
Abstract
Reduced enrichment of short-chain fatty acid (SCFA)-producing pathways in the gut microbiome (GM) and SCFA levels are associated with increased risk of type 1 diabetes (T1D). Free fatty acid receptor 2 (FFA2), an SCFA receptor on pancreatic β-cells, mediates GM and β-cell crosstalk. Here, we examine its T1D-specific role in male mice, using a novel tamoxifen-inducible adult-onset β-cell FFA2 knockout (FFA2 βKO) mouse model and its controls (cre and flox), treated with multiple low-dose streptozotocin (MLDS). FFA2 βKO mice show significantly lower diabetes incidence compared to control mice (57% vs 100%). Early in the MLDS insult (7th day), FFA2 βKO mice show significantly lower β-cell apoptosis and higher β-cell mass, persisting up to 43 days. Mechanistically, we observed higher SOCS1/3 expression and reduced T1-IFN signaling in FFA2 βKO islets. Our data suggest that β-cell FFA2 modulates early islet apoptosis, likely via the T1-IFN-SOCS1/3 pathway and may be a pharmacological target for slowing T1D progression.
Similar content being viewed by others

Introduction
Type 1 diabetes mellitus (T1D) is an autoimmune disease where the immune system targets the pancreatic β-cells. Once T1D begins, the abnormal immune response leads to the destruction or dysfunction of pancreatic β-cells by cytotoxic T-cells1. The progression of T1D to clinical symptoms can take months to years, eventually resulting in hyperglycemia, when T1D patients require exogenous insulin therapy1. Multiple genes and environmental modifiers contribute to and trigger T1D2, with one recently recognized key modulator being the gut microbiome (GM)3. Studies from rodent models suggest that gut microbial modulation of T1D susceptibility is mainly mediated immunologically, as the GM is critical for immune cell development, differentiation, and gut homeostasis4. Gut dysbiosis (functional and compositional changes in the GM) is a prominent feature in rodent models of T1D and genetically susceptible individuals who later develop the disease5. This gut dysbiosis is characterized by increased representation of pathobionts and reduced abundance of beneficial gut bacteria, specifically, short-chain fatty acid (SCFA) producers6,7,8,9. The gut metagenomes of T1D and at-risk T1D individuals have fewer genes associated with fermentation and biosynthesis of SCFAs10,11,12. Blood and fecal SCFA (in particular, acetate and butyrate) levels correlate inversely with T1D progression in nonobese diabetic (NOD) mice, and their diminished levels are reported in T1D subjects compared to healthy controls10,13,14. Through rodent models, it has been shown that SCFAs promote a tolerogenic environment, for example, by reducing the frequency of cytotoxic T-cells and expanding/inducing regulatory T-cells4,7. Immune response modulation by SCFA-producing diet has been shown to ameliorate T1D progression in preclinical studies7,8,9,13,15.
Mechanistically, inhibition of histone deacetylases (HDAC) or activation of cognate GPCRs (free fatty acid receptor (FFA) 2 and FFA3) underlie these effects of SCFAs. SCFA-mediated FFA2 activity represents a means by which the GM interacts directly with the endocrine pancreas16,17. In rodent islets, Ffa2 mRNA expression increases under insulin resistance, and FFA2 activity has been shown to regulate pancreatic β-cell function, mass, and apoptosis16,17. Evidence indicates that FFA2 can modulate T1D progression in rodent models through effects on immune cells, gut barrier integrity, and islet antimicrobial peptide production7,8. As follows, in T1D subjects, FFA2 expression increases in peripheral blood mononuclear cells, indicating its role in immune modulation18. FFA2 expression has not been analyzed in T1D human islets. Our secondary analysis of a public RNA-seq dataset19 revealed that human islet FFA2 expression trends higher in the presence of T1D-like stress (Supplementary Fig. 1). These data indicate a need to elucidate the pancreatic β-cell-specific role of SCFA receptors, like FFA2, in modifying T1D pathology.
T1D involves multi-faceted immune factors, where cytokines are known to contribute to the progression of T1D20. In humans, evidence strongly suggests that T1-IFNs are the initiating cytokine in T1D. Multiple GWAS studies have identified genes in T1-IFN production, signaling, or regulation pathways to correlate with T1D risk2,21. In agreement, longitudinal T1D studies have reported transient T1-IFN gene signatures in the peripheral blood samples before seroconversion22,23. Furthermore, T1-IFN therapy can trigger T1D progression in islet autoantibody-positive individuals24,25,26,27. Similarly, in animal models, manipulation of T1-IFN production (genetic knockout of viral RNA recognition receptor, MDA-5) or signaling (neutralization of IFNAR1) delays/blocks the progression of T1D28,29,30. Damaged and dying β cells, plasmacytoid dendritic cells, and episodes of virus infection can induce T1-IFNs (especially IFNα) in the islets31,32,33. When induced, T1-IFNs bind to their heterodimeric receptor, IFNAR1 and IFNAR2, on pancreatic β-cells, leading to JAK and TYK2-mediated phosphorylation of STAT1 and STAT2 and downstream signaling resulting in transcription of chemotactic genes, elevation of endoplasmic reticulum stress, and increased immunogenicity, instigating β-cell damage34,35. Interestingly, FFA2 activity can likely modify T1-IFN signaling, as experimentally demonstrated in infectious disease models36,37. FFA2 activation in lung epithelial cells and macrophages induces inflammasome (NLRP3) and T1-IFN36,37. Importantly, these effects are compromised in the absence of FFA236,37. This suggests that modulation of FFA2 signaling in β-cells under T1D-like insult may affect IFN responses and β-cell survival.
To test this, we used tamoxifen (TMX)-inducible pancreatic β cell-specific FFA2 knockout (KO) mice treated with multiple low-dose streptozotocin (MLDS) to mimic T1D-like pathology. We observed that compared to the floxed control mice, 57% of FFA2 βKO mice were protected from MLDS-induced T1D owing to higher β-cell mass. This preservation of β-cell mass resulted from less β-cell apoptosis in FFA2 βKO mice. Compared to floxed controls, in the β-cells of FFA2 βKO mice, we found markedly reduced T1-IFN signaling and a higher expression of Socs1 and Socs3, its negative regulators. These data suggest that β-cell-specific deletion of FFA2 in adulthood can attenuate T1-IFN signaling and partially protect against the loss of β-cell mass under T1D-like stress.
Results
Specific knockout of FFA2 in β-cells
For these studies, we generated a novel TMX-inducible mouse model that specifically deletes Ffa2 in pancreatic β-cells in adulthood. We induced Ffa2 deletion at 8 weeks of age to prevent initial β-cell mass loss due to the role of Ffa2 in embryonic and fetal pancreatic β-cell development38,39. The Ffa2-expressing tissues were harvested from floxed control and FFA2 βKO mice to examine the specificity of Ffa2 deletion in pancreatic β-cells. As shown, we noted an 83% decrease in Ffa2 mRNA expression in the pancreatic islets of FFA2 βKO mice compared to that in the floxed control (Fig. 1A), while the expression of the closely related SCFA receptor, Ffa3, remained unchanged (Supplementary Fig. 2A). β-cell-specific deletion did not alter the receptor’s expression in other tissues (Fig. 1B).

Male Ffa2fl/fl x MIP-Cre/ERT+/- and Ffa2fl/fl mice were intraperitoneally administered TMX at 8 weeks to generate FFA2 βKO and floxed control, respectively. The tissues were harvested at 10 weeks of age. Ffa2 expression in (A) islets and (B) spleen, visceral fat, ileum, and colon is shown as fold change against floxed control for each tissue. For (A), N = 6 for floxed control, N = 5 for FFA2 βKO, and for (B), N = 4 per genotype. Data are expressed as mean ± standard error and analyzed by Student’s t-test for (A) and two-way ANOVA with Sidak’s correction for (B). ****p < 0.0001. Blue bar = floxed control, green bar = FFA2 βKO.
MIP-Cre/ERT construct contains a growth hormone fragment, due to which MIP-Cre/ERT mice have been documented to show increased β-cell proliferation and mass40,41. To control for the growth hormone variable, for subsequent studies, we also included a Cre control group. As expected, Ffa2 mRNA expression in the pancreatic islets and other Ffa2-expressing tissues of the Cre control group was similar to that in the floxed control (Supplementary Fig. 2B, C). The effect of TMX was also tested by comparing FFA2 βKO to the floxed control. Using two controls, the function of pancreatic β-cell FFA2 in MLDS-induced T1D could be isolated.
Pancreatic β-cell FFA2 plays a detrimental role in MLDS-induced T1D by reducing β-cell mass
We subjected FFA2 βKO and the two control groups of mice to MLDS-induced T1D and monitored them for 43 days. By day 43, 57% of FFA2 βKO mice were diabetes-free (diabetes defined as random blood glucose ≥250 mg/dL on two consecutive measurements), whereas all control mice developed diabetes, with a modest delay in onset in Cre controls compared to floxed controls (Fig. 2A). Correspondingly, FFA2 βKO mice showed lower random blood glucose levels from day 14 (vs. floxed) and day 18 (vs. Cre) (Fig. 2B). Despite elevated plasma glucose levels in control mice from day 10 onward, no significant intra-group or inter-group weight differences were observed (Fig. 2D). Insignificant intra-group weight phenotype could arise due to the procedure used for MLDS-T1D induction, where mice were not fasted before streptozotocin doses42. Weight loss in MLDS models usually occurs during the acute phase, followed by a stable weight or slight weight gain due to hyperphagia43,44. Consistent with previous studies using a similar MLDS induction approach42, all groups in our cohort lost ~2.5–7% of the body weight by day 7 (Fig. 2D and Supplementary Fig. 6B) stabilizing thereafter (Fig. 2D). The absence of significant weight difference between the control and FFA2 βKO groups may reflect the activation of compensatory responses (like hyperphagia) in control mice to offset weight loss. This aligns with an earlier report where MLDS-treated wild-type and macrophage-specific Ubc9 KO mice showed similar weights despite higher diabetes incidence and hyperglycemia in the KO group45.

After TMX-induced Ffa2 deletion, T1D was induced using MLDS, and all mice were monitored for 43 days. Diabetes incidence (diabetes was defined as random blood glucose ≥250 mg/dL on two consecutive measurements) (A), ad lib blood glucose (B), and insulin levels (C), and percent change in body weight (D) were monitored. On the 21st day of the initial STZ injection, IPGTT (E) at 1 g/kg was carried out. The AUC calculated for IPGTT is shown in (F). All mice were euthanized on the 43rd day, and β-cell mass (G) was assessed. Immune cells were examined for CD4+, CD8+, CD4+ Foxp3+, and F4/80+ in pancreatic lymph nodes (H) and spleen (I) by FACS sorting, and their percentages within all immune cells were plotted. For (A), (B), and (D), N = 9, 6, and 7; for 2C, N = 6, 6, and 7; for (E) and (F), N = 6, 5, and 6; for (G), N = 4, 6, and 6, for cre control, floxed control, and FFA2 βKO, respectively. For (H) and (I), N = 6 and 7 for floxed control and FFA2 βKO. For (G), 1 FFA2 βKO was identified as an outlier and excluded based on the ROUT test, Q = 1%. Data are expressed as mean ± standard error and analyzed by Log-rank (Mantel–Cox) test (2A) or by one-way ANOVA with Tukey multiple comparisons (F and G), or two-way ANOVA with Tukey correction (B, C, D, E, H, and I). *p < 0.05, **p < 0.01, and ***p < 0.001. In (A), (B), (C), and (E), the blue asterisk or number indicates the statistical significance and P-values of FFA2 βKO compared with the floxed control. The gray asterisk and number indicate the statistical significance and P-values of the FFA2 βKO group compared with the Cre control. Blue bar and line = floxed control, gray bar and line = Cre control, green bar and line = FFA2 βKO.
IPGTT at 1 g/kg on the 21st day showed better glucose tolerance in the FFA2 βKO group than in the two control groups (Fig. 2E), which was also reflected in the area under the curve (AUC) for the IPGTT (Fig. 2F). However, the insulin response during the GTT was not different among the genotypes (Supplementary Fig. 3B). This could be due to the preparatory overnight fast known to exert a significant suppressive effect on insulin levels during GTT46. To understand how pancreatic β-cell FFA2 led to glucose intolerance, we harvested the pancreas of these mice on the 43rd day and measured β-cell mass. A trend toward higher β-cell mass was noted in FFA2 βKO mice compared to the two control groups, indicating that FFA2 may be involved in the negative regulation of β-cell mass under MLDS-induced T1D (Fig. 2G). This suggested that the remaining β-cell mass in FFA2 βKO mice could provide long-term glucose regulation even if acute insulin release in response to glucose is insufficient. In support, ad lib insulin levels trended higher up to day 10 (Fig. 2C) and fasting glucose levels were significantly lower (Supplementary Fig. 3A) in the FFA2 βKO group than in the control groups. In addition to preserved β-cell mass, increased insulin sensitivity may have protected FFA2 βKO mice from MLDS-induced T1D. Although not directly tested, its surrogate measure, the HOMA-IR index was significantly higher in the MLDS-treated floxed and cre control mice compared to FFA2 βKO mice, indicating greater insulin sensitivity in the latter (Supplementary Fig. 3G).
As an immune response is elicited during MLDS-induced T1D, we examined T-cell subpopulations and macrophage differences between FFA2 βKO and floxed control mice in the PLN and spleen. At baseline (in saline-treated mice), a higher proportion of CD8+ cytotoxic T-cells in the PLN and a modest but significantly lower proportion in the spleen were noted in FFA2 βKO mice (Supplementary Fig. 3F). Under MLDS treatment, no difference was observed in the PLN T-cell subpopulations and macrophages (Fig. 2H). However, a significant increase in CD4+ T-cell population was seen in the spleen of FFA2 βKO (Fig. 2I), which could be attributed to an increase in protection-conferring T helper cells, such as Th2 cells.
SCFAs, ligands for FFA2, are primarily produced by the gut microbiota, and T1D associates with changes in gut microbiota3,47. However, GM composition was not assessed as streptozotocin effects would dominate over the genotype effect. Furthermore, prior studies on the FFA2 global knockout model suggested minimal gut microbiota compositional and functional shifts from β-cell FFA2 deletion48,49. Thus, we relied on plasma SCFA levels as surrogate markers for gut microbial activity. As expected, total or individual SCFAs at baseline were comparable across genotypes (Supplementary Fig. 3D). Post-MLDS, only acetate increased significantly in flox control mice (Supplementary Fig. 3D), which could indicate a possibility of altered FFA2 signaling in the wild-type MLDS mice. In summary, pancreatic β-cell FFA2 exacerbates MLDS-induced T1D, possibly through effects on β-cell mass.
FFA2 is involved in the regulation of the T1-IFN signaling
To understand our data, we leveraged the existing RNA sequencing dataset from FFA2 global KO and its control wild-type islets from our laboratory50. Within this dataset, a set of genes related to T1-IFN signaling was upregulated in FFA2 global KO islets compared to its control, including the ISGF3 complex (i.e., Stat2, Stat1, and Irf9), negative regulators (i.e., Socs1 and Socs3), and T1-IFN targeted genes (i.e., Ifit1, Ifit3, Ifit3b, and Oasl2) (Fig. 3A). These data suggested that global FFA2 KO islets have higher T1-IFN signaling under baseline conditions, a pathway indispensable for the host antiviral effect. Gene ontology biological process (GO BP) analysis revealed the enrichment of pathways related to viral infection in global FFA2 KO islets (Fig. 3B). The top two enriched pathways were the “cellular response to interferon β” and the “response to interferon β” which further suggests a role of FFA2 in T1-IFN signaling (Fig. 3B). Interestingly, T1-IFN signaling has been implicated in T1D pathogenesis. Specifically, multiple studies have shown that T1-IFN signaling precedes the detection of islet autoimmunity in T1D development in both human cohorts and mouse models, suggesting that it could be the initiator of the abnormal immune response in T1D22,23,28,29,30. From the β-cell perspective, T1-IFNs induce ER stress and apoptosis in β-cells35. Thus, our data suggest a connection between FFA2, T1-IFN signaling, and T1D. Based on these findings, we developed the hypothesis that the significantly increased T1-IFN concentration, induced by damage of β-cells (in MLDS-induced T1D) or enterovirus infection (in humans), can further increase the expression of Socs1 and Socs3 in FFA2 deleted β-cells, consequently leading to reduction of T1-IFN signaling and improved survival of the β-cells. As this RNA-seq was conducted on islets isolated from male mice under physiologic conditions, and islets are composed of >70% β-cells, the RNA changes will likely be like FFA2 βKO islets, but need validation.

A Differentially expressed genes upregulated in FFA2 KO islets are related to T1-IFN signaling, including transcriptional factors, negative regulators, and T1-IFN signaling target genes. B The top enriched gene ontology biological process (GO BP) pathways in global FFA2 KO islets using the GSEA method. N = 3 per genotype.
FFA2 knockdown in the Min6 cell line increased Socs1 and Socs3 protein levels with treatment of IFNα
To test the effect of FFA2 deletion on Socs1 and Socs3, we generated an FFA2 knockdown Min6 cell line, shFFA2 (Fig. 4A). We incubated shFFA2 and its control Min6 cell line (EV; empty vector) with IFNα for 0 h, 18 h, and 36 h. Without treatment, we observed increased Socs1 and −3 protein levels in shFFA2 compared to EV (Fig. 4B, C). Upon IFNα treatment, Socs1 levels did not change in the EV cells up to 36 h. shFFA2 cells, on the other hand, showed a time-dependent significant increase in Socs1. Socs3 levels only trended high at both time points in shFFA2 cells compared to EV cells. Notably, between the EV and shFFA2 Min6 cells, Socs1 and Socs3 protein levels were consistently higher in the shFFA2 cells. mRNA expression of Ffa3 was reduced in shFFA2 cells (Supplementary Fig. 4A). T1-IFN signaling in shFFA2 cells was like that observed in FFA2 βKO islets, suggesting that this reduction had minimal impact on the results. However, the effects of reduced Ffa3 expression on T1-IFN signaling in shFFA2 Min6 cells can’t be completely excluded and need future exploration. In conclusion, in the IFNα-treated Min6 cell line, deletion of FFA2 led to increased Socs1 expression, which could lead to impaired T1-IFN signaling.

Fold change in Ffa2 expression in FFA2 shRNA (shFFA2) containing virus-infected Min6 cells relative to empty vector (EV) virus-infected Min6 cells (A). Immunoblots for Socs1, Socs3, and β-actin (B) at 0 h, 18 h, and 36 h after 20,000 U/mL IFNα treatment of EV and shFFA2 Min6 cell lines. Densitometric analysis (ImageLab software version 6.0, Bio-Rad) of Socs1 and Socs3 immunoblots normalized with β-actin, expressed in fold change relative to its respective EV at 0 h (C). For (A), N = 6. For (B), the experiment was repeated four times, and representative blots are shown. Data are expressed as mean ± standard error and analyzed by Student’s t-test (A) and two-way ANOVA with Tukey’s multiple comparisons (C). *p < 0.05 and ****p < 0.0001. Blue dotted bar = EV virus-infected Min6 cells, green dotted bar = shFFA2 virus-infected Min6 cells.
FFA2 βKO islets and mice showed reduced T1-IFN signaling
Next, we used islets isolated from floxed control and FFA2 βKO mice to test our hypothesis in a physiological system. After overnight recovery, we challenged these islets with 20,000 U/mL of IFNα treatment for 2 h. When no treatment was given, we did not observe any differences in the mRNA expression of Socs1, Socs3, Ifit1, Ifit3, Ifit3b, and Oasl2 between the two genotypes (Fig. 5A, B), suggesting that the elevated T1-IFN signaling in global FFA2 KO islets may be due to other factors, possibly other cell types. However, when floxed control and FFA2 βKO islets were treated with 20,000 U/mL of IFNα, we observed a 3-fold increase in the expression of Socs1 and Socs3 in FFA2 βKO islets compared to floxed control islets (Fig. 5B). As expected, compared to the floxed control, a 35%–55% decrease in T1-IFN signaling transcribed genes, including Ifit1 (trending lower), Ifit3, Ifit3b, and Oasl2 was observed in FFA2 βKO islets. This difference could not be attributed to the differential expression of IFN receptors on FFA2 βKO and floxed control islets (Supplementary Fig. 5). Thus, FFA2 βKO islets show reduced T1-IFN signaling compared to floxed control when treated with 20,000 U/mL of IFNα.

Islets from floxed control or FFA2 βKO received either no treatment or 20,000 U/mL IFNα treatment for 2 h. The expression of T1-IFN-regulated genes (A), Ifit1, Ifit3, Ifit3b, and Oasl2, and negative regulators of T1-IFN signaling (B), Socs1 and Socs3, are shown in fold change relative to floxed control with matched IFNα treatment. Anti-IFNAR1 antibody was given to floxed control, Cre control, and FFA2 βKO male mice at a dose of 1 mg per mouse on days 0 and 6. Floxed and FFA2 βKO male mice treated similarly with IgG1 isotype served as controls. Both groups were given MLDS treatment to induce T1D. The random blood glucose level on the 7th day after the initial STZ injection (C) and diabetes incidence after the 21st day (D) in all groups are shown. The red dotted line in (D) indicates the 7th day, denoting the diabetes incidence difference between the IgG-injected floxed control and the IFNAR1 antibody-injected cre and floxed control. For (A), no treatment: N = 6 (floxed control) and 4 (FFA2 βKO); IFN α treatment: N = 4 for both. For (B), no treatment: N = 4 (floxed control) and 3 (FFA2 βKO); IFN α treatment: N = 5 (floxed control) and 4 (FFA2 βKO). For (C) and (D), N values are 6, 5, 6, 10, and 8 for IgG-injected floxed control, IgG-injected FFA2 βKO, IFNAR1 antibody-injected cre control, floxed control, and FFA2 βKO, respectively. Data are expressed as mean ± standard error and analyzed by Student’s t-test (A and B), one-way ANOVA with Holm-Šídák’s multiple comparisons (C), and Log-rank (Mantel–Cox) test (D). *p < 0.05, **p < 0.01, and ***p < 0.001. In (C) and (D), statistical significance was calculated compared to the floxed control + IgG group. For genotype and condition, blue bar and line = floxed control, green bar and line = FFA2 βKO, gray bar and line = Cre control. For antibody treatment, solid bar and line = IgG-injected groups, and dotted bar and line = IFNAR1 antibody-injected groups.
To further test our hypothesis in the MLDS-induced T1D mouse model, we used a functional IFNAR1 antibody that can block T1-IFN signaling in vivo. We hypothesized that blocking T1-IFN signaling would confer protection to the two control groups at the early stage of MLDS-induced T1D without perturbing the protection conferred by the β-cell FFA2 deletion. IFNα is induced early in T1D51, preceding disease onset in streptozotocin mouse models31 and autoantibody development in humans22,23. Therefore, day 7 was selected to assess altered IFNα signaling in FFA2 βKO mice. Indeed, on the 7th day, we observed a significant reduction in random blood glucose in the Cre control group injected with the IFNAR1 antibody compared to the floxed group injected with the IgG antibody (Fig. 5C). The floxed group injected with the IFNAR1 antibody also displayed a marginally lower random blood glucose level than the floxed control injected with the IgG antibody (Fig. 5C). This marginal reduction of random blood glucose in the floxed control IFNAR1 antibody-injected group was due to three hyperglycemic mice, suggesting the involvement of factors other than T1-IFN signaling. FFA2 βKO groups, both IFNAR1 antibody and control antibody injected, showed no difference in day 7 random blood glucose, which resembles the initial finding that FFA2 βKO protected mice from MLDS-induced hyperglycemia (Fig. 5C). Not surprisingly, the protection conferred by the IFNAR1 antibody in the two control groups was lost later, which was represented by 100% diabetes incidence in the floxed control group at day 10 and slightly delayed in the Cre control group at day 14 (Fig. 5D). FFA2 βKO mice injected with the control antibody or IFNAR1 antibody demonstrated low diabetes incidence compared to the two control groups (Fig. 5D). The two FFA2 βKO groups showed a higher diabetes incidence than the FFA2 βKO group in the initial assessment (Fig. 2A), likely due to an immune reaction to the injected antibody. To summarize, the deletion of FFA2 in pancreatic β-cells leads to reduced T1-IFN signaling in vivo, which confers partial protection at the early stage of MLDS-induced T1D.
FFA2 βKO promotes β-cell mass preservation in early MLDS-T1D by attenuating T1-IFN signaling via Socs1/3
Our ex vivo and in vivo experiments indicated that FFA2 deletion could attenuate T1-IFN signaling and partially protect MLDS-induced T1D mice on the 7th day (early stage). We harvested the pancreas from MLDS-induced T1D mice from FFA2 βKO and the two control groups on day 7 to assess their β-cell mass. We observed similar diabetes incidence and weight loss phenotype as before (Supplementary Fig. 6A, B) and an expected significant reduction in β-cell mass in the two control groups on day 7 compared to FFA2 βKO mice, with no differences in α-cell mass (Fig. 6A, B).

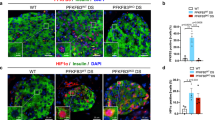
T1D was induced with MLDS, as previously described. The pancreas of the two control groups and the FFA2 βKO group were sectioned and stained. Representative images (10×) of the whole pancreas section scanning (DAPI = nuclei, Fluorescein = Glucagon, Texas Red = Insulin, scale bar 500 µm for Cre and floxed control, 400 µm for FFA2 βKO) and islets are labeled with red arrows (A) for FFA2 βKO and two control groups. The calculated β-cell mass and α-cell mass (B) of FFA2 βKO and the two control groups are shown on the 7th day after the initial STZ injection. Representative islets images (20×, scale bar 20 µm) were shown for cleaved caspase 3 (DAPI = nuclei, Fluorescein = Cleaved caspase 3, Texas Red = Insulin) (C), Ki67 (DAPI = nuclei, Fluorescein = Ki67, Texas Red = Insulin) (D), Socs1 (DAPI = nuclei, Fluorescein = Socs1, Texas Red = Insulin) (E), and Socs3 (DAPI = nuclei, Fluorescein = Socs3, Texas Red = Insulin) (F). Percentage of cleaved caspase 3, Ki67, Socs1, and Socs3 positive β cells within the islets (G). For (A) and (B), N = 5, 7, and 6 for cre control, floxed control, and FFA2 βKO, respectively (1 cre control and 1 FFA2 βKO identified as outliers and excluded based on the ROUT test, Q = 1%). For (C–G), N = 12~16 sections from 4 mice were analyzed per genotype. Data are expressed as mean ± standard error and analyzed by one-way ANOVA with Tukey multiple comparisons (B and G). *p < 0.05, **p < 0.01 and ***p < 0.001. Blue bar = floxed control, gray bar = Cre control, green bar = FFA2 βKO.
To understand the reason for the significantly higher β-cell mass in FFA2 βKO mice on the 7th day, we assessed apoptosis and proliferation by staining the pancreatic sections for cleaved caspase 3 and Ki67, where we observed significantly less cleaved caspase 3 staining in the islets of FFA2 βKO mice than in the two control groups with no difference in Ki67 staining (Fig. 6C, D, G). These data suggest that the higher β-cell mass in FFA2 βKO mice was due to reduced apoptosis.
To verify the reduction in T1-IFN signaling in FFA2 βKO mice, we also stained the pancreatic sections for Socs1 and Socs3. For Socs1, we observed significantly higher Socs1-positive β cells in FFA2 βKO islets than in the floxed or Cre control group (Fig. 6E, G). Significantly higher Socs3 staining was noted in FFA2 βKO islets as compared to the floxed control, and trending higher compared to the Cre control (Fig. 6F, G). Higher Socs1 and Socs3 positive staining in FFA2 βKO islets suggested lower T1-IFN signaling in the FFA2 βKO islets, consistent with our previous finding in both the ex vivo study and the in vivo IFNAR1 antibody neutralization experiment.
The effects of reduced T1-IFN signaling due to increased Socs1/3 were β-cell-specific and not reflected systemically, as T-cell profiling (in PLN, spleen (Fig. 2H, I)); inguinal, and intestinal lymph nodes showed no significant group differences except for increased Cd4+ T-cells in the spleen of MLDS-treated FFA2 βKO mice. Furthermore, all experimental MLDS groups exhibited similar insulitis on day 7 (Supplementary Fig. 6D). These data draw support from other published reports in NOD mice where β-cell-specific Socs1 expression protected against islet damage without affecting peripheral T-cell tolerance or insulitis52.
Discussion
T1D is an autoimmune disease mostly diagnosed in children and adolescents53. The incidence of T1D has increased in recent years54. Currently, the most successful immune modulation therapy can only delay the progression of T1D with other treatments, such as β-cell replacement, which are still in the preliminary phases55,56,57. Thus, diverging focus on potential complementary therapeutic strategies that target endogenous β-cells could be another option. Recently, GM has been implicated in mediating protection against T1D disease progression6,7,8,9. Multiple studies have shown promising results with SCFA supplementation in both humans and animal T1D models, with suppression of aberrant immune response and maintenance of endogenous β-cell function7,58,59,60. However, the individual roles of β-cell SCFA receptors in T1D have not yet been investigated.
In this study, we deleted one of the SCFA receptors, FFA2, in β-cells and used the MLDS-induced T1D model to study β-cell FFA2 function in adulthood during T1D-like challenge. Our earlier studies on FFA2 function in the pancreatic β-cells under physiological and insulin-resistant states like high-fat diet-induced obesity and pregnancy had shown the role of FFA2 in islet insulin secretion and in establishing and maintaining β-cell mass17,48,50. These data indicated that reduced SCFA levels and dampened SCFA-FFA2 signaling may be deleterious to the β-cells in T1D. Surprisingly, we observed fewer diabetes incidences in FFA2 βKO mice than in control mice due to significantly higher β-cell mass on the 7th day of STZ administration that trended higher up to the 43rd day of the initial STZ injection. SCFAs and SCFA-cognate GPCR signaling can exert both pro- and anti-inflammatory effects61. For example, in experimental autoimmune encephalitis, whole-body knockout of FFA2 conferred disease resistance, while SCFAs retained anti-inflammatory effects in specific immune cell types61. Similarly, FFA2 activation on intestinal epithelial cells leads to MAP kinase signaling and increased inflammatory cytokines and chemokines62 production. Furthermore, FFA2-dependent inflammasome activation can also exert inflammatory effects63, as seen in gout and inflammatory bowel disease64,65. These data suggest that FFA2 activation can mount inflammatory responses, which may be required to resolve pathogenic insults and avoid long-term inflammation62,66. Of note, these acute responses are defective in the absence of FFA261,62,66. Consistently, β-cell-specific deficiency of FFA2 in our model attenuated MLDS-induced T1D.
FFA2 expression in other cell types, including gut epithelial cells and immune cells, may impact the immune and metabolic environment in T1D. For example, we recently showed that intestine-specific FFA2 KO male mice on a western diet show reduced insulin response during GTT, indicating a role of intestinal FFA2 in modulating β-cell function67. In our model, receptor expression was not different in non-β-cell tissues with respect to the genotypes at baseline (Fig. 1B) and does not change with MLDS treatment (Supplementary Fig. 6C). While this does not indicate an absence of any role of the receptor in these tissues in T1D development, it does suggest that MLDS-driven T1D models (where β-cell is the primary target) may not highlight these roles. The NOD model with tissue-specific receptor knockout could be more appropriate.
Regarding the islet-specific metabolic effects documented for FFA2, previous studies from our and other laboratories showed that FFA2 promotes islet insulin secretion due to its coupling to Gαq/11; thus, the expected impact on β-cell mass is the opposite of what we observed in our MLDS-induced T1D study, with control groups showing reduced β-cell mass than the FFA2 βKO group17,49. Baseline metabolic function and immune cell profiles in the absence of MLDS treatment were not drastically different among male FFA2 βKO and control (Cre and flox) mice (Supplementary Fig. 2D–F; Supplementary Fig. 3F), indicating no potential contributions of such differences to our current data. However, activation of Gαq/11 by GPCRs can aggravate β-cell ER stress, already induced in T1D68. Though we did not explore this possibility, stress-response pathways (including oxidative stress) could contribute to the phenotype of higher MLDS-induced T1D incidence observed in control (flox and Cre) mice. Interestingly, plasma acetate levels increased significantly in MLDS-treated flox group (Supplementary Fig. 3D), indicating a possibility of higher FFA2 activity in these mice. A recent study has documented the role of SCFAs in maintaining pancreatic β-cell function and reducing stress genes in NOD mice fed with an acetate- and butyrate-releasing diet58. As acetate and butyrate are agonists of FFA2, the results of our study seem to contradict the protective role of SCFA in T1D β-cells. However, this protective effect of SCFAs can result from collective signaling from both SCFA receptors and HDAC inhibition. As FFA2 is only one of the SCFA receptors, other SCFA receptors may play different roles in T1D. For example, FFA3 and HCA2, Gi/o coupling GPCRs, confer protection to β-cells in T1D69,70. Additionally, mixed results have been observed for β-cell function in T1D for HDAC inhibition71,72,73,74. Thus, individually studying these SCFA receptors and HDAC family members in T1D is necessary to understand their collective role in T1D β-cells.
Analysis of untreated global FFA2 KO islet RNA-seq data revealed enrichment of T1-IFN signaling and virus infection-related pathways in the FFA2 KO islets with upregulation of genes related to T1-IFN signaling, especially Socs1 and Socs3, suggesting elevated basal T1-IFN signaling. These data led to our hypothesis that FFA2 deletion in β-cells slightly increases T1-IFN signaling and its negative regulators Socs1 and Socs3 under basal conditions, which may be more pronounced upon T1-IFN exposure as occurs at the initiation of T1D. Thus, a further induction in the expression of Socs1 and Socs3 may occur and attenuate T1-IFN signaling. In isolated FFA2 βKO and floxed control islets ex vivo, no difference in the expression of key genes of the T1-IFN signaling pathway was observed. However, without any treatment, shFFA2 Min6 cells showed higher Socs3 protein levels than EV Min6 cells, supporting the observations from the global FFA2 KO RNA sequencing dataset. After incubation with IFNα, Socs1 and Socs3 were higher at the mRNA level in FFA2 βKO islets ex vivo, and Socs1 at the protein level in shFFA2 Min6 cells compared to their respective controls. Interestingly, IFNα treatment did not induce Socs1 and Socs3 expression, despite upregulation of other downstream targets of IFNα (Fig. 5A, B). Socs1 and Socs3 are key negative regulators of IFNα signaling and are tightly regulated75. In flox and FFA2 βKO islets, Socs1/3 mRNA levels were, in fact, higher in the absence of the treatment (Fig. 5B). This suggests that basal levels of Socs1/3 proteins may already be sufficient to regulate the IFNα pathway, preventing further induction upon treatment, likely avoiding premature termination. A similar observation has been noted in a hepatoma cell line, where pre-stimulation with high doses of IFNα resulted in limited or no induction of Socs1/3 mRNA and protein76. In vivo blocking of IFNAR1 using an antibody in control groups showed partial protection on the 7th day of STZ injection, suggesting that FFA2 deletion impairs T1-IFN signaling, which fits both our hypothesis and the initiator role of T1-IFNs in T1D pathogenesis. The results from Socs1 and Socs3 immunofluorescence staining of pancreas sections harvested on the 7th day of STZ injection agree with our data from ex vivo islet and shFFA2 Min6 cell IFNα treatment. Our study showed that the deletion of adult β-cell FFA2, a nutrient-sensing GPCR, protected mice from MLDS-induced T1D by reducing T1-IFN signaling, potentially through a mechanism involving Socs1 and Socs3. There is a possibility that other pathways, for example, STAT3, inflammasome, and NF-κB, may contribute to Socs1/3 mediated β-cell protection in our model. We did observe significantly reduced expression of NLRP3 in shFFA2 compared to EV cells after IFNα treatment (Supplementary Fig. 4B). Given the potential of SCFAs in regulating β-cell function and β-cell-specific FFA2 receptor deficiency mitigating MLDS-T1D, these possibilities call for further exploration.
In the last decade, T1-IFNs have been identified and considered as the initiation cytokine of T1D, along with the well-recognized enterovirus infection in T1D22,23,77. Here, we found that FFA2 β-cell deletion impairs T1-IFN signaling, potentially by increasing Socs1 and Socs3 levels, and prevents β-cell apoptosis at an early stage in MLDS-induced T1D. These results indicate the potential pharmacological downregulation of FFA2 receptor activity to attenuate T1-IFN signaling in β-cells in T1D treatment. With the growing availability of HLA allele genotyping to assess the risk of T1D and determine T1D progression stage via islet-specific autoimmunity test in at-risk subjects, pharmacologically targeting FFA2 could maintain endogenous β-cell function and slow down T1D progression at an early stage, extending the timeframe for further necessary therapeutic strategies. As the most challenging aspect of studying ubiquitously expressed druggable GPCRs, the broader applicability and safety of FFA2-based approaches require extensive testing. Tissue-specific nano-carrier approaches are being explored for various diseases and can probably be used for targeting β-cell FFA2. Alternatively, if natural antagonists could be identified, diet supplementation could also be achieved, which may limit adverse effects. However, there is a long way to go with testing disease vulnerability in preclinical T1D models with tissue-specific receptor knockout and using agonists/antagonists and natural ligand sources like dietary fibers in these models.
We recognize that our study has limitations. One limitation is that we did not observe complete protection from hyperglycemia in the floxed group injected with IFNAR1 antibody on day 7. It is possible that other cytokines may be involved in MLDS-T1D pathogenesis. Our results showed higher apoptosis in islets in the two control groups than in FFA2 βKO mice. The phagocytic activity of macrophages to clean these apoptotic cells could further initiate adaptive immunity, aggravating MLDS-induced T1D pathogenesis78. Differential cytokine signaling by islet-infiltrating immune cells (not analyzed here due to focus on β-cell intrinsic pathways) could also contribute to the phenotype.
Another open question remains regarding how FFA2 modulates T1-IFN signaling, particularly the specific G protein or GPCR pathway components involved. Our understanding of GPCR regulation of T1-IFN signaling is based on two mechanisms. The first is G protein-mediated modulation of T1-IFN production. FFA2 has been noted to maintain T1-IFN production in lung epithelium and macrophages, which aligns with our MLDS-induced T1D finding, as FFA2 deletion in β cells reduced T1-IFN signaling, potentially by increasing Socs1 and Socs3 expression36,37. Other GPCRs have also been observed to regulate T1-IFN production. For example, TGR5 and GPR146, the Gs-coupled GPCRs, are required for T1-IFN production during viral infection79,80. Gαq/11, the only G protein directly investigated in T1-IFN production, inhibits T1-IFN production81. From these results, although there is a connection between the production of T1-IFN and GPCR signaling, the specific G protein modulation of T1-IFN production is unclear. The second possible mechanism is altered GPCR regulatory protein expression in the disease state, leading to altered T1-IFN production. Specifically, upregulation of RGS8 and RGS16 has been observed in hyperglycemic mouse β-cells in the STZ-induced T1D model and diet-induced hyperglycemia82. RGS2 could keep interferon production in check to prevent inflammation83. In our global FFA2 KO islet RNA sequencing datasets, we observed that RGS4 was upregulated in global FFA2 KO islets (Supplementary Data 3), which could potentially downregulate T1-IFN signaling. Nevertheless, further research is needed to decipher how FFA2 regulates T1-IFN signaling.
Methods
Experimental animals
Ffa2fl/fl mice were generated on a C57BL/6N background. Exon 3 of the Ffa2 gene, where the coding sequence is located, was flanked by the loxP sequence. Homozygous Ffa2fl/fl mice were mated with MIP-Cre/ERT mice84 (Jackson Laboratories, Strain# 024709) to generate Ffa2fl/fl MIP-Cre/ERT+/- mice. Experimental mice were generated by crossing Ffa2fl/fl MIP-Cre/ERT+/- and Ffa2fl/fl. At 8 weeks of age, male Ffa2fl/fl MIP-Cre/ERT+/- mice were intraperitoneally (IP) injected with either 100 mg/kg TMX (Sigma Aldrich, MO, USA) or vehicle (corn oil; Sigma Aldrich) for five consecutive days to generate pancreatic β cell-specific FFA2 KO mice (FFA2 βKO) and Cre control. Male Ffa2fl/fl mice were IP injected with the same dose of TMX for 5 days, which was our floxed control. All mice were housed in a temperature- and humidity-controlled specific pathogen-free barrier facility with ad libitum access to food (Teklad LM-485, IN, USA), water, and 12 h of dark and light cycles. The University of Illinois Chicago Animal Care Committee approved all procedures, and we have complied with all relevant ethical regulations for animal use.
Induction of T1D by MLDS and physiological measurements in FFA2 βKO and control mice
Only male mice were used in the current study, as female mice have been found resistant to STZ-induced diabetes85. At 10 weeks of age, the floxed control, Cre control, and FFA2 βKO mice were administered IP with 50 mg/kg of streptozotocin (STZ, Sigma Aldrich; dissolved in sterile 0.01 M citric buffer, pH 4.5) daily at 16:00 h for 5 days to induce T1D. For mice euthanized on day 43 from the initial STZ injection, random blood glucose and weight were measured on days 0, 7, 10, 14,18, 24, 28, 35, and 43. To avoid confounders such as order of treatment or measurement, mice were followed in a serial fashion at a particular time for each daily measurement. For mice euthanized on day 7, random blood glucose levels and weights were measured on days 0 and 7.
Intraperitoneal glucose tolerance test (IPGTT)
On day 21 from the initial STZ injection, floxed control, cre control, and FFA2 βKO males were fasted overnight (16 h) before the IPGTT. Mice were IP injected with 1 g/kg dextrose. Blood glucose was measured using a OneTouch Ultramini glucometer (LifeScan, CA, USA) via the tail vein for 2 h, and insulin was measured by ELISA (ALPCO)17. Homeostatic Model Assessment for Insulin Resistance (HOMA-IR) was calculated using the formula HOMA-IR = (Fasting Insulin (mU/ml) * Fasting Glucose (mg/dL)) / 22.5.
Fluorescence-activated cell sorting (FACS)
Single-cell suspensions were prepared from the spleen and pancreatic lymph nodes (PLN). Cells were lysed with RBC lysis buffer (BioLegend Cat. 420301) and washed with PBS. Dissociated cells were suspended in PBS containing 0.5% BSA. For surface antigens, cells were labeled with anti-CD4-FITC (11-0041, eBiosciences), anti-CD8-PE-Cy-7 (A15385, Invitrogen), anti-F4/80-PE (12-4801-82, Invitrogen), and anti-CD206-APC (17-2061-82, Invitrogen). For intracellular antigen, cells were fixed and permeabilized using Fix/Perm buffer (eBioscience) and stained with anti-FoxP3 (404-5773-82, Invitrogen). Stained cells were washed three times and analyzed by FACS. Samples were analyzed using a CyAn ADP Analyzer (Beckman and Coulter) and LSR Fortessa (BD Biosciences), and data were analyzed using Summit v4.3 software (Beckman and Coulter) as before86.
Islet isolation
Islets from 10-12 week-old male mice (floxed control, cre control, and FFA2 βKO) were isolated by collagenase (VitaCyte, IN, USA) digestion, as previously described17. Briefly, 3 mL of collagenase (Clzyme-RI, Vitacyte, Cat. 005-1030) in Hank’s buffered salt solution (HBSS) was injected into the pancreas through the bile duct ampullae in the small intestine. The inflated pancreas was excised and incubated for 17 min at 37 °C. The digested pancreas was passed through a 400 μm nylon wire mesh and rinsed with ice-cold HBSS. The islets were separated by Histopaque (Sigma) density gradient. After isolation, mouse islets were left to recover in growth medium containing RPMI 1640 medium (Gibco, MA, USA), 10% FBS (Sigma Aldrich), 1% L-glutamine (Gibco), and 1% penicillin/streptomycin (Gibco) overnight at 37 °C in a 5% CO2, 95% air environment. After overnight recovery, islets were used either for receptor expression analysis or cytokine treatment as indicated below. For expression analysis, 9 floxed control, 5 cre control, and 5 FFA2 βKO mice were used.
Ex vivo islet IFNα treatment
Recovered islets (isolated from 5 floxed control and 5 FFA2 βKO mice) were incubated either in growth medium alone or with 20,000 U/mL IFNα (PBL assay science, NJ, USA) supplemented growth medium for 2 h. The IFNα dose was adopted from Colli et al.19 and modified based on the effectiveness of the cytokine in mouse tissues. The original dose used for human islets is 2000 U/ml. As the human recombinant IFNα (PBL assay science, NJ, USA) used in our study is 10 times less effective in mouse tissues (as measured via L929/EMCV CPE assay by PBL assay science), we used a dose of 20,000 U/mL. Treated and untreated islets were washed with ice-cold PBS and lysed in RLT buffer (Qiagen RNeasy mini kit; Hilden, Germany) and stored at −80 °C freezer for RNA extraction.
In vivo IFNAR1 neutralization
To block IFNα signaling, an anti-IFNAR1 antibody (Leinco Technologies, MO, USA) was given to control (flox and cre) and FFA2 βKO male mice at a dose of 1 mg per mouse28 on days 0 and 6. Flox and FFA2 βKO male mice treated similarly with IgG1 isotype (Leinco Technologies) served as controls. Both groups were given MLDS treatment as described before and monitored for random blood glucose and weights on days 0, 7, 10, 14, 18, and 21.
Pancreata section, immunofluorescence staining, and quantification
The pancreas harvested from the mice was fixed in 4% paraformaldehyde (PFA; Electron Microscopy Science, PA, USA) and dehydrated in a 30% sucrose (Sigma Aldrich) solution. The Optimal cutting temperature compound (OCT; Sakura Finetek, CA, USA) was used to embed the dehydrated pancreas. Each pancreas was continuously sectioned into four groups at a thickness of 7 µm, each consisting of 18 sections. Between groups, 280 µm of the pancreas was discarded. Sections were stored at −80 °C before staining.
For immunofluorescence staining, the sections were thawed in humid chambers at room temperature for 20 min, permeabilized with 0.5% Triton X-100 (Roche Diagnostics, IN, USA) for 10 min, and blocked with goat serum for 1 h. Primary antibodies against insulin (DAKO reagent, 1:10), glucagon (CST-2760S, 1:50), cleaved caspase 3 (CST-9661, 1:400), Ki67 (CST-9129, 1:400), Socs1 (Abcam-9870, 1:200), and Socs3 (Abcam-16030, 1:200) were incubated overnight at 4 °C. Secondary antibodies coupled with Fluorescein or Texas Red (Vector Laboratories, CA, USA) were used at a dilution of 1:200 in PBS for 30 min on the next day. Slides were mounted using Prolong Gold Antifade Reagent with DAPI (Cell Signaling Technology, MA, USA).
Pancreata images were taken at 20× (Leica DMi8; Leica, Wetzlar, Germany). For β-cell mass, the tissue area was scanned at 10× (Akoya PhenoImager HT, Akoya Sciences, MA, USA). QuPath (version 4.0) was used to quantify positively stained cells, β-cell area, and tissue area. β-cell mass was calculated by multiplying the pancreas wet weight by the ratio of the total β-cell area to the total tissue area (four sections per mouse, with one section from each group). The same method was used to calculate α cell mass. For the cleaved caspase 3, Ki67, Socs1, and Socs3 staining, 12–16 pancreas sections from 3–4 mice per genotype were imaged and counted for positively stained β-cells and presented as a percentage of total β-cells.
Min6 cell culture, knockdown of FFA2 in Min6 cells, and IFNα treatment
Min6 cells were cultured in high-glucose DMEM medium with sodium pyruvate (Gibco) supplemented with 15% FBS (Sigma Aldrich), 1% L-glutamine (Gibco), 1% penicillin/streptomycin (Gibco), and 0.1% β-mercaptoethanol (Gibco) in 5% CO2, 95% air environment at 37 °C.
The shRNA specific for FFA2 was chosen from the VectorBuilder library (shRNA sequence GCTGTTGTGACGCTTCTTAAT) and inserted into a Lentivirus U6-based shRNA knockdown vector with the puromycin resistance gene (vector ID VB221122-1200stq). The empty vector also contains the puromycin resistance gene (vector ID VB010000-9298rtf). Lentivirus packaging and production were carried out by VectorBuilder. Min6 cells at passage 10 were infected with either empty vector (EV) or FFA2 knockdown lentivirus (shFFA2) at an MOI of 6 for 20 h. EV and shFFA2-infected Min6 cells were selected to create stable cell lines using puromycin at a concentration of 10 µg/mL. Puromycin selection was performed after every three passages.
EV and shFFA2-infected Min6 cells were treated with 20,000 U/mL IFNα (PBL assay science) for 18 h. Cells were washed with ice-cold PBS and lysed in RIPA buffer (Sigma Aldrich) supplemented with protease and phosphatase inhibitors (Cell Signaling Technology) for immunoblotting.
Immunoblotting
Protein lysates from EV and shFFA2 Min6 cells were quantified using the Pierce™ Rapid Gold BCA Protein Assay Kit (Thermo Fisher Scientific, MA, USA). An equal amount of protein was denatured from each sample, separated by sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE), and transferred to 0.45 µm polyvinylidene difluoride (PVDF) membranes. Primary antibodies against Socs1 (Abcam-Ab9870), Socs3 (Abcam-16030), and β-actin (Sigma Aldrich, A2228) were diluted 1:1000 in 3% bovine serum albumin (BSA; Sigma Aldrich). The secondary antibody was used based on the generating species of the primary antibody at a 1:5000 dilution in 5% milk. The membrane was imaged on ChemiDoc™ MP gel imager (Bio-Rad Laboratories) using SuperSignal™ West Pico PLUS Chemiluminescent Substrate (Thermo Fisher, MA, USA).
RNA isolation and gene expression
RNA was purified using the RNeasy Mini Kit (Qiagen, Hilden, Germany). Two hundred nanograms of RNA from islets or Min6 cells was reverse transcribed using the iScript cDNA synthesis kit (Bio-Rad Laboratories, IL, USA) and transcribed using the iTaq Universal SYBR Green Supermix (Bio-Rad Laboratories). Gene expression was calculated using the 2–∆∆Ct method against the housekeeping gene, mouse 18s, or β-actin. Mouse primer sequences used for amplification are listed below:
Gene | Forward Primer 5′–3′ | Reverse Primer 5′–3′ |
|---|---|---|
Ffa2 | TAATCTGACCCTGGCGGACT | GTAGAAGCCGAAGCCTGTCA |
Ffa3 | TTGCTAAACCTGACCATTTCGG | GATAGGCCACGCTCAGAAAAC |
Nlrp3 | ATTACCCGCCCGAGAAAGG | TCGCAGCAAAGATCCACACAG |
Ifnar1 | AGCCACGGAGAGTCAATGG | GCTCTGACACGAAACTGTGTTTT |
Ifnar2 | TTGTGTCTTGTGGTGTCTGC | TCATCTGGATACCCATCAAAAGC |
Ifit1 | CCAAGTGTTCCAATGCTCCT | GGATGGAATTGCCTGCTAGA |
Ifit3 | AGACAGGGTGTGCAACCAGG | CGACGAATTTCTGATTGATC |
Ifit3b | AGACAGGGTGTGCAACCAGC | CGGCGAATTTCTGCTTGATC |
Oasl2 | CAAACAAACAAACAAACCCTCTC | TCAAGGTGTCACTCTGCAT |
Socs1 | ACCTTCTTGGTGCGCGAC | GGGCCCGAAGCCATCTT |
Socs3 | CGAAGCACGCAGCCAGTT | TCCGTGGGTGGCAAAGAA |
18s | GTAACCCGTTGAACCCCATT | CCATCCAATCGGTAGTAGCG |
β-actin | GTTACAGGAAGTCCCTCACCC | CAGACCTGGGCCATTCAGAAA |
RNA sequencing and analysis
Previously published transcriptome data from our lab50 (GEO: GSE298768) were reanalyzed for pathway enrichment. Sequencing reads were aligned to the mouse genome (genome assembly version GRCm39, annotation version GRCm39.109) by the STAR aligner (version 2.7.10b) and counted by FeatureCounts (version 2.0.1). Differential gene expression was calculated using DESeq2 (version 1.40.2, R package) from raw counts generated by FeatureCounts. The log2 fold change of each gene generated by DESeq2 was input into clusterProfiler (version 4.8.3, R package) to perform weighted gene ontology biological process (GO BP) pathway enrichment analysis (P-value cutoff is 0.05). The top enriched pathways in the islets of global FFA2 KO mice were plotted based on their normalized enrichment scores (NES).
Plasma SCFA measurement
Plasma SCFAs were assayed by liquid chromatography with tandem mass spectrometry (LC-MS/MS) using a targeted approach as previously described87. 20 μL of plasma sample was mixed with 80 μL of acetonitrile (ACN), vortexed thoroughly, and incubated at 4 °C for 45 min. Following the incubation, the samples were vortexed again thoroughly and centrifuged at 4000 × g at 4 °C for 10 min. The supernatant was used for derivatization. Stock solutions of acetate, propionate, and butyrate were freshly and individually prepared in 50% aqueous acetonitrile. Mixed standard solutions containing an equal amount of individual SCFA at the desired concentration were made in 50% aqueous acetonitrile. For derivatization, 60 μL of each standard solution or of each supernatant was mixed with 30 μL of 200 mM 3-NPH in 50% aqueous acetonitrile and 30 μL of 120 mM EDC in the same solution and incubated for 30 min at 40 °C. 0.7 mL 10% ACN was then added to the reaction solution. Diluted reaction solution was mixed with pre-made stable isotope-labeled standards for LC/MS. The LC/MS analysis was performed on AB Sciex Qtrap 5500 coupled to an Agilent UPLC/HPLC system (Agilent Technologies). The quantification was achieved using the area of monitored transitions.
The measurements were performed at the Mass Spectrometry Core in the Research Resources Center of the University of Illinois Chicago.
Statistics and reproducibility
Data was analyzed using GraphPad Prism 9 (GraphPad Software, San Diego, CA, USA). Data are expressed as mean ± standard error, and analyses were performed by Student’s t-test, one-way ANOVA, or two-way ANOVA. Survival curves were compared using the Log-rank (Mantel–Cox) test. The number of biological/experimental replicates (n) is indicated in the figure legends.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
The numerical source data for the main and Supplementary Figs. are available as Supplementary Data 1 and 2. Supplementary Figs., including the unedited blots (Supplementary Fig. 8), are available in the Supplementary Information.
References
Burrack, A. L., Martinov, T. & Fife, B. T. T cell-mediated beta cell destruction: autoimmunity and alloimmunity in the context of type 1 diabetes. Front Endocrinol. 8, 343 (2017).
Steck, A. K. & Rewers, M. J. Genetics of type 1 diabetes. Clin. Chem. 57, 176–185 (2011).
Yau, C. & Danska, J. S. Cracking the type 1 diabetes code: genes, microbes, immunity, and the early life environment. Immunol. Rev. 325, 23–45 (2024).
Rampanelli, E. & Nieuwdorp, M. Gut microbiome in type 1 diabetes: the immunological perspective. Expert Rev. Clin. Immunol. 19, 93–109 (2023).
Knip, M. & Siljander, H. The role of the intestinal microbiota in type 1 diabetes mellitus. Nat. Rev. Endocrinol. 12, 154–167 (2016).
Stewart, C. J. et al. Temporal development of the gut microbiome in early childhood from the TEDDY study. Nature 562, 583–588 (2018).
Marino, E. et al. Gut microbial metabolites limit the frequency of autoimmune T cells and protect against type 1 diabetes. Nat. Immunol. 18, 552–562 (2017).
Sun, J. et al. Pancreatic beta-cells limit autoimmune diabetes via an immunoregulatory antimicrobial peptide expressed under the influence of the gut microbiota. Immunity 43, 304–317 (2015).
Miani, M. et al. Gut microbiota-stimulated innate lymphoid cells support beta-defensin 14 expression in pancreatic endocrine cells, preventing autoimmune diabetes. Cell Metab. 28, 557–572 e556 (2018).
Vatanen, T. et al. The human gut microbiome in early-onset type 1 diabetes from the TEDDY study. Nature 562, 589–594 (2018).
de Goffau, M. C. et al. Aberrant gut microbiota composition at the onset of type 1 diabetes in young children. Diabetologia 57, 1569–1577 (2014).
de Goffau, M. C. et al. Fecal microbiota composition differs between children with beta-cell autoimmunity and those without. Diabetes 62, 1238–1244 (2013).
Huang, J. et al. Gut microbial metabolites alter IgA immunity in type 1 diabetes. JCI Insight. 5, e135718 (2020).
de Groot, P. F. et al. Distinct fecal and oral microbiota composition in human type 1 diabetes, an observational study. PLoS ONE 12, e0188475 (2017).
Zou, J. et al. Inulin fermentable fiber ameliorates type i diabetes via IL22 and short-chain fatty acids in experimental models. Cell Mol. Gastroenterol. Hepatol. 12, 983–1000 (2021).
Priyadarshini, M. et al. FFAR from the Gut microbiome crowd: SCFA receptors in T1D pathology. Metabolites https://doi.org/10.3390/metabo11050302 (2021).
Priyadarshini, M. et al. An acetate-specific GPCR, FFAR2, regulates insulin secretion. Mol. Endocrinol. 29, 1055–1066 (2015).
Shi, G. et al. Free fatty acid receptor 2, a candidate target for type 1 diabetes, induces cell apoptosis through ERK signaling. J. Mol. Endocrinol. 53, 367–380 (2014).
Colli, M. L. et al. An integrated multi-omics approach identifies the landscape of interferon-alpha-mediated responses of human pancreatic beta cells. Nat. Commun. 11, 2584 (2020).
Lu, J., Liu, J., Li, L., Lan, Y. & Liang, Y. Cytokines in type 1 diabetes: mechanisms of action and immunotherapeutic targets. Clin. Transl. Immunol. 9, e1122 (2020).
Todd, J. A. Etiology of type 1 diabetes. Immunity 32, 457–467 (2010).
Ferreira, R. C. et al. A type I interferon transcriptional signature precedes autoimmunity in children genetically at risk for type 1 diabetes. Diabetes 63, 2538–2550 (2014).
Kallionpaa, H. et al. Innate immune activity is detected prior to seroconversion in children with HLA-conferred type 1 diabetes susceptibility. Diabetes 63, 2402–2414 (2014).
Kado, S. et al. Type 1 diabetes mellitus caused by treatment with interferon-beta. Intern. Med. 39, 146–149 (2000).
Oka, R. et al. Type 1 diabetes mellitus associated with pegylated interferon-alpha plus ribavirin treatment for chronic hepatitis C: case report and literature review. Clin. Med. Insights Endocrinol. Diab. 4, 39–45 (2011).
Nakamura, K. et al. Type 1 diabetes and interferon therapy: a nationwide survey in Japan. Diab. Care 34, 2084–2089 (2011).
Uonaga, T., Yoshida, K., Harada, T., Shimodahira, M. & Nakamura, Y. Case of type 1 diabetes mellitus following interferon beta-1a treatment for multiple sclerosis. Intern. Med. 51, 1875–1877 (2012).
Marro, B. S. et al. Progression of type 1 diabetes from the prediabetic stage is controlled by interferon-alpha signaling. Proc. Natl. Acad. Sci. USA 114, 3708–3713 (2017).
Dias Junior, A. G., Sampaio, N. G. & Rehwinkel, J. A balancing Act: MDA5 in antiviral immunity and autoinflammation. Trends Microbiol. 27, 75–85 (2019).
Lincez, P. J., Shanina, I. & Horwitz, M. S. Reduced expression of the MDA5 gene IFIH1 prevents autoimmune diabetes. Diabetes 64, 2184–2193 (2015).
Huang, X., Hultgren, B., Dybdal, N. & Stewart, T. A. Islet expression of interferon-alpha precedes diabetes in both the BB rat and streptozotocin-treated mice. Immunity 1, 469–478 (1994).
Diana, J. et al. Crosstalk between neutrophils, B-1a cells and plasmacytoid dendritic cells initiates autoimmune diabetes. Nat. Med. 19, 65–73 (2013).
Lombardi, A., Tsomos, E., Hammerstad, S. S. & Tomer, Y. Interferon alpha: the key trigger of type 1 diabetes. J. Autoimmun. 94, 7–15 (2018).
Lukhele, S., Boukhaled, G. M. & Brooks, D. G. Type I interferon signaling, regulation and gene stimulation in chronic virus infection. Semin. Immunol. 43, 101277 (2019).
Marroqui, L. et al. Interferon-alpha mediates human beta cell HLA class I overexpression, endoplasmic reticulum stress and apoptosis, three hallmarks of early human type 1 diabetes. Diabetologia 60, 656–667 (2017).
Antunes, K. H. et al. Microbiota-derived acetate protects against respiratory syncytial virus infection through a GPR43-type 1 interferon response. Nat. Commun. 10, 3273 (2019).
Niu, J. et al. Microbiota-derived acetate enhances host antiviral response via NLRP3. Nat. Commun. 14, 642 (2023).
Tang, C. et al. Loss of FFA2 and FFA3 increases insulin secretion and improves glucose tolerance in type 2 diabetes. Nat. Med. 21, 173–177 (2015).
Kimura, I. et al. Maternal gut microbiota in pregnancy influences offspring metabolic phenotype in mice. Science https://doi.org/10.1126/science.aaw8429 (2020).
Ludwig, B. et al. Agonist of growth hormone-releasing hormone as a potential effector for survival and proliferation of pancreatic islets. Proc. Natl. Acad. Sci. USA 107, 12623–12628 (2010).
Wu, Y. et al. Growth hormone receptor regulates beta cell hyperplasia and glucose-stimulated insulin secretion in obese mice. J. Clin. Invest. 121, 2422–2426 (2011).
Chaudhry, Z. Z. et al. Streptozotocin is equally diabetogenic whether administered to fed or fasted mice. Lab Anim. 47, 257–265 (2013).
Norgaard, S. A., Sand, F. W., Sorensen, D. B., Abelson, K. S. & Sondergaard, H. Softened food reduces weight loss in the streptozotocin-induced male mouse model of diabetic nephropathy. Lab Anim. 52, 373–383 (2018).
Motyl, K. & McCabe, L. R. Streptozotocin, type I diabetes severity and bone. Biol. Proced. Online 11, 296–315 (2009).
Wang, F. et al. Loss of ubiquitin-conjugating enzyme E2 (Ubc9) in macrophages exacerbates multiple low-dose streptozotocin-induced diabetes by attenuating M2 macrophage polarization. Cell Death Dis. 10, 892 (2019).
Andrikopoulos, S., Blair, A. R., Deluca, N., Fam, B. C. & Proietto, J. Evaluating the glucose tolerance test in mice. Am. J. Physiol. Endocrinol. Metab. 295, E1323–E1332 (2008).
Priyadarshini, M., Kotlo, K. U., Dudeja, P. K. & Layden, B. T. Role of short chain fatty acid receptors in intestinal physiology and pathophysiology. Compr. Physiol. 8, 1091–1115 (2018).
Fuller, M. et al. The short-chain fatty acid receptor, FFA2, contributes to gestational glucose homeostasis. Am. J. Physiol. Endocrinol. Metab. 309, E840–E851 (2015).
McNelis, J. C. et al. GPR43 potentiates beta-cell function in obesity. Diabetes 64, 3203–3217 (2015).
Villa, S. R. et al. Loss of free fatty acid receptor 2 leads to impaired islet mass and beta cell survival. Sci. Rep. 6, 28159 (2016).
Li, Q. et al. Interferon-alpha initiates type 1 diabetes in nonobese diabetic mice. Proc. Natl. Acad. Sci. USA 105, 12439–12444 (2008).
Flodström-Tullberg, M. et al. Target cell expression of suppressor of cytokine signaling-1 prevents diabetes in the NOD mouse. Diabetes 52, 2696–2700 (2003).
Katsarou, A. et al. Type 1 diabetes mellitus. Nat. Rev. Dis. Prim. 3, 17016 (2017).
Gregory, G. A. et al. Global incidence, prevalence, and mortality of type 1 diabetes in 2021 with projection to 2040: a modelling study. Lancet Diab. Endocrinol. 10, 741–760 (2022).
Sims, E. K. et al. Teplizumab improves and stabilizes beta cell function in antibody-positive high-risk individuals. Sci. Transl. Med. https://doi.org/10.1126/scitranslmed.abc8980 (2021).
Pescovitz, M. D. et al. Rituximab, B-lymphocyte depletion, and preservation of beta-cell function. N. Engl. J. Med. 361, 2143–2152 (2009).
Brusko, T. M., Russ, H. A. & Stabler, C. L. Strategies for durable beta cell replacement in type 1 diabetes. Science 373, 516–522 (2021).
Vandenbempt, V. et al. HAMSAB diet ameliorates dysfunctional signaling in pancreatic islets in autoimmune diabetes. iScience 27, 108694 (2024).
Bell, K. J. et al. Metabolite-based dietary supplementation in human type 1 diabetes is associated with microbiota and immune modulation. Microbiome 10, 9 (2022).
Giacco, R. et al. Long-term dietary treatment with increased amounts of fiber-rich low-glycemic index natural foods improves blood glucose control and reduces the number of hypoglycemic events in type 1 diabetic patients. Diabetes Care 23, 1461–1466 (2000).
Park, J., Wang, Q., Wu, Q., Mao-Draayer, Y. & Kim, C. H. Bidirectional regulatory potentials of short-chain fatty acids and their G-protein-coupled receptors in autoimmune neuroinflammation. Sci. Rep. 9, 8837 (2019).
Kim, M. H., Kang, S. G., Park, J. H., Yanagisawa, M. & Kim, C. H. Short-chain fatty acids activate GPR41 and GPR43 on intestinal epithelial cells to promote inflammatory responses in mice. Gastroenterology 145, 396–406 e391-310 (2013).
Wang, W. et al. Butyrate and propionate are microbial danger signals that activate the NLRP3 inflammasome in human macrophages upon TLR stimulation. Cell Rep. 43, 114736 (2024).
Vieira, A. T. et al. A role for gut microbiota and the metabolite-sensing receptor GPR43 in a murine model of gout. Arthritis Rheumatol. 67, 1646–1656 (2015).
Singh, V. et al. Microbiota fermentation-NLRP3 axis shapes the impact of dietary fibres on intestinal inflammation. Gut 68, 1801–1812 (2019).
Macia, L. et al. Metabolite-sensing receptors GPR43 and GPR109A facilitate dietary fibre-induced gut homeostasis through regulation of the inflammasome. Nat. Commun. 6, 6734 (2015).
Lednovich, K. R. et al. Intestinal FFA2 promotes obesity by altering food intake in Western diet-fed mice. J Endocrinol. 260(2), e230184 (2024).
Gao, Y. et al. ER stress and unfolded protein response (UPR) signaling modulate GLP-1 receptor signaling in the pancreatic islets. Mol. Cells 47, 100004 (2024).
Priyadarshini, M. et al. Free fatty acid receptor 3 differentially contributes to beta-cell compensation under high-fat diet and streptozotocin stress. Am. J. Physiol. Regul. Integr. Comp. Physiol. 318, R691–R700 (2020).
Zhang, Z. et al. GPR109a regulates phenotypic and functional alterations in macrophages and the progression of type 1 diabetes. Mol. Nutr. Food Res. 66, e2200300 (2022).
Remsberg, J. R. et al. Deletion of histone deacetylase 3 in adult beta cells improves glucose tolerance via increased insulin secretion. Mol. Metab. 6, 30–37 (2017).
Kojima, H. et al. Complete remission of diabetes with a transient HDAC inhibitor and insulin in streptozotocin mice. Commun. Biol. 6, 637 (2023).
Bridgeman, S., Ellison, G., Newsholme, P. & Mamotte, C. The HDAC inhibitor butyrate impairs β cell function and activates the disallowed gene hexokinase I. Int J Mol Sci. 22, 13330 (2021).
Lee, H. A. et al. Histone deacetylase inhibitor MGCD0103 protects the pancreas from streptozotocin-induced oxidative stress and beta-cell death. Biomed. Pharmacother. 109, 921–929 (2019).
Alexander, W. S. Suppressors of cytokine signalling (SOCS) in the immune system. Nat. Rev. Immunol. 2, 410–416 (2002).
Kok, F. et al. Disentangling molecular mechanisms regulating sensitization of interferon alpha signal transduction. Mol. Syst. Biol. 16, e8955 (2020).
Lloyd, R. E., Tamhankar, M. & Lernmark, A. Enteroviruses and type 1 diabetes: multiple mechanisms and factors?. Annu. Rev. Med. 73, 483–499 (2022).
Lehuen, A., Diana, J., Zaccone, P. & Cooke, A. Immune cell crosstalk in type 1 diabetes. Nat. Rev. Immunol. 10, 501–513 (2010).
Xiong, Q. et al. Metabolite-sensing g protein coupled receptor TGR5 protects host from viral infection through amplifying type I interferon responses. Front. Immunol. 9, 2289 (2018).
Huang, H. et al. Elimination of GPR146-mediated antiviral function through IRF3/HES1-signalling pathway. Immunology 152, 102–114 (2017).
Wang, N. et al. GNAQ negatively regulates antiviral innate immune responses in a calcineurin-dependent manner. J. Immunol. 203, 1288–1297 (2019).
Villasenor, A. et al. Rgs16 and Rgs8 in embryonic endocrine pancreas and mouse models of diabetes. Dis. Model Mech. 3, 567–580 (2010).
Joshi, J. C. et al. RGS2 is an innate immune checkpoint for suppressing Gαq-mediated IFNγ generation and lung injury. iScience. 28, 111878 (2025).
Wicksteed, B. et al. Conditional gene targeting in mouse pancreatic ss-Cells: analysis of ectopic Cre transgene expression in the brain. Diabetes 59, 3090–3098 (2010).
Saadane, A., Lessieur, E. M., Du, Y., Liu, H. & Kern, T. S. Successful induction of diabetes in mice demonstrates no gender difference in development of early diabetic retinopathy. PLoS ONE 15, e0238727 (2020).
Kumar, P. et al. Critical role of OX40 signaling in the TCR-independent phase of human and murine thymic Treg generation. Cell Mol. Immunol. 16, 138–153 (2019).
Priyadarshini, M. et al. Gestational insulin resistance is mediated by the gut microbiome-indoleamine 2,3-dioxygenase axis. Gastroenterology 162, 1675–1689 e1611 (2022).
Acknowledgements
The work was supported by the National Institutes of Health [R01DK104927 (B.T.L.), P30DK020595 DRTC P&F grant (B.T.L.; M.P.)], and the Department of Veterans Affairs, Veterans Health Administration, Office of Research and Development, VA merit grant [1I01BX003382 (B.T.L.)].
Author information
Authors and Affiliations
Contributions
K.X. (conceptualization, methodology, data curation, analysis, project administration, writing original draft, reviewing and editing); P.K. (methodology); S.C.S. (data curation); H.M.E. (data analysis); B.S.P. (resources, methodology); Irene Corona-Avila (data curation, analysis); Nupur Pandya (data curation, analysis); B.T.L. (conceptualization, formal analysis, validation, funding acquisition, project administration, project supervision, writing, reviewing and editing); M.P. (conceptualization, methodology, data curation, analysis, project administration, project supervision, funding acquisition, writing original draft, reviewing and editing). All the authors approved the final version of the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Communications Biology thanks Li-Ling Wu and the other anonymous reviewer(s) for their contribution to the peer review of this work. Primary Handling Editor: Christina Karlsson Rosenthal.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Xu, K., Kumar, P., Chang Stauffer, S.R. et al. Pancreatic β-cell FFA2 deficiency suppresses multiple low dose streptozotocin induced diabetes in male mice. Commun Biol 8, 1389 (2025). https://doi.org/10.1038/s42003-025-08787-1
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s42003-025-08787-1
