
Abstract
Hepatic fibrosis progression involves complex multicellular crosstalk, highlighting the critical need to identify key therapeutic targets. In this study, we identify insulin-like growth factor binding protein 6 (IGFBP6) as a marker specifically enriched in hepatic stellate cells (HSCs) and upregulated in viral hepatitis-associated fibrosis. Using thioacetamide (TAA)-induced mouse models and transforming growth factor-β (TGF-β)-stimulated cell models, we demonstrate the pro-fibrotic role of IGFBP6. Through network pharmacology screening, pantothenic acid (PA) is identified as a potent compound targeting IGFBP6. PA administration significantly reduces collagen deposition, attenuates HSCs’ activation, and decreases hepatic fibrosis-related markers. Notably, PA maintains efficacy in mouse models with established fibrosis. Mechanistically, PA directly interacts with IGFBP6, inducing ubiquitin-dependent degradation and inhibiting TGF-β/SMADs signaling. This study identifies IGFBP6 as a driver of hepatic fibrosis and validates PA as a potent therapeutic agent. Therefore, targeting IGFBP6 with PA offers a potential clinical treatment strategy for hepatic fibrosis.

Similar content being viewed by others


Introduction
Hepatic fibrosis is a progressive pathological process characterized by excessive extracellular matrix (ECM) deposition following chronic hepatic injury, driven by diverse etiologies including viral hepatitis, alcohol-associated liver disease (ALD), and non-alcoholic steatohepatitis (NASH)1,2. This dysregulated wound-healing response not only promotes cirrhosis but also substantially increases the risk of hepatocellular carcinoma (HCC)3. Thus, targeting fibrogenic pathways represents a critical therapeutic approach to prevent disease progression in chronic hepatic disease4,5. Despite numerous clinical trials evaluating anti-fibrotic agents such as Selonsertib and Cenicriviroc, Phase III trials have demonstrated limited clinical efficacy6,7. Currently, no therapeutic agents specifically targeting hepatic fibrosis have received approval from the U.S. Food and Drug Administration (FDA).
The pathogenesis of hepatic fibrosis involves complex multicellular interactions among hepatocytes (HEPs), endothelial cells (ECs), Kupffer cells (KCs), and hepatic stellate cells (HSCs)2. Central to this process is the activation of HSCs into collagen-secreting myofibroblasts8,9, primarily mediated by transforming growth factor-β (TGF-β) signaling10,11. While TGF-β inhibition demonstrates anti-fibrotic efficacy12,13,14, global suppression of this pleiotropic cytokine causes adverse effects by disrupting physiological homeostasis15,16, including potential progression of some cancers due to the repression of TGF-β-mediated growth inhibition of epithelial cells17. Consequently, targeting critical nodes downstream of TGF-β signaling represents a promising therapeutic strategy for hepatic fibrosis18.
The insulin-like growth factor binding protein (IGFBP) family comprises six secreted members (IGFBP1-6), which modulate cellular processes through both insulin-like growth factor (IGF)-dependent and IGF-independent mechanisms. IGF-independent interactions involve integrins, TGF-β receptors, and other membrane-associated proteins19,20. Accumulating evidence implicates distinct pathological roles of specific IGFBPs in liver disorders: IGFBP3 promotes alcohol-associated steatohepatitis by activating HSCs21, while loss of IGFBP4 exacerbates HCC via dysregulated epigenetic reprogramming22. Although IGFBP6 is constitutively expressed in cardiovascular tissues, the nervous system, and various cancers23,24, its functional contribution to hepatic fibrogenesis remains undefined.
Pantothenic acid (PA, vitamin B5), an essential precursor of coenzyme A (CoA), serves as a key regulator of fundamental metabolic processes including glycolysis, fatty acid synthesis, and protein homeostasis25,26. Emerging evidence reveals context-dependent roles of PA in disease pathogenesis, spanning both pro-tumorigenic and therapeutic effects27. Paradoxically, PA supplementation promotes MYC-driven tumorigenesis in breast cancer28, whereas it improves ineffective erythropoiesis in myelodysplastic syndromes (MDS)29. Despite these dual roles in cancer and hematopoiesis, the effect of PA on fibrotic remodeling remains unexplored.
In this study, we demonstrated that IGFBP6 functions as a fibrosis-specific effector up-regulated in activated HSCs across human clinical specimens and mouse models. Through systematic drug screening, we identified PA as a specific ligand of IGFBP6 that suppresses HSC activation and ECM remodeling. PA-mediated inhibition of IGFBP6 attenuated fibrogenesis both in vivo and in vitro by targeting the TGF-β/SMAD signaling pathway. Collectively, these findings establish IGFBP6 as a therapeutic target for hepatic fibrosis and reveal the anti-fibrotic mechanism of PA through IGFBP6 regulation.
Result
IGFBP6 serves as a critical regulator of hepatic fibrogenesis
To delineate molecular drivers of fibrosis progression in chronic hepatitis, we performed integrative bioinformatics analysis on three Gene Expression Omnibus (GEO) datasets comprising 43 hepatitis samples and 293 hepatic fibrosis specimens. After batch-effect correction, 159 differentially expressed genes (DEGs) were identified (Supplementary Fig. S1a), consisting of 74 up-regulated and 85 down-regulated transcripts (Fig. 1a). Enrichment analysis demonstrated significant DEGs enrichment in fibrosis-related processes, including integrin signaling and metabolic reprogramming (Supplementary Fig. S1b). Notably, IGFBP6 emerged as the most significantly upregulated DEG (Fig. 1b, c).

a Volcano plot showing the distribution of DEGs. Yellow dots indicate significantly up-regulated genes, green dots indicate significantly down-regulated genes, and pink dots indicate non-differentially expressed genes. b Heat map of relevant DEGs between fibrosis and control groups. c Top 20 most significantly differentially expressed genes, with IGFBP6 showing the highest significance. d Validation using dataset GSE130123. CCL4-induced 4 weeks hepatic fibrosis model in mice. (control group n = 4, and fibrosis group n = 5). e IHC staining of COL1A1 and IGFBP6 in hepatic sections from control mice and mice at 4 or 8 weeks after intraperitoneal TAA injection (scale bar: 25 μm). Schematic diagram generated by Biorender (agreement number: AR28HM9B0Q). f Primary HEPs (n = 6) and HSCs (n = 6) were isolated from the same liver. The mRNA expression of mouse Igfbp6. g Primary HSCs were isolated from WT mice treated with three doses of vehicle (PBS, n = 6) or TAA (100 mg/kg, n = 6) every 3 days. Igfbp6 mRNA was measured. h scRNA-seq dataset (GSE221481) from mice with uninjured liver or TAA-induced hepatic fibrosis was analyzed for the expression of Igfbp6. The fraction of cells expressing Igfbp6 in different cell lineages in liver. Data are expressed as mean ± SD, unpaired Student’s t test, two-tailed.
Validation in expanded datasets revealed elevated IGFBP6 expression in both carbon tetrachloride (CCl₄)-induced mouse models (Fig. 1d and Supplementary Fig. S1c, d) and human cirrhotic specimens (Supplementary Fig. S1e). In the TAA-induced hepatic fibrosis model, fibrosis developed at week 4, coinciding with IGFBP6 up-regulation. Extended TAA administration through week 8 sustained elevated IGFBP6 levels concurrent with progressive fibrosis (Fig. 1e). Collectively, these findings demonstrate IGFBP6 up-regulation during hepatic fibrogenesis across multiple etiologies in humans and mice.
IGFBP6 is specifically expressed in HSCs during hepatic fibrosis
Given the critical role of HSCs in hepatic fibrogenesis8, we investigated the cell-type specificity of IGFBP6. Primary HSCs isolated from normal mouse liver tissues exhibited significantly higher Igfbp6 expression compared to HEPs (Fig. 1f). In TAA-induced fibrotic livers, activated HSCs showed marked Igfbp6 enrichment relative to HEPs and KCs (Fig. 1g and Supplementary Fig. S2a). The expression of IGFBP6 and α-SMA increased progressively with prolonged TGF-β stimulation in LX2 cells line, indicating that IGFBP6 expression correlated with HSCs activation (Supplementary Fig. S2b). Analysis of a human primary HSCs sequencing dataset revealed a similar pattern, with elevated IGFBP6 expression in activated versus quiescent HSCs (Supplementary Fig. S2c). Single-cell RNA sequencing (scRNA-seq) data from mice (Fig. 1h) and human samples (Supplementary Fig. S2d, e) confirmed predominant IGFBP6 expression in HSCs, with minimal expression in other hepatic cell types including HEPs, KCs, and ECs. Evaluation of other IGFBPs in mouse and human sequencing data revealed no significant alterations except for IGFBP6 (Supplementary Fig. S3a–e). These findings demonstrate HSC-specific IGFBP6 expression during hepatic fibrosis, supporting its role in modulating the pathological process.
IGFBP6 synergizes with TGF-β in HSCs to drive fibrogenesis
We analyzed mouse hepatic tissues at mRNA and protein levels. Consistent with histopathological findings, TAA administration induced progressive IGFBP6 upregulation concomitant with elevated expression of fibrosis-associated markers30 (Fig. 2a, b). Given the central role of TGF-β in hepatic fibrosis31, primary mouse HSCs were stimulated with TGF-β, revealing concurrent increases in IGFBP6 expression, α-smooth muscle actin (α-SMA, an HSC activation marker)32, collagen type I alpha 1 (COL1A1), and fibronectin (FN) (ECM components)33 (Fig. 2c, d). We generated an IGFBP6-knockout LX2 cell line using CRISPR-Cas9 (Fig. 2e). IGFBP6 ablation significantly attenuated fibrogenic responses (Fig. 2f, g), demonstrating functional synergy between IGFBP6 and TGF-β in promoting hepatic fibrosis.

a qRT-PCR analysis of Acta2, Fn1, Col1a1, and Igfbp6 mRNA levels and b Western blotting analysis of FN, COL1A1, α-SMA, and IGFBP6 protein expression in hepatic tissue homogenates from control and TAA-treated mice at 4 and 8 weeks (n = 6), Data are expressed as means ± SD, One-way ANOVA. c qRT-PCR analysis of Acta2, Fn1, Col1a1 and Igfbp6 mRNA levels in primary mouse HSCs stimulated with TGF-β (10 ng/mL) for 12 h and 24 h or unstimulated (n = 3), Data are expressed as means ± SD, One-way ANOVA. d Western blotting analysis of protein expression levels of FN, COL1A1, α-SMA and IGFBP6 in primary mouse HSCs stimulated with TGF-β for 24 and 48 h or unstimulated. e–g Western blotting confirmed IGFBP6-knockout in LX2 cells line (e). Fibrosis-related gene and protein expression in LX2 cells line stimulated with TGF-β (10 ng/mL) with or without IGFBP6-knockout detected by (f) qRT-PCR and (g) Western blotting (n = 3). Data are expressed as means ± SD, Tow-way ANOVA.
Considering the pathogenic continuum from hepatitis through fibrosis to HCC34, we evaluated the clinical implications of sustained IGFBP6 expression in HCC. Receiver operating characteristic (ROC) analysis (Supplementary Fig. S4a), survival analysis (Supplementary Fig. S4b), and immune-infiltration assessment (Supplementary Fig. S4c) revealed that elevated IGFBP6 expression correlated with poorer prognosis and reduced T helper 17 (Th17) cell infiltration. These integrated findings substantiate the therapeutic hypothesis that IGFBP6 targeting may impede disease progression.
PA is identified as an anti-fibrotic agent targeting IGFBP6
To discover anti-fibrotic compounds targeting IGFBP6, we screened FDA-approved compounds in PubChem, identifying 30 candidates. Subsequent filtering using SwissADME with five drug-likeness rules, eight pharmacokinetic parameters, and bioavailability scores prioritized five compounds for validation (Fig. 3a, b). Quantitative real-time polymerase chain reaction (qRT-PCR) (Fig. 3c) and Western blotting (Fig. 3d) demonstrated that PA effectively suppressed TGF-β-induced fibrotic markers at transcriptional and translational levels, establishing PA (Fig. 3e) as a potent anti-fibrotic candidate. Cell Counting Kit-8 (CCK-8) assays confirmed minimal PA cytotoxicity across physiologically relevant concentrations in multiple hepatic cell types (Fig. 3f and Supplementary Fig. S5a). PA dose-dependently inhibited fibrotic markers in LX2 cells (Fig. 3g, h). Notably, PA reduced IGFBP6 protein expression without altering its mRNA levels, suggesting that PA regulates IGFBP6 in a protein expression-dependent manner.

a SwissADME analysis of 30 FDA-approved compounds related to IGFBP6 showing: Heatmap of five drug-likeness rules (Lipinski, Ghose, Veber, Egan, Muegge) and eight pharmacokinetic parameters (GI absorption, BBB permeation, P-gp substrate, CYP1A2/2C19/2C9/2D6/3A4 inhibition). b Bioavailability scores of the compounds. Effect of five candidate compounds on fibrosis-related indicators in TGF-β (10 ng/mL) stimulated primary mouse HSCs were detected (c) at 24 h for mRNA levels and (d) at 48 h for protein levels (n = 3). e Chemical structural of PA. f The effect of PA on cell viability of LX2 cells viability was measured using the CCK8 assay (n = 5). The effect of concentration gradients (0 μM, 0.1 μM, 1 μM, and10μM) of PA against FN, COL1A1, ACTA2, and IGFBP6 in TGF-β (10 ng/mL) -stimulated LX2 cells was examined using (g) qRT-PCR for mRNA levels and (h) Western blotting for protein levels (n = 3). Data are expressed as means ± SD, Two-way ANOVA.
PA targets IGFBP6 and facilitates its ubiquitin-mediated degradation
Molecular docking analysis revealed PA binding to IGFBP6 at ASN-29 with a binding energy of −4.3 kcal/mol (Fig. 4a). Microscale thermophoresis (MST) assay confirmed direct PA-IGFBP6 interaction (Fig. 4b). Cell thermal shift assay (CETSA) indicated that PA binding to IGFBP6 reduced its thermal stability, rendering IGFBP6 more susceptible to degradation (Fig. 4c). To verify the uniqueness of IGFBP6 as a target for PA efficacy, we performed Western blotting analysis. As shown in Fig. 4d, fibrosis indices decreased following PA treatment in the control group. However, when IGFBP6 was knocked out, PA lost its inhibitory effect. We assessed whether PA affected other members of the IGFBP family and found no effect on IGFBP1-5 (Supplementary Fig. S5b). These results suggest that PA exerts its therapeutic effect primarily by targeting IGFBP6.

a Three-dimensional binding structure of PA (green) with IGFBP6 (gray ribbon). The Pi-H interactions are depicted as magenta dashed lines. b MST analysis of His-tagged IGFBP6 binding to PA (n = 3). c CETSA melting curves and derived thermal stability profiles (n = 3). d Western blotting detection of protein expression levels of FN, COL1A1 and α-SMA in IGFBP6-knockout LX2 cells by PA (10 μM) intervention. The effect of PA’s inhibitory action on TGF-β-induced IGFBP6 overexpression in LX2 cells was assessed by Western blotting in the presence or absence of (e) the proteasome inhibitor MG132 or (f) the autophagy inhibitor 3-MA (n = 3). Data are expressed as means ± SD, Two-way ANOVA. g LX2 cells were treated with 1 µM PA in the presence or absence of MG132 for 12 h. IP detection was performed with anti-IGFBP6 antibody, and expression of ubiquitin-coupled IGFBP6 was detected with anti-UB antibody (n = 3). Data are expressed as means ± SD, Two-way ANOVA.
Mechanistic studies employing the proteasome inhibitor MG132 showed reversal of PA-mediated IGFBP6 reduction (Fig. 4e), whereas autophagy inhibition with 3-MA had no effect (Fig. 4f). Co-immunoprecipitation (Co-IP) assays demonstrated enhanced ubiquitination of IGFBP6 following PA treatment, which was potentiated by MG132 co-treatment and associated with abolished IGFBP6 degradation (Fig. 4g). These results indicate that PA specifically promotes IGFBP6 degradation via the ubiquitin-proteasome pathway.
PA confers protection against TAA-induced hepatic fibrosis in mice
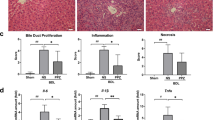
We assessed the antifibrotic efficacy of PA in a TAA-induced mouse model (Fig. 5a). Preliminary safety evaluation revealed no histopathological evidence of hepatotoxicity in PA-treated animals (Supplementary Fig. S5c). PA administration mitigated TAA-induced body weight loss (Fig. 5b) and reduced the liver-to-body weight ratio (Fig. 5c). Furthermore, TAA-treated mice developed hepatic fibrosis with extensive nodule formation, which was attenuated by PA treatment (Supplementary Fig. S6a). These effects were associated with reduced hepatic soluble collagen content (Fig. 5d). PA treatment also reduced inflammatory cell infiltration and collagen deposition in perisinusoidal regions of TAA-exposed livers, as determined by the Ishak scoring system35 (for inflammation and fibrosis) and analysis of positive areas (Fig. 5e–i and Supplementary Fig. S6b–d).

a Schematic representation of prophylactic in vivo studies utilizing a mouse model of TAA-induced hepatic fibrosis. Mice received intraperitoneal injections with gradually increasing concentrations of TAA and were simultaneously treated with PA by gavage (n = 6). Schematic diagram generated by Biorender (agreement number: AR28HM9B0Q). b Weight loss in mice with hepatic fibrosis. Data are expressed as means ± SD, Two-way ANOVA. c The liver-to-body weight ratio. d Soluble collagen in the liver. e H&E staining, Masson’s trichrome staining, and Sirius red staining of hepatic sections (scale bar: H&E staining, 100 μm; Masson’s trichrome staining and Sirius red staining, 200 μm). Three independent pathologists blindly evaluated the effects of TAA and PA on (f) fibrosis and (g) hepatitis using the Ishak scoring system. Mean values from each group were used for statistical analysis. Positive areas of (h) Masson’s trichrome and (i) Sirius red staining. Data are expressed as means ± SD, One-way ANOVA.
Immunohistochemical (IHC) analysis revealed dose-dependent suppression of TAA-induced α-SMA and COL1A1 expression in hepatic tissues (Fig. 6a, b), with maximal effects observed at higher doses. Western blotting (Fig. 6c, d) and qRT-PCR (Fig. 6e) analyses confirmed the dose-dependent antifibrotic effects of PA.

a IHC staining of α-SMA and COL1A1 in mouse liver sections (scale bar: 50 μm). b Quantification of positive staining area (n = 6). c Western blotting of FN, COL1A1, and α-SMA protein expression in hepatic tissue homogenates and d statistical analyses (n = 6). e Detection of Fn1, Col1a1, Col1a2, Acta2 and f p21 expression by qRT-PCR in RNA extracted from mice hepatic tissue homogenate (n = 6). Data are expressed as means ± SD, One-way ANOVA.
Considering the association between fibrogenesis and cellular proliferation/senescence, we examined cyclin-dependent kinase inhibitor 1A (p21) and proliferating cell nuclear antigen (PCNA) expression36,37. While TAA stimulation significantly upregulated both markers in fibrotic livers, PA treatment specifically reduced p21 expression in HSCs without significantly affecting PCNA levels (Fig. 6f and Supplementary Fig. S7a–c). This pattern excludes potential induction of senescence-associated secretory phenotype (SASP) or oncogenic transformation by PA.
PA exerts therapeutic effects on established hepatic fibrosis in mice
Since pharmacological interventions for hepatic fibrosis typically target established fibrosis, we examined PA’s therapeutic efficacy in this context. As shown in Fig. 7a, mice received TAA for 4 weeks to induce fibrosis, followed by daily oral PA treatment with continued TAA administration. After 4 weeks of treatment, PA prevented TAA-induced body weight loss (Fig. 7b) and reduced the elevated liver-to-body weight ratio (Fig. 7c). PA treatment also significantly reduced hepatic nodule formation compared to the TAA group (Supplementary Fig. S8a). Soluble collagen content in hepatic homogenates was reduced by PA treatment (Fig. 7d), further confirming its therapeutic efficacy. Histological analysis revealed severe hepatic structural disruption, extracellular matrix (ECM) deposition, and inflammatory cell infiltration in TAA-treated mice, all of which were attenuated by PA (Fig. 7e–i and Supplementary Fig. S8b–d).

a Schematic of therapeutic intervention in TAA-induced hepatic fibrosis. Mice received intraperitoneal TAA injections at gradually increasing concentrations for 4 weeks, followed by oral gavage of PA for fibrosis treatment (n = 6). Schematic diagram generated by Biorender (agreement number: AR28HM9B0Q). b Trends in body weight of mice in control and PA-treated groups. Data are expressed as means ± SD, Two-way ANOVA. c Liver-to-body weight ratio. d Expression of soluble collagen in hepatic tissues. e Representative H&E staining, Masson’s trichrome staining, and Sirius red staining of hepatic sections (scale bar: H&E staining, 100 μm; Masson’s trichrome staining and Sirius red staining, 200 μm). Positive areas of (f) Masson’s trichrome and (g) Sirius red staining. The effects of TAA and PA on (h) fibrosis and (i) hepatitis using the Ishak scoring system. Data are expressed as means ± SD, One-way ANOVA.
Western blotting (Fig. 8a, b) and IHC (Fig. 8c, d) demonstrated that PA significantly suppressed TAA-induced upregulation of α-SMA and COL1A1. Similarly, PA downregulated mRNA levels of Acta2, Col1a1, Col1a2 and Fn1 (Fig. 8e). These findings demonstrate that PA is a potent therapeutic agent against hepatic fibrosis. Notably, PA limited the progression of established hepatic fibrosis, suggesting therapeutic potential for clinical application.

a The protein levels of mice hepatic tissue homogenates were assayed by Western blotting and b quantitative methods (n = 6). c IHC was performed to detect the expression of α-SMA and COL1A1 in hepatic sections and d analysis of positive area (scale bar: 50 μm) (n = 6). e Detection of mRNA expression levels of fibrosis-related indicators in hepatic tissues by qRT-PCR (n = 6). Data are expressed as means ± SD, One-way ANOVA.
PA ameliorates fibrosis through IGFBP6-mediated suppression of the TGF-β/SMAD signaling pathway
To delineate the antifibrotic mechanism of PA, potential targets were predicted using PharmMapper and GeneCards databases. Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway and Gene Ontology (GO) enrichment analyses identified TGF-β signaling as the predominant pathway (Fig. 9a). Functional clustering analysis further confirmed the centrality of the TGF-β signaling pathway (Supplementary Fig. S9a, b). Experimental validation corroborated these findings. In HSCs stimulated with TGF-β for 12 or 24 h, PA inhibited TGF-β-induced phosphorylation of SMAD2/3 (Fig. 9b), with the inhibitory effect showing concentration dependence (Fig. 9c). Concordant results were obtained in vivo, where PA treatment attenuated TAA-induced elevation of phosphorylated SMAD2/3 levels, as demonstrated by Western blotting and immunohistochemistry (Fig. 9d, e).

a Enrichment analysis results after collection of PA targets via Pharmmapper database and GeneCards database. b Western blotting assay for the influence of PA (10 μM) on different times of TGF-β (10 ng/mL) stimulation-initiated SMAD2/3 phosphorylation levels in primary mouse HSCs (n = 3). c Effects of different concentrations of PA (0 μM, 0.1 μM, 1 μM, and10 μM) on the phosphorylation level of SMAD2/3 induced by TGF-β (10 ng/mL) action in primary HSCs of mice analyzed by Western blotting (n = 3). Effect of PA on phosphorylation levels of SMAD2/3 and IGFBP6 in TAA-induced fibrotic hepatic mice in vivo by (d) Western blotting and (e) IHC (n = 6, scale bar: 50 μm).
Having established IGFBP6 as PA’s molecular target, we investigated whether PA-mediated TGF-β/SMAD inhibition required IGFBP6. PA effectively suppressed SMAD2/3 phosphorylation and nuclear translocation in control cells, whereas this inhibitory effect was abrogated in IGFBP6-knockout cells (Fig. 10a–c). Co-treatment with PA and SIS3 (a selective TGF-β/SMAD inhibitor) did not exhibit synergistic inhibition of TGF-β-induced fibrotic gene expression, despite both agents individually suppressing these markers (Fig. 10d). These data demonstrate that PA primarily targets IGFBP6 to inhibit the TGF-β/SMAD signaling cascade.

Phospho-SMAD2/3 levels detected by Western blotting in (a) control group and (b) IGFBP6-knockout group treated with TGF-β (10 ng/mL) for 15 min, 30 min, or 60 min with or without PA (10 µM) (n = 3). Data are expressed as means ± SD, One-way ANOVA. c Immunofluorescence analysis of TGF-β (10 ng/mL) -induced SMAD2/3 nuclear translocation in LX2 cells with or without IGFBP6 knockout, treated with or without PA (10 µM) (scale bar: 25 μm). d The mRNA expression levels of Acta2, Fn1, and Col1a1 in primary mouse HSCs in the presence or absence of PA or the SMAD3 inhibitor SIS3 were detected by qRT-PCR (n = 3). Data are expressed as means ± SD, Two-way ANOVA.
Discussion
The IGFBP family exerts context-dependent roles in growth, metabolism, and pathogenesis through spatiotemporal regulation of IGF bioavailability19. IGFBP6 displays substantial heterogeneity across fibrotic pathologies in different organ systems38. Specifically, IGFBP6 is significantly downregulated in plasma samples from patients with acute myocardial infarction (AMI) and carotid atherosclerotic plaques and is considered a key biomarker for predicting vulnerable plaques and AMI39. In kidney disease, circulating IGFBP6 concentrations demonstrate disease-stage specificity, with levels reduced in chronic kidney disease (CKD) patients but markedly increased in end-stage renal disease (ESRD) patients40,41.
IGFBP6 exhibits a complex association with hepatic pathology, showing positive correlation with steatosis in non-alcoholic fatty liver disease (NAFLD)42. In chronic hepatitis C (CHC) patients with fibrosis, Reyes et al. correlated serum IGFBP1-7 expression levels with fibrosis stages, reporting elevated concentrations of all IGFBPs during advanced fibrosis stages but with distinct regulatory patterns43. This functional heterogeneity underscores the need for further investigation into IGFBP6’s organ-specific roles. Given potential metabolic alterations affecting housekeeping genes44, vinculin, β-actin and glyceraldehyde-3-phosphate dehydrogenase (GAPDH) expression was evaluated. IGFBP6-associated metabolic changes did not compromise GAPDH’s reliability as a housekeeping gene. Similarly, PA administration did not alter GAPDH expression, confirming its reliability as a housekeeping control under these experimental conditions (Supplementary Fig. S10). Our study demonstrates that IGFBP6 is the most significantly upregulated IGFBP in fibrotic hepatic tissues, suggesting its potential as a stage-specific biomarker for hepatic fibrogenesis.
We demonstrate that IGFBP6 is enriched in quiescent HSCs and exhibits marked upregulation during HSC activation, establishing a mechanistic link between its pathological overexpression and advanced fibrosis stages in both patients and experimental models. These findings establish the HSC-specific profibrotic function of IGFBP6 in hepatic fibrosis. Crucially, genetic ablation or pharmacological inhibition of IGFBP6 significantly attenuated HSC activation and arrested fibrotic progression in vitro and in vivo, demonstrating its indispensable role in hepatic fibrogenesis. Our study establishes IGFBP6 as a regulator of HSC-specific activation programs and direct driver of hepatic fibrogenesis, thereby identifying it as a diagnostic biomarker and therapeutic target for hepatic fibrosis.
Another pivotal finding was the identification of PA as an effective IGFBP6-targeted antifibrotic agent through network pharmacology screening of FDA-approved drugs. Although PA has an established role in coenzyme A biosynthesis25, we uncovered its function in suppressing TGFβ-induced SMAD2/3 phosphorylation and nuclear translocation.
Although direct suppression of the TGF-β/SMAD pathway provides a plausible mechanism for PA’s therapeutic effects, the contribution of non-SMAD regulation roles of TGF-β, such as oxidative stress-related signaling, remains significant in hepatic fibrogenesis45,46. PA has been reported to confer protection against oxidative stress and mitochondrial dysfunction47,48. Thus, whether early PA intervention attenuates hepatic fibrosis through suppression of oxidative stress or other SMAD-independent mechanisms, as well as whether PA’s metabolic regulatory functions synergistically modulate hepatocyte subpopulations to influence fibrotic progression, warrants systematic investigation.
This study has several limitations. While incorporating datasets from human cells and tissues, our mechanistic insights and in vivo validations primarily relied on preclinical mouse models and did not assess diverse pathogenic drivers. PA concentrations for in vivo administration were selected based on previous literature49,50; however, systematic validation of hepatic drug concentrations and their correlation with in vitro dosing regimens remains warranted. Although PA demonstrated therapeutic efficacy in mice with established hepatic fibrosis, its capacity to promote fibrotic regression has not been examined.
In conclusion, our findings demonstrate a cell-type-specific role of IGFBP6 in HSC activation and hepatic fibrosis progression. Pharmacological targeting of IGFBP6 with PA shows potential as a therapeutic strategy for hepatic fibrosis treatment.
Methods
Mouse models of hepatic fibrosis
All animal experiments were approved by the Animal Care and Use Committee of Jinan Central Hospital (No. JNCH-202114) and complied with all relevant ethical regulations for animal use. Eight-week-old C57BL/6J mice (body weight: 20–30 g; randomly grouped and treated; male and female animals; supplied by Jinan Bangyue Laboratory Animal Co.) were housed under a 12-h/12-h light/dark cycle in temperature-controlled rooms with ad libitum access to food and water. No more than five mice were housed per cage. For TAA-induced HSC activation in vivo, mice were intraperitoneally injected with three doses of TAA (MedChemExpress, HY-Y0698; 100 mg/kg body weight, dissolved in PBS) or vehicle every 3 days. HSCs were isolated 24 h after the final TAA injection. To establish the TAA-induced hepatic fibrosis model51, mice received intraperitoneal injections of TAA three times per week with escalating doses: 50 mg/kg in week 1, 100 mg/kg in week 2, and increments of 100 mg/kg every 2 weeks until week 8. During the entire 8-week TAA treatment period, prophylactic and therapeutic studies were conducted using PA (MedChemExpress, HY-B0430) prepared in saline. In the prophylactic study, PA (10 or 50 mg/kg)49 was administered by gavage concurrently with TAA administration for 8 weeks. In the therapeutic study52, PA (50 mg/kg) was administered starting 4 weeks after TAA induction and continued through the eighth week, with ongoing TAA treatment. Saline was administered as a control in both experiments. Mice were suffocated to death by inhaling carbon dioxide (concentration 40%). No animals or data points were excluded from the analysis in any experimental group.
Primary mouse hepatic cells
Mouse HSCs, HEPs, and KCs were isolated from mouse hepatic tissues using a combination of perfusion, collagenase digestion, and density gradient centrifugation. Briefly, mouse livers were perfused with 0.2 mM EGTA (MedChemExpress, HY-D0861) and 5 mg/ml Liberase (Roche, 05401020001) via the inferior vena cava. The livers were then removed and minced under sterile conditions. The cell suspension was filtered through a 100 μm cell strainer, and the cells were washed to remove excess digestive enzymes. HEPs were centrifuged to pellet for seeding, and the supernatant was collected for further density gradient centrifugation to separate different non-parenchymal cell types. For HSC isolation, Nycodenz (NycoPrep, 1002424; Axis-Shield)-cell density gradient centrifugation was performed53. The purity of isolated HSCs was >90% as determined by vitamin A autofluorescence detection. For KC isolation, a discontinuous Percoll (25/50%, Cytiva, 17089101) cell density gradient was used54. Mixtures of LSECs and KCs were collected at the interface between the two density gradients. KCs were separated based on differential adhesion time. Primary HEPs were cultured in HepatoZYME-SFM (Gibco, 17705-021) medium containing 1% penicillin/streptomycin (P/S, Gibco, 15140-122). Primary HSCs were cultured in Dulbecco’s modified Eagle’s medium (DMEM, Gibco, 11995-073) containing 10% fetal bovine serum (FBS, Gibco, 10099-141) and 1% P/S. Primary KCs were cultured in RPMI 1640 medium (Gibco, 11875-093) with 10% FBS and 1% P/S. Primary cells isolated from normal mouse hepatic tissues were passaged no more than twice, whereas those from TAA-stimulated tissues were used at passage 1. All cells were tested with MycAwayTM Plus-Color One-Step Mycoplasma Detection Kit (2G) (Yeasen 40615ES) and confirmed to be free of mycoplasma contamination.
Cell lines and cell-related experiments
LX2 cells (Procell, CL-0560), 293T cells (Procell, CL-0005), and THLE-2 cells (Cell Bank of the Chinese Academy of Sciences, SCSP-5068) were maintained in DMEM containing 10% FBS and 1% P/S. All cell lines were used within 15 passages. PA was dissolved in Dimethyl Sulfoxide (DMSO, MedChemExpress, HY-Y0320) to generate a concentration gradient. TGF-β was diluted to 10 μg/mL according to the manufacturer’s instructions, and 1 μL was added to each milliliter of medium to achieve a final concentration of 10 ng/mL. PA was then added to specified concentrations according to experimental groups, followed by incubation for specified durations. MG132 (MedChemExpress, HY-13259) was diluted in serum-free medium. Cells were divided into groups and treated with fresh medium containing MG132 (final concentration: 15 μM) and TGF-β (10 ng/mL), and incubated at 37 °C for 24 h. Subsequently, PA (1 μM final concentration) was added, and incubation continued for 24 h. After removing the supernatant, cells were collected. 3-MA (MedChemExpress, HY-193123) was dissolved in sterile deionized water (ddH₂O) as a 100 mM stock solution and diluted to 5 mM working concentration in complete medium. Cells were treated with fresh medium containing 5 mM 3-MA and 10 ng/mL TGF-β, and incubated at 37 °C for 4 h. PA was added to a final concentration of 1 μM and incubation continued for 12 h before cell harvest. SIS3 (MedChemExpress, HY-13013) was dissolved in DMSO as a 20 mM stock solution. LX2 cells were treated with complete medium containing 5 μM SIS3. According to experimental design, 10 μM PA and 10 ng/mL TGF-β were added at final concentrations, followed by incubation at 37 °C for 24 h. Control groups were included in all experiments as required. For drug screening, the following compounds were used: biotin (MedChemExpress, HY-B0511), dexamethasone (MedChemExpress, HY-14648), propylthiouracil (MedChemExpress, HY-B0346), and zinc pyrithione (MedChemExpress, HY-B0572). Screening concentrations were 10 μM. Mycoplasma testing using the MycAwayTM Plus-Color One-Step Mycoplasma Detection Kit (2 G) (Yeasen 40615ES) confirmed mycoplasma-free status.
Western blotting
Tissue homogenates or cultured cells were lysed in RIPA buffer (Beyotime, P0013B) and sonicated. Protein concentrations were determined using a BCA assay kit (Beyotime, P0011). After heating at 95 °C for 10 min, samples were separated by SDS-PAGE electrophoresis and transferred onto nitrocellulose (NC) membranes (Servicebio, G6014). The membranes were blocked with 5% skimmed milk in Tris-buffered saline containing 0.1% Tween-20 (TBST) for 1 h at room temperature, followed by overnight incubation with primary antibodies at 4 °C. Primary antibodies included: α-SMA (Proteintech, 14395), FN (Proteintech, 15613), COL1A1 (Proteintech, 66761), IGFBP6 (Proteintech, 18445), GAPDH (Proteintech, 60004), β-tubulin (Cell Signaling Technology, 2146), Phosphorylated SMAD2/3 (p-SMAD2/3, Cell Signaling Technology, 8828), and SMAD2/3 (Cell Signaling Technology, 72255). After washing, membranes were incubated with secondary antibodies (ZSGB-BIO, ZB-2305; ZB-2301) for 1 h at room temperature. Protein bands were visualized using chemiluminescent reagent (Biosharp, BL520B) and imaged with a gel documentation system (ProteinSimple, FluorChem M). Densitometric analysis was performed using ImageJ software (National Institutes of Health, 1.8.0).
qRT-PCR
Total RNA was extracted using the Steady Pure Universal RNA Extraction Kit (Precision Biotechnology, AG21017), followed by reverse transcription with the Evo M-MLV Mix Kit with gDNA Clean for qPCR (Precision Biotechnology, AG11728). qRT-PCR was performed using SYBR Green Premix Pro Taq HS qPCR Kit (Precision Biotechnology, AG11701) on a LightCycler 480 II system (Roche). Reactions were conducted in 20 μL volumes containing 10 μL Premix, 0.4 μL each of forward and reverse primers (final concentration 0.2 μM), 2 μL cDNA template, and nuclease-free water. After brief centrifugation, PCR was performed under the following conditions: initial denaturation at 95 °C for 30 s; 40 cycles of denaturation at 95 °C for 10 s and annealing/extension at 60 °C for 30 s (fluorescence acquisition at each cycle); melting curve analysis with 95 °C for 15 s, 60 °C for 1 min, and 95 °C continuous ramping. Detailed procedures followed manufacturers’ protocols. Target gene mRNA levels were quantified using the ΔΔCT method normalized to GAPDH expression.
CRISPR-Cas9 knockout
The IGFBP6-specific sgRNA (5’-GTGCCCAATTGTGACCATCG-3’) was cloned into the lentiCRISPR v2 vector (Addgene, 52961) downstream of the U6 promoter. The plasmid was co-transfected with packaging plasmids psPAX2 (Addgene, 12260) and pMD2.G (Addgene, 12259) into HEK293T cells (ratio 4:3:1) using polyethylenimine (PEI, Beyotime, C0539)55. Viral supernatant was collected 48 h post-transfection, concentrated by ultracentrifugation (25,000 × g, 2 h), and resuspended in PBS. LX2 cells were infected with lentivirus (MOI 20) in the presence of 5 μg/ml polybrene (MedChemExpress, HY-112735) for 48 h. Stable knockout pools were selected using 2.5 μg/mL puromycin (MedChemExpress, HY-B1743) until untreated control cells completely died. Monoclonal cell lines were isolated via limiting dilution.
CCK-8 assay
Cell viability was assessed using the CCK-8 kit (Beyotime, C0039) following the manufacturer’s protocol. Cells were seeded at 5 × 10³ per well in 96-well plates and incubated for 24 h. Cells were then treated with PA (0.01-1000 μM) for 24 h. Drug-containing medium was replaced with 100 μL fresh medium, and 10 μL CCK-8 reagent was added. After incubation at 37 °C for 1 h protected from light, absorbance was measured at 450 nm using a microplate reader (SpectraMax i3x, Molecular Devices). Cell viability was calculated according to the manufacturer’s instructions.
Molecular docking
Molecular docking of PA with IGFBP6 was performed using MOE software56. The 2D structure of PA was retrieved from PubChem. The IGFBP6 crystal structure was obtained from the RCSB Protein Data Bank (http://www.rcsb.org/). The top-ranked docking conformation based on docking score and confidence score was selected for further analysis. 3D visualization was generated with PyMOL 2.04 (Schrödinger), while 2D interaction analysis including interaction types, bond distances, and frequency statistics was performed using Maestro (academic version, Schrödinger).
MST assay
Recombinant human IGFBP6 protein with His-tag (MCE, HY-P73141) was purified using Ni-NTA affinity chromatography (Thermo Scientific, HisPur). Bacterial lysate was loaded onto a Ni-NTA column after cell lysis. The column was sequentially washed with buffers containing 20 mM and 50 mM imidazole, followed by elution with 250 mM imidazole. The eluate was dialyzed to remove imidazole and concentrated. Purified proteins were labeled using RED-tris-NTA protein labeling kit (NanoTemper) at 4 °C for 30 min in the dark. Free dye was removed by centrifugation. Labeled IGFBP6 was incubated with PA gradient concentrations in PBST buffer (0.05% Tween-20) for 10 min at room temperature. Samples were loaded into Monolith NT capillaries, and MST data were collected at high LED power using a NanoTemper instrument57. Dissociation constants were determined through NanoTemper Analysis software (v1.5.41).
CETSA
LX2 cells were seeded in petri dishes. Upon reaching 80% confluency, cells were treated with either 1 µM PA or DMSO control for 6 h. Cells were then harvested and resuspended in PBS containing protease inhibitors. Aliquots were distributed into PCR tubes for thermal denaturation at graded temperatures (40, 45, 50, 55, 60, 65, 70, 75, 80 °C) for 3 min, followed by immediate cooling in an ice bath for 3 min58. This heating-cooling cycle was repeated three times. After centrifugation at 20,000 × g, supernatants were collected and boiled for 10 min in SDS-PAGE loading buffer. Protein stability was assessed by Western blotting analysis.
Co-IP assay
Cells were harvested at 80–90% confluency and lysed in pre-cooled RIPA buffer on ice for 30 min. Lysates were centrifuged at 12,000 × g for 15 min at 4 °C. The resulting supernatant was incubated overnight at 4 °C with anti-IGFBP6 primary antibody (Proteintech, 32157, 2 μg for 1 mg total protein lysate). Protein A/G agarose beads (Santa Cruz Biotechnology, sc-2003) were then added and incubated for 4 h at 4 °C with rotation to minimize nonspecific binding. Beads were washed 6 times with ice-cold lysis buffer. Bound proteins were eluted by boiling in SDS-PAGE loading buffer for 5 min. Eluates were subjected to Western blotting analysis.
Histology
Hepatic tissues were collected and fixed in 10% neutral buffered formalin for 24 h, then embedded in paraffin. For hematoxylin-eosin (H&E) staining (Beyotime, C0105M), 4μm paraffin sections were deparaffinized, rehydrated, and stained with hematoxylin and eosin. For Sirius Red staining (Solarbio, G1472), dewaxed sections were incubated with 0.1% Sirius Red in saturated picric acid for 1 h, then washed twice with acidified water (0.5% HCl). For Masson’s trichrome staining (Solarbio, G1340), sections were deparaffinized, stained with Weigert’s iron hematoxylin for 10 min, rinsed with distilled water, stained with acid fuchsin for 10 min, rinsed, treated with phosphomolybdic acid for 5 min, and counterstained with aniline blue for 5 min. Sections were incubated in 1% glacial acetic acid for 1 min, dehydrated through graded ethanol and xylene, and mounted with neutral balsam. For IHC staining (ZSGB-BIO, SP9000). Paraffin sections were deparaffinized in xylene, rehydrated through graded ethanol, and washed in PBS. Antigen retrieval was conducted in 10 mM citrate buffer (Solarbio, C1010) at 95 °C for 10 min. Endogenous peroxidase was blocked with peroxidase inhibitor for 10 min at room temperature. After PBS rinsing, sections were blocked with normal goat serum for 30 min. Primary antibodies were applied overnight at 4 °C, followed by PBS washing. Sections were sequentially incubated with biotinylated goat anti-rabbit IgG for 20 min and horseradish peroxidase-conjugated streptavidin for 20 min at room temperature, with PBS washing after each step. Color development was performed using DAB substrate, followed by hematoxylin counterstaining for 20 s, graded ethanol dehydration, xylene clearing, and mounting with neutral balsam. IHC-related primary antibody information: α-SMA (Proteintech, 14395, 1:100 dilution); COL1A1 (Proteintech, 14695, 1:200 dilution,); p-SMAD2/3 (Cell Signaling Technology, 8828, 1:200 dilution) and IGFBP6 (Proteintech, 18445, 1:200 dilution). The sections were observed and captured using a fully automatic pathological section scanner (LogiScience, MSCAN200). ImageJ software was used to analyze and statistically evaluate the area of positive regions.
Sircol soluble collagen assay
Hepatic collagen content was quantified using the Sircol Soluble Collagen Assay (Biocolor, S1000), which specifically binds to collagen types I-V. Procedures followed manufacturer’s instructions. Briefly, hepatic tissues from experimental groups and collagen standards were homogenized. Aliquots (100 mg tissue/sample) were mixed with 1 ml Sircol Dye Reagent, vortexed thoroughly, and incubated at room temperature for 30 min. After centrifugation at 10,000 × g for 10 min at room temperature, supernatants were discarded. Pellets were completely dissolved in 1 ml Alkaline Reagent. Absorbance was measured at 540 nm, and collagen concentrations were calculated using a standard curve with normalization to tissue weight.
Immunofluorescence
Cells were seeded in an encapsulated recapitulation chamber. After PBS washing, cells were fixed in ice-cold methanol for 10 min and blocked with PBS containing 0.25% Triton X-100 and 1% BSA for 30 min. Primary antibody SMAD2/3 (Cell Signaling Technology, 72255, 1:200 dilution) incubation was performed overnight at 4 °C. Following washes, samples were incubated with secondary antibody (Abcam, ab150080, 1:400 dilution) for 1 h and counterstained with 4′,6-diamidino-2-phenylindole (DAPI, SouthernBiotech, 0100-20). Fluorescence visualization was conducted using confocal microscopy.
Sequencing dataset analysis
Nine RNA-seq datasets from the GEO database were analyzed using GEO2R. These included datasets related to hepatitis C virus (HCV) hepatitis and associated hepatic fibrosis59 (GSE182065, GSE85547, GSE85548), hepatic samples from cirrhotic patients60,61 (GSE89377), hepatic tissues from mouse fibrosis models62,63 (GSE130123, GSE34640), and human hepatic stellate cell (HSC) samples64 (GSE68001). scRNA-seq data from GSE13610365,66 were analyzed to characterize gene expression in cells isolated from uninjured and cirrhotic human hepatic tissues. Additionally, scRNA-seq data from GSE22148167 were examined to assess IGFBP6 expression in HSCs and other hepatic cell types in fibrotic hepatic tissues induced by chemical toxicants in mice. All data analyses were performed following established protocols.
Bioinformatics analysis
Enrichment analysis of DEGs and proteins (DEPs) was performed for GO terms (biological processes, cellular components, molecular functions) and KEGG pathways. Statistical significance of enrichment was evaluated using the hypergeometric test, with terms showing p-values < 0.05 considered significant. Protein-protein interactions (PPIs) were predicted using the STRING database, followed by network construction using Cytoscape software. Topological analysis of PPI networks was performed using cytoHubba and MCODE plugins for identification of key functional modules.
Statistical analysis and reproducibility
Data represent mean ± standard deviation (SD) of at least three independent biological experiments (unless stated otherwise), with individual replicates (n) specified in figure legends. Normality was assessed using the Shapiro–Wilk test. Differences between two groups were evaluated by unpaired, two-tailed Student’s t-tests. For comparisons involving multiple groups, one-way or two-way ANOVA was employed, followed by appropriate post hoc tests (Sidak’s, Tukey’s, or Dunnett’s) for multiple comparisons. All statistical analyses were conducted in GraphPad Prism (version 8.0). Statistical significance was defined as p < 0.05.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
Source data for all graphs are available in the Supplementary Data files, with uncropped blots provided in Supplementary Data 1 and statistical source data in Supplementary Data 2. Data related to RNA sequence analysis have been deposited in the GEO, with the analysis code detailed in the Methods section (Sequencing dataset analysis). All other data supporting the findings of this study are available from the corresponding author upon reasonable request.
References
Hernandez-Gea, V. & Friedman, S. L. Pathogenesis of liver fibrosis. Annu Rev. Pathol. 6, 425–456 (2011).
Kisseleva, T. & Brenner, D. Molecular and cellular mechanisms of liver fibrosis and its regression. Nat. Rev. Gastroenterol. Hepatol. 18, 151–166 (2021).
Banerjee, A. & Farci, P. Fibrosis and hepatocarcinogenesis: role of gene-environment interactions in liver disease progression. Int. J. Mol. Sci. 25, 8641 (2024).
Trautwein, C., Friedman, S. L., Schuppan, D. & Pinzani, M. Hepatic fibrosis: concept to treatment. J. Hepatol. 62, S15–S24 (2015).
Zheng, Y., Xie, L., Yang, D., Luo, K. & Li, X. Small-molecule natural plants for reversing liver fibrosis based on modulation of hepatic stellate cells activation: an update. Phytomedicine 113, 154721 (2023).
Harrison, S. A. et al. Selonsertib for patients with bridging fibrosis or compensated cirrhosis due to NASH: results from randomized phase III STELLAR trials. J. Hepatol. 73, 26–39 (2020).
Anstee, Q. M. et al. Cenicriviroc lacked efficacy to treat liver fibrosis in nonalcoholic steatohepatitis: AURORA phase III randomized study. Clin. Gastroenterol. Hepatol. 22, 124–134.e121 (2024).
Tsuchida, T. & Friedman, S. L. Mechanisms of hepatic stellate cell activation. Nat. Rev. Gastroenterol. Hepatol. 14, 397–411 (2017).
Zhang, Z. et al. ANGPTL8 accelerates liver fibrosis mediated by HFD-induced inflammatory activity via LILRB2/ERK signaling pathways. J. Adv. Res. 47, 41–56 (2023).
Zhang, X. et al. GARP on hepatic stellate cells is essential for the development of liver fibrosis. J. Hepatol. 79, 1214–1225 (2023).
Shinn, J. et al. Antioxidative hyaluronic acid-bilirubin nanomedicine targeting activated hepatic stellate cells for anti-hepatic-fibrosis therapy. ACS Nano 18, 4704–4716 (2024).
Hao, M. et al. Atractylodinol prevents pulmonary fibrosis through inhibiting TGF-beta receptor 1 recycling by stabilizing vimentin. Mol. Ther. 31, 3015–3033 (2023).
Yuen, D. A. et al. Recombinant N-terminal Slit2 inhibits TGF-beta-induced fibroblast activation and renal fibrosis. J. Am. Soc. Nephrol. 27, 2609–2615 (2016).
Katanasaka, Y. et al. Fibroblast-specific PRMT5 deficiency suppresses cardiac fibrosis and left ventricular dysfunction in male mice. Nat. Commun. 15, 2472 (2024).
Akhurst, R. J. Targeting TGF-beta Signaling for Therapeutic Gain. Cold Spring Harb. Perspect. Biol. 9, a022301 (2017).
Connolly, E. C., Freimuth, J. & Akhurst, R. J. Complexities of TGF-beta targeted cancer therapy. Int. J. Biol. Sci. 8, 964–978 (2012).
Yingling, J. M., Blanchard, K. L. & Sawyer, J. S. Development of TGF-beta signalling inhibitors for cancer therapy. Nat. Rev. Drug Discov. 3, 1011–1022 (2004).
Jiang, F., Liu, G. S., Dusting, G. J. & Chan, E. C. NADPH oxidase-dependent redox signaling in TGF-beta-mediated fibrotic responses. Redox Biol. 2, 267–272 (2014).
Baxter, R. C. Signaling pathways of the insulin-like growth factor binding proteins. Endocr. Rev. 44, 753–778 (2023).
Hwa, V., Oh, Y. & Rosenfeld, R. G. The insulin-like growth factor-binding protein (IGFBP) superfamily. Endocr. Rev. 20, 761–787 (1999).
Arab, J. P. et al. Hepatic stellate cell activation promotes alcohol-induced steatohepatitis through Igfbp3 and SerpinA12. J. Hepatol. 73, 149–160 (2020).
Lee, Y. Y. et al. Loss of tumor suppressor IGFBP4 drives epigenetic reprogramming in hepatic carcinogenesis. Nucleic Acids Res. 46, 8832–8847 (2018).
Kluge, A. et al. Coordinate expression of the insulin-like growth factor system after microembolisation in porcine heart. Cardiovasc. Res. 33, 324–331 (1997).
Bach, L. A., Fu, P. & Yang, Z. Insulin-like growth factor-binding protein-6 and cancer. Clin. Sci.124, 215–229 (2013).
Spry, C., Kirk, K. & Saliba, K. J. Coenzyme A biosynthesis: an antimicrobial drug target. FEMS Microbiol. Rev. 32, 56–106 (2008).
Nawaz, M. Z. et al. Discovery and characterization of dual inhibitors of human Vanin-1 and Vanin-2 enzymes through molecular docking and dynamic simulation-based approach. Int J. Biol. Macromol. 213, 1088–1097 (2022).
Frost, Z., Bakhit, S., Amaefuna, C. N., Powers, R. V. & Ramana, K. V. Recent advances on the role of B vitamins in cancer prevention and progression. Int. J. Mol. Sci. 26, 1967 (2025).
Kreuzaler, P. et al. Vitamin B(5) supports MYC oncogenic metabolism and tumor progression in breast cancer. Nat. Metab. 5, 1870–1886 (2023).
Mian, S. A. et al. Vitamin B5 and succinyl-CoA improve ineffective erythropoiesis in SF3B1-mutated myelodysplasia. Sci. Transl. Med. 15, eabn5135 (2023).
Lin, C. Y., Adhikary, P. & Cheng, K. Cellular protein markers, therapeutics, and drug delivery strategies in the treatment of diabetes-associated liver fibrosis. Adv. Drug Deliv. Rev. 174, 127–139 (2021).
Meng, X. M., Nikolic-Paterson, D. J. & Lan, H. Y. TGF-beta: the master regulator of fibrosis. Nat. Rev. Nephrol. 12, 325–338 (2016).
Liu, X. et al. Ginsenoside Rg3 promotes regression from hepatic fibrosis through reducing inflammation-mediated autophagy signaling pathway. Cell Death Dis. 11, 454 (2020).
Schuppan, D., Ashfaq-Khan, M., Yang, A. T. & Kim, Y. O. Liver fibrosis: Direct antifibrotic agents and targeted therapies. Matrix Biol. 68-69, 435–451 (2018).
Sankar, K. et al. Recent advances in the management of hepatocellular carcinoma. Clin. Mol. Hepatol. 30, 1–15 (2024).
Desmet, V. J. et al. Formulation and application of a numerical scoring system for assessing histological activity in asymptomatic chronic active hepatitis. J. Hepatol. 38, 382–386 (2003).
Huda, N. et al. Hepatic senescence, the good and the bad. World J. Gastroenterol. 25, 5069–5081 (2019).
Ikeda, H. et al. Sphingosine 1-phosphate regulates regeneration and fibrosis after liver injury via sphingosine 1-phosphate receptor 2. J. Lipid Res. 50, 556–564 (2009).
Liso, A. et al. IGFBP-6: At the crossroads of immunity, tissue repair and fibrosis. Int. J. Mol. Sci. 23, 4358 (2022).
Liu, Y., Huan, W., Wu, J., Zou, S. & Qu, L. IGFBP6 is downregulated in unstable carotid atherosclerotic plaques according to an integrated bioinformatics analysis and experimental verification. J. Atheroscler. Thromb. 27, 1068–1085 (2020).
Jarkovska, Z. et al. Plasma ghrelin levels in patients with end-stage renal disease. Physiol. Res. 54, 403–408 (2005).
Christensson, A. et al. The impact of the glomerular filtration rate on the human plasma proteome. Proteom. Clin. Appl 12, e1700067 (2018).
Stanley, T. L. et al. Relationship of IGF-1 and IGF-binding proteins to disease severity and glycemia in nonalcoholic fatty liver disease. J. Clin. Endocrinol. Metab. 106, e520–e533 (2021).
Martinez-Castillo, M. et al. Differential production of insulin-like growth factor-binding proteins in liver fibrosis progression. Mol. Cell Biochem. 469, 65–75 (2020).
Warrington, J. A., Nair, A., Mahadevappa, M. & Tsyganskaya, M. Comparison of human adult and fetal expression and identification of 535 housekeeping/maintenance genes. Physiol. Genomics 2, 143–147 (2000).
Xu, S. et al. Esculin inhibits hepatic stellate cell activation and CCl(4)-induced liver fibrosis by activating the Nrf2/GPX4 signaling pathway. Phytomedicine 128, 155465 (2024).
Distler, J. H. W. et al. Shared and distinct mechanisms of fibrosis. Nat. Rev. Rheumatol. 15, 705–730 (2019).
Semenovich, D. S., Plotnikov, E. Y., Titko, O. V., Lukiyenko, E. P. & Kanunnikova, N. P. Effects of panthenol and N-acetylcysteine on changes in the redox state of brain mitochondria under oxidative stress in vitro. Antioxidants 10, 1699 (2021).
Depeint, F., Bruce, W. R., Shangari, N., Mehta, R. & O’Brien, P. J. Mitochondrial function and toxicity: role of the B vitamin family on mitochondrial energy metabolism. Chem. Biol. Interact. 163, 94–112 (2006).
Chen, C. et al. Vitamin B5 rewires Th17 cell metabolism via impeding PKM2 nuclear translocation. Cell Rep. 41, 111741 (2022).
Sun, S. et al. Targeting GOLPH3L improves glioblastoma radiotherapy by regulating STING-NLRP3-mediated tumor immune microenvironment reprogramming. Sci. Transl. Med. 17, eado0020 (2025).
Kim, Y. O., Popov, Y. & Schuppan, D. Optimized mouse models for liver fibrosis. Methods Mol. Biol. 1559, 279–296 (2017).
Trebicka, J. et al. Atorvastatin attenuates hepatic fibrosis in rats after bile duct ligation via decreased turnover of hepatic stellate cells. J. Hepatol. 53, 702–712 (2010).
Mederacke, I., Dapito, D. H., Affo, S., Uchinami, H. & Schwabe, R. F. High-yield and high-purity isolation of hepatic stellate cells from normal and fibrotic mouse livers. Nat. Protoc. 10, 305–315 (2015).
Elvevold, K., Kyrrestad, I. & Smedsrod, B. Protocol for Isolation and Culture of Mouse Hepatocytes (HCs), Kupffer Cells (KCs), and Liver Sinusoidal Endothelial Cells (LSECs) in analyses of hepatic drug distribution. Methods Mol. Biol. 2434, 385–402 (2022).
Ran, F. A. et al. Double nicking by RNA-guided CRISPR Cas9 for enhanced genome editing specificity. Cell 154, 1380–1389 (2013).
Ruddarraju, R. R. et al. Design, synthesis, anticancer, antimicrobial activities and molecular docking studies of theophylline containing acetylenes and theophylline containing 1,2,3-triazoles with variant nucleoside derivatives. Eur. J. Med. Chem. 123, 379–396 (2016).
Sedivy, A. Standard operating procedure for NanoTemper Monolith measurements. Eur. Biophys. J. 50, 381–387 (2021).
Jafari, R. et al. The cellular thermal shift assay for evaluating drug target interactions in cells. Nat. Protoc. 9, 2100–2122 (2014).
Qian, T. et al. Molecular signature predictive of long-term liver fibrosis progression to inform antifibrotic drug development. Gastroenterology 162, 1210–1225 (2022).
Son, J. A. et al. Novel gene signatures as prognostic biomarkers for predicting the recurrence of hepatocellular carcinoma. Cancers 14, 865 (2022).
Shen, Q. et al. Barrier to autointegration factor 1, procollagen-lysine, 2-oxoglutarate 5-dioxygenase 3, and splicing factor 3b subunit 4 as early-stage cancer decision markers and drivers of hepatocellular carcinoma. Hepatology 67, 1360–1377 (2018).
Chen, J. et al. Hepatic lipocalin 2 promotes liver fibrosis and portal hypertension. Sci. Rep. 10, 15558 (2020).
Pradere, J. P. et al. Hepatic macrophages but not dendritic cells contribute to liver fibrosis by promoting the survival of activated hepatic stellate cells in mice. Hepatology 58, 1461–1473 (2013).
El Taghdouini, A., Najimi, M., Sancho-Bru, P., Sokal, E. & van Grunsven, L. A. In vitro reversion of activated primary human hepatic stellate cells. Fibrogenes. Tissue Repair 8, 14 (2015).
Ramachandran, P. et al. Resolving the fibrotic niche of human liver cirrhosis at single-cell level. Nature 575, 512–518 (2019).
Regev, A. et al. The human cell atlas. Elife 6, 27041 (2017).
Zhang, L. et al. Disruption of cholangiocyte-B cell crosstalk by blocking the CXCL12-CXCR4 axis alleviates liver fibrosis. Cell Mol. Life Sci. 80, 379 (2023).
Acknowledgements
This work was supported by the National Natural Science Foundation of China (82071383, 82371392), the Natural Science Foundation of Shandong Province (Key Project) (ZR2020KH007), the Natural Science Foundation of Shandong Province (Youth Project) (ZR2022QC222), and the China Postdoctoral Science Foundation Grants (Project No. 2021M691225). We thank the Center for Basic Medical Research, the Center for Translational Medicine Research, and the Laboratory Animal Center of the Central Hospital of Shandong First Medical University for providing experimental support. We thank pathologists Fei Yang, Wei Gao, and Zhengyan Xu of the Department of Pathology at Jinan Central Hospital for blind pathological assessment using the Ishak scoring system. Graphical abstract and the schematic diagrams in Figs. 1e, 5a, and 7a were produced under license from BioRender.com (agreement number: GM28GOFT57, AR28HM9B0Q).
Author information
Authors and Affiliations
Contributions
Zhengxin Jin made major contributions to the experiments and writing. Zhengsen Jin, Yongcheng Yin, Jianning Kang and Zeyu Liu analyzed and interpreted the data related to biosignatures. Yuchen Zhang, Ying Zhang, Yuepeng Fang and Wei Jiang were involved in the preparation of the figures. Bin Ning participated in the design and revision of the article. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Communications Biology thanks Samaneh Siapoush and the other, anonymous, reviewers for their contribution to the peer review of this work. Primary Handling Editor: Joao Valente.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Jin, Z., Jin, Z., Liu, Z. et al. Pantothenic acid ameliorates hepatic fibrosis by targeting IGFBP6 to regulate the TGF-β/SMADs pathway. Commun Biol 8, 1127 (2025). https://doi.org/10.1038/s42003-025-08527-5
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s42003-025-08527-5
