01, ***P < 0.001.
In the wound healing assay conducted 24 hours post-scratch, the cell migration rate was 35.56% in the ULK3 overexpression group compared to 81.66% in the corresponding control group. Similarly, the migration rate was 82.56% in the CSK overexpression group, compared to 95.90% in its control group. These findings further confirmed that overexpression of CSK and ULK3 markedly reduced the migratory ability of MCF-7 cells (P < 0.001) (Supplementary Data 26), consistent with the results of the CCK-8 assay. In conclusion, overexpression of CSK and ULK3 inhibits both the proliferation and migration of MCF-7 breast cancer cells (Fig. 7d–g).
Discussion
This study utilized MR analysis to investigate the potential causality between circulating plasma proteins and the risk of breast cancer and its subtypes. The results revealed 62 circulating proteins, with 33 associated with breast cancer, 24 with Luminal A, 3 with Luminal B, and 2 with triple-negative breast cancer following MR analysis. The circulating proteins, namely ULK3, TLR1, CSK, ASIP, ADH5, SARS2, UBE2N, PEX14, were identified to be highly supported through colocalization analysis. Among them, we identified three plasma proteins with potential druggability, each already associated with specific targeted therapies. Through experimental studies and validation in clinical cohorts, CSK and ULK3 were confirmed as critical targets closely linked to Luminal A breast cancer.
In the pathway enrichment analysis of breast cancer, we identified that these genes were primarily involved in nucleocytoplasmic transport and the HIF-1 signaling pathway. In the context of Luminal A breast cancer, these genes were also enriched in pathways related to nucleocytoplasmic transport. HIF-1 is a critical regulator of cellular adaptation under low-oxygen conditions, driving breast cancer cells to adapt to hypoxic environments and promoting tumor progression. HIF-1 consists of two subunits, HIF-1α and HIF-1β. Under normal oxygen conditions, HIF-1α is rapidly degraded; however, under hypoxic conditions, HIF-1α stabilizes and dimerizes with HIF-1β, thereby activating the transcription of downstream target genes19. The activation of HIF-1α promotes metabolic reprogramming in breast cancer cells, facilitating aerobic glycolysis, increasing glucose uptake, and enhancing lactate production. Moreover, the HIF-1 signaling pathway regulates the expression of genes related to angiogenesis, cell proliferation, invasion, and metastasis, such as vascular endothelial growth factor and glycolytic enzymes. Thus, targeting the HIF-1 signaling pathway could provide new therapeutic opportunities for breast cancer treatment20,21.
Many of the plasma proteins identified in this study were found to play a significant role in regulating hematopoietic and immune system functions. Specifically, these proteins were associated with immunoglobulin levels, cytokine production, immune cell activation, and the functionality of immune organs. Additionally, the observed immune cell infiltration further reinforced the interaction between these plasma proteins and immune cells, suggesting their potential involvement in modulating immune responses. While current researches predominantly focus on the immune landscape of triple-negative breast cancer, our findings indicate that the immune microenvironment in Luminal breast cancer also warrants attention. Future investigations into the potential for immunotherapy in the treatment of Luminal-type breast cancer, particularly in later lines of therapy, would be valuable.
CSK was recognized to participate in environmental information processing, such as the classic pathway for breast cancer, Epidermal growth factor receptor (EGFR) signaling pathway. EGFR is a conserved carboxy-terminal tyrosine kinase, encompassing Src homology domains 2 and 3, along with the Src family catalytic domain SH122. EGFR and Src tyrosine kinase cooperatively regulate EGFR-mediated cell signaling under pathological conditions, facilitating cellular transformation and tumor development. CSK inhibited Src activation, playing a crucial role in downregulating ErbB-2/neu-activated Src kinase and suppressing the tumor invasion of breast cancer23. Src kinase plays a crucial role in promoting hypoxia-induced signaling, leading to the activation of the HIF-1 pathway24. As an inhibitor of Src, CSK may indirectly suppress HIF-1 activation, thereby reducing angiogenesis and invasiveness in breast cancer cells. Additionally, Src collaborates with the Rap1 signaling pathway in regulating cell adhesion and migration25. By negatively regulating Src activity, CSK could indirectly influence integrin-mediated functions within the Rap1 signaling pathway.
Our research findings indicated that CSK protein acted as a protective factor in breast cancer, which aligned with previous research results. CHK, a homologous protein of CSK, has been shown to significantly inhibit tumor size in naked mice implanted with MCF-7 cells transfected with wt-CHK26. These support that CSK is a novel drug targets in breast cancer. Currently, drugs targeting CSK include TG100801 and hydrochlorothiazide. TG100801 is a multi-target inhibitor of vascular endothelial growth factor receptors and Src kinases, designed to induce apoptosis in proliferating endothelial cells involved in neovascularization and to suppress inflammation-mediated processes. It is currently being developed for use in treating macular degeneration27.
ULK3 demonstrated a protective effect in overall breast cancer and luminal A subtype. Furthermore, ULK3 may serve as a potential favorable prognostic marker in RFS in breast cancer, particularly within the luminal A subtype. Our findings reinforce the previous research by Zhang et al., suggesting the significant role in breast cancer28. ULK3 belongs to the ULK family, and its homologous proteins, ULK1 and ULK2, have garnered extensive attention in the pathogenesis of breast cancer recently28,29,30. The ULK3 protein activates glioma-associated oncogene homolog 1 in a dependent manner, upregulating transcription of the DNMT3A gene during autophagy induction, thus stimulating autophagy31. It is also suggested that ULK3 can catalyze the phosphorylation of ESCRT-III proteins CHMP1A/B, CHMP2A, and IST1 in vitro and in situ, leading to subcellular localization changes in ESCRT-III assembly and preventing their aggregation. As a result, catalytically active ULK3 delays the final step of cell division32. Previous literature suggested a connection between Imatinib and the ULK3 protein. Imatinib mesylate is a protein tyrosine kinase inhibitor that selectively inhibits BCR-ABL tyrosine kinase, thereby affecting related downstream pathways such as the Ras/MapK, Src/Pax/Fak/Rac, and PI/PI3K/AKT/BCL-2 pathways33,34. It has been approved for use in various conditions including leukemias, myelodysplastic/myeloproliferative diseases, hypereosinophilic syndrome, and gastrointestinal stromal tumors. Vitro studies have demonstrated that the combination of imatinib with vinorelbine or chemotherapy significantly enhances growth inhibition in breast cancer models35,36. A phase II clinical trial have indicated that imatinib alone shows no clinical activity against metastatic breast cancer (MBC) or MBC with overexpressing platelet-derived growth factor receptor-beta (PDGFR-β)37,38. In cases of MBC expressing c-kit and/or PDGFR-β positive hormone receptors, the combination of imatinib with letrozole resulted in a median RFS of 8.7 months and an OS of 44.3 months39. Another phase I/II trial involving patients with MBC (expressing PDGFR-α and/or -β and/or KIT) demonstrated the fairly low clinical benefits of the combining imatinib with vinorelbine40. Future clinical studies should aim to refine the target population for more precise treatment interventions, also necessitating larger sample sizes for robust conclusions.
MR results revealed significant genetic associations between plasma proteins and breast cancer incidence risk. While these targets may play a role in the initiation of cancer, they may not be suitable for treating cancer progression. The key distinction lies in that proteins associated with cancer risk are more relevant for prevention and early intervention, whereas those linked to cancer progression are better suited for tumor therapies. Experimental validation showed that overexpression of CSK and ULK3 significantly inhibited the proliferation and migration of MCF-7 cells, supporting their potential role in breast cancer progression. These findings suggest that CSK and ULK3 could serve as therapeutic targets for advanced breast cancer. In conclusion, while our study provides insights into the genetic underpinnings of both breast cancer incidence and progression, it is crucial to differentiate between these two mechanisms when evaluating therapeutic targets. Future research should further explore the functional role of these targets in cancer progression and assess their potential in the development of therapeutic agents.
Some plasma proteins were classified as having moderate support in the colocalization analysis, indicating a moderate probability the associations between plasma proteins and breast cancer are driven by the same causal variant. This finding may also be influenced by pleiotropy with a low probability, as certain traits or diseases are affected by multiple genetic loci or complex mechanisms, making it challenging to establish a clear colocalization signal. Such complexity is particularly common in multifactorial diseases41.
There are three main methods available on the market for proteomics analysis: Olink, SomaScan, and direct measurement techniques. Direct measurement methods, being untargeted approaches, offer lower throughput but are applicable to all disease types14,15,16,42. Although both the Olink and SomaScan platforms are affinity-based, they differ in nature: one is antibody-based, while the other is aptamer-based. A recent study comparing the Olink Explore 3072 platform with the SomaScan platform found that the median coefficient of variation for Olink was 16.5%, compared to 9.9% for SomaScan, suggesting that SomaScan provides more accurate measurements43. Therefore, the technology employed in this study, SomaScan, offers greater precision compared to other available techniques.
This study demonstrates significant innovation in methodology, data application, and validation. Unlike Anders Mälarstig et al. and Karl Smith-Byrne et al., who utilized a single MR method to analyze associations between breast cancer and related proteins14,16, this research combines TSMR and SMR methods. This integration of intersectional and colocalization analyses enhances the reliability of the results. While earlier studies by Anders Mälarstig, Keren Papier, and Karl Smith-Byrne have explored breast cancer associations with plasma proteins, these works primarily use MR and colocalization analysis to assess causality and lack functional validation in animal models and substantial investigation of drug effects14,15,16. To bridge this gap, we conducted functional experiments in cell lines and mouse models to investigate the biological mechanisms of breast cancer-related proteins, addressing a key gap in mechanistic validation. Moreover, unlike the study by Keren Papier et al., which was limited by the Olink Explore II panel, our research covers a broader spectrum of proteins, providing a more comprehensive view for the exploration of breast cancer-related biomarkers. In addition to confirming associations with previously identified proteins, such as ULK3, TLR1, and LAYN14,16,28, our cross-analyses identified additional potential biomarkers for breast cancer, including CSK, ASIP, ADH5, SARS2, UBE2N, and PEX14, thus expanding upon findings in the literature. Through validation in functional experiments, this study substantiates the associations between identified proteins and breast cancer, offering crucial support for future research on breast cancer biomarkers.
Several limitations should be noted in this study. Firstly, The GWAS data of breast cancer, deCODE, GEO, EGA, TCGA database predominantly consist of individuals of European ancestries, constraining the generalizability of our conclusions. However, we incorporated a substantial amount of data from non-European populations during the validation phase. Secondly, we cannot mine the relationship between proteins outside the database and breast cancer, due to the limitation of the range of circulating plasma proteins included in deCODE database. Thirdly, despite our efforts to comprehensively search all available drug databases, the exclusion of drugs in the developmental or clinical trial stages may have led to inability to identify drugs targeting the proteins identified. Another limitation of this study is the absence of power calculations specific to breast cancer subtypes. While power analysis was conducted based on the total sample size, the cohort’s limited availability of subtype-specific sample sizes prevented detailed analyses by subtype. However, given the large overall sample size, we believe the statistical power is sufficient to detect meaningful effects across subtypes.
In conclusion, this study identified a total of 9 robust genes and 13 medium genes of plasma protein, which genetically associated with breast cancer and its subtypes. These genes were predominantly enriched in nucleocytoplasmic transport and HIF-1 signaling pathways. Furthermore, our findings underscore the critical role of plasma proteins in regulating hematopoietic and immune functions. Compared to normal tissue, tumor tissue exhibited reduced expression of ULK3 and CSK. Notably, ULK3 expression in both breast cancer and the Luminal A subtype was significantly associated with prolonged recurrence-free survival. Overexpression of CSK and ULK3 was confirmed to significantly inhibit the proliferation and migratory ability of MCF-7 cells, as demonstrated by the CCK-8 assay and wound healing assay, respectively. Moreover, a comprehensive search in drug databases led to the identification of three potential drugs: TG100801, Hydrochlorothiazide, Imatinib. These findings offer genetic evidence that supports the identification of potential therapeutic target drugs for breast cancer, thereby contributing valuable insights for prioritizing drug development in realm of breast cancer.
Method
Study design
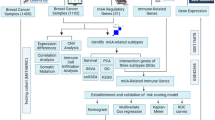
The framework of this study design was elucidated in Fig. 8. Initially, MR analyses, namely SMR and TSMR analysis, were performed on the data of BC and circulating proteins. Subsequently, pathway enrichment analysis and protein-protein interaction (PPI) network analysis were conducted based on the results of SMR and TSMR analyses to further delineate potential mechanisms underlying breast cancer and its subtypes.

GWAS genome-wide association study, pQTL protein quantitative trait loci, cis-pQTL cis-acting protein quantitative trait loci, MHC major histocompatibility complex, LD linkage disequilibrium, MR Mendelian randomization, SMR summary-data-based MR test, HEIDI heterogeneity in dependent instrument, PH4 posterior probability of H4, GEO Gene Expression Omnibus, EGA European Genome-Phenome Archive, TCGA The Cancer Genome Atlas, Metabric Molecular Taxonomy of Breast Cancer International Consortium. (Created in BioRender. quan, l. (2025) https://BioRender.com/ cdz72yh).
To ensure the robustness of the findings, additional colocalization analysis was performed on the results obtained from MR to identify the target circulating protein. In result, the plasma proteins were categorized into three tiers: tier 1 for high-support, tier 2 for medium-support, and tier 3 for all others.
The tissue specimens from patients with breast cancer were collected to perform immunohistochemistry (IHC) to assess the expression of the target protein in tumor tissues and adjacent normal tissues. The relationship between target protein expression and immune cell infiltration in the tumor microenvironment was subsequently analyzed. In addition, survival analysis was performed to investigate the prognostic significance of the target protein in breast cancer. While the mouse knock-out model was performed to validate the result of colocalization analysis. Additionally, functional validation of the target protein was conducted using in vitro experiments in cell line. Ultimately, exploration of drug libraries was undertaken to identify drugs targeting circulating proteins with high support.
Datasets
The genotyping data for BC and its subtypes were gained from a recent publicly available comprehensive study of breast cancer, including 133,384 cases and 113,789 counterparts from over 20 European countries by 82 Breast Cancer Association Consortium studies44. Notably, it is the first study to reveal the GWAS outcomes of BC as well as its four distinct subtypes: HER-2, Luminal A, Luminal B and TNBC subtypes.
The deCODE repository (https://www.decode.com/summarydata/) is a comprehensive genome-wide blood proteome database that contains summary data of 4907 plasma protein. These data were derived from a large-scale pQTL study involving 35,559 individuals8. These individuals were enrolled in two major initiatives: the Icelandic Cancer Project and various genetic programs at deCODE genetics from 2000 to 2019. Egil Ferkingstad et al. employed SomaScan, a multiplexed aptamer assay that measures plasma proteins using 5284 aptamers to assess the relative binding rates of plasma samples to each aptamer. The study measured 27.2 million sequence variants and the levels of 4907 plasma proteins among these participants8. Widely utilized, it has proven instrumental in identifying pivotal genetic risk factors across various prevalent diseases, such as cardiovascular disease and cancer.
MR analysis
For inclusion, pQTLs were required to satisfy these standards: (i) Demonstrate a significant association across the genome (P < 5 × 10−8); (ii) Occur beyond the boundaries of the major histocompatibility complex (MHC) area (chr6, 25.5–34.0 Mb); (iii) Show a distinct association characterized by linkage disequilibrium (LD) clumping r2 being less than 0.01, considering a clumping window of 10000 kb; and (iv) Function as a cis-acting pQTL8. The classification of a pQTL association as cis occurs when its related variant lies within a maximum distance of 1 Mb from the transcription start site of the gene that produces the relevant protein, otherwise it’s categorized as trans8.
MR analyses were performed utilizing R software and TwoSampleMR package. In this study, we used circulating proteins as exposures and breast cancer types as outcomes to perform MR using “TwoSampleMR”. For single pQTL, the Wald ratio was adopted. With multiple pQTLs, we employed the inverse variance weighted (IVW) method and evaluated heterogeneity. Breast cancer risks were considered as OR per standard deviation (SD) increase in plasma protein levels. In addition, the SMR of multi-single nucleotide polymorphisms (SNPs) for every circulating protein was conducted as a sensitivity analysis to corroborate the primary findings. The HEIDI test was utilized to differentiate between two types of genetic effects: pleiotropic models, where a single genetic variant influences multiple traits or outcomes, and linkage models, where a genetic variant is associated with a specific trait due to its proximity to a gene that directly affects that trait. This approach offers advantages over many GWAS and molecular QTL-methods45,46. Associations in the HEIDI test with P values below 0.05, indicative of pleiotropy, were excluded from subsequent analysis. For the SMR analysis, we utilized the SMR software and implemented multiple testing by Benjamini-Hochberg method, setting the false discovery rate (FDR) threshold at α = 0.1. We performed a MR power analysis to assess the statistical power of our study to detect causal effects47. MR analysis was performed following the MR Statement guidelines to ensure robust and transparent methodology48.
Colocalization analysis
Colocalization analysis through the “coloc” R package was performed to determine if the associations between plasma proteins and BC, along with its subtypes, were influenced by LD49. We calculated posterior probabilities by Approximate Bayes Factor computations for the following five hypotheses. H0: No significant association exists between plasma proteins and breast cancer. H1: Only plasma proteins are significantly associated with SNP loci in a specific genomic region. H2: Only breast cancer is significantly associated with SNP loci in a specific genomic region. H3: Both plasma proteins and breast cancer are significantly associated with SNP loci in a specific genomic region, but these associations are driven by different causal variants. H4: Both plasma proteins and breast cancer are significantly associated with SNP loci in a specific genomic region, and these associations are driven by the same causal variant50. The formula for calculating PH4 is as follows:
L0, L1, L2, L3, L4 represent the likelihoods under each corresponding hypothesis. Prior(H0), Prior(H1), Prior(H2), Prior(H3), Prior(H4) represent the prior probabilities for each hypothesis50. The prior probabilities were set as follows: SNP association with only the first trait (p1) at 1 × 10−4, with only the second trait (p2) at 1 × 10−4, and with both traits (p12) at 1 × 10−5. Strong colocalization was inferred when PH4 was å 0.8, while a PH4 between 0.5 and 0.8 indicated medium colocalization3,50. This analysis was only applied to proteins with FDR-corrected P values < 0.1 in TSMR and HEIDI test P values > 0.05 in SMR. Based on the colocalization evidence, we categorized the MR results into three tiers: tier 1 for high-support (PH4 å 0.8), tier 2 for medium-support (0.5 ≤ PH4 ≤ 0.8), and tier 3 for all others.
Protein-Protein interaction network analysis
The protein-protein interactions play a pivotal role in biological functions and processes. It is notable that numerous proteins seem to carry out their functions through these interactions51,52. To further elucidate the interactions among the 62 identified circulating proteins, obtained through Mendelian randomization of drug targets in the initial step. Gene interaction data were obtained from the GeneMANIA website and imported into the R environment53. The graph package was then employed to construct a network graph, with the layout function used to optimize node arrangements for clearer representation of interaction relationships. Finally, the network was visualized using the “ggplot2” package. The interactions were classified into the following aspects: including co-expression, physical interactions, genetic interaction, predicted interaction, shared protein domain and colocation.
Pathway analysis
In this study, we conducted analyses of GO, KEGG, and DO to comprehensively explore the biological functions and relevance of the target genes54,55,56. Initially, we performed GO and KEGG enrichment analyses using the “clusterProfiler” package. Target genes were extracted from the input data, and their corresponding Entrez IDs were obtained from the “org.Hs.eg.db” database. Subsequently, we executed GO and KEGG analyses using the “enrichGO” and “enrichKEGG”functions, respectively, while the “enrichDO”function was employed to identify biological pathways associated with specific diseases. During this process, the gene ratio was calculated to evaluate the enrichment of target genes within specific pathways. P value < 0.05 was considered significant. The dynamic enrichment bubble plot of KEGG analysis and signaling pathways were depicted.
Mouse knock-out models
The Mouse Genome Informatics (MGI, http://www.informatics.jax.org/) is a comprehensive database providing gene function, expression, and phenotypes, as well as genetic maps and molecular markers information of the laboratory mouse57,58,59. Thus, it can serve as a valuable platform to study the gene of identified plasma proteins by comparison phenotypes between gene and its mutations/alleles. The genes of acquires proteins were retrieved in the website and the section “Mutations, Alleles, and Phenotypes” would display phenotype and mutation information of this genes.
Tumor immune cell infiltration
Tumor Immune Estimation Resource (TIMER, https://cistrome.shinyapps.io/timer/), developed by Taiwen Li and colleagues, is an online tool that employs a deconvolution algorithm to estimate the abundance of tumor-infiltrating immune cells based on gene expression data60,61. The expression of selected genes and the infiltration of immune cells in breast cancer were studied using the TIMER. The immune cells consisted of B cells, CD8 + T cells, CD4 + T cells, macrophages, neutrophils, and dendritic cells. The correlation coefficients between the genes and immune cells were denoted by r, which ranged from -1 to 1. A correlation was considered statistically significant if P < 0.05.
Survival analysis
GEO (http://ww.ncbinlm.nih.gov/geo/), TCGA (https://portal.gdc.cancer.gov/), EGA, Metabric (http://molonc.bccrc.ca/aparicio-lab/research/metabric/) are all famous databases of breast cancer, owing a large number of cohorts and patient data. KM-plotter (http://kmplot.m/analysis/) is the most influential survival analysis tool based on the integrative data from Metabric, GEO, TCGA, EGA, encompassing survival information from 7,830 breast cancer patients, with RFS data available for 5,268 patients and OS data for 5,165 patients62,63. Patient classification follows the St. Gallen criteria. To further analyze the prognostic value of the acquired genes of the target proteins, the patients were assigned to two groups accordingly, namely low expression group and high expression group. The OS and RFS were used as primary outcome endpoints in two cohorts, and P value < 0.05 indicated a significant difference.
Druggable protein identification
To evaluate the translational potential of these risk-associated targets, we further prioritized candidates with plausible links to disease progression. While not all identified targets demonstrated significant associations with breast cancer progression, our MR analysis revealed their robust genetic links to breast cancer incidence risk. These risk-associated targets may represent candidates for primary prevention strategies or early-stage therapeutic interventions.
Compared to drugs targeting unsupported targets, medications targeting human genetic-supported targets exhibit a higher probability of therapeutic success. The following database, DrugBank (https://go.drugbank.com/)64, DGIdb (https://dgidb.org/)64, CheMBL (https://www.ebi.ac.uk/chembl/)65,66, Therapeutic Target Database (https://db.idrblab.net/ttd/)67, were searched for potential drugs targeted identified proteins associated with breast cancer. This study prioritized the identification of proteins with substantial evidence (PH4 > 0.8) in the drug databases. We obtained pertinent information for each drug, including details on the protein target, indications, and developmental status.
Immunohistochemistry
We collected tissue samples from 25 patients with Luminal A subtype breast cancer at the Cancer Hospital, Chinese Academy of Medical Sciences and Peking Union Medical College. Tumor and adjacent non-tumor tissues from 25 Luminal A breast cancer patients were analyzed using IHC. Paraffin-embedded sections were dewaxed, rehydrated, and subjected to antigen retrieval in EDTA buffer (pH 9.0) using a microwave. Endogenous peroxidase activity was blocked with 3% hydrogen peroxide, followed by blocking non-specific binding with 3% BSA. Sections were incubated overnight at 4°C with rabbit anti-ADH5 primary antibody (1:200, D225407, Sangon), then with HRP-conjugated secondary antibody (GB23303, Servicebio). DAB was used for chromogenic detection, and nuclei were counterstained with hematoxylin. The slides were dehydrated, mounted, and analyzed under a microscope, with blue nuclei and brown DAB-positive staining indicating expression. Quantification of staining intensity was performed using Image J software. The same protocol was applied for detecting ULK3 (D291625, Sangon), and CSK (D151197, Sangon).
Cell culture and transfection
The MCF-7 cell line was provided by the State Key Laboratory of Biotherapy at Sichuan University (Chengdu, China). MCF-7 cells are derived from human breast cancer and are of female origin. The MCF-7 cells are not listed in the ICLAC register, and therefore, authentication was not performed. MCF-7 cells were cultured in DMEM medium supplemented with 10% fetal bovine serum under sterile conditions at 37°C with 5% CO2. Lentiviral particles for CSK and ULK3 were purchased from GeneChem Co., Ltd (Shanghai, China). Transfection was performed at a multiplicity of infection of 500, with cells incubated with the viral particles for 24 hours. The experiment included four groups: the CSK overexpression group, the ULK3 overexpression group, the empty vector group (Vector), and the blank control group (Control).
qPCR verification
qPCR was performed to verify the expression levels of CSK and ULK3 after transfection. Total RNA was extracted from cells using TRIzol reagent (Invitrogen, CA, USA). The RNA was reverse-transcribed into cDNA using the Prime Script RT reagent kit (TaKaRa, Shanghai, China). qPCR was conducted on a fluorescence-based qPCR system (TaKaRa, Shanghai, China) using the SYBR Premix Ex Taq II reagent kit. The primer sequences used for qPCR were as follows: For the hGAPDH gene, the amplicon size was 101 bp, with the forward primer sequence 5’-ACAACTTTGGTATCGTGGAAGG-3’ and the reverse primer sequence 5’- GCCATCACGCCACAGTTTC-3’. For the hCSK gene, the amplicon size was 126 bp, with the forward primer sequence 5’-AGGACCCCAACTGGTACAAAG-3’ and the reverse primer sequence 5’- CGTGGAACCAAGGCATGAG-3’. For the hULK3 gene, the amplicon size was 102 bp, with the forward primer sequence 5’- GAACGGAATATCTCTCACCTGGA-3’ and the reverse primer sequence 5’-GTGTTGTGCGAAACCAAAGTC-3’.
Cell proliferation and migration analysis
Transfected cells were seeded into 96-well plates, and cell viability was assessed at 24, 48, 72, and 96 hours using CCK-8 assay (MedChemExpress, USA). For the wound healing assay, transfected cells were seeded into 6-well plates. After 24 hours of culture, a uniform scratch was made, and the cells’ migratory ability was evaluated 24 hours post-scratch.
Inclusion and ethics
In this study, local researchers were actively involved throughout the entire research process, including study design, implementation, data ownership, and authorship, to ensure the research addressed the specific needs and context of the community. Roles and responsibilities were clearly defined at the beginning of the study, and capacity-building plans were made for local researchers to ensure equitable involvement. This study was approved by the Ethics Committee of National Cancer Center/Cancer Hospital, Chinese Academy of Medical Sciences and Peking Union Medical College Ethics Committee (Clinical Ethics Approval No. 23/139-3884). Written informed consent was obtained from all participants. All ethical regulations relevant to human research participants were followed.
Statistics and reproducibility
IBM SPSS Statistics (version 21) and GraphPad Prism (version 8.0) were used for statistical analysis and plotting. A paired sample t-test was conducted to assess differences in the expression levels of proteins between tumor tissues and adjacent normal tissues. Group comparisons in qPCR validation, CCK-8 assays, and cell scratch assays were performed using t-tests. Independent sample t-tests or Mann-Whitney U tests were applied based on the distribution of the data, with normality assessed by the Shapiro-Wilk test. Quantification of staining intensity and cell scratch analysis were performed using Image J software (version 1.53a). Statistical significance was defined as a two-sided P-value < 0.05. In vitro experiments were conducted with a minimum of three independent biological replicates.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
Summary statistics for plasma proteins and breast cancer were obtained from the deCODE repository (https://www.decode.com/summarydata/) and the Breast Cancer Association Consortium studies (http://bcac.ccge.medschl.cam.ac.uk/bcacdata/), respectively. Functional genomics data were sourced from GeneMANIA (https://genemania.org/) and MGI database (http://www.informatics.jax.org/). Tumor microenvironment data were accessed via TIMER, while survival analysis data were derived from KM-plotter (http://kmplot.com/analysis/). Drug-related information was retrieved from DrugBank (https://go.drugbank.com/), DGIdb (https://dgidb.org/), ChEMBL (https://www.ebi.ac.uk/chembl/), and TTD.
Code availability
The TSMR, SMR, and colocalization analyses presented in this study were performed on a CentOS Linux release 7.6.1810 platform using R software (version 4.1.3). The R packages employed for statistical analysis included TwoSampleMR (version 0.5.6) (https://github.com/MRCIEU/TwoSampleMR) and MR-PRESSO (version 1.0) (https://github.com/rondolab/MR-PRESSO). Colocalization analysis was conducted using the coloc R package (version 5.2.2) (https://github.com/jerome-f/coloc). The SMR analysis was carried out using the SMR software tool (version 1.3.1) (https://yanglab.westlake.edu.cn/software/smr/#Download). Pathway enrichment analysis was performed using the clusterProfiler (version 4.12.2) (https://github.com/YuLab-SMU/clusterProfiler) and org.Hs.eg.db (version 3.19.1) (https://bioconductor.org/packages/ release/data/annotation/html/org.Hs.eg.db.html). For access to the code used in this study, please contact the corresponding author.
References
Yin, L., Duan, J. J., Bian, X. W. & Yu, S. C. Triple-negative breast cancer molecular subtyping and treatment progress. Breast Cancer Res. : BCR 22, 61 (2020).
Folkersen, L. et al. Genomic and drug target evaluation of 90 cardiovascular proteins in 30,931 individuals. Nat. Metab. 2, 1135–1148 (2020).
Chen, J. et al. Therapeutic targets for inflammatory bowel disease: proteome-wide Mendelian randomization and colocalization analyses. EBioMedicine 89, 104494 (2023).
Tan, J. S., Liu, N. N., Guo, T. T., Hu, S. & Hua, L. Genetic predisposition to COVID-19 may increase the risk of hypertension disorders in pregnancy: A two-sample Mendelian randomization study. Pregnancy Hypertension 26, 17–23 (2021).
Riaz, H. et al. Association Between Obesity and Cardiovascular Outcomes: A Systematic Review and Meta-analysis of Mendelian Randomization Studies. JAMA Netw. Open 1, e183788 (2018).
Bowden, J. & Holmes, M. V. Meta-analysis and Mendelian randomization: A review. Res. Synth. methods 10, 486–496 (2019).
Emdin, C. A., Khera, A. V. & Kathiresan, S. Mendelian Randomization. Jama 318, 1925–1926 (2017).
Ferkingstad, E. et al. Large-scale integration of the plasma proteome with genetics and disease. Nat. Genet. 53, 1712–1721 (2021).
Sun, B. B. et al. Genomic atlas of the human plasma proteome. Nature 558, 73–79 (2018).
Davey Smith, G. & Hemani, G. Mendelian randomization: genetic anchors for causal inference in epidemiological studies. Hum. Mol. Genet. 23, R89–R98 (2014).
Kanduri, C., Bock, C., Gundersen, S., Hovig, E. & Sandve, G. K. Colocalization analyses of genomic elements: approaches, recommendations and challenges. Bioinforma. (Oxf., Engl.) 35, 1615–1624 (2019).
Wang, Q., Shi, Q., Wang, Z., Lu, J. & Hou, J. Integrating plasma proteomes with genome-wide association data for causal protein identification in multiple myeloma. BMC Med. 21, 377 (2023).
Zhang, C., Qin, F., Li, X., Du, X. & Li, T. Identification of novel proteins for lacunar stroke by integrating genome-wide association data and human brain proteomes. BMC Med. 20, 211 (2022).
Mälarstig, A. et al. Evaluation of circulating plasma proteins in breast cancer using Mendelian randomisation. Nat. Commun. 14, 7680 (2023).
Papier, K. et al. Identifying proteomic risk factors for cancer using prospective and exome analyses of 1463 circulating proteins and risk of 19 cancers in the UK Biobank. Nat. Commun. 15, 4010 (2024).
Smith-Byrne, K. et al. Identifying therapeutic targets for cancer among 2074 circulating proteins and risk of nine cancers. Nat. Commun. 15, 3621 (2024).
Iniesta, R. et al. Gene Variants at Loci Related to Blood Pressure Account for Variation in Response to Antihypertensive Drugs Between Black and White Individuals. Hypertension (Dallas, Tex. : 1979) 74, 614–622 (2019).
Dressman, M. A. et al. Correlation of major cytogenetic response with a pharmacogenetic marker in chronic myeloid leukemia patients treated with imatinib (STI571). Clin. Cancer Res. : Off. J. Am. Assoc. Cancer Res. 10, 2265–2271 (2004).
Semenza, G. L. Hypoxia-inducible factors: coupling glucose metabolism and redox regulation with induction of the breast cancer stem cell phenotype. EMBO J. 36, 252–259 (2017).
Dupuy, F. et al. PDK1-dependent metabolic reprogramming dictates metastatic potential in breast cancer. Cell Metab. 22, 577–589 (2015).
Chiavarina, B. et al. Metabolic reprogramming and two-compartment tumor metabolism: opposing role(s) of HIF1α and HIF2α in tumor-associated fibroblasts and human breast cancer cells. Cell Cycle (Georget., Tex.) 11, 3280–3289 (2012).
Schwartzberg, P. L. The many faces of Src: multiple functions of a prototypical tyrosine kinase. Oncogene 17, 1463–1468 (1998).
Zrihan-Licht, S. et al. Csk homologous kinase, a novel signaling molecule, directly associates with the activated ErbB-2 receptor in breast cancer cells and inhibits their proliferation. J. Biol. Chem. 273, 4065–4072 (1998).
Semenza, G. L. HIF-1 and human disease: one highly involved factor. Genes Dev. 14, 1983–1991 (2000).
Bos, J. L., de Rooij, J. & Reedquist, K. A. Rap1 signalling: adhering to new models. Nat. Rev. Mol. Cell Biol. 2, 369–377 (2001).
Bougeret, C., Jiang, S., Keydar, I. & Avraham, H. Functional analysis of Csk and CHK kinases in breast cancer cells. J. Biol. Chem. 276, 33711–33720 (2001).
Ni, Z. & Hui, P. Emerging pharmacologic therapies for wet age-related macular degeneration. Ophthalmologica. J. Int. d’ophtalmologie. Int. J. Ophthalmol. Z. fur Augenheilkd. 223, 401–410 (2009).
Zhang, N., Li, Y., Sundquist, J., Sundquist, K. & Ji, J. Identifying actionable druggable targets for breast cancer: Mendelian randomization and population-based analyses. EBioMedicine 98, 104859 (2023).
Liang, P. & Wang, B. An autophagy-independent role of ULK1/ULK2 in mechanotransduction and breast cancer cell migration. Autophagy, 1-2, https://doi.org/10.1080/15548627.2023.2300916 (2024).
Deng, R. et al. MAPK1/3 kinase-dependent ULK1 degradation attenuates mitophagy and promotes breast cancer bone metastasis. Autophagy 17, 3011–3029 (2021).
González-Rodríguez, P., Cheray, M., Keane, L., Engskog-Vlachos, P. & Joseph, B. ULK3-dependent activation of GLI1 promotes DNMT3A expression upon autophagy induction. Autophagy 18, 2769–2780 (2022).
Caballe, A. et al. ULK3 regulates cytokinetic abscission by phosphorylating ESCRT-III proteins. eLife 4, e06547 (2015).
Cilloni, D. & Saglio, G. Molecular pathways: BCR-ABL. Clin. Cancer Res. : Off. J. Am. Assoc. Cancer Res. 18, 930–937 (2012).
Haguet, H. et al. BCR-ABL Tyrosine Kinase Inhibitors: Which Mechanism(s) May Explain the Risk of Thrombosis? TH Open : Companion J. Thrombosis Haemost. 2, e68–e88 (2018).
Weigel, M. T. et al. Combination of imatinib and vinorelbine enhances cell growth inhibition in breast cancer cells via PDGFR beta signalling. Cancer Lett. 273, 70–79 (2009).
Weigel, M. T. et al. In vitro effects of imatinib mesylate on radiosensitivity and chemosensitivity of breast cancer cells. BMC Cancer 10, 412 (2010).
Modi, S. et al. A phase II trial of imatinib mesylate monotherapy in patients with metastatic breast cancer. Breast Cancer Res. Treat. 90, 157–163 (2005).
Cristofanilli, M. et al. Imatinib mesylate (Gleevec) in advanced breast cancer-expressing C-Kit or PDGFR-beta: clinical activity and biological correlations. Ann. Oncol. : Off. J. Eur. Soc. Med. Oncol. 19, 1713–1719 (2008).
Yam, C. et al. A phase II study of imatinib mesylate and letrozole in patients with hormone receptor-positive metastatic breast cancer expressing c-kit or PDGFR-β. Invest. N. drugs 36, 1103–1109 (2018).
Maass, N. et al. Final safety and efficacy analysis of a phase I/II trial with imatinib and vinorelbine for patients with metastatic breast cancer. Oncology 87, 300–310 (2014).
Hormozdiari, F. et al. Colocalization of GWAS and eQTL Signals Detects Target Genes. Am. J. Hum. Genet. 99, 1245–1260 (2016).
Petrera, A. et al. Multiplatform Approach for Plasma Proteomics: Complementarity of Olink Proximity Extension Assay Technology to Mass Spectrometry-Based Protein Profiling. J. Proteome Res. 20, 751–762 (2021).
Eldjarn, G. H. et al. Author Correction: Large-scale plasma proteomics comparisons through genetics and disease associations. Nature 630, E3 (2024).
Zhang, H. et al. Genome-wide association study identifies 32 novel breast cancer susceptibility loci from overall and subtype-specific analyses. Nat. Genet. 52, 572–581 (2020).
Zhu, Z. et al. Integration of summary data from GWAS and eQTL studies predicts complex trait gene targets. Nat. Genet. 48, 481–487 (2016).
Wu, Y. et al. Integrative analysis of omics summary data reveals putative mechanisms underlying complex traits. Nat. Commun. 9, 918 (2018).
Burgess, S. Sample size and power calculations in Mendelian randomization with a single instrumental variable and a binary outcome. Int. J. Epidemiol. 43, 922–929 (2014).
Skrivankova, V. W. et al. Strengthening the Reporting of Observational Studies in Epidemiology Using Mendelian Randomization: The STROBE-MR Statement. Jama 326, 1614–1621 (2021).
Foley, C. N. et al. A fast and efficient colocalization algorithm for identifying shared genetic risk factors across multiple traits. Nat. Commun. 12, 764 (2021).
Giambartolomei, C., Vukcevic, D., Schadt, E. E., Franke, L. & Plagnol, V. Bayesian Test for Colocalisation between Pairs of Genetic Association Studies Using Summary Statistics. PLoS Genet. 10, e1004383 (2014).
Tomkins, J. E. & Manzoni, C. Advances in protein-protein interaction network analysis for Parkinson’s disease. Neurobiol. Dis. 155, 105395 (2021).
Athanasios, A., Charalampos, V., Vasileios, T. & Ashraf, G. M. Protein-Protein Interaction (PPI) Network: Recent Advances in Drug Discovery. Curr. drug Metab. 18, 5–10 (2017).
David, W. F. et al. The GeneMANIA prediction server: biological network integration for gene prioritization and predicting gene function. Nucleic Acids Res. 38, 214–220 (2010).
Kanehisa, M. & Goto, S. KEGG: kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 28, 27–30 (2000).
Kibbe, W. A., Arze, C., Felix, V., Mitraka, E. & Schriml, L. M. Disease Ontology 2015 update: An expanded and updated database of Human diseases for linking biomedical knowledge through disease data. Nucleic Acids Res. 43, D1071–D1078 (2015).
Ashburner, M. et al. Gene ontology: tool for the unification of biology. The Gene Ontology Consortium. Nat. Genet. 25, 25–29 (2000).
Baldarelli, R. M., Smith, C. L., Ringwald, M., Richardson, J. E. & Bult, C. J. Mouse Genome Informatics: an integrated knowledgebase system for the laboratory mouse. Genetics 227, https://doi.org/10.1093/genetics/iyae031 (2024).
Baldarelli, R. M. et al. The mouse Gene Expression Database (GXD): 2021 update. Nucleic Acids Res. 49, D924–d931 (2021).
Krupke, D. M. et al. The Mouse Tumor Biology Database: A Comprehensive Resource for Mouse Models of Human Cancer. Cancer Res. 77, e67–e70 (2017).
Li, T. et al. TIMER: A Web Server for Comprehensive Analysis of Tumor-Infiltrating Immune Cells. Cancer Res. 77, e108–e110 (2017).
Li, B. et al. Comprehensive analyses of tumor immunity: implications for cancer immunotherapy. Genome Biol. 17, 174 (2016).
Győrffy, B. Integrated analysis of public datasets for the discovery and validation of survival-associated genes in solid tumors. Innov. (Camb. (Mass.)) 5, 100625 (2024).
Győrffy, B. Transcriptome-level discovery of survival-associated biomarkers and therapy targets in non-small-cell lung cancer. Br. J. Pharmacol. 181, 362–374 (2024).
Knox, C. et al. DrugBank 6.0: the DrugBank Knowledgebase for 2024. Nucleic Acids Res. 52, D1265–d1275 (2024).
Zdrazil, B. et al. The ChEMBL Database in 2023: a drug discovery platform spanning multiple bioactivity data types and time periods. Nucleic Acids Res. 52, D1180–d1192 (2024).
Davies, M. et al. ChEMBL web services: streamlining access to drug discovery data and utilities. Nucleic Acids Res. 43, W612–W620 (2015).
Zhou, Y. et al. TTD: Therapeutic Target Database describing target druggability information. Nucleic Acids Res. 52, D1465–d1477 (2024).
Acknowledgements
This study was funded by the National Natural Science Foundation of China (Project Number: 8217102755).
Author information
Authors and Affiliations
Contributions
L.Q.: Concept/design, data analysis/interpretation, and manuscript writing. X.L.: Concept/design, data analysis/interpretation, and manuscript writing. C.M.: Cell line experiment, data analysis/interpretation, and manuscript writing. J.L.: Data analysis, manuscript writing. Z.Y.: Data collection and manuscript writing. J.J.: Data collection and manuscript writing. T.W.: Data interpretation and manuscript writing. Y.S.: Data interpretation and manuscript writing. J M.: Data analysis and manuscript writing. P.Y.: Concept/ design, data analysis/interpretation, financial support, manuscript writing, and final approval of manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Communications Biology thanks the anonymous reviewers for their contribution to the peer review of this work. Primary Handling Editors: Rebecca Richmond & Rosie Bunton-Stasyshyn.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Quan, L., Luo, X., Meng, C. et al. Genetic associations of plasma proteins and breast cancer identify potential therapeutic drug candidates. Commun Biol 8, 610 (2025). https://doi.org/10.1038/s42003-025-08046-3
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s42003-025-08046-3
