
Abstract
Identifying accurate cell markers in single-cell RNA-seq data is crucial for understanding cellular diversity and function. Localized Marker Detector (LMD) is a novel tool to identify “localized genes”—genes exclusively expressed in groups of highly similar cells—thereby characterizing cellular diversity in a multi-resolution and fine-grained manner. LMD constructs a cell-cell affinity graph, diffuses the gene expression value across the cell graph, and assigns a score to each gene based on its diffusion dynamics. LMD’s candidate markers can be grouped into functional gene modules, which accurately reflect cell types, subtypes, and other sources of variation such as cell cycle status. We apply LMD to mouse bone marrow and hair follicle dermal condensate datasets, where it facilitates cross-sample comparisons by identifying shared and sample-specific gene signatures and novel cell populations, without requiring batch effect correction or integration. We also assess the performance of LMD across ten single-cell RNA sequencing datasets, compare it to eight existing methods with similar objectives, and find that LMD outperforms the other methods evaluated.
Similar content being viewed by others
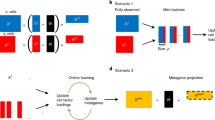
Introduction
Single-cell RNA sequencing can currently reveal the transcriptomes of 103−107 cells simultaneously1. These experimental approaches have enabled a better understanding of heterogeneity, cellular specialization and differentiation, and also provide insights into cell-cell signaling, spatial organization, and temporal dynamics leading to processes such as senescence, apoptosis, or oncogenesis2. An important task in single-cell data analysis is the identification of genes with high specificity to certain cell populations, which may indicate distinct cellular identities or states in this biological system.
Most marker identification methods3,4,5,6,7,8,9,10,11 first cluster cells based on their similarity in the transcriptomic space and then detect genes through differential expression analysis or by evaluating how well a gene’s expression pattern aligns with a given cluster. This approach has been criticized for its unreliable cluster assignments and inability to detect new cell subtypes defined by increased expression of specific genes12,13,14. Several state-of-the-art tools do not rely on an initial clustering step. For instance, singlecellHaystack12 and SEMITONES14 utilize reference cells or grid points in the transcriptomic space to approximate cell geometry. They assess if a gene’s expression aligns with the arrangement of cells within this coordinate system. One limitation of singlecellHaystack and SEMITONES is that they are confined to a specific range of length scales and may therefore fail to detect features at other length scales. SCMarker15 identifies genes with bi- or multi-modal expression patterns that are co-expressed or mutually exclusive with other genes. However, its framework focuses solely on expression patterns without incorporating cell-cell similarity, which results in selecting genes that lack specificity to distinct subsets of transcriptionally similar cells. MarcoPolo13 extends this idea by incorporating cell geometry: in addition to evaluating bimodality and co-expression, it tests whether the expressing cells are proximal in a low-dimensional space. This is done by binarizing the expression level of a gene and measuring the average distance of the expressing cells to their collective centroid. However, the binarization procedure can lead to a loss of quantitative information. Additionally, the positioning of the cell centroid and proximity assessments can be sensitive to outliers. Hotspot16 employs a cell-cell similarity graph to identify informative genes with non-random expression patterns, using a modified Geary’s C. This ‘non-randomness’ is a general term that can include very different patterns: a gene being highly expressed in a certain region of the cell graph or showing a smooth expression gradient across the cell manifold. Although each pattern might suggest different biological interpretations, Hotspot does not provide an automated way to distinguish between these scenarios. This limitation, in turn, means it doesn’t impose constraints to ensure that the genes it reports are exclusively expressed in tight regions comprising highly similar cells. Consequently, the lack of specificity in some of the genes identified by Hotspot limits its effectiveness for clear and definitive cell type classification.
To identify the informative genes that categorize distinct cell subtypes or cellular states, we require that such a gene exhibit a “localized expression pattern” in the cell graph—the graph that represents cell-cell transcriptomic similarities. This is based on the assumption that cells of the same type and state will exhibit highly similar transcriptomic profiles, thereby aggregating in certain neighborhood(s) of the cell graph. To accomplish this goal, we propose a novel algorithm, Localized Marker Detector (LMD), designed to identify all localized gene markers at multi-scale resolutions. Specifically, LMD finds marker genes by (i) constructing a cell-cell affinity graph, (ii) diffusing the gene expression value across the cell graph, and (iii) assigning a score to each gene based on the dynamics of its diffusion process (Fig. 1A). Intuitively, the diffusion process takes longer to spread across the entire cell-cell graph when a gene is active in cells that are closer to each other on the cell-cell graph, or when the cells expressing the gene are a subset of a region that is less connected to the rest of the graph. The LMD’s markers can be used for various downstream applications. For instance, identifying gene modules based on co-localized genes can improve the characterization of cell groups by highlighting genes associated with specific cell types and states, providing deeper insights into their functions. Additionally, our approach allows for cross-sample comparisons to identify sample-specific or universally shared cell populations without the need for batch effect correction or integration methods, which often obscure important biological effects and lack theoretical justification (Fig. 1A).

A Schematic illustration of the workflow of LMD (B) Construction of the cell kNN (k = 5) graph. C Gene diffusion patterns (color-coded by density, computed as the initial or diffused state in each cell) for a localized gene Igll1 (red) and a non-localized gene Tnks1bp1 (cyan) overlaid on the t-SNE cell embedding at different time scales. D Normalized KL-divergence pattern of Igll1 and Tnks1bp1 during the diffusion process, with normalized KL-divergence against the time scale. Time scales in (C) are highlighted in red along the x-axis text labels in (D).
In this work, we benchmark LMD against several state-of-the-art marker identification algorithms on ten real single-cell datasets that span a range of experimental protocols, sample sizes, and biological systems (Supplementary Data 2). Our results show that LMD effectively prioritizes genes that are exclusively expressed in specific cell subpopulations, accurately identifies known markers, and enhances the separation of cell groups with its top candidate markers. We then apply LMD to a mouse bone marrow dataset, grouping markers into distinct functional gene modules based on their co-localization in the cell graph; these modules delineate different cell states at multiple scales. Additionally, we used a mouse embryonic skin dataset to show that gene modules generated from LMD’s markers can be used to compare different conditions of the same system, identifying both condition-specific and universally shared cell populations and their associated genes. Our results highlight LMD’s ability to identify informative markers that enhance various downstream tasks, particularly the identification and characterization of functional cell groups.
Results
Overview of the LMD method
Here, we outline the main three steps of LMD using a single cell RNA-seq mouse bone marrow dataset with FACS-based full length transcript technique from the Tabula Muris consortium17,18 as an example (Fig. 1 and Supplementary Fig. 1A).
Cell graph construction
The first step of LMD involves computing a cell-cell affinity graph that shows which cells are sufficiently similar to each other based on a chosen similarity metric. We construct a k-nearest neighbor (kNN) graph where each cell is represented by a node, and two nodes are connected by an edge if either of their respective transcriptomes is among the closest k neighbors of the other according to a given metric. The metric can be any function that quantifies the similarity of cells, such as the Euclidean distance between their transcription profiles after reducing the dimensionality with Principal Component Analysis (PCA). The graph is undirected and unweighted, i.e., the connections are symmetric and all edges have equal weight. In our experimental setting, we ensure that the graph is connected by linking disconnected components via a minimum spanning tree. Fig. 1B shows a cell kNN (k = 5) graph constructed from the Euclidean distances between the transcription profiles after PCA.
The unweighted graph is not a strict requirement for LMD; it is primarily used to promote computational efficiency. LMD is also robust to alternative weighted kNN-like graph constructions, including shared nearest neighbor (SNN) graphs and α-decaying kernels with adaptive bandwidth (see details in Methods-‘Robustness evaluation’, Supplementary Fig. 9).
Multi-scale diffusion process
In this step, we examine the gene expression pattern over the cell graph. We first normalize the expression of a gene into a probability distribution over cells, which we define as the initial state vector of the corresponding gene. Next, we apply a diffusion process with a series of time scales—a technique widely adopted in various fields19,20,21—and track how this state vector changes as a function of time. If the graph is connected, given enough time, all nodes will reach a non-zero value regardless of initial states, and the equilibrium state vector for our setup is a uniform distribution.
Some initial states are localized, meaning that most nodes in the graph have zero values except for the nodes in a specific subgraph (Fig. 1C, top panel). We observe that for a more localized initial state, it takes a longer time to reach a non-zero value at every node and then reach the equilibrium state (Fig. 1D). To quantify this effect, we measure changes in the state vector as a function of time by computing the Kullback-Leibler divergence (KL-divergence) between the state at time t and the initial state at time t0. To adjust for the effect where initial states distributed across different numbers of nodes result in varying convergent values, we normalize this time-dependent divergence by the KL-divergence between the initial state and the equilibrium state, and term this quantity as normalized KL-divergence. As expected, we observe that the normalized KL-divergence for more localized initial states increases at a slower rate during the early stages of the diffusion process, indicating that the redistribution of values primarily occurs between nodes with similar values. In contrast, for initial states that are not localized, the diffusion process quickly redistributes values from higher-value nodes to neighboring lower-value nodes at the beginning. This results in a rapid increase in normalized KL-divergence.
The LMD-score (LMDS)
To summarize the time-dependent profile of the normalized KL-divergence, we calculate the area under the curve (Fig. 1D), which we term as the LMD-score (LMDS). A lower LMDS indicates a more localized initial state. In the context of gene expression profiles, we find that genes expressed in specific regions of the cell graph are assigned a lower LMDS, implying LMDS can distinguish localized genes from the genes non-specific to any local regions of the cell graph. This is the main idea of LMD.
LMD prioritizes localized markers
To better understand the difference between LMD and the other state-of-the-art marker gene identification methods – standard DEG workflow based on clustering and Wilcoxon rank sum test from Seurat v43, COSG11, Hotspot16, SEMITONES14, Marcopolo13, singleCellHaystack12, and SCMarker15—we compared their gene rankings on the Tabula Muris bone marrow FACS dataset18. Among the methods that provide ranked outputs, Seurat v4 showed the highest concordance with LMD, while singleCellHaystack had the lowest (Fig. 2A). SCMarker exhibited low correlation, as it returns only a binary classification of marker genes rather than a ranked list. Candidate markers selected by LMD were typically expressed in one or several compact cell neighborhoods, whereas other methods either prioritized markers lacking locality or overlooked markers expressed in a subset of a cluster (Fig. 2B). In detail, Seurat v4 identifies genes through differential expression and Hotspot prioritizes genes with global variation, thus both methods may overlook the confinement of markers to specific cell groups where their candidate genes can be expressed at high levels in a cluster but also at lower levels in dispersed cells outside the cluster; COSG introduces this confinement by measuring the cosine similarity between gene expression and a given cell cluster, favoring genes that are tightly aligned with that cluster, but this cluster-dependency limits its ability to identify genes jointly expressed across multiple cell subsets or within groups of cells that are not well captured by the clustering; the markers identified by SCMarker or Marcopolo often exhibit noisy and less localized expression patterns; SEMITONES and SingleCellHaystack missed markers expressed in cell subsets smaller than their pre-defined fixed length scale. The same ranking comparison analysis performed on the Azimuth 10X human kidney demo dataset22 revealed similar trends, with LMD showing the highest concordance with Seurat v4 and the lowest with singleCellHaystack, and gene expression patterns reflecting the same method-specific limitations discussed above (Supplementary Fig. 2).

A Comparison of gene rankings by LMD against Seurat v4, COSG, Hotspot, Marcopolo, SCMarker, SEMITONES, and SingleCellHaystack, with concordance measured using the Pearson correlation coefficient of log10-transformed rankings. Highlighted genes fall within LMD's top 100 ranked genes but are outside the top 500 ranked genes of the other method (red), and vice versa (blue). B Top localized genes selected by LMD but not highly ranked by other methods and vice versa. The rankings of these genes across all methods can be found in Supplementary Data 1.
LMD identifies known markers with high accuracy
We evaluated LMD on its ability to identify known markers using ten real scRNA-seq datasets: four datasets from the Tabula Muris consortium17,18 and six datasets from the Azimuth demo datasets22. Detailed biological systems, experimental protocols, and sample sizes are provided in Supplementary Data 2. We compared LMD performance to a suite of marker gene identification algorithms, including singleCellHaystack, Hotspot, SEMITONES, MarcoPolo, SCMarker, Seurat v4, Highly Variable Gene (HVG) implemented in Seurat v4 with VST option and COSG. To validate performance, we used two distinct true marker sets as suggested in13. The first set was derived from marker databases, and the second set consisted of the top 100 genes with the largest expression fold change across cell types (see Methods-‘Gold-standard cell-type marker sets’). We measured the AUROC of the methods for each dataset and found that LMD outperforms all other methods in most cases (Fig. 3). For the first true marker set, LMD ranked first in 5 out of 10 datasets, and for the second true marker set, it also ranked first in 5 out of 10 datasets. Overall, LMD achieved the best performance for both true marker sets based on the median rank across all datasets.

Each block represents the AUROC score for a method on a dataset, with the number showing the AUROC value and the color indicating the method’s rank. Marcopolo (CPU) was only run on the Tabula Muris dataset due to runtime and hardware restrictions. Two true marker sets: 1 genes listed in marker databases (CellMarkerDB and Azimuth marker list); 2 the top 100 genes with the maximum log fold change across pre-annotated cell types.
We also compared the ability of markers from each method to enhance separability between cell groups on the Tabula Muris bone marrow FACS dataset using the density index23. The density index measures ‘clumpiness’ in cell distribution by comparing the average closeness of nearest neighbors to the average distance between random cell pairs. A higher value indicates a more compact and clumped cell distribution. LMD achieved a higher density index than other methods (Supplementary Fig. 3A). The density index for LMD peaked around the top 300 genes, suggesting that even a small number of LMD genes is informative for separating diverse cell populations. Furthermore, when focusing on the granulocyte subset of this dataset, LMD still maintained a higher density index, demonstrating its ability to identify informative markers that help distinguish cell subpopulations, even in more homogeneous cases (Supplementary Fig. 3B).
LMD also demonstrated reasonable runtime performance (details in Methods-‘Scalability’, Supplementary Data 3). For example, it processed 1564 cells in ~30 s and 30,651 cells in about 44 min. For large datasets, we adapted a cell graph coarse-graining strategy24 to improve computational efficiency (also described in Methods-‘Scalability’).
LMD markers unveil functional modules and cell states across varied length scales in mouse bone marrow
Gene modules
We demonstrate the flexibility of LMD markers in characterizing functional cell groups using the Tabula Muris bone marrow FACS dataset, ranging from broad gene expression across multiple regions to fine-grained expression within a single cluster. First, we selected the top 1741 LMD candidate markers based on the knee point of the gene LMDS curve. To characterize various expression patterns, we grouped these markers into 34 distinct gene modules (Methods-‘Gene modules’, Supplementary Fig. 5A, B): of these, 20 were enriched for at least one GO molecular function term (Benjamini-Hochberg adjusted p-value < 0.05), and 24 were enriched for at least one pathway from the Reactome Pathway Database25 (Benjamini-Hochberg adjusted p-value < 0.05). For detailed information on each module, see Supplementary Data 4. As expected, given the locality of the underlying markers, each module was associated with specific regions of the cell-cell affinity graph, which enables the characterization of the data at different length scales (Supplementary Fig. 5C). At a broader length scale, we found several modules that were jointly expressed across multiple local regions, for instance, modules associated with progression through the cell cycle. At cell-type resolution, LMD identified several modules that aligned well with known annotations. Finally, at a finer length scale, LMD revealed several gene modules, each associated with a specific subset in a pre-annotated cell type, suggesting the potential of using LMD to define novel cell types at a high resolution. Moreover, inspecting certain gene modules may elucidate the different cell states along a dynamic biological process (e.g., B lymphocyte differentiation) and facilitate the discovery of novel cell subtypes in this system.
Broad length-scale, larger data structures
LMD identified three gene modules significantly enriched for cell cycle phases based on Reactome pathway analysis. Specifically, modules 22, 20, and 19 were significantly enriched for the S phase, S/M phase, and M phase, respectively (Benjamini-Hochberg adjusted p-value < 0.05, Fig. 4A). We confirmed this result using an alternative Tabula Muris bone marrow dataset with microfluidic droplet-based 3′-end counting technique17 (Supplementary Fig. 1B). In both datasets (bone marrow FACS and droplet), the depiction of these gene modules on the cell t-SNE embedding closely matches cell annotations derived from Seurat cell cycle scores, which rely on established cell cycle markers (Fig. 4B). The modules derived from LMD show incremental activation of markers, revealing the dynamics of cell-cycle progression (Fig. 4B and Supplementary Fig. 7).

A, B Gene modules associated with the cell cycle. Gene modules were identified from the Tabula Muris bone marrow FACS dataset, and their function was classified by Reactome pathway analysis. Modules 22, 20, and 19 were associated with S phase, S/G2/M and G2/M phases, respectively. A Bubble plot of the top 5 significantly enriched Reactome pathways (Benjamini-Hochberg adjusted p-value < 0.05) for each module. Bubble color indicates -log10-adjusted p-value, and the size represents the fraction of module genes associated with the pathway. B Module scores in two Tabula Muris bone marrow datasets, and reference cell-cycle annotation from Seurat (left). C Gene modules associated with B cells at different stages of maturation. Module 7 is associated with pro-B cells, module 6 with pre-pro-B cell, module 2 with immature and mature B cells, and module 3 with naive B cells. D Gene modules associated with the production of granulocytes. Module 8 contains genes expressed during granulocytopoiesis, module 15 contains genes common for granulocytopoietic cells and granulocytes, module 11 contains genes involved in granulocyte maturation, and module 10 may represent a rare granulocyte subgroup.
Cell-type resolution
LMD identified several modules that demonstrated specificity to pre-annotated cell types. The strength of this association was quantified using the Jaccard Index (see Methods-‘Cell type-specific activated modules’). Due to limited cell numbers, we merged T cell subtypes into “T cell” and NK cell subtypes into “NK cell”. Of the resulting 18 cell types, 11 contained at least one distinctly activated module (Supplementary Fig. 6A, B). The relationship between these modules and cell types was further validated as the modules incorporated canonical cell type markers used for annotating this Tabula Muris bone marrow dataset (see figure notes of Supplementary Fig. 6). We performed cross-dataset verification on the droplet dataset and found that modules identified as specifically activated in particular cell types in the FACS dataset also showed high specificity for the same cell types in the droplet dataset (Supplementary Fig. 6C).
High resolution, beyond cell types
LMD identified gene modules that trace the B lymphocyte differentiation process (Fig. 4C) or various developmental stages and subtypes of granulocytes (Fig. 4D). Modules associated with B lymphocytes are substantiated by the inclusion of genes with well-established roles in this differentiation process: module 7 (Lef1 and Enpep (also known as BP-1)) distinguish pro-B cells26,27; module 6 (Rag2 and Il7r) represents pre-pro-B cell stage28,29; module 2 (Ms4a1 (also known as Cd20) and Cd22) pertains to B cell activation and regulation, marking both immature and mature B cells in the bone marrow30,31; module 3 (Fcer2a (also known as Cd23) and Cxcr5) characterizes naive B cells32,33. We similarly identified at least 4 modules associated with granulopoiesis: module 8, exclusively expressed in granulopoietic cells, including Ms4a3, a marker gene used to label bone marrow granulocyte-monocyte progenitors (GMP) and GMP progenies34; module 15, featuring secondary granules genes Camp and Ltf, expressed in both granulocyte and granulopoietic cells35; module 11, specific to a subset of granulocytes and containing Retnlg and Cxcr2, which suggests a potential role in neutrophil mature and release36,37; lastly, module 10, characterized by genes implicated in pyroptosis and antigen processing such as Stfa2l1, Il1b, Gm5483, and H2-Q1038,39,40, which may represent a rare granulocyte subgroup engaged in the immune response against intracellular pathogens.
LMD characterizes shared and mutant-specific cell populations in mouse dermal condensate genesis
Mouse hair follicle dermal condensates (DCs) appear in the dermis of the skin around embryonic day 14.5 (E14.5) and play a critical role in hair follicle development41. The differentiation of DC cells is influenced by the cooperative actions of Wnt/β-catenin and sonic hedgehog (SHH) signaling pathways. In wild type (WT) mice, Wnt signaling is activated prior to E14.5 as a gradient spanning the upper dermis, while SHH is expressed by hair follicle epithelial cells around E14.5, inducing SHH activation in the Wnt-active dermal cells beneath the hair follicle epithelium. Together, they regulate the emergence and differentiation of dermal condensates41,42,43. The SmoM2YFP mutant (SmoM2) induces uniformly high SHH activation across the dermis, resulting in the early appearance of DC-like structures at E13.541.
We collected mouse skin samples from E13.5 and E14.5 WT and paired SmoM2 mutant embryos for 10X scRNA-seq. We used LMD to examine the functional cell groups that are conserved between SmoM2 and WT, as well as those that reflect SmoM2-specific changes. We focused on the E13.5 SmoM2 sample and identified 17 gene modules (Supplementary Fig. 17, Supplementary Data 5). Among them, we identified modules that: (1) reveal consistent fine-grained cell cycle phases across SmoM2 and WT (Fig. 5A), (2) reflect different cell stages in DC genesis of SmoM2 mutant (Fig. 5B, C), and (3) a novel module that highlights a special cell subset with a consistent transcriptomic profile in both E13.5 and E14.5 mutants but is absent in the WT condition (Fig. 6A and Supplementary Fig. 23).

A Module scores of three cell cycle-associated gene modules on E13.5 SmoM2 and paired WT, with cell cycle annotations obtained from Seurat for reference (left). Different UMAP embeddings: by top 2000 HVGs of E13.5 SmoM2 (first row), top 2000 HVGs of E13.5 WT (second row), and merged embeddings of E13.5 SmoM2 and E13.5 WT by Seurat cell cycle genes (third row: SmoM2; fourth row: WT). B Module scores of three Wnt-associated gene modules on E13.5 SmoM2 (first row), paired E13.5 WT (second row) and E14.5 WT (third row). C Left: Cells categorized by three Wnt-associated gene modules (module score > 0.5, mutually exclusive: cells assigned to innermost module). Right: Lef1 (Wnt) level in three modules: The box represents the interquartile range (IQR), with the line inside the box indicating the median. Whiskers extend to a maximum of 1.5 × IQR beyond the box, with outliers represented as individual points. A Wilcoxon test was performed between adjacent boxes, with “NS” for p > 0.05 and “***” for p < 0.001.

A Module scores of module 12 on E13.5 SmoM2 (first row, columns 2 and 4) and paired WT (first row, column 5), and E14.5 SmoM2 (second row, columns 2 and 4) and paired WT (second row, column 5), with cell cycle annotations from Seurat (columns 1 and 3). Activated cells (module score > 0.5) in E13.5 SmoM2: 114 G1, 34 G2M, 9 S cells; in E14.5 SmoM2: 44 G1, 16 G2M, 4 S cells. UMAP embeddings: standard scaling (columns 1, 2 and 5) and scaling regressing out cell cycle scores (columns 3 and 4). B FISH images (scale bar = 50 μm) showing the spatial distribution of Trp53inp1 in the upper dermis of SmoM2 pulsed with EdU at E14.5 (first row) and 24 h prior at E13.5 (second row). EdU is a nucleotide that is incorporated by cells in the S phase. C %EdU+ of Trp53inp1+ cells at E13.5 and E14.5, pulsed with EdU (EdU 1hr) and 24 h prior (EdU 24hr). Data as mean ± SEM (n = 3), one-way ANOVA with Tukey’s HSD test was performed between groups, with “NS” for p > 0.05, “*” for p < 0.05, “**” for p < 0.01.
LMD reveals functional similar cell populations in mutant and wild-type samples
We identified three distinct gene modules that capture fine-grained phases along the cell cycle procession: module 1 is characterized by genes typical of the S to G2/M transition, including Hist1h2ap, Hist2h2bb, Hist1h2ae44; module 3 exhibits genes expressed in the G2/M phase, such as Cenpf, Cenpe and Cdk145,46,47; while module 2 is enriched for genes prevalent during late G2 and M phases, such as Nek2, Kif14, and Kif18b46,48 (Fig. 5A, first row). We also observed the localized pattern of these three modules in the paired E13.5 WT sample (Fig. 5A, second row). To investigate the functional connections of these localized patterns between SmoM2 and WT, we merged two samples and re-embedded cells by cell cycle marker genes (Methods-‘Cell cycle annotation, embedding, and regression’). Notably, despite the drastic transcriptome-wide differences between SmoM2 and WT, the new embedding generated using cell cycle marker genes aligns well between the two conditions (Supplementary Fig. 16). In this new embedding, we noted that cells highlighted by these modules demonstrated good overlap between SmoM2 and WT (Fig. 5A, third and fourth rows), indicating the presence of these cell cycle phases across both samples.
The functional connections of localized patterns between two samples can also be validated by independent gene modules in each sample that share similar functions. Specifically, we apply LMD to each sample separately to identify gene modules (see detailed information in Supplementary Figs. 17–20 and Supplementary Data 5). To pair gene modules between samples, we project the gene modules from one sample onto the other and match them based on their co-localization (Methods-‘Measuring co-localization between two gene modules’). We then inspect whether the paired gene modules share similar biological functions through pathway analysis. For the three CC-associated modules identified from SmoM2, we projected them onto the WT sample and consistently found that each module co-localized with a corresponding module identified from the WT, with the minimum Jaccard index > 0.8 (Supplementary Fig. 21A). Additionally, these paired modules are significantly enriched in common cell cycle-related pathways from the Molecular Signatures Database (MsigDB)49 (Supplementary Fig. 21B).
With the aforementioned points, LMD identifies conserved biological functions across different samples by projecting gene module expression patterns from one sample onto another. This approach detects localized patterns and corresponding cell subsets in the second sample without requiring cell integration or alignment between samples, eliminating reliance on “batch correction” methods that often fail to differentiate between batch effects and true biological variation.
LMD captures different cell states in the DC genesis mutant model
Previous work showed that higher levels of Wnt signaling correlate with increased DC formation in response to high SHH activation41. We identified three modules in E13.5 SmoM2–modules 14, 16, 15–that represent cell subsets with incremental activation of genes within the Wnt signaling gradient (Fig. 5B, C). Module 14, which includes Lef1, a direct downstream target of Wnt/b-catenin signaling50,51, captures the broadest group of cells engaged in Wnt signaling. This is corroborated by its co-localization with two other modules from the E13.5 WT and E14.5 WT samples, with all three modules significantly enriched for the Wnt beta-catenin signaling pathway (Supplementary Fig. 21). Module 15, containing the DC marker Sox252, captures the narrowest subset of cells representing DCs. It co-localizes with module 1 from the E14.5 WT sample, which also contains Sox2 (Supplementary Fig. 21, Supplementary Data 5). However, the localized pattern is absent in the E13.5 WT sample (Fig. 5B). This is consistent with the biological observation that early DC-like structures begin to form in the SmoM2 mutant at around E13.5, while DC emergence in WT happens around E14.5. Module 16 represents an intermediate stage in this activation process; this module is expressed by some Sox2-negative cells, but these cells express all the genes from the Wnt module (module 14). Similar to the DC module (module 15), module 16 co-localizes with module 1 from the E14.5 WT sample, while the localized pattern is absent in the E13.5 WT sample (Fig. 5B). RNA fluorescent in situ hybridization (FISH) validated this observation, showing that Gal, a marker featured in this module, is present in the upper dermis region in E13.5 SmoM2 but not in the paired WT, while Gal-positive cells are concentrated in the DC in E14.5 WT (Supplementary Fig. 22A, C). We also noticed that the Wnt level (represented by Lef1 expression levels) of this group of cells is comparable to that of DC cells, which are significantly higher than other cells involved in Wnt signaling (Fig. 5C, Supplementary Fig. 22B, D). This suggests that module 16 may highlight cells committed to transitioning to DC states, potentially induced by high Wnt and SHH levels.
Using this example, we demonstrate the ability of LMD to capture gene modules that depict different cell states within a biological process that is represented by incremental activation of genes.
LMD detects a novel mutant-specific cell state
We identified a gene module from E13.5 SmoM2, module 12, showing a localized pattern in both E13.5 and E14.5 SmoM2, which is absent in their paired WT samples (Fig. 6A). The functional connection between two localized patterns is further validated by the co-localization of module 12 with module 15 from E14.5 SmoM2 when projecting module 12 onto E14.5 SmoM2, with a Jaccard index of 0.872. They also share 3 of the top 5 significantly enriched pathways from MsigDB, including p53 pathway (Supplementary Fig. 21). Additionally, we validate this finding using FISH, demonstrating that Trp53inp1, a marker in both E13.5 SmoM2 module 12 and E14.5 SmoM2 module 15 (Supplementary Data 5), is present in the upper dermis region in E13.5 and E14.5 SmoM2 but expressed at a negligible level in the paired WT (Supplementary Fig. 22A, C).
We observed that module 12 delineates two distinct cell populations in E13.5 SmoM2: one predominantly in the G1 phase and the other in the G2M phase, with few cells in the S phase. When we regressed out the cell cycle score (see Methods-‘Cell cycle annotation, embedding, and regression’), these two populations merged, indicating that their separation was primarily driven by cell cycle differences (Fig. 6A, first row). A similar result is observed in E14.5 SmoM2, where module 12 also consists of two distinct cell populations (one in G1 and the other in G2M) that merge after regressing out the cell cycle score (Fig. 6A, second row). Given the phase composition and the significant enrichment of the p53 pathway in module 12 (Supplementary Fig. 21), we hypothesize that module 12 delineates a quiescent cell state with G1 cells being the immediate progeny of a cell division (G2M). We test this hypothesis using FISH and EdU pulse-chase analysis. We show that most E14.5 Trp53inp1-positive cells do not incorporate EdU nucleotide, indicating they are not in the S phase of the cell cycle and largely quiescent. However, when pulsed with EdU 24 h prior, we found that many precursors are labeled with EdU, suggesting that these cells are the progeny of proliferating cells (Fig. 6B). Additionally, the EdU percentage of Trp53inp1-positive cells is significantly higher when pulsed with EdU 24 h prior either at E12.5 (chase to E13.5) and E13.5 (chase to E14.5) (Fig. 6C), indicating that most Trp53inp1-positive cells are quiescent but represent immediate progeny of a cell division. Thus, module 12 represents a mutant-specific quiescent cell state that represents the transition from proliferation to quiescence, a process that is normally coupled to DC differentiation. This transition from proliferation to quiescence occurs continuously between embryonic day 13.5 and 14.5. Since this module is specific to SmoM2, it may suggest a regulatory mechanism of cell cycle exit influenced by SHH pathway.
In this instance, we show that LMD can capture gene modules delineating condition-specific cell subsets without needing sample integration or differential abundance testing, effectively avoiding the need to use “batch correction” methods that cannot truly separate batch effects from biological effects and thus are not reliable.
Discussion
We present LMD, a novel approach for identifying localized markers at multiple length scales in single-cell RNA sequencing data. The core idea of LMD is that the dynamics of a diffusion process for each gene on a given cell-cell affinity graph reflects certain properties of the gene expression pattern, such as locality. We have shown that expression patterns concentrated over a compact region of the cell-cell affinity graph yield a slower progression of diffusion at the beginning of this process. This property can be represented by LMD-score for each gene, and it enables the prioritization of localized expression patterns.
LMD consistently recovered known cell-type markers from several mouse and human scRNA-seq data, often outperforming other marker identification algorithms, which do not necessarily prioritize the localization of candidate markers. This success implies that many known markers are inherently localized, highlighting the necessity for algorithms like LMD that can distinguish between local and global expression profiles. Additionally, LMD’s top markers also enhance the separation of cells into distinct clumps. We further demonstrate the advantages of markers exhibiting localized patterns through two biological examples: LMD’s markers can identify compact groups of similar cells regardless of scale. Additionally, LMD’s markers can be used to match functionally similar cell populations between samples and detect condition-specific cell subsets without requiring sample integration, effectively avoiding the need to use “batch correction” methods that cannot truly separate batch effects from biological effects and thus are not reliable. LMD achieves these results without relying on existing cell annotations or gene functions. Its candidate markers are selected solely based on their localization within the cell-cell affinity graph.
In our second example, the mouse hair follicle dermal condensates system, we identified a previously uncharacterized potential intermediate state committed to DC, with Gal as a featured marker for this cell state. We also discovered a novel mutant-specific cell population located in two distinct regions within a single cluster in the UMAP visualization, with Trp53inp1 as a marker. We demonstrated that this separation is driven by cell cycle differences and confirmed the biological connection through FISH and EdU pulse-chase experiments. These results suggest that this population may represent a quiescent cell state, potentially regulated by the SHH pathway. This example highlights the advantage of identifying markers in a cluster-independent manner, as traditional methods based on clustering and differential expression are less effective in single-cluster cases and in capturing connections between different cell groups.
However, a limitation of LMD is its potential bias introduced by the cell graph. Genes expressed in well-connected neighborhoods diffuse faster than those in more isolated neighborhoods of the same size. Further improvement of LMD will focus on better balancing gene distribution on the cell graph with the graph’s geometry. Additionally, using gene expression data for both constructing the cell graph and ranking the genes may introduce a bias toward highly variable genes. This bias arises because the PC coordinates used in graph construction tend to preserve information depicting the large variability of the data.
In this study, we demonstrated the effectiveness of LMD with scRNA-seq data. We expect that LMD can be adapted to other single-cell techniques, such as scATAC-seq and spatial transcriptomics. Beyond data modality, LMD also holds potential for post-transcriptional regulatory analysis. As LMD gene modules reflect groups of co-localized genes, they may represent targets of shared regulatory mechanisms. By intersecting these modules with known or predicted miRNA or transcription factor target sets, one could gain insights into regulator activity patterns confined to subtle subpopulations or fine-grained cellular states.
Methods
Localized marker detector (LMD)
Input data
The input to LMD is a gene by cell log-normalized count matrix \(X\in {{\mathbb{R}}}^{m\times n}\), where G is the set of all genes (∣G∣ = m), C is the set of all cells (∣C∣ = n), and the element xi,j is the measured expression level of gene i in cell j.
Cell-cell affinity matrix P
First, we identify the k-nearest neighbors for each cell from the Euclidean distance between the transcription profiles after applying dimensional reduction, e.g. PCA (see Methods-‘Dataset and Processing’). Next, the adjacency matrix A is constructed such that the element ai,j is 1 if cell i is a k-nearest neighbor of cell j and 0 otherwise. If disconnected components are detected, we connect them by identifying the shortest inter-component edges and adding a set of minimal connections using Prim’s algorithm for constructing a minimum spanning tree53. Finally, the doubly-stochastic cell-cell affinity matrix P is obtained by applying the Sinkhorn-Knopp algorithm54 to the symmetrized matrix \({({A}_{sym})}_{ij}=max({a}_{ij},{a}_{ji})\).
Diffusion process
Given an initial state over the cells, propagate it over the nodes of the graph using a diffusion process. The initial state for gene i on the cell-cell affinity graph is the (n × 1) vector ρ(i, 0) whose elements are \({\rho }_{j}^{(i,0)}={x}_{i,j}/{\sum }_{j\in C}{x}_{i,j}\). The state of gene i at time t is estimated using a diffusion process: \({\rho }^{(i,t)}={({P}^{t})}^{{\prime} }{\rho }^{(i,0)}\). Note that for t → ∞, ρ(i, t) → 1/n ⋅ 1, ∀ i ∈ G.
LMD score
For a given gene i, the Kullback-Leibler divergence (KL-divergence) between the initial state and the state at time t is \(\delta (i,t)={D}_{KL}({\rho }^{(i,0)}| | {\rho }^{(i,t)})={\sum }_{j\in C}{\rho }_{j}^{(i,0)}\log (\,{\rho }_{j}^{(i,0)}/{\rho }_{j}^{(i,t)})\). The Normalized KL-divergence is computed as d(i, t) = δ(i, t)/δ(i, ∞). We define the LMD score as LMDS = ∑td(i, t). In LMD, we compute LMDS over t = 2τ, τ = 1…T for a T where the diagonal entries of \({P}^{{2}^{T}}\) converge to 1/n with a maximum relative error of 1%. We rank candidate genes by ascending values of LMDS.
Gene modules
Identification
To identify gene modules, we selected the top candidate genes based on their LMDS. Since gene “localization” is a continuous property, there is no universal cutoff to distinguish localized from non-localized genes. Instead, users are encouraged to define a threshold based on their specific analysis goals –for example, by selecting a fixed number of top-ranked genes. As a general recommendation, we suggest using the knee point of the gene LMD score curve, which reflects the transition from a rapid increase to a gradual rise. The knee point can be identified as the data point with the maximum perpendicular distance from the line connecting the first and last points55. For these selected genes, we computed the gene-gene pairwise distance matrix using the Jaccard distance56 between denoised gene expression profiles across all cells, with denoising performed by ALRA57. A small Jaccard distance suggests that these two genes are co-localized in the cell graph. We then adopted a widely accepted strategy for gene module identification58 where we processed the gene-gene pairwise distance matrix with hierarchical clustering with average linkage (hclust function from stats R package (v. 4.1.3)59) and we determined gene modules using the dynamic tree-cutting algorithm (cutreeDynamic function from dynamicTreeCut R package (v.1.63.1)60). We then removed outlier genes following this spectral filtering procedure61:
-
1.
compute the gene-gene Jaccard Index matrix;
-
2.
compute the first eigenvector and the associated eigenvalue λ1 of the Jaccard Index matrix;
-
3.
compute the absolute values of the entries in the first eigenvector of the Jaccard Index matrix;
-
4.
scale the absolute values by \(\sqrt{{\lambda }_{1}}\);
-
5.
discard genes corresponding to scaled entries smaller than 0.5.
Only modules containing at least 10 genes were retained for downstream analysis.
Module activity scores
The module activity score (or module score) represents the probability of a module being expressed in a given cell, ranging from 0 to 1. To estimate this score, we performed the following sampling procedure 100 times to ensure stability: 1. Randomly select half of the genes from the module without replacement. 2. Calculate the average expression of these genes in each cell and cluster the cells using GMM function from ClusterR R package (v. 1.3.1)62, with the optimal number of clusters estimated by Optimal_Clusters_GMM from ClusterR using the BIC criterion. 3. Assign the resulting clusters into two categories (0—not expressing the module; 1—expressing the module) by performing hierarchical clustering on the cluster centroids with average linkage (hclust function from stats R package), then cutting the tree into two clusters (cutree function from stats R package). Finally, determine the module activity score for each cell as the proportion of times it was assigned to category 1 over 100 iterations.
Cell type-specific activated modules
Cell type-specific activated modules are those for which the Jaccard index between the module score and the one-hot vector representation of a given cell type exceeds 0.4.
Measuring co-localization between two gene modules
The degree of co-localization between two gene modules is quantified using the Jaccard index, which compares their module score vectors. A higher Jaccard index indicates greater co-localization between the two modules.
Experiments and analyses
Dataset and processing
Tabula Muris
We extracted publicly available scRNA-seq mouse bone marrow (FACS and droplet-based), lung (FACS-based), and pancreas (FACS-based) datasets (see Data availability). For LMD input, we constructed a cell-cell kNN (k = 5) graph using the top 20 PCs coordinates provided in the original datasets. Genes expressed in more than 10 cells but fewer than 50% of cells were included for analysis. The raw count matrix was log-normalized using the Seurat v4 R package3. For gene module identification in the Tabula Muris bone marrow FACS dataset, we used the cutreeDynamic function with the parameters minClusterSize = 10 and deepSplit = 1. For visualization, we use t-SNE coordinates provided in the original datasets. For benchmark methods (see Methods-‘Gene Ranking by Competing Method’), we used the same data, scaled or transformed as required by each method’s input specifics, e.g. either raw count matrix, log-normalized count matrix, or PC coordinates.
Azimuth demo dataset
We extracted publicly available scRNA-seq datasets provided by Azimuth3 and annotated them using the Azimuth app at a reasonable resolution level (10~50 cell types). The datasets include: human motor cortex (annotated by subclass), mouse motor cortex (subclass), human pancreas (annotation.l1), human kidney (annotation.l2), human bone marrow (celltype.l2), and human lung v2 (ann_level_3) (see Data availability). We removed cell types containing fewer than 10 cells. Following standard normalization, selection of the top 2, 000 highly variable genes, and scaling with the Seurat v4 R package, we performed PCA and retained the top 20 PCs. For LMD input, we constructed a cell-cell kNN (k = 5) graph using the top 20 PCs coordinates. Genes expressed in more than 10 cells but fewer than 50% of cells were included for analysis. The raw count matrix was log-normalized using the Seurat v4 R package. For benchmark methods, we used the same data, scaled or transformed as required by each method’s input specifics, e.g. either raw count matrix, log-normalized count data, or PC coordinates.
Mouse embryo skin data
We separated dermal cell populations from newly collected mouse embryo skin samples (see Methods-’Experimental details of mouse embryo skin sample preparation’; aligned to the mouse genome mm10 using CellRanger (v.5.0.1)). To avoid batch effects from pooling or integrating, we analyzed each condition separately: E13.5 SmoM2, E13.5 WT, E14.5 SmoM2, and E14.5 WT. For each condition, quality control was performed by removing cells with fewer than 500 or more than 5,000 detected genes, or with a mitochondrial ratio exceeding 10%. Dermal cells were extracted and retained using markers Dkk1, Dkk2, Lef1 and Sox2. In mutant samples, dermal cells were further extracted using the marker eYFP to exclude cells resembling the wildtype phenotype. Then we performed standard normalization, selected the top 2,000 highly variable genes, and scaled the data using the Seurat v4 R package. We then applied PCA, retaining the number of PCs determined by the elbow plot63: E13.5 SmoM2 (14 PCs), E13.5 WT (12 PCs), E14.5 SmoM2 (12 PCs), and E14.5 WT (11 PCs). For LMD input in each condition, we constructed a cell-cell kNN (k=5) graph using the retained PC coordinates. Genes expressed in more than 10 cells but fewer than 50% of cells were included for analysis. The count data was log-normalized using the Seurat v4 R package. For gene module identification, we used cutreeDynamic with minClusterSize=10 and deepSplit=2 for E13.5 SmoM2, and deepSplit=1 for E13.5 WT, E14.5 SmoM2, and E14.5 WT. For visualization, we used UMAP coordinates generated from the retained PC coordinates.
Gold-standard cell-type marker sets
We employ two distinct gold-standard marker sets, as suggested in ref. 13. The first set includes marker genes listed in marker databases: for the Tabula Muris dataset, we use the CellMarker database64 specific to the relevant tissue; for the Azimuth dataset, we use the marker list22 used to annotate the reference dataset at the annotation level described in Methods-’Dataset and Processing’. The second set consists of the top 100 genes with the maximum differential expression across cell types, using cell-type annotations provided by the original studies (Tabula Muris) or annotated by Azimuth (Azimuth dataset).
Gene ranking by competing method
LMD was qualitatively compared to other marker gene identification methods: singleCellHaystack, Hotspot, SEMITONES, MarcoPolo, SCMarker, Seurat v4, COSG, and HighlyVariableGenes. Each method uses the same gene list as input. The gene rankings for each method are summarized below:
-
singleCellHaystack: We use singleCellHaystack1.065 with the advanced mode of the highD method. Genes are ranked in ascending order of log(adjusted p-value).
-
Hotspot: Genes are ranked in ascending order of FDR.
-
SEMITONES: Genes are ranked in ascending order of their minimum adjusted p-value in any reference cell. If two genes had the same adjusted p-value, we prioritize genes with larger absolute enrichment scores in any reference cell.
-
MarcoPolo: The gene rankings are given by MarcoPolo_rank.
-
SCMarker: No ranked outputs were produced; all identified marker genes were included for downstream analyses.
-
Seurat v4: We used Louvain clustering with resolution = 5 and applied the wilcoxauc.Seurat function from presto R package (v.1.0.0)66 to perform the fast Wilcoxon rank sum test among clusters. Genes were ranked in ascending order based on their minimum adjusted p-value among clusters. In cases where two genes had the same adjusted p-value, genes with larger fold-change were prioritized.
-
COSG: We used Louvain clustering with resolution = 5, and genes were ranked in descending order based on their maximum COSG score across clusters.
-
HighlyVariableGenes: Genes are ranked by FindVariableFeatures function with the VST option implemented in Seurat v4.
Note: For cluster-dependent methods (Seurat v4 and COSG), we set the Louvain clustering resolution to 5, as performance was largely consistent across different resolutions (Supplementary Fig. 4), and this value yielded the best overall performance across datasets for both methods.
Robustness evaluation
We evaluated the robustness of LMD to variations in graph construction, data quality, technical noise, and cell type marker bias. Depending on the test, robustness was assessed using rank consistency, measured by the Jaccard index of top-ranked genes or the correlation of selected gene rankings, and performance consistency, measured by AUROC using the two gold-standard gene sets described in Methods-‘Gold-standard cell-type marker sets’.
Graph construction
LMD relies on a cell-cell graph constructed from k-nearest neighbors in a dimensionality-reduced space. By default, we use the top 20PCs and set k = 5. To evaluate the sensitivity of graph construction, we examined three components: the number of nearest neighbors (k), the input space, and the type of graph. To assess the effect of k, we varied it from 3 to 50 while keeping the input space fixed at 20PCs. To assess the effect of the input space, we tested top 10–100 PCs and a 2D t-SNE embedding while fixing k = 5. Both experiments were performed on Tabula Muris bone marrow FACS dataset18. For each setting, we evaluated rank stability using the Jaccard index of top N genes and performance using AUROC based on two gold-standard marker sets (Supplementary Fig. 8). To assess the effect of graph type, we replaced the default kNN graph with either a shared nearest neighbor (SNN) or a Gaussian kernel graph, and evaluated performance across six datasets: Tabula Muris bone marrow FACS, Droplet, and pancreas FACS datasets17,18, and Azimuth human kidney, lung, and pancreas demo datasets22. Evaluation used the same criteria described above (Supplementary Fig. 9). The SNN graph was constructed from a kNN graph (k = 5) by computing the neighborhood overlap (Jaccard index) between each cell and its k nearest neighbors, as implemented in Seurat3. The Gaussian kernel graph was constructed using an α-decaying kernel with k = 5 and α = 10, following recommendations for graph construction in single-cell data analysis from ref. 67.
Data quality
We assessed the robustness of LMD under different levels of data sparsity, sample size, and expression scaling. For all tests, rank consistency was measured by the Jaccard index of top-ranked genes (compared to the original, unperturbed data), and performance was evaluated using AUROC on two gold-standard marker sets. To assess sensitivity to the number of cells, we selected three representative datasets covering different technologies and sizes: the Tabula Muris bone marrow FACS and Droplet datasets, and the Azimuth human kidney dataset. We downsampled the dataset to 50 cells and up to the full size and observed that AUROC performance reached a plateau between 500 and 1000 cells, with a Jaccard index above 0.5 (Supplementary Fig. 10). Based on this, we recommend using datasets with more than 1000 cells for stable performance. To evaluate robustness to sequencing depth, we selected three datasets with high coverage, each with more than 500,000 UMIs per cell on average: the Tabula Muris bone marrow FACS and lung FACS datasets, and the Azimuth human pancreas dataset. UMI downsampling was performed using the downsampleMatrix function from the scuttle R package (v.1.4.0)68, varying the proportion of retained UMIs from 10−4 to 10−1. In all three datasets, performance stabilized around a proportion of 0.005 with the Jaccard index above 0.5 (Supplementary Fig. 11). This corresponds to approximately 3000 UMIs per cell–a typical depth for 10X data. To assess the effect of expression scaling, we tested binarizing expression values by setting all non-zero entries to one, using the Tabula Muris bone marrow FACS dataset (Supplementary Fig. 12). While AUROC decreased slightly, we recommend using log-normalized data to retain quantitative expression information.
Technical noise: ambient RNA contamination
We assessed the robustness of LMD to ambient RNA contamination using an artificially contaminated pancreas dataset69. The dataset includes simulated contamination affecting 500 genes at three levels: low, medium, and high. Robustness was evaluated by assessing the preservation of top-ranked genes using the Jaccard index, and by measuring rank consistency of all 500 perturbed genes via the Pearson correlation of their log10-transformed ranks between contaminated and uncontaminated data (Supplementary Fig. 13). Even at the highest contamination level, the Jaccard index remained above 0.75 for top 1000 genes, and the Pearson correlation was above 0.9, indicating that LMD is robust to ambient RNA contamination. Nonetheless, we recommend applying ambient RNA removal tools such as scCDC69 or DecontX70 when substantial contamination is suspected.
Cell type marker bias
To evaluate whether LMD exhibits bias toward specific cell types—either due to differences in marker detectability or imbalanced cell numbers—we investigated its behavior under two scenarios on the Tabula Muris bone marrow FACS dataset: variation in marker gene expression across cell types and the effects of downsampling specific populations. First, we examined whether LMD’s top-ranked genes were disproportionately associated with specific cell types. We compared the top LMD markers with two gold-standard gene sets (see Methods-‘Gold-standard cell-type marker sets’), matching the number of genes in each set to ensure a fair comparison. Then, for each gene set, we calculated the proportion of genes expressed per cell and visualized the resulting distributions across all pre-annotated cell types (Supplementary Fig. 14A). The overall patterns for LMD markers were comparable to those of the gold-standard sets, suggesting that the observed variability is inherent to the dataset rather than being specific to LMD. Notably, most cells expressed a higher proportion of top LMD markers compared to those from the CellMarkerDB-derived gene set, suggesting that LMD may capture cell identity more effectively in this dataset than curated reference markers. Next, we assessed the robustness of LMD to cell type imbalance by downsampling two representative populations-granulocytes and progenitor cells. For each cell type, we defined a reference marker set: LMD gene modules that showed specificity to the respective cell type (as described in Supplementary Fig. 6). Preservation of the reference marker set among top-ranked genes at each downsampling level was measured using AUROC from the Wilcoxon rank-sum test, comparing reference marker genes against all other genes (Supplementary Fig. 14B). Marker gene prioritization remained highly consistent, with AUROC values staying above 0.8 even after downsampling to 5% of its original size.
Scalability
We assessed the scalability of LMD by running it on ten datasets, as listed in Supplementary Data 2, with cell numbers ranging from 1500 to 30,000 and gene numbers from 13,000 to 34,000 on a Linux operating system with 72 2.70 GHz cores (Supplementary Data 3). The runtime was primarily dependent on the number of cells rather than the number of genes. For a fixed cell number, runtimes were similar whether gene counts were around 14,000 or 34,000. The runtime increased with the number of cells: ~30 s for 1500 cells and 44 min for 30,000 cells. This trend aligns with the theoretical time complexity of LMD, which involves repeated multiplication of the diffusion operator and matrix-matrix products with the gene expression matrix, resulting in an maximal time complexity of \({\mathcal{O}}(t({n}^{3}+{n}^{2}m))\), where n is the number of cells, m is the number of genes, and t is the number of dyadic diffusion steps which typically remains below 15.
For large datasets with more than 20,000 cells, we recommend an optional cell graph coarse-graining strategy adapted from ref. 24 to further reduce computational cost. This approach aggregates cells into a smaller number of “meta-cells" using k-means clustering, and defines the expression profile of each meta-cell by summing the expression values of its constituent cells. The one-time cost of this coarse-graining step is \({\mathcal{O}}(nmk+{k}^{2}n+k{n}^{2})\), where k is the number of “meta-cells". This reduces the size of the cell graph from n to k, effectively lowering the downstream maximal complexity to \({\mathcal{O}}(t({k}^{3}+{k}^{2}m))\), where k ≪ n.
We evaluated the effect of coarse-graining on two large datasets with more than 20,000 cells: the Azimuth human lung v2 and human bone marrow demo datasets22. The cell-cell graph was coarse-grained to various levels, ranging from 500 to 10,000 cells, and robustness was assessed using both rank stability and performance consistency (Supplementary Fig. 15). Coarse-graining did not compromise the accuracy of cell type marker identification and, in some cases, improved performance by enhancing the aggregation of neighborhood information—particularly in datasets with low UMI coverage (Supplementary Data 2, Supplementary Fig. 15A). We recommend coarse-graining a typical large dataset (10,000 to 50,000 cells) to ~5000 cells. In this range, performance remains stable, the rank stability of the top 1000 genes exceeds 0.75 (Jaccard index) relative to the original graph, and runtime remains reasonable (under 5 min). However, coarse-graining may reduce resolution and accuracy for detecting genes with small cell populations, and may bias results toward larger clusters (Supplementary Fig. 15C). Users are advised to select the degree of coarse-graining judiciously, balancing computational constraints with data quality and the desired level of resolution.
GO and reactome enrichment analysis
We perform Gene Ontology (GO) enrichment analysis using the clusterProfiler R package (v.4.2.2)71, Reactome Pathway Database enrichment analysis with the ReactomePA R package (v.1.38.0)72 and Molecular Signatures Database (MsigDB) enrichment analysis with msigdbr R package (v.7.5.1)73 and clusterProfiler R package (v.4.2.2). The background gene set comprises all genes that are applied during the localized marker ranking process. We regard GO terms and pathways with Benjamini-Hochberg adjusted p-values smaller than 0.05 as significantly enriched.
Cell cycle annotation, embedding, and regression
We first extract the 2019-updated human cell cycle gene set74 from cc.genes.updated.2019 in Seurat v4 R package3 and transform it to mouse genes using the gprofiler2 R package (v.0.2.3)75. This reference cell cycle gene list, containing 54 G2/M phase markers and 41 S phase markers, is then used for annotation and embedding. Then we perform cell cycle scoring and annotation using the Seurat v4 R package. The CellCycleScoring function assigns cell cycle scores based on the reference cell cycle gene list, classifying cells into G1, S, or G2/M phases. To create UMAP/t-SNE embeddings that preserve only the heterogeneity from the cell cycle, we first run PCA on the reference cell cycle gene list, retaining the number of PCs determined by the elbow plot63. We then apply UMAP/t-SNE on the retained PCs. To create UMAP/t-SNE embeddings that subtract the heterogeneity from the cell cycle, we follow the Seurat workflow. We regress out cell cycle scores during data scaling, then perform dimensional reduction on the scaled data.
Experimental details of mouse embryo skin sample preparation
Mice
Axin2CreER76 mice were bred to Rosa-lox-STOP-lox-SmoM2YFP77 mice. Details of the mouse lines are as follows:
-
Axin2CreER
-
Official allele name: Axin2tm1(cre/ERT2)Rnu
-
MGI ID: MGI:5433373
-
Original citation: van Amerongen et al., Cell Stem Cell, 2012 (PMID: 22863533)
-
Source: Jackson Laboratory (JAX Stock #018867)
-
Provenance: Developed by Dr. Roel Nusse (Stanford University)
-
Background: Primarily CD1
-
-
Rosa-lox-STOP-lox-SmoM2YFP
-
Official allele name: Gt(ROSA)26Sortm1(Smo/EYFP)Amc
-
MGI ID: MGI:3576373
-
Original citation: Jeong et al., Genes & Development, 2004 (PMID: 15107405)
-
Source: Jackson Laboratory (JAX Stock #005130)
-
Provenance: Developed by Dr. Andrew P. McMahon (University of Southern California)
-
Background: Primarily CD1
-
A random population of both male and female embryos was used for all experiments. All procedures involving animal subjects were performed under the approval of the Institutional Animal Care and Use Committee of the Yale School of Medicine.
Ethical compliance
All procedures involving animal subjects were performed under the approval of the Institutional Animal Care and Use Committee of the Yale School of Medicine. We have complied with all relevant ethical regulations for animal use.
EdU and Tamoxifen administration
Embryos were staged as days post coitum, with embryonic day E0.5 considered as noon of the day a vaginal plug was detected after overnight mating. To induce expression of SmoM2YFP, pregnant dams were given one dose of tamoxifen dissolved in corn oil (20 mg/ml, Sigma Aldrich Cat#T5648) at 60 μg/g body weight by oral gavage at E11.5. To assess proliferation, EdU (5-ethynyl-2-deoxyuridine; Thermo Fisher Scientific Cat#E10187) was dissolved in PBS at 5 mg/mL and administered to pregnant mice intraperitoneally (25 μg/g) and embryos were harvested after either 1.5 h or 24 h.
In vivo procedures and sample collection
Pregnant moms were sacrificed by CO2 asphyxiation according to the IACUC approved protocol. Upon confirmation of death, uterine horns were removed from the euthanized moms and placed in cold sterile PBS. E13.5 or E14.5 embryos were released from uterine horns using sterile forceps under a stereomicroscope and placed into individual wells containing cold PBS. A limb was taken for genotyping by PCR before harvesting dorsolateral skin from each embryo under a stereomicroscope, using sterile microdissection scissors and forceps. Skin was placed in individual Eppendorf containing ice-cold sterile 2% FBS in PBS. Cells were prepared separately as described and then pooled by condition before FACS sorting.
In situ hybridization
Formalin-fixed paraffin-embedded (FFPE) whole embryos were used for histological analysis. FFPE specimens were sectioned at 10 μm thickness. The RNAscope Multiplex Fluorescent Detection Kit v2 (ACDBio, 323110) was used for single-molecule fluorescence in situ hybridization (FISH) according to the manufacturer’s protocol. Briefly, sections were deparaffinized and permeabilized with hydrogen peroxide followed by antigen retrieval and protease treatment before probe hybridization. After hybridization, amplification and probe detection were done using the Amp 1–3 reagents. Probe channels were targeted using the provided HRP-C1-3 reagents and tyramide signal amplification VIVID fluorophores-650 and 570 (ACD biotechne, 323271). EdU staining was done using the Click-it EdU Imaging Kit Alexa 488 (Life Technologies, c10338) according to the manufacturer’s instructions. Nuclear counter-stain was done using Hoechst 33342 (Invitrogen, H3570) before mounting with SlowFade Mountant. RNA scope probes used (ACDBio)-Mm-Lef1 (441861), Mm-Gal (400961) and Mm-Trp53inp1 (1161531).
Microscopy
FISH paraffin-embedded images were acquired using the Leica Stellaris 8 DMi8 confocal microscope with a × 40 oil immersion (Numerical Aperture 1.3) objective lens, scanned at 5 μm thickness, 1024 × 1024 pixel width, 400Hz.
Single-cell dissociation
Dorsolateral/flank skin was microdissected from E13.5 or E14.5 littermate control and mutant embryos and dissociated into a single-cell suspension using 0.25% trypsin (Gibco, Life Technologies) for 20 min at 37 °C. After genotyping, two to three embryos were pooled by condition. Single-cell suspensions were then stained with DAPI (Thermo Fisher Scientific, NBP2-31156) before fluorescence-activated cell sorting.
Fluorescence-activated cell sorting
DAPI-excluded live skin cells were sorted on a BD FACS Aria II (BD Biosciences) sorter with a 100 μm nozzle. Cells were sorted in bulk and submitted for 10X Genomics library preparation at 0.75−1.0 × 106 ml−1 concentration in 4% fetal calf serum/phosphate-buffered saline (FCS/PBS) solution.
H-score quantification
For quantification based on FISH, cells with 4–5 dots were considered positive (according to the RNAScope manufacturer’s instructions), and subsections from a total of n = 4 different embryos were examined. To measure RNA expression levels, H scores were calculated according to ACDBio manufacturer’s instructions: a cell with 0 dot is scored 0, 1–3 dots is scored 1, 4–9 dots is scored 2, 10–15 dots and/or less than 10% clustered dots is scored 3 and more than 15 dots and/or more than 10% clustered dots is scored 4; then the final H score of a given cell type A is calculated by summing the (% cells scored B within all cells in A) × B for score B in 0–4. Bin 4 Trp53inp1+ cells were quantified for %EdU.
scRNA-seq and library preparation
Chromium Single Cell 3′ GEM Library and Gel Bead Kit v3.1 (PN-1000121) were used according to the manufacturer’s instructions in the Chromium Single Cell 3′ Reagents Kits V3.1 User Guide. After cDNA libraries were created, they were subjected to Novaseq 6000 (Illumina) sequencing. For each scRNA-seq experiment, control and littermate mutant samples were prepared in parallel at the same time, pooled and sequenced on the same lane.
Statistics and Reproducibility
Statistical analyses were performed using R (v 4.1.3) as specified in the Methods. Performance metrics for evaluating gene ranking and marker identification included Jaccard index, AUROC, and Pearson correlation. One-way ANOVA with Tukey’s HSD test was applied to FISH validation data across sample groups. For mouse embryonic skin experiments, no statistical methods were used to predetermine sample sizes. The phenotype and findings were repeated n = 3 for biological experiments, and at least two embryos per condition (from the same litter) were pooled for scRNA-seq experiments.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
The Tabula Muris scRNA-seq datasets are available at ref. 18. These include FACS-based full-length transcript datasets for bone marrow78, pancreas79 and lung80, as well as a microfluidic droplet-based 3′-end counting dataset for bone marrow81. The Azimuth demo datasets are available at ref. 22. These include human motor cortex82, mouse motor cortex83, human pancreas84, human kidney85, human bone marrow86, and human lung v287. Raw sequencing data for E14.5 mouse embryonic skin are available at GSE198487, and the newly generated E13.5 mouse embryonic skin data are available at GSE280825. The processed Seurat data objects (Tabula Muris bone marrow dataset and mouse embryonic skin dataset) are available at Figshare: https://doi.org/10.6084/m9.figshare.26507098. Source data used to generate the graphs and charts in the main figures are available at https://doi.org/10.5281/zenodo.15644954.
Code availability
The R package of LMD and the code used for data analysis are available on GitHub (https://github.com/KlugerLab/LocalizedMarkerDetector). A snapshot of the exact version used in this study has been archived on Zenodo and is available at https://doi.org/10.5281/zenodo.15597475. The analysis code for reproducing the main figures is available at https://doi.org/10.5281/zenodo.15644954.
References
Fleck, J. S., Camp, J. G. & Treutlein, B. What is a cell type? Science 381, 733–734 (2023).
Cheng, C., Chen, W., Jin, H. & Chen, X. A review of single-cell RNA-seq annotation, integration, and cell–cell communication. Cells 12, 1970 (2023).
Hao, Y. et al. Integrated analysis of multimodal single-cell data. Cell 184, 3573–3587 (2021).
Patrick, R. et al. Sierra: discovery of differential transcript usage from polya-captured single-cell RNA-seq data. Genome Biol. 21, 1–27 (2020).
Svensson, V., Teichmann, S. A. & Stegle, O. Spatialde: identification of spatially variable genes. Nat. Methods 15, 343–346 (2018).
Miao, Z., Deng, K., Wang, X. & Zhang, X. Desingle for detecting three types of differential expression in single-cell rna-seq data. Bioinformatics 34, 3223–3224 (2018).
Nabavi, S., Schmolze, D., Maitituoheti, M., Malladi, S. & Beck, A. H. Emdomics: a robust and powerful method for the identification of genes differentially expressed between heterogeneous classes. Bioinformatics 32, 533–541 (2016).
Korthauer, K. D. et al. A statistical approach for identifying differential distributions in single-cell RNA-seq experiments. Genome Biol. 17, 1–15 (2016).
Robinson, M. D., McCarthy, D. J. & Smyth, G. K. edger: a bioconductor package for differential expression analysis of digital gene expression data. bioinformatics 26, 139–140 (2010).
Qiu, X. et al. Reversed graph embedding resolves complex single-cell trajectories. Nat. Methods 14, 979–982 (2017).
Dai, M., Pei, X. & Wang, X.-J. Accurate and fast cell marker gene identification with COSG. Brief. Bioinforma. 23, bbab579 (2022).
Vandenbon, A. & Diez, D. A clustering-independent method for finding differentially expressed genes in single-cell transcriptome data. Nat. Commun. 11, 4318 (2020).
Kim, C., Lee, H., Jeong, J., Jung, K. & Han, B. Marcopolo: a method to discover differentially expressed genes in single-cell RNA-seq data without depending on prior clustering. Nucleic acids Res. 50, e71–e71 (2022).
Vlot, A. H. C., Maghsudi, S. & Ohler, U. Cluster-independent marker feature identification from single-cell omics data using semitones. Nucleic Acids Res. 50, e107–e107 (2022).
Wang, F., Liang, S., Kumar, T., Navin, N. & Chen, K. Scmarker: ab initio marker selection for single cell transcriptome profiling. PLoS Comput. Biol. 15, e1007445 (2019).
DeTomaso, D. & Yosef, N. Hotspot identifies informative gene modules across modalities of single-cell genomics. Cell Syst. 12, 446–456 (2021).
Consortium, T. M. et al. Single-cell transcriptomics of 20 mouse organs creates a Tabula Muris. Nature 562, 367–372 (2018).
Tabula Muris Consortium. Robject files for tissues processed by Seurat, https://figshare.com/articles/Robject_files_for_tissues_processed_by_Seurat/5821263/3 (2018).
Coifman, R. R. et al. Geometric diffusions as a tool for harmonic analysis and structure definition of data: Multiscale methods. Proc. Natl. Acad. Sci. USA 102, 7432–7437 (2005).
Coifman, R. R. & Maggioni, M. Diffusion wavelets. Appl. Comput. Harmonic Anal. 21, 53–94 (2006).
Dsilva, C. J., Talmon, R., Rabin, N., Coifman, R. R. & Kevrekidis, I. G. Nonlinear intrinsic variables and state reconstruction in multiscale simulations. J. Chem. Phys. 139, 184109 (2013).
Lab, S. & Consortium, H. Azimuth demo dataset repository, https://azimuth.hubmapconsortium.org/references/ Accessed June 4, 2025 (2021).
Ranjan, B. et al. Dubstepr is a scalable correlation-based feature selection method for accurately clustering single-cell data. Nat. Commun. 12, 5849 (2021).
Qu, R. et al. Gene trajectory inference for single-cell data by optimal transport metrics. Nat. Biotechnol. 43, 1–11 (2024).
Gillespie, M. et al. The Reactome Pathway Knowledgebase 2022. Nucleic Acids Res. 50, D687–D692 (2022).
Tandon, B. et al. Nuclear overexpression of lymphoid-enhancer-binding factor 1 identifies chronic lymphocytic leukemia/small lymphocytic lymphoma in small B-cell lymphomas. Mod. Pathol. 24, 1433–1443 (2011).
Lu, L.-S. et al. Identification of a germ-line pro-B cell subset that distinguishes the fetal/neonatal from the adult B cell development pathway. Proc. Natl. Acad. Sci. USA 99, 3007–3012 (2002).
Li, Y.-S., Hayakawa, K. & Hardy, R. R. The regulated expression of B lineage associated genes during B cell differentiation in bone marrow and fetal liver. J. Exp. Med. 178, 951–960 (1993).
Peschon, J. J. et al. Early lymphocyte expansion is severely impaired in interleukin 7 receptor-deficient mice. J. Exp. Med. 180, 1955–1960 (1994).
Jensen, M., Tan, G., Forman, S., Wu, A. M. & Raubitschek, A. Cd20 is a molecular target for scfvfc: zeta receptor redirected t cells: implications for cellular immunotherapy of cd20+ malignancy. Biol. Blood Marrow Transplant. 4, 75–83 (1998).
Tedder, T. F., Poe, J. C. & Haas, K. M. Cd22: a multifunctional receptor that regulates B lymphocyte survival and signal transduction. Adv. Immunol. 88, 1–50 (2005).
Bonnefoy, J.-Y. et al. Structure and functions of CD23. Int. Rev. Immunol. 16, 113–128 (1997).
Sáez de Guinoa, J., Barrio, L., Mellado, M. & Carrasco, Y. R. Cxcl13/cxcr5 signaling enhances bcr-triggered b-cell activation by shaping cell dynamics. Blood, J. Am. Soc. Hematol. 118, 1560–1569 (2011).
Liu, Z. et al. Fate mapping via MS4A3-expression history traces monocyte-derived cells. Cell 178, 1509–1525 (2019).
Xie, X. et al. Single-cell transcriptome profiling reveals neutrophil heterogeneity in homeostasis and infection. Nat. Immunol. 21, 1119–1133 (2020).
Calcagno, D. M. et al. Siglecf (hi) marks late-stage neutrophils of the infarcted heart: a single-cell transcriptomic analysis of neutrophil diversification. J. Am. Heart Assoc. 10, e019019 (2021).
Eash, K. J., Greenbaum, A. M., Gopalan, P. K. & Link, D. C. et al. Cxcr2 and Cxcr4 antagonistically regulate neutrophil trafficking from murine bone marrow. J. Clin. Investig. 120, 2423–2431 (2010).
Goodall, K. J. et al. Multiple receptors converge on h2-q10 to regulate NK and γ δt-cell development. Immunol. Cell Biol. 97, 326–339 (2019).
Kwack, K. H. et al. Discovering myeloid cell heterogeneity in mandibular bone–cell by cell analysis. Front. Physiol. 12, 731549 (2021).
Ping, S., Qiu, X., Gonzalez-Toledo, M. E., Liu, X. & Zhao, L.-R. Stem cell factor in combination with granulocyte colony-stimulating factor protects the brain from capillary thrombosis-induced ischemic neuron loss in a mouse model of Cadasil. Front. Cell Dev. Biol. 8, 627733 (2021).
Qu, R. et al. Decomposing a deterministic path to mesenchymal niche formation by two intersecting morphogen gradients. Dev. Cell 57, 1053–1067 (2022).
Gupta, K. et al. Single-cell analysis reveals a hair follicle dermal niche molecular differentiation trajectory that begins prior to morphogenesis. Dev. Cell 48, 17–31 (2019).
Myung, P., Andl, T. & Atit, R. The origins of skin diversity: lessons from dermal fibroblasts. Development 149, dev200298 (2022).
Wu, R. S. & Bonner, W. M. Separation of basal histone synthesis from S-phase histone synthesis in dividing cells. Cell 27, 321–330 (1981).
Liao, H., Winkfein, R., Mack, G., Rattner, J. & Yen, T. Cenp-f is a protein of the nuclear matrix that assembles onto kinetochores at late G2 and is rapidly degraded after mitosis. J. Cell Biol. 130, 507–518 (1995).
Zhu, C. et al. Functional analysis of human microtubule-based motor proteins, the kinesins and dyneins, in mitosis/cytokinesis using RNA interference. Mol. Biol. cell 16, 3187–3199 (2005).
Diril, M. K. et al. Cyclin-dependent kinase 1 (cdk1) is essential for cell division and suppression of DNA re-replication but not for liver regeneration. Proc. Natl. Acad. Sci. USA 109, 3826–3831 (2012).
Hames, R. S. & Fry, A. M. Alternative splice variants of the human centrosome kinase Nek2 exhibit distinct patterns of expression in mitosis. Biochem. J. 361, 77–85 (2002).
Liberzon, A. et al. The molecular signatures database hallmark gene set collection. Cell Syst. 1, 417–425 (2015).
Hovanes, K. et al. β-catenin–sensitive isoforms of lymphoid enhancer factor-1 are selectively expressed in colon cancer. Nat. Genet. 28, 53–57 (2001).
Filali, M., Cheng, N., Abbott, D., Leontiev, V. & Engelhardt, J. F. Wnt-3a/β-catenin signaling induces transcription from the lef-1 promoter* 210. J. Biol. Chem. 277, 33398–33410 (2002).
Driskell, R. R., Giangreco, A., Jensen, K. B., Mulder, K. W. & Watt, F. M. Sox2-positive dermal papilla cells specify hair follicle type in mammalian epidermis. Development 136, 2815–2823(2009).
Prim, R. C. Shortest connection networks and some generalizations. Bell Syst. Tech. J. 36, 1389–1401 (1957).
Sinkhorn, R. & Knopp, P. Concerning nonnegative matrices and doubly stochastic matrices. Pac. J. Math. 21, 343–348 (1967).
Satopaa, V., Albrecht, J., Irwin, D. & Raghavan, B. Finding a" kneedle" in a haystack: Detecting knee points in system behavior. In 2011 31st international conference on distributed computing systems workshops, 166–171 (IEEE, 2011).
Cha, S.-H. Comprehensive survey on distance/similarity measures between probability density functions. City 1, 1 (2007).
Linderman, G. C. et al. Zero-preserving imputation of single-cell RNA-seq data. Nat. Commun. 13, 192 (2022).
Langfelder, P. & Horvath, S. Wgcna: an R package for weighted correlation network analysis. BMC Bioinforma. 9, 1–13 (2008).
R Core Team. R: A Language and Environment for Statistical Computing. R Foundation for Statistical Computing, Vienna, Austria https://www.R-project.org/ (2022).
Langfelder, P., Zhang, B., & with contributions from Steve Horvath. dynamicTreeCut: Methods for Detection of Clusters in Hierarchical Clustering Dendrograms.https://CRAN.R-project.org/package=dynamicTreeCut. R package version 1.63-1 (2016).
Parisi, F., Strino, F., Nadler, B. & Kluger, Y. Ranking and combining multiple predictors without labeled data. Proc. Natl. Acad. Sci. USA 111, 1253–1258 (2014).
Mouselimis, L.ClusterR: Gaussian Mixture Models, K-Means, Mini-Batch-Kmeans, K-Medoids and Affinity Propagation Clustering.https://CRAN.R-project.org/package=ClusterR. R package version 1.3.1 (2023).
Harvard Chan Bioinformatics Core. Elbow plot: quantitative approach. https://hbctraining.github.io/scRNA-seq/lessons/elbow_plot_metric.html (2021).
Zhang, X. et al. Cellmarker: a manually curated resource of cell markers in human and mouse. Nucleic Acids Res. 47, D721–D728 (2019).
Vandenbon, A. & Diez, D. A universal tool for predicting differentially active features in single-cell and spatial genomics data. Sci. Rep. 13, 11830 (2023).
Korsunsky, I., Nathan, A., Millard, N. & Raychaudhuri, S. presto: Fast Functions for Differential Expression using Wilcox and AUC R package version 1.0.0 (2024).
Moon, K. R. et al. Visualizing structure and transitions in high-dimensional biological data. Nat. Biotechnol. 37, 1482–1492 (2019).
McCarthy, D. J., Campbell, K. R., Lun, A. T. & Wills, Q. F. Scater: pre-processing, quality control, normalization and visualization of single-cell RNA-seq data in R. Bioinformatics 33, 1179–1186 (2017).
Wang, W. et al. sccdc: a computational method for gene-specific contamination detection and correction in single-cell and single-nucleus rna-seq data. Genome Biol. 25, 136 (2024).
Yang, S. et al. Decontamination of ambient RNA in single-cell RNA-seq with decontx. Genome Biol. 21, 1–15 (2020).
Wu, T. et al. clusterprofiler 4.0: A universal enrichment tool for interpreting omics data. Innovation 2, 100141 (2021).
Yu, G. & He, Q.-Y. Reactomepa: an r/bioconductor package for reactome pathway analysis and visualization. Mol. Biosyst. 12, 477–479 (2016).
Dolgalev, I. msigdbr: MSigDB Gene Sets for Multiple Organisms in a Tidy Data Format.https://igordot.github.io/msigdbr/. R package version 7.5.1.9001 (2022).
Tirosh, I. et al. Dissecting the multicellular ecosystem of metastatic melanoma by single-cell RNA-seq. Science 352, 189–196 (2016).
Kolberg, L., Raudvere, U., Kuzmin, I., Vilo, J. & Peterson, H. gprofiler2–an R package for gene list functional enrichment analysis and namespace conversion toolset g: Profiler. F1000Research 9, ELIXIR-709 (2020).
Van Amerongen, R., Bowman, A. N. & Nusse, R. Developmental stage and time dictate the fate of wnt/β-catenin-responsive stem cells in the mammary gland. Cell Stem Cell 11, 387–400 (2012).
Jeong, J., Mao, J., Tenzen, T., Kottmann, A. H. & McMahon, A. P. Hedgehog signaling in the neural crest cells regulates the patterning and growth of facial primordia. Genes Dev. 18, 937–951 (2004).
Tabula Muris Consortium. Bone marrow FACS dataset, https://figshare.com/ndownloader/files/13092380 (2018).
Tabula Muris Consortium. Pancreas FACS dataset, https://figshare.com/ndownloader/files/13092386 (2018).
Tabula Muris Consortium. Lung FACS dataset, https://figshare.com/ndownloader/files/13092194 (2018).
Tabula Muris Consortium. Bone marrow droplet dataset, https://figshare.com/ndownloader/files/13089821 (2018).
Azimuth Demo Datasets. Human—Motor Cortex demo dataset, https://seurat.nygenome.org/azimuth/demo_datasets/allen_m1c_2019_ssv4.rds (2021).
Azimuth Demo Datasets. Mouse—Motor Cortex demo dataset, https://seurat.nygenome.org/azimuth/demo_datasets/allen_mop_2020.rds (2021).
Azimuth Demo Datasets. Human—Pancreas demo dataset, https://seurat.nygenome.org/azimuth/demo_datasets/enge.rds (2021).
Azimuth Demo Datasets. Human—Kidney demo dataset, https://seurat.nygenome.org/azimuth/demo_datasets/kidney_demo_stewart.rds (2021).
Azimuth Demo Datasets. Human—Bone Marrow demo dataset, https://seurat.nygenome.org/azimuth/demo_datasets/bmcite_demo.rds (2021).
Azimuth Demo Datasets. Human—Lung v2 (HLCA) demo dataset, https://seurat.nygenome.org/hlca_ref_files/ts_opt.rds (2021).
Acknowledgements
The authors thank Ronald Coifman, Junchen Yang, Will Yaochen Zhu for fruitful discussions. Y.K. is supported by NIH [R01GM131642, UM1PA051410, U54AG076043, U54AG079759, U01DA053628, P50CA121974, and R33DA047037]. X.C. is partially supported by NSF [DMS-2237842, DMS-2007040] and Simons Foundation [MPS-MODL-00814643]. P.M. is supported by NIH/NIAMS [R01AR076420] and LEO Foundation [LF-OC-23-001347].
Author information
Authors and Affiliations
Contributions
R.L., R.Q., F.P., F.S., X.C., and Y.K. designed research; R.L., R.Q., F.P., F.S., and X.C. developed the method; R.L. performed data analysis; R.L., R.Q., and F.S. implemented the software; H.L. and P.M. performed the experiments and contributed to the biological interpretation; R.L., F.P., and F.S. wrote the paper; R.Q., J.S.S., X.C., P.M., Y.K. offered vital insights into improving the work and assisted in writing.
Corresponding authors
Ethics declarations
Competing interests
F.P. and F.S. are employed as directors by PCMGF Limited. All other authors declare no competing interests.
Peer review
Peer review information
Communications Biology thanks Andrea Calabria and the other anonymous reviewer(s) for their contribution to the peer review of this work.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Li, R., Qu, R., Parisi, F. et al. Cluster-independent multiscale marker identification in single-cell RNA-seq data using localized marker detector (LMD). Commun Biol 8, 1058 (2025). https://doi.org/10.1038/s42003-025-08485-y
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s42003-025-08485-y
