
Abstract
Urination, a fundamental physiological process, is intricately regulated by central neural circuits. The periaqueductal gray (PAG) is a critical hub for urination control, yet the specific neuronal subtypes involved remain undefined. Here, we identified somatostatin (SST)-expressing neurons in the lateral and ventrolateral PAG (l/vlPAG) as key regulators of urination. SST+ neurons in the l/vlPAG exhibited activity closely aligned with urination onset in freely moving animals. Optogenetic activation of these neurons reliably triggered urination and bladder contraction, whereas their acute inhibition at urination onset halted ongoing urination. Moreover, activation of l/vlPAG SST+ neurons projecting to the pontine micturition center (PMC) elicited urination and bladder contraction, effects that were absent following pelvic nerve transection. These findings reveal that l/vlPAG SST+ neurons regulate urination through the PAG-PMC pathway, providing new insights into bladder control and potential therapeutic targets for bladder dysfunction.
Similar content being viewed by others
Introduction
Urination serves dual roles as a physiological necessity for waste elimination and a socially significant behavior. Mammals integrate internal cues, such as bladder sensation, with external factors and subsequently determine whether to initiate urination1,2,3,4. Humans typically develop voluntary control over urination at socially acceptable times and places in early childhood5, while certain animals deliberately release urine into the environment to convey social signals through pheromones in urine, a behavior known as urine scent-marking6,7. This behavioral complexity underscores the tight regulation of urination by both peripheral and central nervous systems. However, the cellular and circuit-level mechanisms underpinning central control remain incompletely understood.
The periaqueductal gray matter (PAG) has emerged as a pivotal structure in regulating lower urinary tract function8. Electrical stimulation at distinct PAG sites has been shown to induce bladder contraction, evoke neuronal firing at bladder postganglionic nerves, or suppress bladder contraction9,10,11. Lesions in the PAG result in acute urinary retention, altered bladder sensation, or enhanced bladder activity12,13. Single-unit recordings in animal studies have identified that intermixed neurons in the PAG display either increased or decreased firing rates during urination and bladder contraction14. These opposite results possibly indicate functional diversity among neuronal subtypes in distinct PAG columns. Notably, the PAG receives direct input from sacral dorsal horn neurons that process bladder afferent signals and has extensive connections with brain regions involved in urination control15,16,17. Given the complex circuitry and cellular heterogeneity of the PAG, there is a pressing need to identify specific neuronal subsets and their precise contributions to urination control.
The lateral and ventrolateral columns of the PAG (l/vlPAG) are particularly important in urination control8,17. These regions harbor diverse neuronal subtypes defined by molecular markers18. To date, only the roles of excitatory neurons expressing vesicular glutamate transporter 2 (Vglut2) within the l/vlPAG in urination control have been partially characterized. These Vglut2+ neurons promote urination via excitatory monosynaptic connections with neurons in the pontine micturition center (PMC), and optogenetic stimulation of their axon terminals in the PMC elicits bladder contraction and urination19. Somatostatin (SST)-positive neurons are widely distributed throughout the mammalian brain20 and participate in regulating various innate behaviors like sleep, feeding, and defensive behaviors21,22,23. Their function and importance in the control of urination, which is also an innate behavior24, have not yet been demonstrated. Notably, SST+ neurons in the l/vlPAG are a specific subpopulation of glutamatergic neurons, in contrast to SST+ neurons in most other brain regions being inhibitory interneurons25,26. Moreover, SST+ neurons in the l/vlPAG and Vglut2+ neurons in the same area exhibit distinct roles in regulating some physiological functions (e.g., pain and itch processing)25,27,28,29. Taking into account the PAG’s essential role in urination control and the unique characteristics of l/vlPAG SST+ neurons, we propose that these neurons may be key regulators of urination.
In this study, we first verified that PAG neurons involved in controlling the bladder and external urethral sphincter (EUS) were distributed in the l/vlPAG using retrograde trans-multisynaptic tracing. We then focused on the involvement of l/vlPAG SST+ neuronal population in the regulation of urination and bladder function, employing a combination of cell type-specific neuronal activity recording, optogenetic stimulation, and chemogenetic manipulation alongside cystometrograms and EUS electromyograms (EMG). Our findings demonstrate that l/vlPAG SST+ neurons are essential for controlling urination and bladder function via the PAG-PMC pathway. This work provides novel insights into the cellular and circuit mechanisms underlying urination control, offering a foundation for future therapeutic interventions.
Results
l/vlPAG neurons innervate the bladder and EUS
The bladder and external urethral sphincter (EUS) are essential components of the lower urinary tract, requiring coordinated bladder contraction and EUS relaxation for proper urination5. This coordination is mediated by both peripheral and central nervous systems. To visualize the peripheral nerve distribution within the intact bladder and EUS, we applied the FDISCO clearing method30, a three-dimensional (3D) imaging approach that preserves fluorescence in cleared tissues (Fig. 1a, b). Using light-sheet microscopy, we imaged the cleared bladder and EUS (Fig. 1a), which were immunolabeled with PGP9.5, a pan-neuronal marker, and α-SMA, a marker for smooth muscle cells (Fig. 1c–f). We observed an intricate network of nerve fibers spanning the bladder and EUS, particularly concentrated in the muscular layers (Fig. 1d–f). Thick nerve fiber bundles were prominently located at the base of the bladder and within the EUS (Fig. 1f). These data provide detailed information on the nerve fiber distribution in the bladder and EUS, which is critical for guiding pseudorabies virus (PRV) injection into the specific sites (muscular layers) in the following retrograde trans-synaptic tracing experiments.

a Schematic representation of tissue clearing and 3D imaging to visualize bladder and EUS innervation. b Transparent bladder and EUS following FDISCO clearing. c Images of the entire FDISCO-cleared bladder stained with PGP9.5 and α-SMA. d Nerve fibers in the muscular layer of the bladder. e Image of the entire FDISCO-cleared EUS stained with PGP9.5. f 3D images of the entire bladder and EUS stained with PGP9.5 and α-SMA. d: dorsal; v: ventral; r: rostral; c: caudal. g Schematic diagram of bladder wall injection with PRV-EGFP (PRV531). Representative image of EGFP expression in the bladder wall. h Representative images showing PRV-labeled neurons in the MPG, SPN, PMC, and l/vlPAG at 96 hours after PRV injection. The same experiments were performed on three male mice. i Schematic diagram of EUS injection with PRV-EGFP (PRV531). Expression of EGFP in the EUS. j Representative images showing PRV-labeled neurons in the On, DGC, PMC, and l/vlPAG at 96 h after PRV injection. The same experiments were performed on three male mice.
To identify central PAG neurons involved in controlling the bladder and EUS, we injected the trans-synaptic retrograde tracer PRV-EGFP (PRV531) into the muscular layers of the bladder and EUS (Fig. 1g–j). At 48 h post-injection into the bladder, PRV-labeled neurons were primarily found in the major pelvic ganglion (MPG) of the peripheral nervous system and the sacral parasympathetic nucleus (SPN) within the spinal cord (Supplementary Fig. 1a). Neurons in the SPN send parasympathetic preganglionic fibers to MPG parasympathetic postganglionic cells, which subsequently innervate the bladder5,31. At 48 h post-PRV injection into the EUS, PRV-labeled neurons were only observed in the Onuf’s nucleus (On) and the dorsal gray commissure (DGC) in the spinal cord (Supplementary Fig. 1b). Somatic motor neurons in the Onuf’s nucleus (On) receive dense inputs from the interneurons in the DGC and then send their axons to directly control EUS activity3,32. Based on our results and these previous reports, the spinal cord neurons in the SPN and DGC act as the second-order neurons for controlling the bladder and EUS, respectively. By 72 h post-injection, viral labeling had progressed to the pontine micturition center (PMC), as seen in both bladder and EUS PRV injection groups (Supplementary Fig. 1c, d). It is known that PMC neurons directly project to the SPN and DGC32,33, indicating that PMC neurons are the third-order neurons. At 96 hours post-injection into the bladder and EUS, PRV-labeled neurons appeared in the l/vlPAG (Fig. 1h, j), while such neurons were not present at earlier time points (Supplementary Fig. 1a-d). These observations suggest retrograde trans-synaptic transport of PRV through at least four synaptic connections from the bladder or EUS to the l/vlPAG. These results are consistent with prior studies identifying regions that innervate the bladder and EUS34,35. Collectively, these findings demonstrate that l/vlPAG neurons are trans-synaptically upstream of the bladder and EUS.
Activity of SST+ neurons in the l/vlPAG increases during urination
To explore the role of l/vlPAG SST+ neurons in the process of urination, we measured their Ca2+ activity alongside real-time urine deposition in freely moving mice. An adeno-associated virus (AAV) expressing the Cre-dependent Ca2+ indicator GCaMP6f (AAV-DIO-GCaMP6f) was infused into the l/vlPAG of SST-Cre mice, and an optical fiber was implanted above the l/vlPAG three weeks later (Fig. 2a). Fiber photometry was used to monitor Ca2+ signals, while urination events were recorded using a camera following intraperitoneal (i.p.) administration of the diuretic furosemide, which increased the number of urination events during the recording period (Fig. 2b).

a Schematic of AAV-DIO-GCaMP6f injection (left) and optical fiber implantation (middle) in SST-Cre mice. Representative image of GCaMP6f-labeled SST+ neurons in the l/vlPAG (right). b Schematic of fiber photometry for Ca2+ signal detection and real-time urination tracking. c Example Ca2+ fluorescent signal trace in experimental mice expressing GCaMP6f in l/vlPAG SST+ neurons during urination event (left). Averaged Ca2+ signals (middle) and heatmaps of individual traces (right) from experimental mice expressing GCaMP6f (n = 86 trials from 8 mice) aligned to each urination event. Black arrows and dotted bars indicate urination onsets. d Example fiber photometry trace from control mice expressing GFP in l/vlPAG SST+ neurons during urination (left). Averaged fiber photometry traces (middle) and heatmaps of individual traces (right) from control mice expressing GFP (n = 82 trials from 7 mice) aligned to each urination event. e Quantification of peak amplitudes of Ca2+ signals from all fiber photometry traces across experimental mice expressing GCaMP6f (n = 8 mice) and control mice expressing GFP (n = 7 mice). **P = 0.001 (Wilcoxon rank-sum test). f Quantification of the percentage of Ca2+ signals that correlated with each urination. ***P < 0.001 (GCaMP6f group versus GFP group, Wilcoxon rank-sum test). g Onset of each urination-associated Ca2+ signal from experimental mice expressing GCaMP6f plotted to the onset of urination event at time = 0 s. h Averaged Ca2+ signals (red) from experimental mice expressing GCaMP6f aligned to the onset of each urination event or to shuffled urination events (black). All data are presented as mean ± SEM.
In both the average and individual urination events, the Ca2+ fluorescence intensity from GCaMP6f-expressing l/vlPAG SST+ neurons in experimental mice showed a robust increase during urination, and the fluorescence increase began prior to the onset of each urination event (Fig. 2c). No changes in fluorescence were observed in control mice expressing GFP in l/vlPAG SST+ neurons during each urination event (Fig. 2d, e; GCaMP6f group, max Δf/f = 10.83% ± 1.15%; GFP group, max Δf/f = 0.83% ± 0.20%; Wilcoxon rank-sum test, P = 0.001), confirming that the Ca2+ signals were not movement artifacts. Each urination event coincided with an increase in the GCaMP6f fluorescence in experimental mice (Fig. 2f). Temporal alignment of Ca2+ signals with urination onset revealed that l/vlPAG SST+ neuron activity began increasing approximately 1.6 s before urination (Fig. 2g). This activity was absent in shuffled data (Fig. 2h). These results indicate that l/vlPAG SST+ neurons are highly active during urination and exhibit temporally correlated activity with its initiation.
Activation of l/vlPAG SST+ neurons triggers urination in awake and anesthetized mice
We hypothesized that activating l/vlPAG SST+ neurons would be sufficient to evoke urination. To test this, we selectively expressed channelrhodopsin-2 (ChR2) in l/vlPAG SST+ neurons using Cre-dependent AAV (AAV-DIO-ChR2-mCherry) injections in SST-Cre mice (Fig. 3a). Four weeks post-injection, optical fibers were implanted above the l/vlPAG to deliver 473 nm excitation light (5 mW, 25 Hz for 5 s, 10 ms duration for each pulse) every 2–3 min. Light-induced urination was monitored using a camera positioned beneath the test chamber (Fig. 3b). To confirm neural activation, we analyzed c-Fos expression, a marker of neuronal activation, in ChR2-expressing l/vlPAG SST+ neurons following photostimulation. The majority of ChR2-labeled neurons co-expressed c-Fos (Fig. 3c; 93.70% ± 2.29%), demonstrating successful activation. In freely moving mice, light stimulation consistently induced urination in 94% of trials, with an average latency of 2 s (Fig. 3d, e). In contrast, control mice expressing mCherry (AAV-DIO-mCherry) in l/vlPAG SST+ neurons showed no light-induced urinations (Fig. 3d, e; SST-ChR2, 94.17% ± 2.65%; SST-mCherry, 0.00% ± 0.00%; Wilcoxon rank-sum test, P < 0.001). Thus, these findings demonstrate that activation of l/vlPAG SST+ neurons is sufficient to drive urination in freely moving mice.

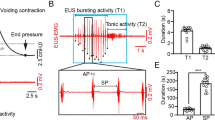
a Schematic for AAV-DIO-ChR2-mCherry injection into the l/vlPAG of SST-Cre mice (left) and optical fiber implantation above the l/vlPAG four weeks following viral injection (right). Representative image of ChR2 expression in l/vlPAG SST+ neurons (right). b Schematic diagram of optogenetic activation and real-time tracking of urination events in a freely moving mouse. c Representative images of c-Fos expression in the l/vlPAG following photostimulation of ChR2-expressing l/vlPAG SST+ neurons (left). Percentage of ChR2-positive neurons in the l/vlPAG co-expressing c-Fos protein (right). n = 3 mice. d Raster plot of urination duration (yellow bars) of each trial during and after photostimulation of l/vlPAG SST+ neurons expressing ChR2 or mCherry (n = 120 trials from 8 mice for each group). White bars indicate periods without urination. e Quantification of the percentage of photostimulation trials inducing urination (left). n = 8 mice for each group. ***P < 0.001 (ChR2 group versus mCherry group, Wilcoxon rank-sum test). Summary of light-evoked urination latency after photostimulation of ChR2-expressing l/vlPAG SST+ neurons (right). f Schematic of optogenetic stimulation in conjunction with cystometry (saline infusion into the bladder, filling bladder) and EUS-EMG recording in anesthetized mice. g Representative time-locked bladder pressure (black) and EUS-EMG (red) traces from ChR2-expressing and mCherry-expressing l/vlPAG SST+ neurons throughout photostimulation. Blue bars are light-stimulation periods. h Averaged bladder pressure traces from experimental mice expressing ChR2 (red) and control mice expressing mCherry (black) aligned to light stimulation. i Heatmaps of individual bladder pressure and EUS-EMG traces from experimental mice expressing ChR2 (n = 105 trials from 7 mice), aligned to photostimulation. j Heatmaps of individual bladder pressure and EUS-EMG traces from control mice expressing mCherry (n = 105 trials from 7 mice), aligned to photostimulation. k Quantification of pressure changes (∆P) between the end of the 5-s photostimulation and the pressure before stimulation (left), as well as the pressure ratio (calculated as the ratio of the end pressure of the 5-s photostimulation over the pressure before stimulation, right). n = 7 mice for each group. Left, ***P < 0.001 (ChR2 group versus mCherry group, unpaired t test); right, ***P < 0.001 (ChR2 group versus mCherry group, unpaired t test). l Quantification of the percentage of photostimulation trials inducing bladder contractions (left) and EUS bursting activity (right). n = 7 mice for each group. Left, **P = 0.001 (ChR2 group versus mCherry group, Wilcoxon rank-sum test); right, **P = 0.002 (ChR2 group versus mCherry group, Wilcoxon rank-sum test). All data are presented as mean ± SEM.
To evaluate whether activating l/vlPAG SST+ neurons could directly elicit reflex voiding, we performed optogenetic stimulation in anesthetized mice while recording bladder pressure (cystometry) and external urethral sphincter (EUS) electromyography (EMG) (Fig. 3f). Reflex voiding was triggered by slowly perfusing saline into the bladder (filling bladder) to establish regular urination cycles, characterized by elevated bladder pressure and associated EUS-EMG bursting activity followed by a decrease in bladder pressure. After the formation of regular urination cycles, the excitation light (5 mW, 25 Hz for 5 s, 10 ms duration for each pulse) was delivered via optical fibers above the l/vlPAG at randomized intervals (20 s or 40 s stimulation intervals in a random sequence). Photostimulation reliably induced bladder contractions (pressure increases) in every trial, with reflex voiding occurring in some trials when the bladder was sufficiently filled (Fig. 3g–i). Importantly, no bladder contractions or urinations were observed in mCherry-expressing control mice under identical conditions (Fig. 3g–j). Aligning bladder pressure data to the onset of light stimulation revealed that bladder contractions were tightly time-locked to the stimulation and persisted beyond the light pulse (Fig. 3h, i). The magnitude of pressure increase between the end of the 5-s light stimulation and the pre-stimulation baseline was significantly higher in ChR2-expressing mice compared to controls (Fig. 3k; SST-ChR2, ∆P = 3.59 ± 0.51 cmH2O; SST-mCherry, ∆P = −0.03 ± 0.15 cmH2O; unpaired t test, P < 0.001; SST-ChR2, Pressure ratio = 1.28 ± 0.04; SST-mCherry, Pressure ratio = 1.06 ± 0.02; unpaired t test, P < 0.001). Across all the trials of ChR2-expressing mice, light-induced bladder contractions were observed in 99% of trials, while light-evoked urination (bladder contractions accompanied by EUS bursting activity) occurred in 48% of trials (Fig. 3l). These findings confirm that activation of l/vlPAG SST+ neurons triggers bladder contractions but does not affect the EUS unless the bladder is partially filled.
l/vlPAG SST+ neurons control bladder contraction
To determine whether l/vlPAG SST+ neurons directly regulate bladder activity or the external urethral sphincter (EUS) relaxation, we performed bladder pressure recordings without saline infusion and monitored EUS-EMG activity in anesthetized mice (Fig. 4a). After the bladder was emptied, as indicated by minimal bladder pressure at the end of a urination cycle36, saline perfusion was halted to prevent refilling (not filling bladder). When the bladder pressure no longer changed, optogenetic stimulation was then applied. Interestingly, light stimulation elicited bladder pressure increases without accompanying EUS-EMG activity (Fig. 4b). Alignment of bladder pressure traces to the onset of photostimulation revealed that light reliably triggered time-locked bladder contractions in ChR2-expressing mice, whereas no such responses were observed in mCherry-expressing control mice (Fig. 4b–e). The pressure difference (∆P) between the end of the 5-s light stimulation and the pre-stimulation baseline, as well as the pressure ratio, was significantly higher in ChR2-expressing mice than in controls (Fig. 4f; SST-ChR2, ∆P = 1.99 ± 0.38 cmH2O; SST-mCherry, ∆P = -0.02 ± 0.01 cmH2O; unpaired t test, P = 0.002; SST-ChR2, Pressure ratio = 1.30 ± 0.06; SST-mCherry, Pressure ratio = 1.02 ± 0.002; unpaired t test, P = 0.004). In all trials involving ChR2-expressing mice, light-induced bladder pressure increases occurred in 100% of trials, but no light-evoked EUS-EMG bursting activities were observed (Fig. 4g). These findings demonstrate that activation of l/vlPAG SST+ neurons induces time-locked bladder contractions independently of EUS activity.

a Schematic of optogenetic stimulation in conjunction with cystometry (no saline infusion into the bladder, not filling bladder) and EUS-EMG recording in anesthetized mice. b Representative time-locked traces of bladder pressure (black) and EUS-EMG (red) from ChR2-expressing and mCherry-expressing l/vlPAG SST+ neurons throughout light-stimulation. Blue bars are light-stimulation periods. c Averaged bladder pressure traces from experimental mice expressing ChR2 (red) or control mice expressing mCherry (black) aligned to light stimulation. d Heatmaps of individual bladder pressure and EUS-EMG traces from experimental mice expressing ChR2 (n = 70 trials from 7 mice), aligned to photostimulation. e Heatmaps of individual bladder pressure and EUS-EMG traces from mice expressing mCherry (n = 70 trials from 7 mice), aligned to photostimulation. f Quantification of pressure changes (∆P, calculated as the difference between the end pressure of 5-s photostimulation and pre-stimulation baseline, left) and pressure ratio (calculated as the ratio of end pressure to baseline pressure, right). n = 7 mice for each group. Left, **P = 0.002 (ChR2 group versus mCherry group, unpaired t test); right, **P = 0.004 (ChR2 group versus mCherry group, unpaired t test). g Quantification of the percentage of photostimulation trials inducing bladder contraction (left) and EUS bursting activity (right). n = 7 mice for each group. Left, ***P < 0.001 (ChR2 group versus mCherry group, Wilcoxon rank-sum test); right, P = 1.00 (ChR2 group versus mCherry group, Wilcoxon rank-sum test). All data are presented as mean ± SEM.
Inhibition of l/vlPAG SST+ neurons terminates the ongoing urination
To investigate whether l/vlPAG SST+ neuronal activity is necessary for urination, we examined the effects of inhibiting these neurons during ongoing urination events. A Cre-dependent AAV encoding inhibitory protein Guillardia theta anion conducting channelrhodopsin 1 (GtACR1)37 was bilaterally injected into the l/vlPAG of SST-Cre mice (Fig. 5a). Four weeks post-injection, optical fibers were implanted above the l/vlPAG to deliver continuous blue light (3 mW, 473 nm) for optogenetic inhibition (Fig. 5a, b). After intraperitoneal injection of the diuretic furosemide, mice were placed in a custom test chamber lined with filter paper. A camera beneath the chamber monitored urine deposition (Fig. 5b). We recorded a spontaneous urination event without light stimulation (light off), followed by a subsequent urination event where optogenetic inhibition was applied for 30 s, starting within 2 s of urination onset (Fig. 5b). Optogenetic inhibition of l/vlPAG SST+ neurons disrupted ongoing urination, resulting in smaller urine deposition areas and shorter urination durations compared to light-off trials (Fig. 5c–e; urine area: hGtACR1 light off, 39.30 ± 4.35 cm2; hGtACR1 light on, 12.25 ± 0.75 cm2; paired t test, P < 0.001; total duration: hGtACR1 light off, 5.10 ± 0.25 s; hGtACR1 light on, 1.37 ± 0.26 s; Wilcoxon signed-rank test, P = 0.012). The latency to terminate urination after light onset was approximately 0.6 s (Fig. 5f). In contrast, photoinhibition had no effect on mCherry-expressing control mice, with no changes in urine deposition area or urination duration observed between light-off and light-on conditions (Fig. 5c–e; urine area: mCherry light off, 34.57 ± 2.93 cm2; mCherry light on, 35.17 ± 2.71 cm2; paired t test, P = 0.568; total duration: mCherry light off, 4.97 ± 0.20 s; mCherry light on, 5.21 ± 0.21 s; paired t test, P = 0.281). These results indicate that l/vlPAG SST+ neuronal activity is required for sustained urination.

a Schematic for AAV-DIO-hGtACR1-mCherry injection into the l/vlPAG of SST-Cre mice (left) and two optical fibers implantation above the bilateral l/vlPAG four weeks after viral expression (right). b Each mouse was placed in a custom-made test chamber lined with filter paper after intraperitoneal (i.p.) injection of the diuretic furosemide. Spontaneous urination without light stimulation (light off) was recorded, followed by a urination event with 30-s optogenetic inhibition (light on). The light was delivered within 2 s of urination onset. In the subsequent session, each mouse was tested again. c Representative trials showing the urine spots deposited on the filter paper with or without optogenetic inhibition from a single mouse of hGtACR1 group or mCherry group. d Quantification of urine area on the filter paper of the light-off trial and the subsequent light-on trial. n = 8 mice for hGtACR1 group, n = 7 mice for mCherry group. Left, ***P < 0.001 (paired t test); right, P = 0.568 (paired t test). e Quantification of total urination duration of the light-off trial and subsequent light-on trial. n = 8 mice for hGtACR1 group, n = 7 mice for mCherry group. Left, *P = 0.012 (Wilcoxon signed-rank test); right, P = 0.281 (paired t test). f Summary of the latency of urination suspension following photostimulation of hGtACR1-expressing l/vlPAG SST+ neurons. n = 8 mice for hGtACR1 group. All data are presented as mean ± SEM.
Activation of PMC-projecting l/vlPAG SST+ neurons promotes urination in awake mice
To identify downstream regions receiving projections from l/vlPAG SST+ neurons, we unilaterally injected an AAV expressing Cre-dependent GFP (AAV-DIO-GFP) into the l/vlPAG of SST-Cre mice and analyzed projections four weeks post-injection (Fig. 6a). Besides the PMC, dense projections were found in the bed nucleus of stria terminalis (BNST), medial preoptic area (MPA), lateral preoptic area (LPO), periventricular area (PVA), paraventricular nucleus (PVN), zona incerta (Zi) lateral hypothalamic area (LHA), dorsomedial hypothalamus (DMH), ventral tegmental area (VTA), and rostral ventromedial medulla (RVM) (Fig. 6b, c). To further confirm the existence of projections from l/vlPAG SST+ neurons to the PMC, AAV2/2-Retro-DIO-Tdtomato was injected into the PMC, revealing retrogradely labeled neurons in the l/vlPAG (Fig. 6d). Notably, TdTomato-labeled PAG neurons were predominantly located in the l/vlPAG (Fig. 6e–g, Bregma: −4.24 to −4.84 mm), confirming a PMC-directed projection.

a Schematic of AAV-DIO-GFP injection into the unilateral l/vlPAG of SST-Cre mice (left). Representative image of GFP-labeled somas in the l/vlPAG (right). b, c These l/vlPAG SST+ neurons mainly sent descending outputs to the PMC, BNST, MPA, LPO, PVA, PVN, Zi, LHA, DMH, VTA, and RVM. n = 3 mice. d Schematic of AAV-Retro-DIO-Tdtomato injection into the unilateral PMC of SST-Cre mice. n = 3 mice. e, f Serial coronal brain slices from a single mouse with Tdtomato-labeled neurons in the whole l/vlPAG. g Summary of Tdtomato-labeled neurons in the ipsilateral l/vlPAG (Bregma: -4.24 to -4.84 mm, n = 3 mice). Black circles indicate the average number of Tdtomato-labeled l/vlPAG neurons in each brain section.
l/vLPAG neurons are known to regulate urination through a descending PAG-PMC pathway19. To evaluate whether activation of l/vlPAGSST-PMC projections triggers urination, we bilaterally injected Retro-AAV (AAV2/2-Retro-FLEX-Flpo) into the PMC and AAV-fDIO-hM3D-mCherry into the l/vlPAG of SST-Cre mice, enabling expression of the chemogenetic tool hM3D in PMC-projecting l/vlPAG SST+ neurons (Fig. 7a). During a 4-day behavioral test, each mouse received intraperitoneal (i.p.) injections of either saline (two trials) or clozapine-N-oxide (CNO; two trials; Fig. 7b). Pharmacogenetic activation of PMC-projecting l/vlPAG SST+ neurons significantly increased urination events, total urine area and shortened intervals between consecutive urinations during the 2-hour observation window compared to saline trials (Fig. 7c–f; number: day 1, 2.9 ± 1.0; day 2, 24.9 ± 2.7; day 3, 2.5 ± 0.5; day 4, 25.0 ± 4.1; Friedman’ s test, P < 0.001; interval: saline, 50.4 ± 9.3 min; CNO, 4.7 ± 0.5 min; paired t test, P = 0.002; urine area: saline, 68.2 ± 25.8 cm2; CNO, 131.2 ± 27.0 cm2; Wilcoxon signed-rank test, P = 0.012). Control mice expressing mCherry in PMC-projecting l/vlPAG SST+ neurons exhibited no changes in urination patterns following CNO administration (Supplementary Fig. 2a–d; number: day 1, 3.1 ± 0.9; day 2, 3.3 ± 1.3; day 3, 2.4 ± 0.8; day 4, 3.1 ± 0.8; Friedman’ s test, P = 0.285; interval: saline, 49.1 ± 11.9 min; CNO, 50.7 ± 14.0 min; paired t test, P = 0.824; urine area: saline, 94.8 ± 15.8 cm2; CNO, 105.3 ± 20.3 cm2; paired t test, P = 0.524). To further explore this pathway, we used optogenetics to manipulate PMC-projecting l/vlPAG SST+ neurons. AAV2/2-Retro-FLEX-Flpo was injected into the PMC, and AAV-fDIO-ChR2-mCherry was delivered to the l/vlPAG of SST-Cre mice (Fig. 7g). Optogenetic stimulation (5 s, 473 nm, 25 Hz, 10 ms duration for each pulse) of these neurons via an optical fiber implanted above the l/vlPAG induced urination events with a 90% success rate and an average latency of 1.9 s (Fig. 7g–j). In contrast, light stimulation in control mice expressing mCherry did not elicit urination (Fig. 7i, j; ChR2, 90.00% ± 3.56%; mCherry, 0.00% ± 0.00%; Wilcoxon rank-sum test, P = 0.001). These findings underscore the critical role of l/vlPAG SST+ neurons in regulating urination through the PAG-PMC pathway.

a Schematic for AAV-Retro-FLEX-Flpo injection into the PMC and AAV-fDIO-hM3D-mCherry injection into the l/vlPAG of SST-Cre mice (left). Representative image of hM3D-expressing somas in the l/vlPAG (right). b Experimental timeline for investigating the effects of CNO-induced activation on urination. c Representative urine spots deposited on filter papers from hM3D-expressing mice treated with saline or CNO. d Raster plots showing urination events during the 4-day behavioral test. e Daily quantification of urination events on filter paper from hM3D-expressing mice (n = 8 mice). ***P < 0.001 (Friedman’ s test); day 1 vs. day 2, *P = 0.016; day 3 vs. day 4, **P = 0.008. f Quantification of the urination interval (left) and total urine area (right) following saline or CNO administration from hM3D-expressing mice (n = 8 mice). Left, **P = 0.002 (saline versus CNO, paired t test); right, *P = 0.012 (saline versus CNO, Wilcoxon signed-rank test). g Schematic of AAV-Retro-FLEX-Flpo injection into the PMC and AAV-fDIO-ChR2-mCherry injection into the l/vlPAG of SST-Cre mice (left) and optical fiber implantation above the l/vlPAG four weeks following viral expression (right). Representative images of ChR2 expression in PMC-projecting l/vlPAG SST+ neurons (right). h Schematic of optogenetic activation experiment and real-time tracking of urination events in freely moving mice. i Raster plot of urination duration (yellow bars) in each trial during and after photostimulation of PMC-projecting l/vlPAG SST+ neurons that express ChR2 (n = 120 trials from 8 mice) or mCherry (n = 105 trials from 7 mice). White bars indicate periods without urination and blue bars indicate light-stimulation periods. j Quantification of the percentage of photostimulation trials inducing urination (left). n = 8 mice for ChR2 group, n = 7 mice for mCherry group. **P = 0.001 (ChR2 group versus mCherry group, Wilcoxon rank-sum test). Summary of the latency of light-evoked urination following photostimulation of ChR2-expressing PMC-projecting l/vlPAG SST+ neurons (right). All data are presented as mean ± SEM.
Activation of PMC-projecting l/vlPAG SST+ neurons induces bladder contraction that pass through the pelvic nerve
We hypothesized that l/vlPAG SST+ neurons regulate bladder contraction through the PMC. To test this, we examined the effect of optogenetic activation of PMC-projecting l/vlPAG SST+ neurons on bladder pressure during continuous saline perfusion into the bladder (Supplementary Fig. 3a). Photostimulation of these PMC-projecting neurons induced bladder contraction and triggered reflex voiding (Supplementary Fig. 3b–d), while control mice expressing mCherry exhibited no changes in bladder pressure during light stimulation (Supplementary Fig. 3b–e), similar to the results obtained by photoactivating axon terminals of the entire l/vlPAG Vglut2+ neurons in the PMC in previous work19. ChR2-expressing mice showed significantly greater bladder pressure increases compared to controls (Supplementary Fig. 3f; ChR2, ∆P = 4.09 ± 0.45 cmH2O; mCherry, ∆P = -0.03 ± 0.04 cmH2O; unpaired t test, P < 0.001; ChR2, Pressure ratio = 1.31 ± 0.07; mCherry, Pressure ratio = 1.03 ± 0.01; Wilcoxon rank-sum test, P = 0.002). The success rates of light-induced bladder contraction and voiding were 99% and 59%, respectively (Supplementary Fig. 3g). To eliminate spontaneous reflex voiding caused by continuous bladder infusion, we conducted optogenetic activation of PMC-projecting l/vlPAG SST+ neurons without bladder filling and simultaneously monitored bladder pressure and EUS-EMG activity in anesthetized mice (Fig. 8a). Photostimulation elicited sustained bladder pressure increases without altering EUS-EMG activity (Fig. 8b–d). Control mice expressing mCherry showed no bladder contractions or EUS changes during light stimulation (Fig. 8b–e). The pressure difference (ΔP) and the pressure ratio between the end of photostimulation and baseline were significantly higher in ChR2-expressing mice than in controls (Fig. 8f; ChR2, ∆P = 2.96 ± 0.39 cmH2O; mCherry, ∆P = 0.007 ± 0.01 cmH2O; unpaired t test, P < 0.001; ChR2, Pressure ratio = 1.28 ± 0.03; mCherry, Pressure ratio = 1.02 ± 0.005; unpaired t test, P < 0.001). Light-induced bladder pressure increases were observed in 100% of trials, while no EUS bursting activity occurred during photostimulation (Fig. 8g). These results align with observations of l/vlPAG SST+ neuron activation experiments (Fig. 4).

a Schematic illustrating optogenetic stimulation combined with cystometry (without saline infusion, mimicking an empty bladder) and EUS-EMG recording in anesthetized mice. b Representative traces of bladder pressure (black) and EUS-EMG activity (red) from ChR2-expressing and mCherry-expressing PMC-projecting l/vlPAG SST+ neurons during light stimulation. Blue bars mark the duration of photostimulation. c Averaged bladder pressure traces from experimental mice expressing ChR2 (red) and control mice expressing mCherry (black), aligned with the onset of light stimulation. d Heatmaps of individual bladder pressure and EUS-EMG traces aligned with photostimulation from experimental mice expressing ChR2 (n = 105 trials from 7 mice). e Heatmaps of individual bladder pressure and EUS-EMG traces aligned with photostimulation from control mice expressing mCherry (n = 105 trials from 7 mice). f Quantification of the pressure change (∆P) between the end pressure of the 5-s photostimulation and the pressure before stimulation (left), as well as the pressure ratio (calculated as the ratio of the end pressure of the 5-s photostimulation over the pressure before stimulation, right). n = 7 mice for each group. Left, ***P < 0.001 (ChR2 group versus mCherry group, unpaired t test); right, ***P < 0.001 (ChR2 group versus mCherry group, unpaired t test). g Proportion of light-stimulation trials resulting in bladder contraction (left) and EUS bursting activity (right), with data from 7 mice per group. Left, ***P < 0.001 (ChR2 group versus mCherry group, Wilcoxon rank-sum test); right, P = 1.00 (ChR2 group versus mCherry group, Wilcoxon rank-sum test). h Schematic diagram of cutting off the pelvic nerve (PELx). i Overlaid bladder pressure traces (gray) and their averages (red or black) during light stimulation in intact mice (43 trials from 6 mice, left) and after PELx (50 trials from 6 mice, right). j Quantification of the percentage of light-stimulation trials inducing bladder contraction (left) and the difference (∆P) between the end pressure of the 5-s photostimulation and the pre-stimulation pressure (right). n = 6 mice. Left, *P = 0.014 (intact versus PELx, Wilcoxon signed-rank test); right, **P = 0.009 (intact versus PELx, paired t test). All data are presented as mean ± SEM.
The glutamatergic neurons within the PMC form direct projections to the sacral parasympathetic nucleus of the spinal cord33. Parasympathetic motoneurons located in the SPN densely innervate the bladder via fibers traveling through the pelvic nerve3,24. To further elucidate the role of PMC-projecting l/vlPAG SST+ neurons in bladder contraction, we examined the impact of their optogenetic activation before (intact) and after pelvic nerve transection (PELx; Fig. 8h). In intact mice, photostimulation consistently induced significant increases in bladder pressure (Fig. 8i). However, after PELx, these photostimulation-evoked responses were absent (Fig. 8i, j; Intact, ∆P = 2.39 ± 0.58 cmH2O; PELx, ∆P = 0.004 ± 0.018 cmH2O; paired t test, P = 0.009). Collectively, these findings support a mechanistic model wherein l/vlPAG SST+ neurons regulate bladder function and urination via the PAG-PMC pathway, with signal transmission being critically dependent on the integrity of the pelvic nerve (Fig. 9).

Schematic illustration depicting the model for l/vlPAG SST+ neurons controlling urination and bladder function via the PAG-PMC pathway.
Discussion
The periaqueductal gray matter (PAG) is a key midbrain structure essential for regulating various physiological processes, including pain perception, cough reflex, sleep, and urination17,28,38,39. Despite its broad involvement in these functions, the specific mechanisms by which distinct neuronal subpopulations in the PAG contribute to urination control have remained largely unexplored. In this study, we identified a subset of glutamatergic neurons (SST+ neurons) in the lateral and ventrolateral PAG (l/vlPAG) that play a critical role in bladder function in mice. The activity of these l/vlPAG SST+ neurons was strongly correlated with urination. Optogenetic activation of these neurons elicited urination in awake mice and bladder contraction but did not affect the EUS unless the bladder was partially filled in anesthetized mice. Furthermore, acute silencing of l/vlPAG SST+ neurons disrupted ongoing urination. These neurons project to the pontine micturition center (PMC), and chemogenetic or optogenetic manipulation of the l/vlPAGSST-PMC pathway was sufficient to trigger both urination and bladder contraction. Importantly, bladder contraction induced by the activation of PMC-projecting l/vlPAG SST+ neurons was absent following pelvic nerve transection. Taken together, these findings establish that l/vlPAG SST+ neurons regulate urination and bladder function through a neuronal circuit linking the PAG to the PMC (Fig. 9).
The regulation of bladder sensory signals and the voiding reflex are two fundamental roles of the l/vlPAG in urination control8,16,40. Retrograde tracing studies in cats have shown that the l/vlPAG directly connects with spinal cord neurons distributed in laminae I, V–VII, and X at the lumbosacral segments, which receive primary afferent inputs from the bladder41. Functional neuroimaging in humans has further demonstrated that bladder distention activates the vlPAG42. In conscious rats, noxious bladder stimulation (e.g., chemical irritation) induces c-Fos expression in a significant number of l/vlPAG neurons43. Additionally, Vglut2+ neurons in the vlPAG are activated by bladder electrical stimulation44. These findings support the theory that bladder sensation is relayed via the l/vlPAG. However, unlike previous studies, our results suggest that the l/vlPAG plays an active role in regulating the voiding reflex, as evidenced by the following points: First, our transsynaptic retrograde tracing data indicated that a subset of l/vlPAG neurons participated in controlling the motor functions of the bladder and EUS via descending neural pathways, probably sending out signals that promote urination. This is in contrast to the previous notion that l/vlPAG neurons receive ascending inputs from the spinal cord, which might carry sensory information from the bladder41. Second, fiber photometry recordings during urination in freely moving mice revealed that excitatory SST+ neurons in the l/vlPAG were active only during the voiding phase, with no increase in activity during the storage phase (Fig. 2). This suggests the presence of another subset of l/vlPAG glutamatergic neurons responsive to bladder sensory input. Third, optogenetic activation of l/vlPAG SST+ neurons was sufficient to trigger the voiding reflex and bladder contraction (Fig. 3). Fourth, acute inhibition of l/vlPAG SST+ neuron activity during the voiding phase interrupted ongoing urination (Fig. 5), underscoring the necessity of these neurons for the proper execution of the voiding reflex.
The PMC is the most important downstream region of the PAG and has been recognized as a “command center” for urination control13,45,46,47. The PMC is a functionally conserved area across vertebrate species, including cats, rodents, and humans, and its neurons project directly to the lumbosacral spinal cord to regulate bladder contraction, EUS relaxation, and the voiding reflex5,48,49,50. Classic studies have demonstrated that damage to the PMC or inhibition of PMC neurons results in the loss of the urination reflex and urinary retention19,51,52, whereas lesions in forebrain regions or decerebration do not impair reflexive urination13,53,54. The PMC consists of a variety of cell subtypes that express different kinds of marker genes55. Recent research has further revealed that PMC neurons expressing corticotropin-releasing hormone (CRH) or estrogen receptor 1 mediate distinct aspects of urination control32,33. CRH+ PMC neurons drive bladder contraction by exciting autonomic motoneurons in the sacral parasympathetic nucleus, while ESR1+ PMC neurons regulate EUS relaxation by activating spinal interneurons in the dorsal gray commissure, which inhibit somatic motoneurons in Onuf’s nucleus32,33. Our findings support the idea that the roles of PMC CRH+ neurons and l/vlPAG SST+ neurons in urination control are highly similar. Specifically, the activity of both l/vlPAG SST+ neurons (Fig. 2) and PMC CRH+ neurons correlates with urination, and both can trigger the voiding reflex when the bladder is partially full33,56. Moreover, photostimulation of l/vlPAG SST+ neurons (Fig. 4) or PMC CRH+ neurons induces bladder contraction in the absence of EMG bursting when the bladder is empty32. Given that l/vlPAG SST+ neurons control bladder contraction via the PAG-PMC pathway, we hypothesize that PMC CRH+ neurons may receive direct input from l/vlPAG SST+ neurons, an idea that warrants further investigation.
The current model of neuronal control of the lower urinary tract posits that the PAG relays bladder sensory information from the spinal cord to higher forebrain regions while also receiving descending signals from forebrain that modulate the voiding reflex through the PMC1,13. Our data suggest that l/vlPAG SST+ neurons may act as an “on-switch” for reflexive urination by exciting PMC neurons via the PAG-PMC pathway (Fig. 9). In contrast, inhibitory PAG neurons likely function as an “off-switch”, suppressing the voiding reflex by inhibiting excitatory PMC neurons8. Future studies will aim to identify subtypes of l/vlPAG neurons responsible for transmitting bladder sensory information or inhibiting PMC activity. Furthermore, functional neuroimaging studies in humans suggest that the location and timing of voluntary urination are tightly regulated by decision-making forebrain regions that act on the PAG16,40. In the absence of forebrain input, however, the PAG can directly activate the PMC to initiate the voiding reflex once bladder volume exceeds a certain threshold13,14. Thus, the PAG serves as a crucial link between the lower urinary tract and brain regions that regulate urination, and it deserves in-depth study.
In conclusion, our study provides new insights into the role and descending pathways of l/vlPAG SST+ neurons in regulating urination and bladder contraction. These findings contribute to a deeper understanding of the central cellular and circuit mechanisms underlying the control of urination. The l/vlPAG may serve as a potential target for diagnosing and treating urination dysfunctions, such as urinary retention, through deep brain stimulation.
Methods
Animals
All experimental protocols and animal care were approved by the Third Military Medical University (TMMU) Animal Care and Use Committee. Experiments were conducted following the TMMU Animal Welfare Guidelines and the National Institutes of Health Guidelines for the Care and Use of Laboratory Animals. SST-IRES-Cre mice (JAX: 013044) and wild-type C57BL/6 J mice (3-month-old to 5-month-old) used in this study were purchased from the Jackson Laboratory and SPF (Beijing) Biotechnology Co., Ltd., respectively. All experimental mice were group-housed under a 12-hour light/dark cycle at a temperature of 21-23 °C and had free access to mouse chow and water. Mice were housed in single cages after receiving optical fiber implantation surgery. The mouse sample size for each experiment is indicated in the respective figure legends.
PRV-based retrograde tracing
For retrograde trans-synaptic tracing experiments, the transsynaptic retrograde tracer pseudorabies virus encoding EGFP (PRV-EGFP, titer: 2×10⁹ pfu/mL, BrainVTA Technology Co., Ltd., Wuhan, China) was used, as described previously17,35,57. Adult male mice were anesthetized with 1% sodium pentobarbital (i.p., 10 ml/kg). A midline incision in the lower abdomen was made to expose the bladder and external urethral sphincter (EUS). A glass micropipette (tip diameter 20 µm) loaded with PRV-EGFP was connected to an injection pump. The micropipette was carefully inserted into the muscular layer of the bladder dome (one injection site) or EUS (left and right sides). A volume of 1 μL PRV-EGFP was delivered into each injection site at a rate of 200 nL/min. The micropipette was held in place for 10 minutes to ensure adequate infusion before being withdrawn. The incision was sutured, and the mice were perfused at different time points (48, 72, and 96 hours) following PRV-EGFP injection.
3D immunolabeling and FDISCO tissue clearing
For tissue clearing and 3D immunolabeling, adult male mice were deeply anesthetized with 1% sodium pentobarbital (i.p., 10 ml/kg) and transcardially perfused with phosphate-buffered saline (PBS), followed by paraformaldehyde (PFA, 4%, Sigma). The bladder and EUS were harvested and post-fixed overnight in 4% PFA. Whole-mount staining and tissue clearing were performed by Jarvis Biological Pharmaceutical Co., Ltd. (Wuhan, China). The organs were permeabilized for 3 days and subjected to an overnight treatment with hydrogen peroxide (H₂O₂) solution to facilitate decolorization. They were then permeabilized with 0.2% Triton X-100 solution for 4 days and blocked with a blocking solution containing 10% normal donkey serum for an additional 4 days. Primary antibodies were incubated for 7 days at room temperature, followed by incubation with secondary antibodies for 2 days at room temperature. The primary antibodies used included mouse anti-α-SMA (1:500, Jarvis Biological Pharmaceutical Co., Ltd, A101001) and rabbit anti-PGP9.5 (1:500, Abcam, ab108986). Secondary antibodies included donkey anti-mouse Alexa Fluor 555 (1:500, Invitrogen, A32773) and donkey anti-rabbit Alexa Fluor 647 (1:500, Invitrogen, A32795). Tissue clearing was conducted using the FDISCO tissue clearing reagent kit (JA11012, Jarvis Biological Pharmaceutical Co., Ltd, Wuhan, China) following the manufacturer’s instructions. The cleared tissues were imaged using a light-sheet microscope (LiToneXL, Light Innovation Technology, Hong Kong, China).
Stereotaxic injection and fiber implantation
Adult SST-Cre mice were anesthetized using isoflurane in oxygen (4% induction, 1–2% maintenance) and placed on a stereotaxic frame (RWD Technology Corp., Ltd.; Shenzhen, China). A heating pad was used to maintain body temperature at ~37 °C. After performing a midline scalp incision to expose the skull, a craniotomy was carried out above the target brain area using a hand-held drill, resulting in a craniotomy window approximately 1.5 mm in diameter. A volume of 100–250 nL of AAV suspension was injected into the l/vlPAG or the PMC using a glass micropipette. The injection rate was set at 20 nL/min. After viral injection, the glass pipette was left in place for 10 min before being withdrawn. Following surgery, mice were returned to their home cages and allowed to recover for 3-4 weeks before subsequent experiments. Coordinates for l/vlPAG (AP: -4.72 mm; ML: ±0.5 mm; DV: -1.9 mm from dura) and PMC (AP: -5.45 mm; ML: ±0.7 mm; DV: -3.14 mm from dura) were based on the Paxinos and Franklin Mouse Brain Atlas (second edition).
For fiber photometry experiments, 200 nL of either AAV-DIO-GCaMP6f (AAV2/9, titer: 1.2 × 10¹² v.g./mL, Obio Biotechnology Co., Ltd., Shanghai, China) or AAV-DIO-GFP (AAV2/9, titer: 4.2 × 10¹² v.g./mL, BrainVTA Technology Co., Ltd., Wuhan, China) as control, was injected into the right l/vlPAG of SST-Cre mice. Three weeks post-injection, a second surgery was performed where a 200-µm diameter optic fiber (NA 0.48, Doric lenses, Quebec City, QC, Canada) with an attached metal cannula was inserted ~50 µm above the viral injection site. The fiber was secured to the skull with dental cement. Mice were housed individually and allowed to recover for one week following fiber implantation.
For optogenetic activation experiments, 200 nL of either AAV-DIO-ChR2-mCherry (AAV2/9, titer: 3.67 × 10¹³ v.g./mL, Taitool Bioscience Co., Ltd., Shanghai, China) or AAV-DIO-mCherry (AAV2/5, titer: 5.0 × 10¹² v.g./mL, BrainVTA Technology Co., Ltd., Wuhan, China) as control, was injected into the right l/vlPAG of SST-Cre mice. To activate PMC-projecting SST+ neurons in the l/vlPAG, 100 nL of AAV2/2-Retro-FLEX-Flpo (AAV2/2-Retro, titer: 1.0 × 10¹³ v.g./mL, Taitool Bioscience Co., Ltd., Shanghai, China) was injected into the right PMC. Additionally, 250 nL of either AAV-fDIO-ChR2-mCherry (AAV2/9, titer: 1.0 × 10¹³ v.g./mL, Taitool Bioscience Co., Ltd., Shanghai, China) or AAV-fDIO-mCherry (AAV2/9, titer: 1.0 × 10¹³ v.g./mL, Taitool Bioscience Co., Ltd., Shanghai, China) as control, was injected into the right l/vlPAG. Three to four weeks post-injection, a second surgery was conducted, where a 200-µm diameter optic fiber (NA 0.48, Doric lenses, Quebec City, QC, Canada) with an attached metal cannula was inserted ~200 µm above the viral injection site in the right l/vlPAG and fixed to the skull with dental cement. Mice were housed individually and allowed to recover for one week following fiber implantation.
For optogenetic inhibition experiments, 200 nL of either AAV-DIO-hGtACR1-mCherry (AAV2/9, titer: 1.72 × 10¹³ v.g./mL, Taitool Bioscience Co., Ltd., Shanghai, China) or AAV-DIO-mCherry (AAV2/5, titer: 5.0 × 10¹² v.g./mL, BrainVTA Technology Co., Ltd., Wuhan, China) as control, was bilaterally injected into the l/vlPAG of SST-Cre mice. After 3-4 weeks, a second surgery was performed, in which two 200-µm diameter optic fibers (NA 0.48, Doric lenses, Quebec City, QC, Canada) were bilaterally inserted into the l/vlPAG (the coordinates for left l/vlPAG: AP: -4.72 mm; ML: -0.5 mm; DV: -1.6 mm from dura, the coordinates for right l/vlPAG: the angle is 45 degrees; AP: -4.72 mm; ML: 1.62 mm; DV: -3.2 mm from dura). The fibers were secured to the skull with dental cement. Mice were housed individually and allowed to recover for one week following fiber implantation.
For chemogenetic activation of PMC-projecting SST+ neurons in the l/vlPAG, 100 nL of AAV2/2-Retro-FLEX-Flpo (AAV2/2-Retro, titer: 1.0 × 10¹³ v.g./mL, Taitool Bioscience Co., Ltd., Shanghai, China) was bilaterally injected into the PMC. Additionally, 250 nL of either AAV-fDIO-hM3D-mCherry (AAV2/9, titer: 1 × 10¹³ v.g./mL, Taitool Bioscience Co., Ltd., Shanghai, China) or AAV-fDIO-mCherry (AAV2/9, titer: 1.0 × 10¹³ v.g./mL, Taitool Bioscience Co., Ltd., Shanghai, China) as control, was bilaterally injected into the l/vlPAG of SST-Cre mice.
For anterograde tracing of l/vlPAG-PMC projections, 200 nL of AAV-DIO-GFP (AAV2/9, titer: 4.2 × 10¹² v.g./mL, BrainVTA Technology Co., Ltd., Wuhan, China) was injected into the right l/vlPAG of SST-Cre mice. For retrograde tracing of l/vlPAG-PMC projections, 100 nL of AAV2/2-Retro-DIO-Tdtomato (AAV2/2-Retro, titer: 2.5 × 10¹³ v.g./mL, Obio Biotechnology Co., Ltd., Shanghai, China) was injected into the right PMC of SST-Cre mice.
Fiber photometry recordings
Fiber photometry experiments were conducted as described previously17,35,57. A fiber photometry system (FOM-02M FiberOptoMeter) was employed to detect Ca²⁺ fluorescent signals at a sampling rate of 2000 Hz. Light intensity at the fiber tip was set to 0.22 mW/mm². In order to increase the number of urination events during the recording period, each mouse was administered furosemide (40 mg/kg) via intraperitoneal injection and subsequently placed in a test chamber. The implanted recording fiber was connected to the fiber photometry system. Urination events were captured by a video recorder (30 Hz, 1280 × 720 resolution, Sony, Japan) positioned beneath the test chamber, while activity of l/vlPAG SST+ neurons was recorded simultaneously. Each session lasted 40–60 min. Ca²⁺ signal changes (Δf/f) were calculated using the formula (f − fbaseline)/fbaseline). The fbaseline represents the minimum fluorescent signal during the entire recording. To minimize noise, the Ca²⁺ signal data (Δf/f) were smoothed using a Savitzky-Golay filter (window size 50, corresponding to 25 ms). Data were aligned to individual urination onsets, which were defined as the appearance of urine at the urinary meatus. For data shuffling, the GCaMP6f fluorescence data were divided into ten segments, which were then randomly assigned to detected urination events.
Cystometric measurement and EUS-EMG recordings
Cystometric and electrode implantation procedures were performed as previously described17,35,58,59. Fiber-implanted mice were anesthetized with isoflurane in oxygen (4% induction, 1–2% maintenance) and placed on a heating pad. The bladder and external urethral sphincter (EUS) were exposed via a midline abdominal incision (2–3 cm). A 7 cm length of PE10 tubing was inserted into the bladder using a 20 G guiding needle and secured with 6-0 non-absorbable sutures. Three silver wire electrodes (36 AWG) were stripped of insulation at the ends (1–2 mm). Two electrodes were implanted into the same side of EUS using a 30 G needle, and the third electrode was placed under the skin as a ground wire. The electrodes and tubing were pulled through a small (1 cm) incision at the back of the neck. Incisions were sutured, and mice were allowed to recover for one day before the subsequent experiments.
For optogenetic stimulation during cystometry and EUS-EMG recordings, mice were anesthetized with urethane (1 g/kg, i.p.). A pressure transducer (YPJ01H; Chengdu Instrument Factory, China), syringe pump (RWD404; RWD Technology Corp., Shenzhen, China), and appropriate adapters were connected via a three-way valve. The PE10 tubing was connected to the syringe pump via PE50 tubing. Electrodes and the pressure transducer were linked to a multi-channel physiological recording system (RM6240E, Chengdu Instrument Factory, China) for bladder pressure and EUS-EMG data acquisition. Bladder pressure was recorded with a sensitivity of 12.5 cm H₂O, a sample rate of 8 kHz, and a low-pass filter at 30 Hz. EUS-EMG was recorded with a sensitivity of 1 mV and a sample rate of 8 kHz. Saline was infused into the bladder at 30–40 μL/min using the syringe pump, inducing a regular urination cycle, marked by rising bladder pressure and associated EUS-EMG bursting activity, followed by a decrease in bladder pressure. For analysis, the differences (ΔP) in bladder pressure before and after 5-s photostimulation, pressure ratios (end pressure vs. pre-stimulation), and the root mean square (RMS) of EMG traces were calculated.
Optogenetic manipulations
For optogenetic activation of l/vlPAG SST+ neurons or PMC-projecting l/vlPAG SST+ neurons in freely moving mice, fiber-implanted animals were placed in a test chamber lined with filter paper in the darkroom following furosemide injection (i.p.). Each animal was treated with furosemide before optogenetic stimulation to ensure that a larger volume of urine could be stored in the bladder within a short period. They underwent light stimulation over two consecutive days, with one 40-min photostimulation session per day (no more than 10 stimulations per day). The implanted fibers were connected to a solid-state laser (473 nm, MBL-III-473, Changchun New Industries, China) controlled by custom LabView software. Light intensity at the fiber tip was set to 5 mW. Excitation light was delivered for 5 s at 25 Hz (10 ms pulse duration) every 2–3 min. Light-induced urination events were monitored at 30 Hz via an infrared camera placed beneath the test chamber, featuring a resolution of 1920 × 1080 pixels.
For optogenetic activation of l/vlPAG SST+ neurons or PMC-projecting l/vlPAG SST+ neurons during cystometry and EUS-EMG recordings in anesthetized mice, each mouse was placed on a heating pad. Excitation light (5 mW, 25 Hz for 5 s, 10 ms pulse) was delivered at randomized intervals (20 s or 40 s stimulation intervals in a random order) during bladder filling or while the bladder was not filling to avoid the potential formation of bladder contracture, and changes in bladder pressure and EUS activity were simultaneously recorded.
For optogenetic inhibition of l/vlPAG SST+ neurons during ongoing urination in freely moving mice, animals were placed in the test chamber lined with filter paper in the darkroom after furosemide injection (i.p.). Light intensity at the fiber tip was adjusted to ~3 mW. Continuous 473 nm blue light was delivered within 2 s of the initiation of the urination event and lasted for 30 s. A spontaneous urination event without light stimulation (light-off trial) and a subsequent event with optogenetic inhibition (light-on trial) were recorded at 30 Hz via an infrared camera placed beneath the test chamber, with a resolution of 1920 × 1080 pixels. We took screenshots of the filter paper that absorbed urine after each urination event in the video, and then used Image J software to manually outline the edge of the urine spot and calculate its area. The exact time points of urination onset and the total urination duration for each urination event were analyzed by reviewing the video. Urine area and total urination duration in the light-on trial were compared with the light-off trial.
Chemogenetic manipulations
For chemogenetic activation of PMC-projecting l/vlPAG SST+ neurons in freely moving mice, each mouse received intraperitoneal (i.p.) injections of either saline (0.1 mL) or clozapine-N-oxide (CNO, 0.1 mL) for two trials during a 4-day urination behavior assay. Mice were first injected with saline or CNO and returned to their home cages. Thirty minutes later, all mice received a second saline injection (1.5 mL) and were placed in a test chamber lined with filter paper in the darkroom. Urine spots were recorded at 30 Hz over 2 h using an infrared camera placed beneath the test chamber, with a resolution of 1920 × 1080 pixels. The number of urination events on the filter paper and the interval between consecutive urinations within the 2-h observation window were analyzed by reviewing the video. If the mouse did not urinate within the 2-h observation window, we recorded the inter-urination interval as 120 min. We also took screenshots of the filter paper that absorbed urine after each urination event in the video, and then used Image J software to manually outline the edge of the urine spot and calculate its area. Prior to testing, mice were habituated to the test chamber for 3 days.
Histology and immunohistochemistry
Mice were deeply anesthetized with 1% sodium pentobarbital (i.p., 10 mL/kg) and perfused intracardially with PBS, followed by 4% paraformaldehyde (PFA, Sigma). Brains were extracted, postfixed overnight in 4% PFA, and then incubated in a 30% sucrose solution at 4 °C. Coronal brain sections (40 μm) were obtained using a freezing microtome (CryoStar NX50, Thermo). Sections were stained with DAPI (Beyotime, Shanghai, China, C1006) for nuclei labeling and imaged using either a fluorescence microscope (Olympus, BX51) or a confocal microscope (TCS SP5, Leica). For immunohistochemistry, brain sections were permeabilized with 1% Triton X-100/PBS (PBST) for 40 min and blocked with 10% normal donkey serum for 2 h. Sections were then incubated with primary antibodies overnight at 4 °C and secondary antibodies for 2 h at room temperature. Primary antibodies included chicken anti-GFP (1:200, Abcam, ab13970) and rabbit anti-c-Fos (1:200, Abcam, ab190289). Secondary antibodies included donkey anti-chicken CF488 (1:200, Sigma-Aldrich, cat. no. SAB4600031) and donkey anti-rabbit Alexa Fluor 488 (1:200, Invitrogen, A21206).
Statistics and reproducibility
Summary data are presented as means ± standard error of the mean (mean ± SEM). No statistical methods were used to predetermine sample sizes, similar to those reported in previous studies. Parametric or nonparametric statistical analyses were conducted using MATLAB and SPSS 21.0 software. The Wilcoxon rank-sum test or unpaired t test was used for comparisons between two unpaired groups. The Wilcoxon signed-rank test or paired t test was conducted for comparisons of two paired groups. Friedman’ s test was conducted for comparisons of the 4-day behavioral data from chemogenetic experiments. Statistical significance is denoted as follows: ***P < 0.001, **P < 0.01, *P < 0.05; ns indicates no significant difference. Statistical tests and significance are provided in the Figures and Figure Legends.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
All data supporting the findings of this study are available in the Supplementary data file. All other data are available from the corresponding author on reasonable request.
References
Griffiths, D. Neural control of micturition in humans: a working model. Nat. Rev. Urol. 12, 695–705 (2015).
Griffiths, D. J. Cerebral control of bladder function. Curr. Urol. Rep. 5, 348–352 (2004).
Holstege, G. Micturition and the soul. J. Comp. Neurol. 493, 15–20 (2005).
Mukhopadhyay, S. & Stowers, L. Choosing to urinate. Circuits and mechanisms underlying voluntary urination. Curr. Opin. Neurobiol. 60, 129–135 (2020).
Fowler, C. J., Griffiths, D. & de Groat, W. C. The neural control of micturition. Nat. Rev. Neurosci. 9, 453–466 (2008).
Kaur, A. W. et al. Murine pheromone proteins constitute a context-dependent combinatorial code governing multiple social behaviors. Cell 157, 676–688 (2014).
Hurst, J. L. & Beynon, R. J. Scent wars: the chemobiology of competitive signalling in mice. Bioessays 26, 1288–1298 (2004).
Zare, A., Jahanshahi, A., Rahnama’i, M. S., Schipper, S. & van Koeveringe, G. A. The Role of the Periaqueductal Gray Matter in Lower Urinary Tract Function. Mol. Neurobiol. 56, 920–934 (2019).
Taniguchi, N. et al. A study of micturition inducing sites in the periaqueductal gray of the mesencephalon. J. Urol. 168, 1626–1631 (2002).
Noto, H., Roppolo, J. R., Steers, W. D. & de Groat, W. C. Electrophysiological analysis of the ascending and descending components of the micturition reflex pathway in the rat. Brain Res. 549, 95–105 (1991).
Numata, A. et al. Micturition-suppressing region in the periaqueductal gray of the mesencephalon of the cat. Am. J. Physiol. Regul. Integr. Comp. Physiol. 294, R1996–R2000 (2008).
Yaguchi, H. et al. A case of acute urinary retention caused by periaqueductal grey lesion. J. Neurol. Neurosurg. Psychiatry 75, 1202–1203 (2004).
de Groat, W. C., Griffiths, D. & Yoshimura, N. Neural control of the lower urinary tract. Compr. Physiol. 5, 327–396 (2015).
Liu, Z. et al. Micturition-related neuronal firing in the periaqueductal gray area in cats. Neuroscience 126, 1075–1082 (2004).
Kuipers, R. & Klop, E. M. Neurons in the guinea pig (Cavia porcellus) lateral lumbosacral spinal cord project to the central part of the lateral periaqueductal gray matter. Brain Res. 1101, 43–50 (2006).
Fowler, C. J. & Griffiths, D. J. A decade of functional brain imaging applied to bladder control. Neurourol. Urodyn. 29, 49–55 (2010).
Rao, Y. et al. Ventrolateral periaqueductal gray neurons are active during urination. Front Cell Neurosci. 16, 865186 (2022).
Yu, H. et al. Social touch-like tactile stimulation activates a tachykinin 1-oxytocin pathway to promote social interactions. Neuron 110, 1051–1067.e1057 (2022).
Verstegen, A. M. J. et al. Non-Crh Glutamatergic Neurons in Barrington’s nucleus control micturition via glutamatergic afferents from the midbrain and hypothalamus. Curr. Biol. 29, 2775–2789.e2777 (2019).
Martel, G., Dutar, P., Epelbaum, J. & Viollet, C. Somatostatinergic systems: an update on brain functions in normal and pathological aging. Front Endocrinol.3, 154 (2012).
Tossell, K. et al. Somatostatin neurons in prefrontal cortex initiate sleep-preparatory behavior and sleep via the preoptic and lateral hypothalamus. Nat. Neurosci. 26, 1805–1819 (2023).
Luo, S. X. et al. Regulation of feeding by somatostatin neurons in the tuberal nucleus. Science 361, 76–81 (2018).
Yu, K., Garcia da Silva, P., Albeanu, D. F. & Li, B. Central amygdala somatostatin neurons gate passive and active defensive behaviors. J. Neurosci. 36, 6488–6496 (2016).
Xiao, W. et al. Neural circuit control of innate behaviors. Sci. China Life Sci. 65, 466–499 (2022).
Gao, Z. R. et al. Tac1-expressing neurons in the periaqueductal gray facilitate the itch-scratching cycle via descending regulation. Neuron 101, 45–59.e49 (2019).
Urban-Ciecko, J. & Barth, A. L. Somatostatin-expressing neurons in cortical networks. Nat. Rev. Neurosci. 17, 401–409 (2016).
Zhang, Y. et al. Somatostatin neurons from periaqueductal gray to medulla facilitate neuropathic pain in male mice. J. Pain. 24, 1020–1029 (2023).
Samineni, V. K. et al. Divergent modulation of nociception by glutamatergic and GABAergic neuronal subpopulations in the periaqueductal gray. eNeuro 4, https://doi.org/10.1523/ENEURO.0129-16.2017 (2017).
Samineni, V. K., Grajales-Reyes, J. G., Sundaram, S. S., Yoo, J. J. & Gereau, R. W. t. Cell type-specific modulation of sensory and affective components of itch in the periaqueductal gray. Nat. Commun. 10, 4356 (2019).
Qi, Y. et al. FDISCO: Advanced solvent-based clearing method for imaging whole organs. Sci. Adv. 5, eaau8355 (2019).
Berigan, B. R., Wang, J., Schulz, D. J. & Ozden, I. Imaging neuro-urodynamics of mouse major pelvic ganglion with a micro-endoscopic approach. J. Neurophysiol. 129, 479–493 (2023).
Keller, J. A. et al. Voluntary urination control by brainstem neurons that relax the urethral sphincter. Nat. Neurosci. 21, 1229–1238 (2018).
Hou, X. H. et al. Central control circuit for context-dependent micturition. Cell 167, 73–86.e12 (2016).
Marson, L. Identification of central nervous system neurons that innervate the bladder body, bladder base, or external urethral sphincter of female rats: a transneuronal tracing study using pseudorabies virus. J. Comp. Neurol. 389, 584–602 (1997).
Yao, J. et al. A corticopontine circuit for initiation of urination. Nat. Neurosci. 21, 1541–1550 (2018).
Uvin, P. et al. The use of cystometry in small rodents: a study of bladder chemosensation. J. Vis. Exp., e3869, (2012).
Govorunova, E. G., Sineshchekov, O. A., Janz, R., Liu, X. & Spudich, J. L. NEUROSCIENCE. Natural light-gated anion channels: a family of microbial rhodopsins for advanced optogenetics. Science 349, 647–650 (2015).
Chen, Z. et al. A descending pathway emanating from the periaqueductal gray mediates the development of cough-like hypersensitivity. iScience 25, 103641 (2022).
Weber, F. et al. Regulation of REM and Non-REM Sleep by Periaqueductal GABAergic Neurons. Nat. Commun. 9, 354 (2018).
Kitta, T. et al. Brain-bladder control network: the unsolved 21st century urological mystery. Int J. Urol. 22, 342–348 (2015).
Mouton, L. J. & Holstege, G. Segmental and laminar organization of the spinal neurons projecting to the periaqueductal gray (PAG) in the cat suggests the existence of at least five separate clusters of spino-PAG neurons. J. Comp. Neurol. 428, 389–410 (2000).
Matsuura, S. et al. Human brain region response to distention or cold stimulation of the bladder: a positron emission tomography study. J. Urol. 168, 2035–2039 (2002).
Mitsui, T. et al. Chemical bladder irritation provokes c-fos expression in the midbrain periaqueductal gray matter of the rat. Brain Res. 967, 81–88 (2003).
Zare, A., Jahanshahi, A., Meriaux, C., Steinbusch, H. W. & van Koeveringe, G. A. Glutamatergic cells in the periaqueductal gray matter mediate sensory inputs after bladder stimulation in freely moving rats. Int J. Urol. 25, 621–626 (2018).
Drake, M. J. et al. Neural control of the lower urinary and gastrointestinal tracts: supraspinal CNS mechanisms. Neurourol. Urodyn. 29, 119–127 (2010).
Benarroch, E. E. Neural control of the bladder: recent advances and neurologic implications. Neurology 75, 1839–1846 (2010).
de Groat, W. C. & Wickens, C. Organization of the neural switching circuitry underlying reflex micturition. Acta Physiol. (Oxf.) 207, 66–84 (2013).
Satoh, K., Shimizu, N., Tohyama, M. & Maeda, T. Localization of the micturition reflex center at dorsolateral pontine tegmentum of the rat. Neurosci. Lett. 8, 27–33 (1978).
Verstegen, A. M. J., Vanderhorst, V., Gray, P. A., Zeidel, M. L. & Geerling, J. C. Barrington’s nucleus: Neuroanatomic landscape of the mouse “pontine micturition center. J. Comp. Neurol. 525, 2287–2309 (2017).
Fowler, C. J. Neurological disorders of micturition and their treatment. Brain 122, 1213–1231 (1999).
Lachkepiani, G., Giorgadze, G. & Mania, M. Acute urine retention as a clinically isolated syndrome (a case report). Georgian Med News, 54–57 (2011).
Mallory, B. S., Roppolo, J. R. & de Groat, W. C. Pharmacological modulation of the pontine micturition center. Brain Res. 546, 310–320 (1991).
Tish, M. M. & Geerling, J. C. The brain and the bladder: forebrain control of urinary (In)Continence. Front Physiol. 11, 658 (2020).
Kuru, M. Nervous control of micturition. Physiol. Rev. 45, 425–494 (1965).
Nardone, S. et al. A spatially-resolved transcriptional atlas of the murine dorsal pons at single-cell resolution. Nat. Commun. 15, 1966 (2024).
Ito, H. et al. Probabilistic, spinally-gated control of bladder pressure and autonomous micturition by Barrington’s nucleus CRH neurons. Elife 9, https://doi.org/10.7554/eLife.56605 (2020).
Yao, J. et al. Simultaneous measurement of neuronal activity in the pontine micturition center and cystometry in freely moving mice. Front Neurosci. 13, 663 (2019).
Li, J. et al. A wireless, battery-free device for electrical neuromodulation of bladder contractions. Mater. Today Bio. 28, 101233 (2024).
Li, J. et al. An innovative electrical neurostimulation approach to mimic reflexive urination control in spinal cord injury models. Sci. Rep. 14, 25305 (2024).
Acknowledgements
This work was supported by grants from the National Natural Science Foundation of China (32100912 to J.Ya., 31970946 to J.Y.), China Postdoctoral Science Foundation (2021M700602 to J.Ya.), Talent Project of Chongqing (4246ZP1252 to J.Y.).
Author information
Authors and Affiliations
Contributions
J.Ya., H.Z., and J.Y. designed the experiments, interpreted the data, and wrote the manuscript with the help of all authors. Z.G. and X.L. conducted PRV tracing experiments, fiber photometry recordings, optogenetic experiments, and chemogenetic experiments. X.L., J.L. (from Guangxi University), and C.Y. performed cystometry with EUS-EMG recordings and acquired the corresponding data. Z.G., X.L., J.L. (from Third Military Medical University), C.Y., L.Y., and W.S. contributed to viral injections and post-hoc histology. J.Ya., C.Y., S.L., J.L. (from Guangxi University), G.D., S.P., and X.X. analyzed the data.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Communications Biology thanks Mark Zeidel, Luke Grundy and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. Primary Handling Editor: Benjamin Bessieres. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Yan, J., Gao, Z., Li, X. et al. SST neurons in the periaqueductal gray regulate urination and bladder function. Commun Biol 8, 639 (2025). https://doi.org/10.1038/s42003-025-08069-w
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s42003-025-08069-w
