
Abstract
Plasmodium vivax malaria is a major public health problem outside sub-Saharan Africa. However, an increasing number of P. vivax infections in Duffy-negative individuals has been reported across Africa in recent years, raising concerns that the parasites may have evolved alternative pathways to invade reticulocyte and overcome Duffy-negativity. Here, we investigated the global genetic structure and diversity of sub-Saharan African P. vivax populations, exploring possible molecular signatures of adaptation to Duffy-negative hosts. We analyzed 204 previously published P. vivax genome sequences from Africa, Southeast Asia, the Pacific Coral Triangle, and South America and generated whole-genome sequences of 133 P. vivax field isolates collected from 10 sub-Saharan African countries. Our analysis revealed four distinct geographic clusters, with clear contrasts between East/West Africa and the Indian Ocean populations. Despite the limited number of interpretable sequences from Duffy-negative individuals - attributable to low parasitemia - and the lack of clear evidence of selective pressure acting on invasion-related genes of the P. vivax parasite populations circulating in sub-Saharan Africa, our study offers valuable insights into the genetic diversity of P. vivax and lays the groundwork for future research exploring parasite adaptation to Duffy-negative hosts.
Similar content being viewed by others
Introduction
Plasmodium vivax malaria remains a major public health concern in South America, Southeast Asia, the Middle East, the Pacific Coral Triangle, and Eastern and Southern Africa1. Historically, the predominance of Duffy-negative human populations in sub-Saharan Africa has led to the belief that P. vivax transmission is limited in this region2. This paradigm stems from evidence that P. vivax merozoites rely on the interaction between the Duffy Antigen Receptor for Chemokines (DARC) and the P. vivax Duffy Binding Protein (PvDBP) to invade erythrocytes3. However, recent studies have reported cases of P. vivax infections in Duffy-negative individuals across Africa4,5,6,7,8,9,10,11,12,13,14, challenging the conventional understanding and raising questions about potential alternative invasion pathways.
Despite these intriguing observations, robust genomic data from Duffy-negative individuals remain limited due to the technical challenges of obtaining high-quality sequences from low-parasitemia infections. Consequently, the molecular mechanisms underlying P. vivax invasion into Duffy-negative reticulocytes remain poorly understood. Previous studies have described duplications of the pvdbp gene15,16 in regions such as Madagascar, where Duffy-positive and Duffy-negative populations coexist, and Southeast Asia17,18. Additional work has identified homologs such as the P. vivax Erythrocyte Binding Protein (PvEBP/PvDBP2)19, which preferentially binds immature (CD71high) reticulocytes in Duffy-positive individuals but shows minimal binding to Duffy-negative reticulocytes20. In parallel, other ligands like PvRBP2a and PvRBP2b have been identified as key players in reticulocyte recognition prior to invasion21,22. However, no conclusive evidence has demonstrated that these genes facilitate Duffy-independent invasion.
Here, we focus on analyzing the global genetic diversity and population structure of P. vivax across Africa and other endemic regions to better understand the selective pressures shaping parasite populations. We provide whole-genome sequences of 133 P. vivax field isolates from 10 African countries (Angola, Burundi, Comoros, Djibouti, Egypt, Eritrea, Ethiopia, Madagascar, Mauritania, and Sudan) using selective whole-genome amplification (sWGA) and next-generation sequencing. Our dataset was enriched with 204 publicly available sequences from Africa, Southeast Asia, the Pacific Coral Triangle, and South America23, for a total of 337 samples across 18 countries.
To ensure a robust analysis, we assessed within-host diversity by calculating Fws values to distinguish monoclonal infections from polyclonal infections and to estimate heterozygosity. Our study primarily explores the global genetic structure of P. vivax, identifies regions under selection, and examines population dynamics in areas with a high prevalence of Duffy-negativity. While the limited availability of high-quality Duffy-negative sequences prevents definitive conclusions on parasite adaptation, this work lays the foundation for future studies with larger and more balanced datasets.
Results
Samples
A total of 133 P. vivax field isolates were obtained between 2016 and 2021 from symptomatic patients originating from ten sub-Saharan African countries. Samples were collected locally at health centers in Ethiopia (N = 66), Madagascar (N = 27), Mauritania (N = 2), and Angola (N = 12). Additional DNA extracts were obtained from symptomatic travelers returning to France from the Comoros (N = 8), Mauritania (N = 5), Djibouti (N = 3), Sudan (N = 3), Madagascar (N = 2), Eritrea (N = 2), Ethiopia (N = 1), Burundi (N = 1), and Egypt (N = 1).
We amplified P. vivax DNA using sWGA to address the challenge of low parasitemia in clinical samples and successfully sequenced 72/133 P. vivax genomes using high-throughput sequencing. Among the successfully sequenced genomes, 18/72 (25%) were from homozygous Duffy-positive patients, 52/72 (72.3%) were from heterozygous Duffy-positive patients, and 2/72 (2.7%) had undetermined Duffy status. Despite our efforts, P. vivax genome sequences from 18 homozygous Duffy-negative patients could not be properly exploited due to insufficient parasitemia levels, which resulted in low DNA amplification and inadequate sequencing quality (see “Methods” for details). This limitation highlights the ongoing technical challenges in obtaining high-quality genomic data from Duffy-negative individuals, an issue that remains a bottleneck for understanding potential adaptations of P. vivax to these hosts.
To expand the scope of our analysis, we enriched our dataset with 204 publicly available P. vivax genome sequences from prior studies. These included sequences from Southeast Asia (Cambodia, Thailand, Vietnam), the Pacific Coral Triangle (Indonesia, Malaysia, Papua New Guinea), South America (Brazil, Colombia), and Africa (Ethiopia, Madagascar, Mauritania). Full details, including sample metadata and references, are provided in Table 1 and Supplementary Information (Table S1).
Genomic data
We aligned the sequencing reads to the PvP01 (v.48) reference genome using bwa mem and used GATK 4.0 (Genome Analysis Toolkit) following best practices guidelines to identify Single Nucleotide Polymorphisms (SNPs). Genotypes were defined in diploid mode. To ensure robust variant calling, we applied hard filtering based on several summary statistics. Variants were excluded if they met any of the following criteria: QD < 2.0, QUAL < 30.0, SOR > 3.0, FS > 60.0, MQ < 40.0, MQRankSum < −12.5, ReadPosRankSum < −8.0. Further details of these filters and an overview of the workflow are provided in Fig. S1.
The final dataset retained >300,000 SNPs across 276 samples (72 newly sequenced samples from this study and 204 sequences from previously published datasets). To assess the presence of polyclonal infections, we evaluated within-host diversity by calculating Fws coefficients24. Empirically, isolates with Fws < 0.95 are generally considered as polyclonal, while those with Fws > 0.95 are expected to be monoclonal. This analysis revealed that approximately one-third of the isolates (32.4%) could be classified as polyclonal (Tabel S2 and Fig. S2). This finding corroborates previous studies showing that P. vivax infections in Southeast Asia and South America are frequently characterized by multiple clones8,25,26,27,28. For subsequent analyses, polyclonal infections were excluded to avoid biases in heterozygosity, while complementary analyses including both monoclonal and polyclonal infections were conducted and reported in the Supplementary Information to assess the robustness of the results (Fig. S3).
Global genetic diversity
The population structure of P. vivax isolates was characterized by low levels of admixture overall, with clear geographic genetic clusters identified (Fig. 1A). Isolates from South America (Brazil and Colombia) exhibited more ambiguous clustering profiles, suggesting some degree of admixture that requires further investigation with a larger sample size.

A Structural analysis of chromosomes. B Principal Coordinate analysis (PCO). C Genome-wide genetic differentiation with genomic islands.
The Principal Coordinate Analysis (PCO) corroborated the genetic clustering and revealed four distinct geographic groups (Fig. 1B): an East/West African cluster (Ethiopia, Djibouti, Mauritania), an Indian Ocean cluster (Madagascar, Comoros), an Asian/Pacific Coral Triangle cluster (Cambodia, Indonesia, Malaysia, Papua New Guinea, Thailand, and Vietnam), and a South American cluster (Brazil, Colombia), which appeared centrally positioned relative to the other three clusters.
Among the sub-Saharan African samples, we observed a notable distinction between the two African subpopulations: the East/West African cluster (Ethiopia, Djibouti, Mauritania) and the Indian Ocean cluster (Madagascar, Comoros). Notably, the Indian Ocean cluster was genetically closer to the East/West African cluster than to the Asian/Pacific cluster, suggesting significant historical or contemporary gene flow between these African populations.
This observation contrasts with the assumption of a unique Indonesian origin for the P. vivax population in Madagascar29. While past human migrations have been proposed as a major driver of P. vivax population structure6,30, our results suggest that the genetic landscape in Madagascar may reflect a more complex history, potentially involving multiple introductions and subsequent gene flow between African and Indian Ocean populations. These findings align partially with previous studies but highlight the need for larger sample sizes and deeper analyses to refine our understanding of the genetic origins and population dynamics of P. vivax in Madagascar and the surrounding regions.
Genomic islands of genetic differentiation
We investigated whether the low frequency of Duffy antigens in human populations across sub-Saharan Africa might exert selective pressure on P. vivax parasites, potentially favoring adaptations enabling Duffy-independent invasion pathways. To test this hypothesis, we performed a genome-wide analysis of genetic differentiation using pairwise comparisons of P. vivax sequences from multiple regions. We identified eight genomic regions with clear peaks of differentiation on chromosomes 4, 5, 7, 10, 11, 12, 13, and 14 (Fig. 1C). These regions contained genes with diverse functions, including loci involved in metabolic pathways, cellular components, and genes of unknown function (Table 2). Interestingly, we observed signals in three genes previously associated with antimalarial drug resistance: mdr-1 (PVP01_1010900), dhfr (PVP01_0526600), and dhps (PVP01_1429500). These findings likely reflect differential drug pressures acting on P. vivax populations worldwide, consistent with observations from other studies25,26,27. Importantly, no significant signals were detected in genes encoding parasite ligands directly implicated in erythrocyte invasion pathways, such as PvDBP or members of the PvRBP family, which would have suggested adaptations for Duffy-independent invasion. However, one notable exception was a signal detected in the thrombospondin-related anonymous protein gene (TRAP) (PVP01_1218700), located on chromosome 12. This protein is known to be expressed in P. vivax sporozoites and plays a critical role in the invasion of human hepatocytes31,32. While this signal may not directly relate to erythrocyte invasion, it highlights the potential involvement of P. vivax genes in other stages of the parasite’s lifecycle. Taken together, our results suggest that the global genomic structure of P. vivax populations reflects consistent patterns of differentiation, likely driven by geographic and environmental pressures, including drug pressure. Although no evidence was found for specific genetic adaptations to Duffy-independent invasion in the regions examined, these findings underscore the complexity of P. vivax evolution in regions where Duffy negativity predominates. Future studies incorporating larger datasets of high-quality genomes from Duffy-negative individuals will be essential to conclusively identify loci under selection for alternative invasion pathways.
Genetic diversity of genes associated with drug resistance
In the African P. vivax population, we detected four single non-synonymous mutations (P33L, C49R, N130K, A255T) in the bifunctional dihydrofolate reductase-thymidylate synthase gene (PVP01_0526600) in samples from Madagascar, Comoros, and Mauritania. The C49R mutation, only found in Madagascar (18/23), was the most common mutation (26%). The P33L mutation was detected in samples from the Comoros (1/6), the N130K mutation in samples from Madagascar (3/23) and the A255T mutation in samples from Mauritania (1/3). No such mutations were found in samples from Ethiopia and Djibouti.
Analysis of mutation points in the hydroxymethyldihydropterin pyrophosphokinase-dihydropteroate synthase gene (PVP01_1429500), likely associated with sulfadoxine resistance, revealed the presence of five different non-synonymous mutations (E142G, M205I, G383A, I545T and A647V) in varying proportions. The three most common mutations were G383A (36/68) from 4 countries, followed by M205I (30/68) from 3 countries, and E142G (25/68) from 2 countries. The E142G, I545T and A647V mutations were country-specific: E142G in Ethiopia and Djibouti (24/34 and 1/1, respectively), I545T in Mauritania (1/3) and A647V in the Comoros (1/6) and Ethiopia (10/34). The M205I and G383A mutations were found in varying proportions in all countries except in samples from Madagascar in which we did not find M205I mutations. Three triple mutant alleles were observed including the E142G/M205I/G383A (15/68, mainly in Ethiopia 14/15), the E142G/M205I/A647V (10/68, only in Ethiopia 10/34) and the M205I/G383A/I545T (1/68, Mauritania). We also found two double mutant alleles, the M205I/G383A (4/68) and the G383A/A647V (1/68).
For the ABC transporter B family member 1 - multidrug resistance protein 1 gene (PVP01_1010900), which is thought to modulate parasite susceptibility to amino-4 and amino-alcohol quinolines, five-point mutations were found (F194Y, S698G, L845F, F976Y and T1269S). The F976Y mutation was the most common mutation (34/68) and was found in high proportions in Mauritania (3/3), in Ethiopia (30/34) and in Djibouti (1/1). The F194Y mutation was found only in samples from Madagascar (1/24), the S698G mutation was observed in samples from Mauritania (1/3) and Ethiopia (1/34) and the L845F mutation only in Mauritania (2/3). Two double mutant alleles were observed including the S698G/F976Y (7/68, mainly in Ethiopia 6/34) and the L845F/F976Y (2/68, Mauritania 2/3). More details are given in the Supplementary Information (Table S3).
Genetic diversity of invasion-related genes
We then restricted our analysis to regions containing genes validated or suspected to encode parasite ligands involved in reticulocyte invasion pathways25,26,27,33, such as the pvdbp, pvebp/pvdbp2, pvrbp2a, and pvrbp2b genes (Table S4). Only two mutations were found in the P. vivax Duffy binding gene (pvdbp, PVP01_0623800), the K277T (2/68) and the G830V (1/68). The K277T mutation was specific to samples from the Comoros (1/6) and Madagascar (1/24), while the G830V mutation was detected only in Mauritania (1/3). No mutations were detected in Ethiopia and Djibouti. Analysis of mutation points in the P. vivax erythrocyte binding gene (pvebp/pvdbp2, PVP01_0102300) revealed 7 non-synonymous mutations: D268N, E341K, K595N, I611F, E660K, V705L and F746I. The most common mutation was the I611F (8/68), found only in Comoros (1/6) and Madagascar (7/24). In Ethiopia, only the E660K mutation was detected at a low proportion (1/34), while no mutation was found in samples from Djibouti. The K595N mutation was specific for Madagascar (1/24) and the V705L mutation for the Comoros (1/6). The D268N mutation was found in Comoros (1/6), in Madagascar (1/24) and in Mauritania (1/3). Two double mutant alleles were found in Comoros and Madagascar (D268N/E341K) and in Mauritania (D268N/F746I).
No mutations were found in the reticulocyte binding protein 2a (pvrbp2a, PVP01_1402400) and the reticulocyte binding protein 2b (pvrbp2b, PVP01_0800700) genes in samples from Mauritania, Ethiopia, and Djibouti. In samples from the Comoros, the K112I mutation in the pvrbp2a gene and the S186N and D461G mutations in the pvrbp2b gene were detected once (1/6). The same mutations were found in samples from Madagascar, once for the K112I mutation in the pvrbp2a gene (1/24) and at higher frequencies for the S186N (6/24) and D461G (3/24) mutations in the pvrbp2b gene. The L84F in the pvrbp2b gene was unique to samples from Madagascar (5/24). More details can be found in the Supplementary Information (Table S4).
We also analyzed genetic differentiation by estimating Tajima’s D values at loci associated with invasion-related genes and comparing these values with those obtained for all individual genes across the genome. This analysis revealed that invasion-related genes from P. vivax populations in all regions fell well within the boundaries defined by other genes (Fig. 2A). In addition, a country-level comparison of Tajima’s D values across 13 populations confirmed that invasion-related genes did not exhibit patterns of differentiation distinct from the genome-wide average. Interestingly, values of Tajima’s D across all populations appeared shifted toward negative values, suggesting an excess of low-frequency alleles. This trend may indicate ongoing geographic differentiation at a finer scale, possibly driven by local demographic events, such as population expansions or recent bottlenecks, rather than by selective pressure on invasion-related loci.

A Differentiation of invasion-related genes in all P. vivax populations. B Analysis of Tajima’s D within all countries. C Analysis of Tajima’s D in Ethiopia, Madagascar, and Thailand.
Overall, we found no evidence of selective pressure specifically acting on invasion-related genes in sub-Saharan P. vivax strains (Fig. 2B, C). These results suggest that while geographic structure may influence allele frequency patterns, it does not appear to impose measurable selection on genes linked to erythrocyte invasion pathways.
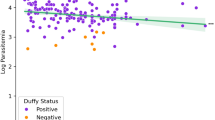
Genetic diversity associated with Duffy genotype in human hosts
We hypothesized that the Duffy status of patients (homozygous and heterozygous Duffy-positives, and homozygous Duffy-negative) might influence the diversity and selection of invasion-related genes in African P. vivax genomes. To test this, we conducted a Genome-Wide Association Study (GWAS) on sequences obtained from 18 isolates infecting homozygous Duffy-positive patients and 52 isolates from heterozygous Duffy-positive patients. The results of the GWAS showed no significant associations between the patient’s Duffy status and parasite genotypes at loci encoding invasion-related genes. Specifically, we did not observe any molecular signatures suggestive of adaptive evolution in these genes that could facilitate infection in Duffy-negative hosts (Fig. 3). These findings suggest that the evolution of African P. vivax populations may not be primarily driven by diversification or selection of invasion-related genes, at least within the resolution of our current analysis. Alternatively, adaptations to infect Duffy-negative individuals, if they exist, may involve molecular mechanisms too complex or subtle to be detected by broad-scale genomic association methods, particularly given the constraints imposed by the limited availability of high-quality Duffy-negative sequences.

A GWAS analysis of sequences obtained from 18 P. vivax African isolates infecting homozygous Duffy-positive patients and 52 P. vivax African isolates infecting heterozygous Duffy-positive patients for invasion-related genes. Manhattan plot displaying the genome-wide association results. Each dot represents a single nucleotide polymorphism (SNP), plotted as the negative log10-transformed p value (−log10(P))(−log_{10}(P))(−log10 (P)) of the association test. The x-axis represents genomic regions, labeled as PvP01_01_v1 through PvP01_14_v1, corresponding to different chromosomes or contigs of the Plasmodium vivax genome. The dashed horizontal line indicates the genome-wide significance threshold. No SNPs surpassed the genome-wide significance threshold, suggesting that no variants showed statistically significant associations after multiple testing correction. B Association of genetic variants in selected genes with the phenotype of interest. Genes are displayed on the x-axis, and the dashed horizontal line indicates the genome-wide significance threshold. Genes include AMA1, DBP, EBP, ETRAMP, GAMA, MSA180, MSP1, MSP1P, P12, RAMA, RBP2a, RBP2b, and RON. No SNPs reached the genome-wide significance threshold in this analysis.
Overall, while our data do not rule out the possibility of Duffy-independent invasion pathways, they underscore the need for future studies incorporating higher-resolution genomic data from larger numbers of Duffy-negative infections to identify potential loci under selection and fully elucidate the evolutionary dynamics of P. vivax populations in Africa.
Discussion
To date, the global genetic diversity and population structure of P. vivax remain poorly understood, particularly in Africa, where vivax malaria is rare and characterized by low parasitemia. While previous studies have demonstrated distinct population structures in Southeast Asia and South America, limited genomic data exist for P. vivax populations circulating in Africa, except for southern Ethiopia. Here, using genomic data from African and global isolates, we provide a comparative analysis of P. vivax populations and explore the potential impact of Duffy negativity on parasite evolution.
Our results confirm the clear geographic genetic structure of P. vivax, distinguishing African, Asian, and South American clusters, consistent with prior studies. Within Africa, we observed two distinct sub-clusters: the East/West African cluster (Ethiopia, Djibouti, Mauritania) and the Indian Ocean cluster (Madagascar and Comoros). Notably, the Indian Ocean cluster was genetically closer to East Africa than to Asia, challenging previous assumptions of a strictly Indonesian origin for the Malagasy P. vivax population. While earlier work suggested single origins, our findings, combined with the complex demographic history of Madagascar, indicate multiple introductions likely shaped by human migrations6,30.
Our findings align with previous observations reported by Hupalo et al.26, which demonstrated a clear geographic genetic structure of P. vivax populations worldwide, including low levels of admixture. Similarly, our analysis revealed well-defined clusters for African, Asian, and South American populations, with limited gene flow between regions. In addition, the work of Benanvente et al.25 identified loci under selective pressure, particularly in drug resistance genes (pvkelch10, pvmrp1), which differ from the loci identified in our study (pvmdr1, pvdhfr, pvdhps). This discrepancy likely reflects regional variations in drug use and the selective pressures acting on P. vivax populations in Africa compared to other regions.
A key hypothesis motivating this study was that Duffy-negativity might impose selective pressure on invasion-related genes, driving adaptations in P. vivax African populations. Genetic diversity within major invasion-related loci did not deviate significantly from the genome-wide background, and Tajima’s D analyses confirmed a lack of positive selection. For example, the pvdbp gene showed only two novel non-synonymous mutations (K277T and G830V), while mutations in pvebp/pvdbp2 and pvrbp2b were observed at low frequencies without clear evidence of selection.
As a result, potential adaptive signals may remain undetected, particularly if they involve subtle or complex evolutionary mechanisms.
Recent studies suggest that Duffy-negative erythroblasts transiently express DARC during terminal differentiation, providing a limited opportunity for P. vivax merozoites to invade15,34. This biological constraint could explain the low parasitemia consistently observed in Duffy-negative individuals and the apparent lack of strong selection signals in invasion-related genes. If alternative invasion pathways exist, they may rely on molecular mechanisms that are currently undetectable with our analytical approach or require larger, more balanced datasets for validation.
While no evidence of selection was observed in invasion-related genes, we identified strong signals of selection in genes associated with antimalarial drug resistance, including pvdhfr, pvdhps, and pvmdr1. These results are consistent with earlier studies, such as Benavente et al.25, which reported signals of antifolate resistance markers. However, our findings differ in specific loci under selection, as we detected positive selection in pvmdr1 (not observed by Benavente et al.), while signals in pvkelch10 and pvmrp1 were absent in our dataset25. These discrepancies likely reflect region-specific drug pressures and variations in antimalarial use across African and global populations. Notably, the detection of previously unreported mutations, such as I545T in pvdhps and F194Y in pvmdr1, highlights the ongoing evolution of drug resistance in African P. vivax populations.
Despite these advances, analysis of the P. vivax genome remains technically challenging due to field samples with low parasitemia and high human DNA contamination. While sWGA prior to WGS improved parasite genome recovery, it was insufficient to obtain high-quality sequences from Duffy-negative patients. Moreover, the sWGA approach precluded the detection of copy number variations in key invasion-related genes, such as pvdbp and pvebp/pvdbp2, which may play critical roles in alternative invasion pathways35.
Despite increasing evidence of P. vivax infections in Duffy-negative individuals, our study was unable to identify clear genomic signatures of parasite adaptation. This limitation primarily stems from the low parasitemia observed in these individuals, which restricted the availability of high-quality genomic sequences. As a result, potential adaptive mechanisms may remain undetected, particularly if they involve complex or subtle evolutionary processes. Overcoming these challenges will require larger, well-balanced datasets, improved parasite enrichment techniques, and functional studies to elucidate alternative invasion pathways.
In summary, this study provides new insights into the genetic diversity and structure of African P. vivax populations and places them within a global context. We confirm the distinct clustering of African P. vivax populations and highlight their divergence from Asian and South American populations. However, we found no evidence of selective pressure on invasion-related genes, suggesting that adaptations to Duffy-negative hosts, if they exist, may involve mechanisms too complex to be detected by our analysis. These results emphasize the biological and technical challenges of studying P. vivax in Duffy-negative individuals.
Methods
Biological samples
All biological samples used in this study were obtained from Plasmodium vivax-infected blood samples collected between 2016 and 2021 after obtaining informed consent from all participants. The study protocol and sample collection were approved by the Institutional Review Board of Addis Ababa University (Ethiopia), the Comité d’Éthique Biomédicale de Madagascar (Madagascar), the Comité National d’Éthique pour la Recherche en Santé de Mauritanie (Mauritania), and the Comité de Protection des Personnes Île-de-France (France) for samples from returning travelers. All ethical regulations relevant to human research participants were followed. Ethics approval was obtained from an ethics committee located in Angola for the sample collection that took place in Angola. Dried blood spots and/or veinous blood samples were collected from symptomatic patients at health centers in Ethiopia (Addis Ababa University), Madagascar (Institut Pasteur Madagascar), and Mauritania (Université de Nouakchott) and from symptomatic travelers returning from Comoros, Djibouti, Madagascar, Mauritania, and Ethiopia (French National Reference Center for malaria). Blood samples from Madagascar were leukodepleted using CF11-packed columns to minimize the amount of human DNA36. We enriched our dataset with 204 published genomic sequences of P. vivax strains circulating in Southeast Asia (Cambodia, Thailand, Vietnam), Pacific Coral Triangle (Indonesia, Malaysia, Papua New Guinea), South America (Brazil, Columbia), and Africa (Ethiopia, Madagascar, Mauritania) (Table 1 and Table S1).
DNA extraction
Genomic DNA was extracted from 100 µL of red blood cell pellets or from a 6-mm dried blood spot punch using the QIAamp mini-blood DNA kit (Qiagen), according to the supplier’s instructions. The total DNA concentration was quantified using a dsDNA HS assay kit and a Qubit fluorometer (Invitrogen).
Screening of Plasmodium species
The Plasmodium species screening of P. vivax isolates was performed by real-time PCR as previously described37 (Table S5).
Duffy genotyping
The Duffy genotype of the patients was determined by PCR and Sanger sequencing as previously described6 (Table S5).
Selective whole-genome amplification
sWGA was performed to enrich P. vivax DNA as previously described35 (Table S5). PCR products obtained from sWGA were diluted (1:1) with DNase-free and RNase-free water (ThermoFisher Scientific) and purified with AMPure XP beads (Beckman Coulter) according to the supplier’s recommendations.
Whole genome sequencing
Whole genome sequencing was performed as previously described5. Sample libraries were prepared using a Truseq Nano Kit (Illumina). Briefly, 100–150 ng of DNA was subjected to shearing, end repair, A-tailing, and adapter ligation. When necessary, they were enriched using 10–15 cycles of PCR. The sample libraries were then multiplexed and loaded. Paired-end sequencing was performed with 2 × 150 base reads kit on a NovaSeq6000. Illumina sequence reads were submitted to an archived system.
Variant calling
Sequencing reads were aligned to the Plasmodium vivax PvP01 reference genome (version 48, PlasmoDB), which is publicly available at https://plasmodb.org/plasmo/38.
Low-quality reads (MAPQ < 30), secondary alignments, and anomalous insert sizes (>1000 bp) were excluded. Duplicates were marked using Picard. SNPs were called with GATK HaplotypeCaller (v4.1.7.0) in diploid mode, and joint genotyping was performed using GenotypeGVCFs39. Variants were filtered using VariantFiltration with the following parameters: QUAL < 30, QD < 2.0, MQ < 40, MQRankSum <–12.5, SOR > 3.0, FS > 60.0, and ReadPosRankSum <–8.0. Genotypes with depth ≤4 were considered missing. SNPs with >10% missing data or not biallelic were excluded using VCFtools, and final VCFs were merged with Picard GatherVcfs. Additional technical details are provided in the Supplementary Methods.
Analysis of genotypic data
Infection complexity and filtering
To distinguish between mono- and polyclonal P. vivax infections, we computed the within-host diversity index Fws across loci using the formula Fws = 1 − Hw/Hs, where Hw is the observed heterozygosity within samples and Hs is the expected heterozygosity at the population level. Given the bias introduced by mixed infections in measures of diversity, structure, and genotype–phenotype associations, we restricted the main analyses to monoinfections. To evaluate the robustness of this choice, we conducted parallel analyses including all infections, whose results were consistent with those from the monoinfection subset (Fig. S3).
Population structure and genetic clustering
Genetic population structure was investigated using principal coordinate analysis (PCoA) based on pairwise genetic distances between samples, implemented in R (v4.3.0) with the ade4 package. Clustering analysis was performed using fastSTRUCTURE, a variational Bayesian algorithm, across a range of K values (K = 2–10)40. The most likely number of clusters was inferred using the chooseK function. These analyses were restricted to monoinfections. Further methodological details are provided in the Supplementary Methods.
Genomic differentiation and selection
Genomic islands of differentiation were identified by computing Nei’s coefficient of genetic differentiation (GST) for each SNP across populations. To explore selective signatures, we calculated Tajima’s D for all genes and focused on loci involved in reticulocyte invasion pathways. Gene-level estimates of genetic differentiation were also computed using GST. These analyses aimed to identify regions potentially under selection in association with host Duffy phenotype or geography.
Genotype–phenotype association analysis
To test for associations between parasite genotypes and host Duffy phenotype, we applied a generalized linear model (GLM) with a binomial distribution: Duffy_status = parasite_genotype + country_of_origin. P values associated with genotype effects were computed and corrected for multiple comparisons using the Bonferroni method. Only monoinfections with available Duffy genotyping were included in this analysis.
Statistics and reproducibility
This study is based on 133 P. vivax field isolates collected from ten sub-Saharan African countries and 204 publicly available genomes from other endemic regions. All analyses were conducted on monoinfections (as defined by Fws > 0.95), unless otherwise specified. Replicates correspond to independent patient-derived parasite isolates. No experimental replication was possible due to the observational and retrospective nature of the dataset. Statistical analyses were performed in R (v4.3.0), using appropriate packages and functions as described in the corresponding sections. Significance thresholds and multiple-testing corrections (Bonferroni) were applied where relevant. Genetic diversity, population structure, differentiation, and genotype–phenotype association were assessed with standard population genetics metrics and GLMs. Consistency of results was assessed by including and excluding polyclonal infections (Fig. S3). Full details of the analytical pipelines are provided in the Supplementary Methods.
Data availability
All raw sequence data is available from the European Nucleotide Archive (www.ebi.ac.uk/ena; see Supplementary Data 1 for accession numbers). These data include samples generated as part of this study (PRJEB81739) as well as publicly available sequences. Specifically, the dataset incorporates sequences from the LSHTM returning travelers project (PRJEB44419) and the MalariaGEN P. vivax Genome Variation project23,27. It is important to clarify that while our study includes sequences from the publicly available MalariaGEN dataset, our samples were independently collected and were not generated as part of MalariaGEN activities41.
Code availability
All custom R and SLURM scripts used for the analysis of *Plasmodium falciparum* WGS data (BioProject PRJEB81739) are available on Zenodo: https://doi.org/10.5281/zenodo.15391778. The archive includes all scripts used for variant filtering, diversity metrics (e.g., Tajima’s D), population structure, GWAS, and HPC workflows (SLURM submission). All files are versioned and released under the MIT license.
References
World Health Organization. World Malaria Report 2022 (ed. IGO. GWHOLCB-N-S) (World Health Organization, 2022).
Miller, L. H., Mason, S. J., Clyde, D. F. & McGinniss, M. H. The resistance factor to Plasmodium vivax in blacks. N. Engl. J. Med. 295, 302–304 (1976).
Chitnis, C. E. & Miller, L. H. Identification of the erythrocyte binding domains of Plasmodium vivax and Plasmodium knowlesi proteins involved in erythrocyte invasion. J. Exp. Med. 180, 497–506 (1994).
Albsheer, M. M. A. et al. Distribution of Duffy phenotypes among Plasmodium vivax infections in Sudan. Genes 10, 437 (2019).
Baird, J. K. African Plasmodium vivax malaria improbably rare or benign. Trends Parasitol. 38, 683–696 (2022).
Ménard, D. et al. Plasmodium vivax clinical malaria is commonly observed in Duffy-negative Malagasy people. Proc. Natl. Acad. Sci. USA 107, 5967–5971 (2010).
Mendes, C. et al. Duffy negative antigen is no longer a barrier to Plasmodium vivax-molecular evidences from the African West Coast (Angola and Equatorial Guinea). PLoS Negl. Trop. Dis. 5, e1192 (2011).
Ngassa Mbenda, H. G. & Das, A. Molecular evidence of Plasmodium vivax mono and mixed malaria parasite infections in Duffy-negative native Cameroonians. PloS ONE 9, e103262 (2014).
Niangaly, A. et al. Plasmodium vivax Infections over 3 Years in Duffy Blood Group Negative Malians in Bandiagara, Mali. Am. J. Trop. Med. Hyg. 97, 744–752 (2017).
Poirier, P. et al. The hide and seek of Plasmodium vivax in West Africa: report from a large-scale study in Beninese asymptomatic subjects. Malar. J. 15, 570 (2016).
Rubio, J. M. et al. Semi-nested, multiplex polymerase chain reaction for detection of human malaria parasites and evidence of Plasmodium vivax infection in Equatorial Guinea. Am. J. Trop. Med. Hyg. 60, 183–187 (1999).
Russo, G. et al. Molecular evidence of Plasmodium vivax infection in Duffy negative symptomatic individuals from Dschang, West Cameroon. Malar. J. 16, 74 (2017).
Ryan, J. R. et al. Evidence for transmission of Plasmodium vivax among a Duffy antigen negative population in Western Kenya. Am. J. Trop. Med. Hyg. 75, 575–581 (2006).
Wurtz, N. et al. Vivax malaria in Mauritania includes infection of a Duffy-negative individual. Malar. J. 10, 336 (2011).
Bouyssou, I. et al. Unveiling P. vivax invasion pathways in Duffy-negative individuals. Cell Host Microbe 31, 2080–2092.e2085 (2023).
Menard, D. et al. Whole genome sequencing of field isolates reveals a common duplication of the Duffy binding protein gene in Malagasy Plasmodium vivax strains. PLoS Negl. Trop. Dis. 7, e2489 (2013).
Gunalan, K. et al. Role of Plasmodium vivax Duffy-binding protein 1 in invasion of Duffy-null Africans. Proc. Natl. Acad. Sci. USA 113, 6271–6276 (2016).
Hostetler, J. B. et al. Independent Origin and Global Distribution of Distinct Plasmodium vivax Duffy Binding Protein Gene Duplications. PLoS Negl. Trop. Dis. 10, e0005091 (2016).
Hester, J. et al. De novo assembly of a field isolate genome reveals novel Plasmodium vivax erythrocyte invasion genes. PLoS Negl. Trop. Dis. 7, e2569 (2013).
Ntumngia, F. B. et al. A novel erythrocyte binding protein of Plasmodium vivax suggests an alternate invasion pathway into Duffy-positive reticulocytes. mBio 7, e01261-16 (2016).
Gruszczyk, J. et al. Transferrin receptor 1 is a reticulocyte-specific receptor for Plasmodium vivax. Science 359, 48–55 (2018).
Malleret, B. et al. Plasmodium vivax binds host CD98hc (SLC3A2) to enter immature red blood cells. Nat. Microbiol. 6, 991–999 (2021).
MalariaGen et al. An open dataset of Plasmodium vivax genome variation in 1895 worldwide samples. Wellcome Open Res. 7, 136 (2022).
Auburn, S. et al. Characterization of within-host Plasmodium falciparum diversity using next-generation sequence data. PLoS ONE 7, e32891 (2012).
Benavente, E. D. et al. Distinctive genetic structure and selection patterns in Plasmodium vivax from South Asia and East Africa. Nat. Commun. 12, 3160 (2021).
Hupalo, D. N. et al. Population genomics studies identify signatures of global dispersal and drug resistance in Plasmodium vivax. Nat. Genet. 48, 953–958 (2016).
Pearson, R. D. et al. Genomic analysis of local variation and recent evolution in Plasmodium vivax. Nat. Genet. 48, 959–964 (2016).
Siegel, S. V. et al. Lineage-informative microhaplotypes for recurrence classification and spatio-temporal surveillance of Plasmodium vivax malaria parasites. Nat. Commun. 15, 6757 (2024).
Goodman, S. M. & Benstead, J. P. (eds) The Natural History of Madagascar (University of Chicago Press, 2003).
Pierron, D. et al. Genomic landscape of human diversity across Madagascar. Proc. Natl. Acad. Sci. USA 114, E6498–E6506 (2017).
Kappe, S. et al. Conservation of a gliding motility and cell invasion machinery in apicomplexan parasites. J. Cell Biol. 147, 937–944 (1999).
Sultan, A. A. et al. TRAP is necessary for gliding motility and infectivity of plasmodium sporozoites. Cell 90, 511–522 (1997).
Bouyssou, I. et al. Plasmodium vivax blood stage invasion pathways: contribution of omics technologies in deciphering molecular and cellular mechanisms. C. R. Biol. 345, 91–133 (2022).
Dechavanne, C. et al. Duffy antigen is expressed during erythropoiesis in Duffy-negative individuals. Cell Host Microbe 31, 2093–2106.e2097 (2023).
Cowell, A. N. et al. Selective whole-genome amplification is a robust method that enables scalable whole-genome sequencing of Plasmodium vivax from unprocessed clinical samples. mBio 8, e02257-16 (2017).
Venkatesan, M. et al. Using CF11 cellulose columns to inexpensively and effectively remove human DNA from Plasmodium falciparum-infected whole blood samples. Malar. J. 11, 41 (2012).
Canier, L. et al. An innovative tool for moving malaria PCR detection of parasite reservoir into the field. Malar. J. 12, 405 (2013).
Auburn, S. et al. Genomic analysis of Plasmodium vivax in southern Ethiopia reveals selective pressures in multiple parasite mechanisms. J. Infect. Dis. 220, 1738–1749 (2019).
Van der Auwera G. & O’Connor B. D. Genomics in the Cloud (O’Reilly Media, Inc., 2020).
Raj, A., Stephens, M. & Pritchard, J. K. fastSTRUCTURE: variational inference of population structure in large SNP data sets. Genetics 197, 573–589 (2014).
Ménard, D. et al. Custom R scripts and SLURM workflows for analysis of PRJEB81739 Illumina WGS data. Zenodo. https://doi.org/10.5281/zenodo.15391778 (2025).
Acknowledgements
We thank all patients who contributed blood samples, their guardians, and all team members in the respective health centers of the participating countries. We acknowledge the help of the HPC Core Facility of the Institut Pasteur for this work as well as the Biomics Platform, C2RT, Institut Pasteur, Paris, France (supported by France Génomique, ANR-10-INBS-09, and IBISA). This study was supported by grants overseen by the Human Heredity and Health in Africa (H3Africa, grant H3A/18/002), the French National Research Agency (VIPeRs, ANR-18-CE15-0026, VICTOR ANR- ANR-21-CE35-0006 and PvINV ANR-21-CE15-0013), the Institut Pasteur, Paris, the Laboratoire d’Excellence (LabEx) ‘French Parasitology Alliance for Health Care’ (ANR-11-15 LABX-0024-PARAFRAP). IB received financial support from the French National Research Agency (VIPeRs, ANR-18-CE15-0026), the FRM (Fondation pour la Recherche Médicale, FRM FDT202204014872), and the Fondation Thérèse Lebrasseur (DM was the winner of the Fondation Thérèse Lebrasseur award in 2021).
Author information
Authors and Affiliations
Contributions
I.B., P.C., F.L., C.C., and D.M. conceived and directed the project. M.S., I.V.-W., A.R., L.G., A.O.M.S.B., S.H., and M.F.F.C. organized the sample collection and processing. I.B. and L.M. performed the laboratory work, including sequencing. I.B. performed bioinformatic analysis under the supervision of P.C., I.B., P.C., and D.M. together interpreted the results. Additional advice from F.L. and C.C. was sought during the analysis. I.B. wrote the first draft of the manuscript under guidance from P.C. and D.M. All the authors commented on the version of the manuscript and approved the final manuscript. I.B., P.C., and D.M. compiled the final version of the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Communications Biology thanks Hai-Mo Shen and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. Primary Handling Editors: Johannes Stortz. [A peer review file is available].
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Bouyssou, I., Golassa, L., Vigan-Womas, I. et al. Genomic analysis of Plasmodium vivax field isolates circulating in sub-Saharan Africa. Commun Biol 8, 1012 (2025). https://doi.org/10.1038/s42003-025-08276-5
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s42003-025-08276-5
