
Abstract
Biobank data provide a rich source for studying the coheritability of multiple disease phenotypes, which can provide information on shared genetic etiology. However, the large number and heterogeneous types of phenotypes (e.g., continuous, discrete, time-to-event) pose significant statistical and computational challenges for estimating coheritability. In this work, we propose a unified modeling framework with latent random effects distinguishing genetic and family-shared environmental contributions to variation across multi-type phenotypes. To avoid high-dimensional integrals over many phenotypes and family members in joint likelihood approaches, we develop a computationally efficient procedure by first maximizing the marginal likelihood function for each individual phenotype and then estimating the coheritability using only pairs of phenotypes. We apply our method to analyze the heritability and coheritability of 290 phenotypes obtained from the UK Biobank. We find that a substantial number of phenotype pairs present statistically significant genetic coheritability.
Similar content being viewed by others
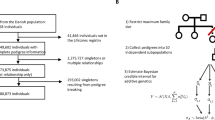

Introduction
Coheritability refers to the extent to which multiple phenotypes share a genetic basis, reflecting the genetic contribution to phenotypic correlations. Understanding coheritability is the key to uncovering the genetic architecture of complex diseases and traits, particularly those that co-occur in genetically related individuals1,2. The joint modeling of a diverse range of phenotypes allows investigation of the genetic and environmental interdependencies that drive comorbidity3,4,5,6. In addition, quantifying coheritability can inform which traits should be jointly tested in a downstream PheWAS study and inform better treatment strategies for individuals with multiple comorbidities7. Traditional study designs to assess heritability have been based on twin studies or family studies, which are resource-intensive and generally focus on a limited number of phenotypes2,8,9. In contrast, large-scale population-based biobank studies, such as the UK Biobank, allow the estimation of coheritability across hundreds of phenotypes, providing the statistical power needed to explore shared genetic risks in diverse populations10,11. These datasets offer a unique opportunity to investigate the genetic contribution to comorbidity on a scale previously unattainable.
Large biobank studies collect comprehensive data on a wide range of phenotypes, including continuous measures (e.g., biomarkers measured in laboratories), discrete traits (e.g., participants’ disease diagnoses via ICD codes, family history reports of a family member’s disease status), and time-to-event outcomes (e.g., time to onset of dementia). However, estimating coheritability across these diverse phenotypes presents statistical and computational challenges due to heterogeneity in data types and the large scale of biobank studies. Commonly used methods for estimating coheritability include Genomic Restricted Maximum Likelihood (GREML)12, Linkage Disequilibrium Score Regression (LDSC)13,14, and the closed-form Haseman-Elston estimator (HEc)15. GREML uses individual-level genotype data to estimate the proportion of variance explained by genetic factors for multiple phenotypes. Still, it is computationally intensive, especially for large datasets such as biobanks, due to the high dimensionality of the kernel matrices of the genetic relationship. LDSC, which uses GWAS summary statistics to estimate genetic correlations, is computationally more feasible for larger datasets. However, it requires reliable reference panels and can be inaccurate when there is genetic heterogeneity16. Although HEc is computationally efficient due to closed-form solutions, none of these existing methods can account for differential types of phenotypes (e.g., continuous, discrete, time-to-event traits) that must be modeled accordingly to capture true shared genetic effects.
Furthermore, biobank and community cohort studies have shown multiple levels of significant covariation between traits, demonstrating that shared genetic factors can contribute to the correlation of multiple phenotypes within individuals and familial clusters in a hierarchical manner13,17,18. At the individual level, the risks of multiple diseases may be correlated due to shared genetic variants. At the family level, phenotypic correlation between multiple diseases may be attributed to both the genetic effect and the common environmental effect or shared lifestyle (e.g., diet) in the family. This hierarchical structure, where genetic and environmental factors operate at both individual and familial levels, presents additional challenges for coheritability analysis, particularly in studies involving related individuals.
Current statistical methods for estimating genetic risk primarily focus on single or bivariate traits within a single data type (e.g., bivariate continuous traits, bivariate censored traits)15,18,19,20. Phenotypic covariation is modeled for continuous traits using random effects within a structural equation framework21,22. Genetic effects and different types of shared environmental effects can be distinguished by considering various covariance structures. On the other hand, binary traits are typically modeled using a probit model with a similar structure of random effects23. For time-to-event traits, such as the time to onset of a disease, semiparametric transformation models can be used to study the dependence of the event time on genetic and environmental factors in single-trait analysis24. For multiple time-to-event outcomes, shared frailty models have been proposed, particularly in case-control family designs, to account for shared genetic and environmental effects25,26,27. Additionally, copula models have been suggested for multivariate failure times28, but these approaches do not separate genetic and environmental risks, leading to overestimating heritability. More importantly, none of these methods scale efficiently to handle the large size of data typical of biobank studies.
Further statistical and computational challenges complicate the application of existing methods to phenome-wide coheritability analysis. First, phenotypes are measured on a heterogeneous scale (e.g., continuous, binary, ordinal, and time-to-event data), each with different magnitudes of variation, making it challenging to incorporate them into a single unified model. Second, coheritability analysis must account for the multi-level structure of dependence–both the relationships between phenotypes within a subject and the genetic or environmental correlations between individuals within families. Third, the large number of phenotypes and sample sizes in biobank studies typically results in a high-dimensional covariance matrix for the random effects. While maximum likelihood estimation (MLE) based on a joint likelihood is theoretically the most efficient approach for estimating coheritability parameters, it is computationally prohibitive because it requires high-dimensional numerical integration over many phenotypes and family members in the data likelihood function. For example, with 300 phenotypes and an average family size of 3, the dimension for the integration is 300 × 3 + 1 = 901, which is infeasible.
To overcome these challenges, we propose a semiparametric joint modeling approach with latent random effects. Applying appropriate transformations, phenotypic measures are modeled using the exponential distribution family for continuous, binary, or ordinal traits and the proportional hazards model for time-to-event traits. Importantly, our unified framework allows modeling phenotypes’ dependencies due to genetic and environmental factors across different data types. Furthermore, our approach distinguishes between genetic random effects and unobserved family-shared environmental effects, ensuring that both sources of variability are appropriately accounted for. To address the challenge of high-dimensional integrals over many phenotypes and family members (roughly, the dimension is the number of phenotypes times the family size) in traditional joint likelihood approaches, we propose a computationally efficient procedure called the Multi-type Phenotype CoHeritability (MPCH). Our method begins by estimating heritability and environmental correlations by maximizing the marginal likelihood for individual traits, followed by coheritability estimation with pairwise traits. This leads to a much lower dimension of numerical integration (at most five dimensions) than the joint estimation of coheritability. Thus, our method is computationally more efficient than the traditional joint likelihood approaches and can be scaled up for analyzing large studies such as the UK Biobank. Furthermore, we establish the asymptotic properties of the estimators. Simulation studies show that our estimators are consistent, that confidence intervals have coverage rates close to the nominal level, and that computation cost is significantly less than the joint likelihood approaches. Finally, we apply our approach to 290 selected phenotypes in the UK Biobank, where our method gives shorter confidence intervals for coheritability than the HEc for continuous traits and, for the first time in the literature, provides coheritability for discrete and time-to-event traits. Our analysis reveals that most phenotypes exhibit small to moderate heritability and coheritability, with genetic factors contributing more to phenotypic correlation than family-shared environmental factors. These results provide information on the relative contribution of shared genetic and environmental factors to comorbidity and have implications for precision medicine by improving the accuracy of genetic coheritability estimation.
Results
Simulation
In this section, we conduct a simulation study to assess the performance of estimating coheritability parameters. Suppose there are four types of families: (1) one parent and one child, (2) two parents and one child, (3) one parent and two children, (4) two parents and two children. The family structures with kinship matrices are illustrated in Panel (A) of Fig. 1. We generate 300 families for each type, so there are 3600 individuals in total.

A Structure of families to generate simulated data. B Bias of the estimated heritability, family-shared environmental effect, coheritability, and environmental correlation. Red lines represent mean ± 1.96 standard deviation, and blue lines represent mean ± 1.96 standard error.
We generate three independent baseline covariates \({\widetilde{{{\bf{X}}}}}_{ij}={({X}_{ij1},{X}_{ij2},{X}_{ij3})}^{{{\rm{T}}}}\), each following a uniform distribution in [ − 1, 1]. We consider six phenotypes, including two continuous (No. 1 and 2), two ordinal (No. 3 and 4), and two time-to-event (No. 5 and 6). The parameters are set to be
The censoring time is generated to follow a uniform distribution in [5, 10] for the time-to-event outcomes. The event rate is about 0.25 for the 5th and 0.3 for the 6th outcomes. The off-diagonal elements of Γ are set to be \({\gamma }_{k{k}^{{\prime} }}=0.5 \sqrt{{\gamma }_{kk}{\gamma }_{k{k}^{{\prime} }}}\) (\(k\, \ne \,{k}^{{\prime} }\)). To generate the outcomes, we first generate the environmental factor \({\theta }_{k}{e}_{i} \sim N(0,{\theta }_{k}^{2})\) and the genetic factor ϵi~N(0, Γ ⊗ Gi) in each family, and then generate outcomes according to the exponential distribution family or proportional hazards model.
We replicate the estimation procedure in 1000 independently generated datasets. In estimating the single-trait fixed effects and random effects, the computation for a single-trait phenotype takes about 3.1 min (standard deviation [SD]: 1.1 min) for continuous outcomes, 2.9 h (SD: 2.9 h) for binary outcomes, 2.2 h (SD: 1.6 h) for ordinal outcomes and 46.3 min (SD: 51.9 min) time-to-event outcomes using R version 4.4.0 on a multi-core CPU server (Intel Xeon E5-2450 0 @2.10GHz). Computing continuous outcomes takes the least time because numerical integration is avoided in estimating the parameters by utilizing the explicit form of conditional distributions in multivariate normal distributions23. The computation for binary outcomes takes a very long time because the convergence of estimated parameters is slow. It is also time-consuming for ordinal and time-to-event outcomes because the model has more parameters. In estimating the off-diagonal elements of Γ, the computation takes no more than 20 seconds for each pair.
Panel (B) of Fig. 1 shows the estimation results. The first column displays the bias of the estimated genetic and family-shared environmental effects for six phenotypes in black points among these 1000 replications. The standard deviation of the estimated random effects for binary outcomes is generally larger than that for other types of outcomes, indicating that binary outcomes have little information about the genetic effect. There are 15 phenotypic pairs generated from the six phenotypes in total. The second column displays the bias of the estimated coheritability and environmental correlation for these 15 pairs. The red lines represent the average bias ±1.96 standard deviation, and the blue lines (average confidence intervals) represent the average bias ±1.96 average standard errors. The average bias is generally close to zero, and all the intervals cover zero. The estimates show a small variation for continuous phenotypes but a larger variation involving binary and time-to-event outcomes. Detailed numerical results are provided in Supplementary Tables 1–2. In the “Method” section, we conduct additional simulation studies to examine the impact of varied family sizes, the trade-off between the efficiency gain and computation cost when maximizing the joint likelihood function, and the bias in estimation when ignoring the random effects due to shared environment.
Protocol
The UK Biobank is a large-scale biomedical database containing de-identified genetic, lifestyle, and health information along with biological samples from over 500,000 participants in the UK. Figure 2 illustrates the data analysis process. For our analysis, we first constructed family genetic relationships from all participants using the ‘KING’ toolset, which was designed to explore genetic relatedness through the genetic relationship matrices (GRM) from whole-genome single nucleotide polymorphisms (SNPs) data in a genome-wide association study29. Genetically related pairs were selected up to the second degree of relationship, defined by the kinship coefficient greater than 0.0884. This process identified 5091 families of two members, 632 families of three members, and 114 families with more than three members. Among the 19 families with more than four members, we only retained four members with the largest genetic relatedness for subsequent analysis. The genetic correlation between the fifth member and other members (mean 0.06) is much smaller compared with the genetic correlation between the other four members (mean 0.47), and restricting the sample size can reduce the computational burden. In total, our sample includes 12,534 participants with derived familial relationships based on GRM and 489,621 unrelated individuals without familial relationships. Our analysis adjusted for baseline covariates, including sex, age at recruitment, education, and income. We also adjusted for the top 10 genetic principal components to account for multiple ancestries in our sample.

Workflow of MPCH analysis for the UK Biobank data.
We extracted phenotype data from Tier-1 UK Biobank data, including measurements at each assessment center, biological samples, online follow-up, and health-related outcomes as listed in Supplementary Data 1. In addition, we obtained ICD-10 codes from health records and mapped them to 199 PHEcodes with observed occurrences. For quality control, we removed phenotypes considered not biologically meaningful, such as ‘noisy workplace’ and ‘time since last prostate-specific antigen test’. When multiple data fields measured the same phenotype (e.g., basophill count and basophill percentage), we only kept one for the analysis. We excluded continuous or binary phenotypes with an attrition rate higher than 95% and time-to-event phenotypes with an event rate less than 1%. After applying these quality control measures, we obtained 290 phenotypes for analysis. The composition of the families with observed measurements and summary statistics for each phenotype are listed in Supplementary Data 2. All continuous phenotypes are normalized using the rank-based inverse normal transformation30.
We first applied MPCH to estimate the heritability and coheritability using family data, removing the phenotypes whose estimates did not converge in 5000 iterations. Our results include the heritability and coheritability estimates from 288 phenotypes: 152 continuous, 95 binary, 27 ordinal, and 14 time-to-event phenotypes. The mean computation time for estimating heritability was 3.1 min (SD: 1.1 min) for continuous, 2.5 h (SD: 1.9 h) for binary, 2.9 h (SD: 4.4 h) for ordinal, and 3.7 h (SD: 5.3 h) for time-to-event phenotypes. The mean computation time for estimating coheritability was 1.0 min (SD: 1.0 min) for each pair of phenotypes. Next, we estimated the heritability and coheritability using all data, with the estimates based on family data as initial values in the algorithm. The mean computing time for estimating heritability was 21.8 min (SD: 14.2 min) for continuous, 4.2 h (SD: 3.5 h) for binary, 7.2 h (SD: 19.5 h) for ordinal, and 50.2 h (SD: 29.7 h) for time-to-event phenotypes. The computational time for estimating coheritability was approximately 2.1 h (SD: 1.0 h) for each pair of phenotypes. Due to the increase in sample size, the estimation using all data involves a higher computational cost.
Single-trait heritability
Figure 3 presents the histograms of the estimated single-trait genetic heritability and family-shared environmental effect. Overall, the estimated heritability is small or moderate, with most phenotypes (94.1%) having heritability below 50%. A notable proportion (66.3%) of phenotypes have heritability in the range of 10–30%. Among the 288 phenotypes under consideration, 84.4% have significant heritability after Bonferroni correction for multiple comparisons using one-sided Z-tests. The family-shared environmental effect in phenotypic variation is skewed toward very low values, with most examined phenotypes showing near-zero environmental contribution.

A Histogram of estimated genetic heritability among 288 phenotypes. B Histogram of estimated family-shared environmental effect among 288 phenotypes. C Q-Q plot of P values of significance tests on heritability. The sample size is 502,155, including 12,534 individuals in families and 489,621 unrelated individuals.
In Table 1, we highlight five phenotypes with the highest heritability for each data type (continuous, binary, ordinal, time-to-event) and list the estimated heritability (\({\widehat{h}}_{kk}^{2}\)). To ensure that heritability estimates remain within the [0, 1] range, 95% confidence intervals (CIs) are given based on the logit transformation using asymptotic variance. Among all traits, standing height has the highest heritability, suggesting that genetic factors predominantly determine standing height. The five continuous traits, including standing height, lipoprotein A, and 3mm weak meridian (left and right) and avMSE (refractive error) show high heritability, with values ranging from 58.0 to 74.8%, suggesting that genetic factors strongly influence traits related to physical measurements (e.g., eye metrics). In contrast, phenotypes related to behaviors and complex diseases and behaviors have moderate heritability (e.g., smoking 40.2%, CI: 34.2–46.6%; hypertensive disease 35.7%, CI: 31.5–40.1%), suggesting a significant role for environmental or lifestyle factors. Some time-to-event phenotypes, such as age asthma diagnosed (31.7%, CI: 30.8–32.6%) and age diabetes diagnosed (25.7%, CI: 24.7–26.7%), generally have lower heritability, which suggests that environmental and lifestyle factors likely play a larger role in these conditions.
Our single-trait heritability estimates are consistent with those reported in the literature31,32. For example, we estimate the heritability of standing height at 74.8% (CI: 74.3–75.2%), which falls within the widely reported range (68–95%33, 85%34, 81%35, 60–70%36). Our heritability estimate for body mass index (BMI) is 49.9% (CI: 47.3–52.5%), consistent with prior findings (49–78%37, 30–40%36, 39%38). For diastolic blood pressure (DBP) and systolic blood pressure (SBP), we estimate heritability at 25.5% (CI: 21.8–29.6%) and 25.6% (CI: 12.5–45.4%), respectively, which are well within the literature reported ranges of 19–60% for DBP and 17–54% for SBP39,40,41,42. Additionally, we estimate the heritability of smoking at 40.2% (CI: 34.2–46.6%), aligning with the literature (37%43, 75%44, 44%45). Our estimate for diagnosed asthma is 31.7% (CI: 30.8–32.6%), while the previous estimates in the literature vary widely from 25 to 95%46,47,48,49. Supplementary Data 3 provides detailed information on the estimation results, including genetic heritability and family-shared environmental effect for all phenotypes.
Next, we compared the MPCH with the closed-form Haseman-Elston (HEc) estimator, a moment-based estimator for the heritability15,20, for continuous phenotypes since HEc can only be applied for such phenotypes. The 95% confidence intervals in the HEc are obtained via bootstrapping. For comparison, in Fig. 4, we present the results of 21 common phenotypes available in the UK Biobank and reported in literature15. The heritability estimates from both methods generally follow similar trends across phenotypes. However, the MPCH-estimated heritability is lower than that obtained by HEc. The HEc estimate for standing height is unreliable, outside the range of [0,1]. We conducted an additional analysis where we did not model the environmental effect by assuming θk = 0 in MPCH, and the results are shown in Supplementary Data 3. We found estimates to be close to those given by HEc, which confirms that the proposed MPCH can distinguish genetic heritability from family-shared environmental effect. Furthermore, MPCH uses all available data, while HEc can only use family data, so MPCH yields estimates with generally smaller standard errors. Based on the estimates given by MPCH, the heritability for blood-cell traits is generally small to moderate. Basophill count has the lowest heritability (\({\widehat{h}}_{kk}^{2}=15.9 \%\), CI: 12.8–19.4%) and platelet count has the highest heritability (\({\widehat{h}}_{kk}^{2}=43.0 \%\),CI: 41.0–45.0%) among these blood-cell traits using MPCH. This pattern is consistent with existing literature, e.g., the heritability was estimated at 3.1% for basophill count and 21.8% for platelet count50.

The confidence intervals corresponding to MPCH are given based on the logit transformation using asymptotic variance. The confidence intervals corresponding to HEc are given by bootstrap.
Coheritability and environmental correlation
Figure 5 shows the histograms of genetic coheritability and environmental correlations for all 288 × (288−1)/2 = 41,328 pairs of phenotypes. Positive coheritability indicates that the underlying genetic factors influence two phenotypes in the same direction. Most estimated genetic coheritability values are small in magnitude, with 94.4% of the coheritability having an absolute value of smaller than 20%. The estimated environmental correlation is also concentrated at around zero. While the environmental correlation is almost negligible, genetic coheritability shows a wider spread, with some pairs of phenotypes presenting modest shared genetic influence. Among the pairs of phenotypes under consideration, 61.6% have significant coheritability after Bonferroni correction for multiple comparisons using two-sided Z-tests. The highest coheritability is observed in the phenotypes measuring similar traits, such as in 3 mm strong/weak meridian and various measures of body size metrics (e.g., leg-predicted mass, whole body fat-free mass, whole body water mass, and weight). Note that we also identify significant coheritability in different types of phenotypes. For instance, diabetes derived from PHEcodes has high coheritability with boss mass index (\({\widehat{h}}_{k{k}^{{\prime} }}^{2}=31.4 \%\), CI: 29.0–33.8%), glycated hemoglobin (HbA1c) (\({\widehat{h}}_{k{k}^{{\prime} }}^{2}=31.0 \%\), CI: 24.5–37.5%), hypertensive disease (\({\widehat{h}}_{k{k}^{{\prime} }}^{2}=30.6 \%\), CI: 24.5–34.7%), disorders of lipoid metabolism (\({\widehat{h}}_{k{k}^{{\prime} }}^{2}=26.6 \%\), CI: 15.6–37.6%) and ischemic heart disease (\({\widehat{h}}_{k{k}^{{\prime} }}^{2}=23.2 \%\), CI: 12.8–33.6%).

A Histogram of estimated genetic coheritability among all pairs of phenotypes. B Histogram of environmental correlation among all pairs of phenotypes. C Q-Q plot of P values of significance tests on coheritability. The sample size is 502,155, including 12,534 individuals in families and 489,621 unrelated individuals.
Panel (A) of Fig. 6 shows the coheritability estimates among the same set of phenotypes (21 continuous phenotypes related to anthropometry and blood tests). The coheritability of BMI and diastolic blood pressure (DBP) is estimated at 20.2% (CI: 18.4–22.0%), whereas the phenotypic correlation is 28.9%. The coheritability of BMI and systolic blood pressure (SBP) is estimated at 24.6% (CI: 16.6–32.6%), whereas the phenotypic correlation is 21.9%. These results are similar to previous findings using UK Biobank data51. The coheritability of platelet count and platelet crit is estimated at 40.4% (CI: 38.4–42.4%), whereas the phenotypic correlation is 88.3%. The coheritability of red blood (erythrocyte) count and hemoglobin concentration is estimated at 31.3% (CI: 24.4–38.2%), whereas the phenotypic correlation is 40.1%. Clustering patterns suggest a common genetic etiology for several phenotypes. A cluster is formed by white blood cell (leukocyte) count, neutrophil, lymphocyte count, and monocyte and eosinophill count. In contrast, another cluster is formed by triglycerides, red blood cell (erythrocyte) count, BMI, and reticulocyte count.

A The estimated coheritability among 21 selected phenotypes by MPCH. The diagonal elements are heritability. B The estimated coheritability among 21 selected phenotypes by HEc. The diagonal elements are heritability. C The squared coheritability estimates are rescaled to a range from 0 to 0.1. The diagonal elements are squared heritability. The sample size is 502,155, including 12,534 individuals in families and 489,621 unrelated individuals.
In comparison, Panel (B) in Fig. 6 displays the estimated coheritability using the HEc method. While the overall patterns are similar across methods, HEc yields larger standard errors because it only uses paired phenotypes across individuals and does not fully leverage the information available from two phenotypes within the same individual. This limitation contributes to less precise estimates compared to the MPCH. Panel (C) displays the heat map of estimated squared coheritability among 288 phenotypes. Five coheritability clusters were identified: body composition (e.g., obesity, impedance of arm/leg, arm/leg mass), metabolic syndrome (e.g., HDL cholesterol, diabetes, reticulocyte count), bone density (e.g., heel bone mineral density, speed of sound through heel), mental health (e.g., irritability, nervous feelings, felt distant from other people, neuroticism score), and organ/chronic diseases (e.g., neoplasms, esophageal disorders, disorders of stomach, bacteria infection, abdominal pain). Supplementary Figs. 1–2 present the heat map of estimated squared coheritability with phenotype names on the graph by MPCH, using family data and all data, respectively. To compare MPCH with HEc, Supplementary Figs. 3–5 present the estimated coheritability for continuous phenotypes by MPCH using family data, MPCH using all data, and HEc, respectively. Supplementary Data 4 provides further details on coheritability and environmental correlations with standard error for all phenotype pairs.
Discussion
In this work, we proposed a computationally efficient method to estimate the coheritability across a wide range of phenotypes, including continuous, binary, ordinal, and time-to-event traits. Based on a joint Gaussian model, we integrated fixed effects, genetic effects, and family-shared environmental effects into a unified modeling framework to accommodate various data types, which is not available for existing methods. Through appropriate transformation, our unified modeling framework applies to conventional linear models, probit models, and proportional hazards models. Our method distinguishes genetic effects on comorbidity from environmental influences. One of the key advantages of the unified linear model is that we can define heritability and coheritability for different types of phenotypes within this framework. Furthermore, by leveraging a joint framework, we can borrow information reflecting a large population’s distant common ancestry to analyze each phenotype separately.
Given the large number of phenotypes and the extensive sample size typical of biobank datasets, conventional methods relying on joint likelihood estimation become computationally infeasible. We adopted a two-stage estimation procedure to address this challenge, effectively avoiding the need for computationally expensive numerical integration over high-dimensional covariance matrices. While this approach incurs a minimal loss of statistical efficiency, it significantly improves computational efficiency, allowing for parallel processing and large-scale biobank analyses. Specifically, the computational complexity is on a polynomial order of K for MPCH, much lower than the exponential order for maximizing the joint likelihood. We estimated heritability and coheritability by using family data first and then all data. The statistical efficiency improves 196 times for heritability and 3.2 times for coheritability by incorporating unrelated individual data, but the computation time increases dramatically due to the large sample size. Compared to the HEc, MPCH is more efficient in estimating coheritability since HEc does not use correlation information from phenotypes from the same individual.
It is well known that confounding threatens the validity of genome-wide association studies (GWAS). The most common approach to control confounding is to control population substructure52, which reflects a large population’s distant common ancestry. In contrast, family-level substructure describes recent common ancestry among smaller groups of individuals defined by observed pedigrees or estimated from the GRM. MPCH provides several predicted random effects (e.g., ei and ϵijk), which may reflect hidden confounding due to shared recent ancestry and shared environment (e.g., lifestyle) specific to a disease of interest to be included in the downstream GWAS tests to gain better control of confounding. We can predict these confounders (family effect representing the shared recent ancestry and shared environmental effect) by the posterior mean of the random effects. More precise control of confounding specific to the disease of interest is desirable and achievable by using predicted random effects to adjust the observed family substructures in the sample.
In addition to heritability and coheritability, we may also wish to examine the relative magnitude of the genetic coheritability and environmental correlation. Noticing that the total variation contributed by genetic and environmental factors \({\widetilde{\sigma }}_{k}^{2}={\theta }_{k}^{2}+{\gamma }_{kk}\), we define the fraction of genetic effect \({\rho }_{kk}={\gamma }_{kk}/{\widetilde{\sigma }}_{k}^{2}\) and the fraction of environmental effect \({\zeta }_{kk}={\theta }_{k}^{2}/{\widetilde{\sigma }}_{k}^{2}\) in single-trait phenotypic correlation. For a pair of phenotypes k and \({k}^{{\prime} }\), the fraction of genetic effect in coheritability \({\rho }_{k{k}^{{\prime} }}={\gamma }_{k{k}^{{\prime} }}/{\widetilde{\sigma }}_{k}{\widetilde{\sigma }}_{{k}^{{\prime} }}\) and the fraction of environmental correlation \({\zeta }_{k{k}^{{\prime} }}={\theta }_{k}{\theta }_{{k}^{{\prime} }}/{\widetilde{\sigma }}_{k}{\widetilde{\sigma }}_{{k}^{{\prime} }}\). Since the family-shared environmental effect does not interact between phenotypes, the environmental correlation and fraction of environmental correlation can be directly inferred from single-trait parameters. Panel (A) in Fig. 7 presents the fraction of the genetic effect in the total variation of genetic and family-shared environmental effects. The family-shared environmental effect for most phenotypes is small compared to genetic heritability. Panel (B) in Fig. 7 presents the fraction of the genetic effect standardized by the total variation of genetic and family-shared environmental effects for the pairwise phenotypic correlation. Supplementary Data 3 shows the estimated fraction of the genetic effect in the total genetic/environmental effect for single traits. Supplementary Data 4 shows the estimated fraction of the genetic effect in the total genetic/environmental correlation for pairwise phenotypes.

A Histogram of the estimated fraction of genetic effect in the single-trait phenotypic correlation. B Histogram of the estimated fraction of genetic effect in the pairwise phenotypic correlation. The sample size is 502,155, including 12,534 individuals in families and 489,621 unrelated individuals.
Our results show a strong genetic contribution to several traits, consistent with previous findings, particularly in anthropometric, blood-related, and metabolic-related phenotypes. For example, the heritability for most blood-cell traits was estimated to range from 50 to 90% in twin studies and 30–40% in population-based studies53,54,55,56. However, many other phenotypes, such as blood pressure and cholesterol levels, show moderate genetic influences, suggesting a substantial contribution of environmental factors to these traits. These findings are relevant in precision medicine as they suggest that interventions targeting modifiable lifestyle factors may play a role in managing conditions related to cardiovascular and metabolic health.
The coheritability estimates from MPCH reveal five distinct clusters: body composition, metabolic syndrome, bone density, mental health, and organ/chronic diseases. These clusters offer information on how seemingly disparate phenotypes may share common genetic underpinnings, which has implications for future research on pleiotropy. MPCH finds that the genetic contribution to phenotype correlation is generally stronger than the environmental contribution, such as family lifestyle or common exposures. However, for most phenotype pairs, neither genetic nor family-shared environmental factors contribute strongly to the phenotypic correlation. This suggests other factors, such as personal lifestyle choices, unique environmental exposures, or individual stress levels, could play a substantial role in phenotype correlations.
There are some limitations in our work. First, families with various genetic correlations between members are required to distinguish the genetic and family-shared environmental effects. Larger families help increase statistical efficiency but require a higher computational burden as more numerical integration is needed for larger families. Second, other unobserved factors may co-varying with kinship or genetic relationships. Third, we only consider the family-shared environmental effect without distinguishing other environmental effects, like household-structured or community environmental effects. Fourth, the family relation in the UK Biobank data is derived. Genetic correlation can represent common genetic characteristics, but not household information. Thus, the estimated family-shared environmental effect may underestimate the true environmental effect. In studies with known family structure, the estimation of environmental effects may be larger and more precise.
Methods
Models
Suppose the study sample consists of n independent families. In the ith family (i = 1, …, n), there are ni members with a known kinship matrix defined by familial relationships (e.g., parents, siblings, children) or a genetic relationship matrix (GRM) defined by genome-wide single nucleotide polymorphisms (SNPs). For each member j = 1, …, ni, the study collects baseline covariates Xij (including constant 1) and measurements of at most K phenotypes. The phenotypes can be continuous, binary, ordinal, or time-to-event. To model the dependence among different phenotypes for each member in the same family, we introduce a family-specific random effect to account for family-shared environmental factors (e.g., lifestyle). Additionally, we incorporate subject- and phenotype-specific random effects to capture genetic factors, with correlations determined by the kinship matrix or the GRM. Specifically, for the kth continuous phenotype Yijk such as body mass index, we assume a linear mixed effects model as follows:
where ei is an unobserved environmental factor, ϵijk is an unobserved genetic factor, and \({u}_{ijk} \sim N(0,{\sigma }_{uk}^{2})\) is an independent error term with unknown variance. For the kth binary or ordinal phenotype Yijk with Lk levels such as smoking status, we assume that Yijk arises from an underlying latent trait Zijk modeled as
where uijk ~ N(0, 1) and \({\widetilde{{{\bf{X}}}}}_{ij}\) is the covariates matrix excluding the column of constant 1. The observed phenotype Yijk is then determined by unknown thresholds \(-\infty ={\delta }_{k0} < {\delta }_{k1} < \cdots < {\delta }_{k{L}_{k}}=\infty\), such that Yijk = l if the latent trait Zijk falls within the interval (δk,l−1, δkl). Equivalently, we assume an ordinal probit model for Yijk, given by
where Φ( ⋅ ) denotes the cumulative distribution function of the standard normal distribution. Specially, for ordinal phenotypes, \(P({Y}_{ijk}=1| {{{\bf{X}}}}_{ij},{e}_{i},{\epsilon }_{ijk})=\Phi ({\delta }_{1}-{{{\boldsymbol{\alpha }}}}_{k}^{{{\rm{T}}}}{\widetilde{{{\bf{X}}}}}_{ij}-{\theta }_{k}{e}_{i}-{\epsilon }_{ijk})\). Finally, when the phenotype is a time-to-event variable, such as age at disease onset, let \({\widetilde{T}}_{ijk}\) denote the kth time to the event of interest. We assume the following transformation model:
where Λk is an increasing, unknown transformation function with Λk(0) = 0, and uijk follows the extreme value distribution whose variance is π2/657. This transformation model is equivalent to the commonly used proportional hazards model, where the conditional hazard rate for \({\widetilde{T}}_{ijk}\) is \({\Lambda }_{k}(t)\exp ({{{\boldsymbol{\alpha }}}}_{k}^{{{\rm{T}}}}{\widetilde{{{\bf{X}}}}}_{ij}+{\theta }_{k}{e}_{i}+{\epsilon }_{ijk})\). Subject to random right censoring, let Cijk be the censoring time, so the observed outcome on the time-to-event phenotype is Yijk = (Tijk, Δijk), where the follow-up time \({T}_{ijk}=\min \{{\widetilde{T}}_{ijk},{C}_{ijk}\}\) and censoring indicator \({\Delta }_{ijk}=I\{{\widetilde{T}}_{ijk}\le {C}_{ijk}\}\).
Thus, regardless of whether the phenotype is continuous, ordinal, or time-to-event, the underlying model for generating these phenotypes takes the form
where \({\widetilde{Y}}_{ijk}\) represents Yijk for continuous phenotypes, Zijk for ordinal (binary) phenotypes, and \(-\log {\Lambda }_{k}({\widetilde{T}}_{ijk})\) for time-to-event phenotypes, and \({{{\bf{X}}}}_{ijk}^{* }\) is Xij with or without constant term. We assume var(ei) = 1 for the identifiability of \({\theta }_{k}^{2}\).
The advantage of this unified model is its ability to separate the sources of variability, distinguishing between family-shared environmental effect (through θkei), genetic effect (through ϵijk), and measurement error (through uijk). Furthermore, it is natural to model the correlations among the genetic effects for the same type of phenotype across all family members using the kinship coefficient or GRM and to model the correlation between two types of phenotypes through their coheritability parameters. In particular, we let Gi denote the kinship matrix or GRM for the ith family. We assume that \({{{\boldsymbol{\epsilon }}}}_{ik}={({\epsilon }_{i1k},\ldots ,{\epsilon }_{i{n}_{i}k})}^{{{\rm{T}}}}\) has a covariance matrix of γkkGi, where γkk represents the genetic inheritance (related to heritability) for this particular phenotype. For two different phenotypes k and \({k}^{{\prime} }\), we model the covariance between their genetic effects as \({{\rm{Cov}}}({\epsilon }_{ijk},{\epsilon }_{i{j}^{{\prime} }{k}^{{\prime} }})={\gamma }_{k{k}^{{\prime} }}{g}_{ij{j}^{{\prime} }}\) where \({g}_{ij{j}^{{\prime} }}\) is the \((j,{j}^{{\prime} })\)-entry in Gi and \({\gamma }_{k{k}^{{\prime} }}\) relates to the coheritability between the two phenotypes. In other words, we assume
where ‘⊗’ denotes the Kronecker product. Here, the matrix Γ captures the genetic variances and covariances of interest, with \({\gamma }_{k{k}^{{\prime} }}\) representing genetic covariation between traits k and \({k}^{{\prime} }\).
The existing literature estimates heritability for continuous phenotypes through a linear mixed effect model, with narrow-sense heritability defined as the proportion of phenotypic variance due to additive genetic variation58,59. Our unified modeling framework extends this definition to accommodate additional types of phenotypes, including ordinal and time-to-event data. Under this framework, the total variation of the phenotype k is \({\sigma }_{k}^{2}={\theta }_{k}^{2}+{\gamma }_{kk}+{{\rm{var}}}({u}_{ijk})\), which includes contributions from the genetic effect, family-shared environmental effect, and measurement error. Note that \({{\rm{var}}}({u}_{ijk})={\sigma }_{uk}^{2}\) for continuous phenotypes, var(uijk) = 1 for ordinal or binary phenotypes, and var(uijk) = π2/6 for time-to-event phenotypes. Therefore, the single-trait heritability \({h}_{kk}={\gamma }_{kk}/{\sigma }_{k}^{2}\) reflects the proportion of the total variability that can be explained by genetic factors. Similarly, \({\xi }_{kk}={\theta }_{k}^{2}/{\sigma }_{k}^{2}\) is the proportion from the family-shared environmental effect within families. Furthermore, for a pair of different phenotypes, say k and \({k}^{{\prime} }\), the genetic coheritability \({h}_{k{k}^{{\prime} }}={\gamma }_{k{k}^{{\prime} }}/{\sigma }_{k}{\sigma }_{{k}^{{\prime} }}\) quantifies the proportion of covariance attributable to shared genetic factors, while \({\xi }_{k{k}^{{\prime} }}={\theta }_{k}{\theta }_{{k}^{{\prime} }}/{\sigma }_{k}{\sigma }_{{k}^{{\prime} }}\) is the proportion of covariance explained by family-shared environmental factors.
Estimation
Let f(ei), f(ϵi; Γ) and f(Yijk∣Xij, ei, ϵijk; ηk) be the density function of ei, ϵi and Yijk, respectively. The full likelihood function for all observed data \({{\mathcal{O}}}=\{({{{\bf{X}}}}_{ij},{Y}_{ijk}):i=1,\ldots ,n,j=1,\ldots ,{n}_{i},k=1,\ldots ,K\}\)
When considering a large number of phenotypes, the genetic covariance matrix Γ becomes high-dimensional, making its estimation computationally challenging. Estimating Γkk by the likelihood-based method is computationally prohibitive because evaluating the likelihood function requires numerical integration over the random effects ϵi with covariance matrix Γ ⊗ Gi. In the UK Biobank data, we have 290 phenotypes to analyze. In a family of 4 members, we need to perform over 290 × 4 + 1 times of integration to calculate the contribution of the ith family to the full likelihood.
To improve the computational efficiency, we propose a two-stage estimation procedure called the Multi-type Phenotype CoHeritability (MPCH), which maintains the efficiency of the likelihood-based method and significantly reduces the computational burden. Let \({{{\mathcal{O}}}}_{k}\) denote the observed data on the kth phenotype, and ηk denote the single-trait parameters including αk, \({\theta }_{k}^{2}\), γkk and nuisance parameters such as \({\sigma }_{uk}^{2}\), δkl or Λk( ⋅ ). In the first stage, we maximize the marginal likelihood for a single phenotype,
allowing us to estimate single-trait parameters such as heritability and family-shared environmental effect using the EM algorithm60. In a family with at most four members, we need to perform numerical integration up to five dimensions.
In the second stage, we maximize the pairwise pseudo-likelihood for each pair of phenotypes, which can be either from the same individual or two different individuals in the same family \({{{\mathcal{J}}}}_{i}=\{(j,{j}^{{\prime} }):{g}_{ij{j}^{{\prime} }}\ne 0\}\), to estimate the genetic coheritability and environmental correlation, after plugging in the single-trait estimates \({\widehat{{{\boldsymbol{\eta }}}}}_{k}\) already obtained in the first stage,
This pseudo-likelihood-based approach mitigates computational burden by reducing a high-dimensional numerical integration to at most three dimensions. Numerical integration over the inter-phenotypic and inter-subject covariance matrix is avoided. The optimization in this step is performed for a single parameter for each coheritability, which can be achieved by a bisection search.
Algorithm 1
Two-stage estimation procedure for MPCH 
The two-stage estimation procedure of MPCH is summarized in Algorithm 1. Let m be the maximum family size, p be the dimension of covariates, and b be the number of Gaussian quadrature knots in numerical integration. We note that the complexity of evaluating the likelihood by MPCH is no more than \(O(Kn\log (n){p}^{2}{m}^{3}{b}^{m})\) in the first stage and \(O({K}^{2}n\log (n){p}^{2}{m}^{2}{b}^{3})\) in the second stage. In contrast, the complexity of evaluating the full likelihood of K phenotypes is \(O({K}^{2}n\log (n){p}^{2}{m}^{2}{b}^{Km})\). When the number of phenotypes K is large, the complexity of maximizing the full likelihood is much higher and usually infeasible. For model identifiability, we assume that there must be families with distinct genetic correlation matrices or more than three members, and the covariates should not be completely linearly dependent on the latent genetic factors in a family. The regularity conditions are formally presented below.
Variance estimation and inference
We let \({{{\mathcal{K}}}}_{1}\) denote the labels for continuous, binary, and ordinal phenotypes and write βk = ηk as the parametric part. For the time-to-event phenotypes, denoted as \(k\in {{{\mathcal{K}}}}_{2}\), we write ηk = (βk, Λk), where \({{{\boldsymbol{\beta }}}}_{k}={({{{\boldsymbol{\alpha }}}}_{k}^{{{\rm{T}}}},{\theta }_{k}^{2},{\gamma }_{kk})}^{{{\rm{T}}}}\) is the parametric part and Λk is the baseline cumulative hazard function. Let \({{\boldsymbol{\beta }}}={({{{\boldsymbol{\beta }}}}_{k}^{{{\rm{T}}}},{\gamma }_{k{k}^{{\prime} }}:k = 1,\ldots ,K,{k}^{{\prime} }\ne k)}^{{{\rm{T}}}}\) be the vector of all parametric parts. Denote the true values of β and Λk be β0 and Λk0, respectively. We need the following conditions to establish the asymptotic properties for the estimated parameters24.
Condition 1
The true value β0 is an interior in a known compact set in the domain of β. The true function Λk0( ⋅ ) for \(k\in {{{\mathcal{K}}}}_{2}\) is strictly increasing and continuously differentiable in [0, τ], where τ is the end time of study.
Condition 2
With probability one, Xij is bounded, and there exists a positive constant δ such that \({\sum }_{j = 1}^{{n}_{i}}P({T}_{ijk}\ge \tau | {{{\bf{X}}}}_{ij})\, > \,\delta\) for each i = 1, …, n and \(k\in {{{\mathcal{K}}}}_{2}\), where τ is the end of study.
Condition 3
There exists a constant n0 such that the family size satisfies P(1 ≤ ni ≤ n0) = 1 and P(ni ≥ 2) > 0.
Condition 4
Conditional on ni, for \(k\in {{{\mathcal{K}}}}_{1}\), if two vectors ηk and \({{{\boldsymbol{\eta }}}}_{k}^{* }\) satisfy
with probability one, then \({{{\boldsymbol{\eta }}}}_{k}={{{\boldsymbol{\eta }}}}_{k}^{* }\). let eij = (1, 0, …, 0, 1, 0, …, 0)T be an (ni + 1)-dimensional vector, with only the first and the jth element equal to one. For \(k\in {{{\mathcal{K}}}}_{2}\), if \({({{{\boldsymbol{\gamma }}}}^{{{\rm{T}}}},{c}_{1},{c}_{2})}^{{{\rm{T}}}}\) is a constant vector satisfying
for \(j\, \ne \, {j}^{{\prime} }\) with probability one, then c1 = c2 = 0 and γ = 0.
Condition 1 assumes that the effects are bounded. Condition 2 implies that individuals are still at risk at the end of the study. Condition 3 indicates that family data are necessary to identify the genetic and environmental effects that can capture the correlations among family members. Condition 4 ensures that the parameters are identifiable from the marginal likelihood function. Essentially, the covariates from all family members cannot be completely linearly dependent. For example, if all families have two members for continuous outcomes and the genetic correlation matrices are identical across families, then Condition 4 will fail. Under these conditions, we have the following results on the asymptotic convergence of estimators.
Theorem 1
Under Conditions 1–4, we have \(\parallel \widehat{{{\boldsymbol{\beta }}}}-{{{\boldsymbol{\beta }}}}_{0}\parallel \to {{\boldsymbol{0}}}\) and \({\sum }_{k\in {{{\mathcal{K}}}}_{2}}\parallel {\widehat{\Lambda }}_{k}-{\Lambda }_{k0}{\parallel }_{\infty }\to 0\), where ∥ ⋅ ∥ is the Euclidean norm and ∥ ⋅ ∥∞ is the supreme norm in [0, τ].
This theorem implies that the estimates by our method are asymptotically unbiased. Even though we estimated the single-trait parameters ηk and coheritability parameters \({\gamma }_{k{k}^{{\prime} }}\) separately, we can study the joint distribution of \(\widehat{{{\boldsymbol{\beta }}}}\) by deriving the influence functions of these parameters. Let
be the log-likelihood contributed by the ith family. In the exponential distribution family \(k\in {{{\mathcal{K}}}}_{1}\), the score function Sk(Oik; ηk) = ∂ℓik(Oik; ηk)/∂ηk in the ith family and the information matrix \({{\bf{{{\mathcal{I}}}}}}({{{\boldsymbol{\eta }}}}_{k})=-E\{{\partial }^{2}{\ell }_{ik}({O}_{ik};{{{\boldsymbol{\eta }}}}_{k})/\partial {{{\boldsymbol{\eta }}}}_{k}\partial {{{\boldsymbol{\eta }}}}_{k}^{{{\rm{T}}}}\}\). Let ηk0 be the true value of ηk. The influence function of ηk is given by
For the proportional hazards model, \(({\widehat{{{\boldsymbol{\beta }}}}}_{k}-{{{\boldsymbol{\beta }}}}_{k0},{\widehat{\Lambda }}_{k}-{\Lambda }_{k0})\) converges weakly to a Gaussian process in \({{\mathbb{R}}}^{d}\times {l}^{\infty }({{\mathcal{L}}})\) where d is the dimension of the parametric part βk61. The influence function of βk is given in Supplementary Methods.
For the coheritability parameters \({\gamma }_{k{k}^{{\prime} }}\) (\(k\, \ne \,{k}^{{\prime} }\)), the asymptotic property of \({\widehat{\gamma }}_{k{k}^{{\prime} }}\) can be established from the theory of Z-estimation. We consider ηk as a function of βk, so \({U}_{i,k{k}^{{\prime} }}({\gamma }_{k{k}^{{\prime} }};{{{\boldsymbol{\eta }}}}_{k},{{{\boldsymbol{\eta }}}}_{{k}^{{\prime} }})\) becomes a function of βk and \({{{\boldsymbol{\beta }}}}_{{k}^{{\prime} }}\). Let
\(l=k,{k}^{{\prime} }\), and
Let \({\gamma }_{k{k}^{{\prime} }0}\) be the true value of \({\gamma }_{k{k}^{{\prime} }}\). Then the influence function of \({\gamma }_{k{k}^{{\prime} }}\) is given by
We summarize the asymptotic result in the following theorem.
Theorem 2
Under Conditions 1–4, \(\widehat{{{\boldsymbol{\beta }}}}\) is regular and asymptotically linear,
where \({{\boldsymbol{\varphi }}}({{{\mathcal{O}}}}_{i})={({{{\boldsymbol{\varphi }}}}_{k}({{{\mathcal{O}}}}_{i}),{{{\boldsymbol{\varphi }}}}_{k{k}^{{\prime} }}({{{\mathcal{O}}}}_{i}):k,{k}^{{\prime} } = 1,\ldots ,K;k \,\ne \, {k}^{{\prime} })}^{{{\rm{T}}}}\) is the influence function of β, and \(\sqrt{n}(\widehat{{{\boldsymbol{\beta }}}}-{{{\boldsymbol{\beta }}}}_{0})\) converges to a normal distribution,
Furthermore, the estimators of heritability and coheritability follow an asymptotic normal distribution by the delta method.
This theorem implies that the estimates have root-n convergence rates, and we can make inferences based on the normal approximation using influence functions. To facilitate inference for the estimated parameters, we can plug the estimates \({\widehat{{{\boldsymbol{\eta }}}}}_{k}\) and \({\widehat{\gamma }}_{k{k}^{{\prime} }}\) in the influence functions, with the information matrix estimated by Louis’ formula62. For time-to-event phenotypes, although we can estimate the asymptotic variance of estimators using the observed information matrix, the dimension of this information matrix is too large63,64. Evaluating each entry of the information matrix requires multiple numerical integrations, severely limiting computational efficiency. Therefore, we only consider the inference for the parametric part βk, \(k\in {{{\mathcal{K}}}}_{2}\). The profile log-likelihood
where \({{{\mathcal{S}}}}_{k}\) is the set of step functions with jumps at {Tijk: i = 1, …, n, j = 1, …, ni}. Let \(p{l}_{i}({{{\mathcal{O}}}}_{ik};{{{\boldsymbol{\beta }}}}_{k})\) be the contribution to \(pl({{{\mathcal{O}}}}_{k};{{{\boldsymbol{\beta }}}}_{k})\) by the ith family. Then, the score function of the parametric part can be evaluated by
where ej is a vector with 1 in the jth entry and 0 in other entries, and pk is the dimension of βk. We take \({h}_{n}=0.03/\sqrt{n}\) in our algorithm. The information matrix of the parametric part \({{\bf{{{\mathcal{I}}}}}}({{{\boldsymbol{\beta }}}}_{k})\) is then estimated by
In summary, we can simplify the estimated influence function of βk (\(k\in {{{\mathcal{K}}}}_{1}\cup {{{\mathcal{K}}}}_{2}\)) as
The influence function of \({\gamma }_{k{k}^{{\prime} }}\) can be similarly estimated by plug-in estimators. Finally, the asymptotic variances of the estimated parameters can be estimated by plug-in estimators according to Theorem 2.
Additional simulation studies
Effect of family size
In our previous data-generating process, the family size was moderate. To assess the effect of family size, we consider two additional settings for generating family data. In the setting of small families, we let the number of families with two members be 1300 and the number of families with four members be 100. In the setting of large families, we let the number of families with two members be 100 and the number of families with four members be 700. The total number of individuals is 3600, identical in these three settings. Panel (A) of Fig. 8 shows the bias of estimates in these three settings. We find that larger families help reduce the variation of estimates for continuous and ordinal traits. However, the computation time for moderate families is 1.78 times that of small families, and the computation time for large families is 1.63 times that of moderate families.

A Bias of the estimated heritability, family-shared environmental effect, coheritability, and environmental correlation under three settings of family sizes. B Bias of the estimated heritability, family-shared environmental effect, coheritability, and environmental correlation by MPCH and joint MLE. C Bias of the estimated heritability and coheritability by MPCH, MPCH_NE, and HEc.
Efficiency loss of MPCH compared with joint MLE
To assess the loss of statistical efficiency for our proposed method compared to jointly maximizing the full likelihood, we compare the estimates of parameters (heritability, family-shared environmental effect, coheritability, and environmental correlation) given by MPCH and joint maximum likelihood estimation (MLE). We consider the first two continuous outcomes we generated. Panel (B) of Fig. 8 shows the bias of the estimates. The average bias is close to zero by both methods. The standard deviation by joint MLE is about 10% lower for estimated heritability and 14% lower for estimated coheritability than that by MPCH. The gain of statistical efficiency by joint MLE is not large. Note that although it is possible to find the solution of the joint MLE for two continuous phenotypes using the properties of the multivariate normal distribution, it is almost computationally impossible to solve the joint MLE for other data types. If the joint likelihood for many continuous phenotypes is maximized, the estimation would be computationally much less efficient since optimization should be performed over high-dimensional parameters. Due to the unified framework to deal with multiple data types and the computational efficiency of MPCH, it is reasonable to use MPCH to handle multiple phenotypes.
Effect of not modeling the environmental effect
The closed-form Haseman-Elston (HEc) estimator does not model the environmental effect. To assess the effect of not modeling the environmental effect, we compare the proposed MPCH, MPCH_NE (where we always set \({\widehat{\theta }}_{k}=0\)), and HEc for estimating heritability and coheritability. Panel (C) of Fig. 8 shows the bias of the estimated heritability and coheritability by MPCH, MPCH_NE, and HEc. The true within-family correlation matrix of the two outcomes is \({{{\bf{G}}}}_{i}\otimes {{{\mathbf{\Gamma }}}}_{12}+{{{\boldsymbol{J}}}}_{{n}_{i}}\otimes {{\rm{diag}}}({\theta }_{1},{\theta }_{2})\), where Γ12 is the upper-left 2 × 2 submatrix of Γ and \({{{\boldsymbol{J}}}}_{{n}_{i}}\) is an ni × ni matrix with all entries equal to 1. MPCH_NE and HEc do not consider the correlation \({{{\boldsymbol{J}}}}_{{n}_{i}}\otimes {{\rm{diag}}}({\theta }_{1},{\theta }_{2})\) resulting from the family-shared environmental effect in estimating the heritability and coheritability, so the estimates are biased. This result explains the overestimation of heritability in the UK Biobank data.
Statistics and reproducibility
Significance tests are based on one-sided Z-tests for heritability and two-sided Z-tests for coheritability. Estimates with standard error are presented in Supplementary Data 3 for heritability and Supplementary Data 4 for coheritability. The data processing procedure was illustrated in Protocol Section.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
The data that support the findings of this study are available from the UK Biobank, which were used under license for the current study. The UK biobank data via application through access@ukbiobank.ac.uk.
Code availability
The computer codes programmed in R (version 4.4.0) are available on GitHub https://github.com/naiiife/MPCH.
References
Marees, A. T. et al. A tutorial on conducting genome-wide association studies: Quality control and statistical analysis. Int. J. Methods Psychiatr. Res. 27, e1608 (2018).
Baselmans, B. M., Yengo, L., van Rheenen, W. & Wray, N. R. Risk in relatives, heritability, SNP-based heritability, and genetic correlations in psychiatric disorders: a review. Biol. Psychiatry 89, 11–19 (2021).
Yang, J. et al. Common SNPs explain a large proportion of the heritability for human height. Nat. Genet. 42, 565–569 (2010).
Chen, G.-B. et al. Estimation and partitioning of (co) heritability of inflammatory bowel disease from GWAS and immunochip data. Hum. Mol. Genet. 23, 4710–4720 (2014).
Armbruster, W. S., Pélabon, C., Bolstad, G. H. & Hansen, T. F. Integrated phenotypes: Understanding trait covariation in plants and animals. Philos. Trans. R. Soc. B: Biol. Sci. 369, 20130245 (2014).
Zhou, J. J. et al. Integrating multiple correlated phenotypes for genetic association analysis by maximizing heritability. Hum. Heredity 79, 93–104 (2015).
Polubriaginof, F. C. et al. Disease heritability inferred from familial relationships reported in medical records. Cell 173, 1692–1704 (2018).
Johnson, W., Turkheimer, E., Gottesman, I. I. & Bouchard Jr, T. J. Beyond heritability: Twin studies in behavioral research. Curr. Dir. Psychol. Sci. 18, 217–220 (2009).
Hallmayer, J. et al. Genetic heritability and shared environmental factors among twin pairs with autism. Arch. Gen. Psychiatry 68, 1095–1102 (2011).
Zaitlen, N. & Kraft, P. Heritability in the genome-wide association era. Hum. Genet. 131, 1655–1664 (2012).
Lakhani, C. M. et al. Repurposing large health insurance claims data to estimate genetic and environmental contributions in 560 phenotypes. Nat. Genet. 51, 327–334 (2019).
Yang, J., Lee, S. H., Goddard, M. E. & Visscher, P. M. GCTA: a tool for genome-wide complex trait analysis. Am. J. Hum. Genet. 88, 76–82 (2011).
Bulik-Sullivan, B. et al. An atlas of genetic correlations across human diseases and traits. Nat. Genet. 47, 1236–1241 (2015).
Bulik-Sullivan, B. K. et al. LD score regression distinguishes confounding from polygenicity in genome-wide association studies. Nat. Genet. 47, 291–295 (2015).
Elgart, M. et al. Correlations between complex human phenotypes vary by genetic background, gender, and environment. Cell Rep. Med. 3, 100844 (2022).
Ni, G. et al. Estimation of genetic correlation via linkage disequilibrium score regression and genomic restricted maximum likelihood. Am. J. Hum. Genet. 102, 1185–1194 (2018).
Zhang, X. et al. Phenome-wide association study (PheWAS) of colorectal cancer risk SNP effects on health outcomes in UK Biobank. Br. J. Cancer 126, 822–830 (2022).
Gao, F., Zeng, D. & Wang, Y. Semiparametric regression analysis of bivariate censored events in a family study of Alzheimer’s disease. Biostatistics 24, 32–51 (2023).
Lee, S. H., Yang, J., Goddard, M. E., Visscher, P. M. & Wray, N. R. Estimation of pleiotropy between complex diseases using single-nucleotide polymorphism-derived genomic relationships and restricted maximum likelihood. Bioinformatics 28, 2540–2542 (2012).
Sofer, T. Confidence intervals for heritability via Haseman-Elston regression. Stat. Appl. Genet. Mol. Biol. 16, 259–273 (2017).
van den Oord, E. J. Estimating effects of latent and measured genotypes in multilevel models. Stat. Methods Med. Res. 10, 393–407 (2001).
Muñoz, M. et al. Evaluating the contribution of genetics and familial shared environment to common disease using the uk biobank. Nat. Genet. 48, 980–983 (2016).
Rabe-Hesketh, S., Skrondal, A. & Gjessing, H. K. Biometrical modeling of twin and family data using standard mixed model software. Biometrics 64, 280–288 (2008).
Liang, B., Wang, Y. & Zeng, D. Semiparametric transformation models with multilevel random effects for correlated disease onset in families. Stat. Sin. 29, 1851 (2019).
Chen, L., Hsu, L. & Malone, K. A frailty-model-based approach to estimating the age-dependent penetrance function of candidate genes using population-based case-control study designs: an application to data on the BRCA1 gene. Biometrics 65, 1105–1114 (2009).
Graber-Naidich, A., Gorfine, M., Malone, K. E. & Hsu, L. Missing genetic information in case-control family data with general semi-parametric shared frailty model. Lifetime Data Anal. 17, 175–194 (2011).
Gorfine, M., Hsu, L. & Parmigiani, G. Frailty models for familial risk with application to breast cancer. J. Am. Stat. Assoc. 108, 1205–1215 (2013).
Hsu, L., Gorfine, M. & Zucker, D. On estimation of the hazard function from population-based case–control studies. J. Am. Stat. Assoc. 113, 560–570 (2018).
Manichaikul, A. et al. Robust relationship inference in genome-wide association studies. Bioinformatics 26, 2867–2873 (2010).
Millard, L. A., Davies, N. M., Gaunt, T. R., Davey Smith, G. & Tilling, K. Software application profile: PHESANT: A tool for performing automated phenome scans in UK Biobank. Int. J. Epidemiol. 47, 29–35 (2018).
McEvoy, B. P. & Visscher, P. M. Genetics of human height. Econ. Hum. Biol. 7, 294–306 (2009).
Min, J., Chiu, D. T. & Wang, Y. Variation in the heritability of body mass index based on diverse twin studies: A systematic review. Obes. Rev. 14, 871–882 (2013).
Silventoinen, K. et al. Heritability of adult body height: a comparative study of twin cohorts in eight countries. Twin Res. Hum. Genet. 6, 399–408 (2003).
Macgregor, S., Cornes, B. K., Martin, N. G. & Visscher, P. M. Bias, precision and heritability of self-reported and clinically measured height in australian twins. Hum. Genet. 120, 571–580 (2006).
Perola, M. et al. Combined genome scans for body stature in 6,602 european twins: evidence for common caucasian loci. PLoS Genet. 3, e97 (2007).
Yang, J. et al. Genetic variance estimation with imputed variants finds negligible missing heritability for human height and body mass index. Nat. Genet. 47, 1114–1120 (2015).
Haworth, C. M. et al. Increasing heritability of BMI and stronger associations with the FTO gene over childhood. Obesity 16, 2663–2668 (2008).
Chodick, G. et al. Heritability of body mass index among familial generations. JAMA Netw. Open 7, e2419029–e2419029 (2024).
Kolifarhood, G. et al. Heritability of blood pressure traits in diverse populations: a systematic review and meta-analysis. J. Hum. Hypertens. 33, 775–785 (2019).
Tegegne, B. S. et al. Heritability and the genetic correlation of heart rate variability and blood pressure in > 29 000 families: the lifelines cohort study. Hypertension 76, 1256–1262 (2020).
Chen, J. et al. Heritability and genome-wide association study of blood pressure in chinese adult twins. Mol. Genet. Genom. Med. 9, e1828 (2021).
Kandasamy, S. & Chanchlani, R. Familial aggregation of blood pressure and the heritability of hypertension. Pediatric Hypertens. 159–167 (2023).
Li, M. D., Cheng, R., Ma, J. Z. & Swan, G. E. A meta-analysis of estimated genetic and environmental effects on smoking behavior in male and female adult twins. Addiction 98, 23–31 (2003).
Maes, H. H. et al. A twin study of genetic and environmental influences on tobacco initiation, regular tobacco use and nicotine dependence. Psychol. Med. 34, 1251–1261 (2004).
Vink, J. M., Willemsen, G. & Boomsma, D. I. Heritability of smoking initiation and nicotine dependence. Behav. Genet. 35, 397–406 (2005).
Thomsen, S. F., Van Der Sluis, S., Kyvik, K., Skytthe, A. & Backer, V. Estimates of asthma heritability in a large twin sample. Clin. Exp. Allergy 40, 1054–1061 (2010).
Ober, C. & Yao, T.-C. The genetics of asthma and allergic disease: a 21st century perspective. Immunol. Rev. 242, 10–30 (2011).
Vinkhuyzen, A. A., Wray, N. R., Yang, J., Goddard, M. E. & Visscher, P. M. Estimation and partition of heritability in human populations using whole-genome analysis methods. Annu. Rev. Genet. 47, 75–95 (2013).
Han, Y. et al. Genome-wide analysis highlights contribution of immune system pathways to the genetic architecture of asthma. Nat. Commun. 11, 1776 (2020).
Kachuri, L. et al. Genetic determinants of blood-cell traits influence susceptibility to childhood acute lymphoblastic leukemia. Am. J. Hum. Genet. 108, 1823–1835 (2021).
Handelman, S. K. et al. Population-based meta-analysis and gene-set enrichment identifies fxr/rxr pathway as common to fatty liver disease and serum lipids. Hepatol. Commun. 6, 3120–3131 (2022).
Price, A. L. et al. Principal components analysis corrects for stratification in genome-wide association studies. Nat. Genet. 38, 904–909 (2006).
Evans, D. M., Frazer, I. H. & Martin, N. G. Genetic and environmental causes of variation in basal levels of blood cells. Twin Res. Hum. Genet. 2, 250–257 (1999).
Garner, C. et al. Genetic influences on f cells and other hematologic variables: a twin heritability study. Blood J. Am. Soc. Hematol. 95, 342–346 (2000).
Pilia, G. et al. Heritability of cardiovascular and personality traits in 6,148 sardinians. PLoS Genet. 2, e132 (2006).
Astle, W. J. et al. The allelic landscape of human blood cell trait variation and links to common complex disease. Cell 167, 1415–1429 (2016).
Cheng, S., Wei, L. J. & Ying, Z. Analysis of transformation models with censored data. Biometrika 82, 835–845 (1995).
Visscher, P. M., Hill, W. G. & Wray, N. R. Heritability in the genomics era—concepts and misconceptions. Nat. Rev. Genet. 9, 255–266 (2008).
Tenesa, A. & Haley, C. S. The heritability of human disease: estimation, uses and abuses. Nat. Rev. Genet. 14, 139–149 (2013).
Dempster, A. P., Laird, N. M. & Rubin, D. B. Maximum likelihood from incomplete data via the EM algorithm. J. R. Stat. Soc.: Ser. B (Methodol.) 39, 1–22 (1977).
Van Der Vaart, A. W. & Wellner, J. A.Weak Convergence and Empirical Processes (Springer, 1996).
Louis, T. A. Finding the observed information matrix when using the em algorithm. J. R. Stat. Soc. Ser. B: Stat. Methodol. 44, 226–233 (1982).
Zeng, D. & Lin, D. Maximum likelihood estimation in semiparametric regression models with censored data. J. R. Stat. Soc. Ser. B: Stat. Methodol. 69, 507–564 (2007).
Zeng, D. & Lin, D. A general asymptotic theory for maximum likelihood estimation in semiparametric regression models with censored data. Stat. Sin. 20, 871–910 (2010).
Acknowledgements
This research has been conducted using the UK Biobank Resource under Application Number 177198. We thank the UK Biobank participants and coordinators for their invaluable contributions. We thank the journal editors and reviewers for their valuable comments. This work is partially supported by NIH grants NS073671, MH123487, and GM124104.
Author information
Authors and Affiliations
Contributions
Y.D., Data curation, formal analysis, investigation, methodology, writing–original draft preparation. D.Z., Conceptualization, funding acquisition, methodology, writing–review & editing. Y.W., Conceptualization, funding acquisition, methodology, writing–review & editing.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Communications Biology thanks Hannah Klinkhammer and the other, anonymous, reviewers for their contribution to the peer review of this work. Primary Handling Editors: Qiao Fan and Aylin Bircan.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Deng, Y., Zeng, D. & Wang, Y. Computationally efficient methods for estimating phenome—wide coheritability of multi-type phenotypes using biobank data. Commun Biol 8, 1460 (2025). https://doi.org/10.1038/s42003-025-08180-y
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s42003-025-08180-y
