
Abstract
Automation has revolutionized many fields by improving efficiency, accuracy, and reproducibility. However, in organic chemistry, automating key tasks such as reaction optimization and analysis remains a significant challenge. To accelerate advancements in organic chemistry research and development, we propose a fully automated system based on real-time inline analysis performed by Fourier-transform infrared spectroscopy and assisted by a neural network model. To rapidly collect data, a linear combination of spectral intensities was used as training data for a yield prediction model. Using this model, we demonstrated real-time yield prediction of Suzuki–Miyaura cross-coupling with remarkable accuracy. By combining this yield prediction model with real-time inline analysis and a flow chemistry setup, we have developed a fully automated system for the rapid and efficient optimization of reaction conditions and process analysis.

Similar content being viewed by others
Introduction
Many industries are actively pursuing or have already implemented automation systems where machines replace humans in labor-intensive tasks for product manufacturing. Just as Henry Ford revolutionized industrial production in the 20th century with the introduction of the assembly line, commonly referred to as Fordism, the concept of fully automated workflows may similarly drive innovation in today’s industrial systems. Organic synthesis, which involves the creation of molecular-scale products, also stands to benefit from automation1,2. Machines can now perform various synthetic operations such as reagent addition3,4, reaction monitoring5, and product purification6,7, thereby reducing or eliminating the need for human intervention. Since organic synthesis is often labor-intensive and requires delicate manipulation, the introduction of machines into the process can significantly enhance both efficiency and reproducibility.
In addition to large-scale chemical manufacturing, automation also holds significant promise for synthetic chemists working in research and development (R&D)8. R&D frequently demands the synthesis of novel compounds with specific functions or biological activities, which requires performing a large number of reactions and optimizing reaction conditions. Automation can greatly accelerate these demanding workflows. In this context, flow reactors9,10 are especially well-suited for automated synthesis because of their inherent compatibility with high-throughput experimentation11. Furthermore, flow systems offer precise control over time-12,13,14, temperature-15,16, and concentration17,18 to perform precisely controlled reactions19,20,21,22,23. In contrast, conventional batch reactors often suffer from variability and limited control, which can reduce reproducibility.
Although automated flow systems can significantly accelerate the execution of reactions, R&D in organic synthetic chemistry remains a time-consuming process. Synthesis represents only one aspect of the workflow. The most time-consuming tasks, such as analyzing reaction products and designing new reaction conditions, still largely depend on human expertise. Automation of analysis and condition design can, in principle, be achieved using autosamplers to perform automatic measurements. For instance, Jamison and Jensen demonstrated a strategy in which inline sampling was coupled with HPLC analysis to optimize reaction conditions by maximizing the peak area of the desired product24. However, chromatographic methods lack immediacy and are therefore not ideally suited for fully exploiting the advantages of flow chemistry. In contrast, spectroscopic techniques such as infrared (IR) spectroscopy allow real-time, inline analysis. This makes them highly attractive for rapid optimization and process monitoring. Jamison and Jensen also performed automatic optimization by minimizing the carbonyl peak intensity using inline IR analysis24. Knight extended the applicability of inline IR analysis to reaction systems lacking distinctive spectral features—an area where traditional peak-based approaches are not easily applicable25. However, their method provided only qualitative insights and could not support accurate yield prediction. Notably, these limitations are not resolved even by incorporating internal standards or calibration curves, as accurate quantification still requires clearly assignable spectral features. In systems such as ours—where peaks are weak, overlapping, or entirely absent—traditional quantification methods are inherently limited. Therefore, an alternative approach is needed to achieve quantitative analysis under these conditions.
In cases where characteristic peaks are absent—or when spectral differences are too subtle for human interpretation—machine learning becomes especially powerful. It offers the potential to extract meaningful patterns from nuanced spectral variations and enable quantitative predictions that would otherwise be inaccessible through conventional analysis. However, machine learning typically requires large, high-quality datasets derived from a wide range of experimentally measured reaction mixtures. This demand poses a substantial experimental burden, especially when the spectral data lack distinct, easily identifiable features. Additionally, existing methods for automated reaction analysis based on repeated inline measurements still require a vast number of experiments to achieve precise yield prediction.
To address this, we propose a streamlined approach in which IR spectra are generated by linearly combining the spectra of the reactants and products to produce “mimicked spectra” that approximate those of actual reaction mixtures. These mimicked spectra serve as effective substitutes for experimentally measured data, enabling the rapid and efficient construction of training datasets without exhaustive experimentation. Using this method, we demonstrate a fully automated system for organic synthesis that integrates real-time inline analysis with a neural network-based predictive model. This system enables closed-loop reaction optimization with minimal experimental burden (Fig. 1).

A schematic illustration of the proposed strategy integrating machine-controlled flow synthesis, machine learning-based analysis, and reaction design.
Results and discussion
We began this study using an existing flow system equipped with an inline monitoring unit26 capable of real-time, high-accuracy analysis. Real-time inline analysis, where the reaction solution is continuously analyzed without manual sampling, is widely used to monitor reaction progress27,28. Among the available analytical techniques29, Fourier-transform infrared spectroscopy (FTIR) is one of the most commonly employed because of its speed, sensitivity, and resolution30. Historically, FTIR analysis has been extensively used for the characterization of organic molecules, as it provides valuable information linking specific vibrational frequencies to molecular substructures (e.g., carbonyl groups typically exhibit a strong peak near 1715 cm−1). As a result, changes in reagent consumption or product formation be tracked by monitoring specific peaks in the spectrum. However, this peak-based approach is limited to reactions that involve significant changes in functional groups31,32. Although the fingerprint region of the spectrum, typically from 1500 to 650 cm−1, is unique to each molecule, its complexity often makes it difficult for humans to interpret all of the peaks. Machine learning, when combined with multivariate data analysis, can help address this issue by interpreting complex spectral features. However, building an accurate model still requires a large and diverse training dataset33.
To address this challenge, we focused on the fundamental principles of FTIR spectroscopy. The intensity of each peak in an FTIR spectrum reflects the abundance of the corresponding molecular species. When molecules are mixed, the resulting spectrum can be approximated as a linear combination of the spectra of the individual components. We hypothesized that it would be possible to simulate the spectrum of a reaction mixture by linearly combining the spectra of the materials involved, including the products. This approach would allow for rapid generation of a large training dataset using only a small number of measured spectra34.
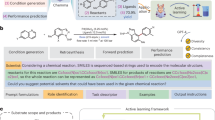
As a model reaction, we selected the Suzuki–Miyaura cross-coupling (SMC) reaction due to its wide applicability in organic synthesis. SMC forms a carbon–carbon bond between the organic groups from a boronic ester and a (pseudo)halide, typically without significant changes in the FTIR spectrum. This is characteristic of many cross-coupling reactions, especially those in diluted conditions35. Indeed, the FTIR spectra of representative reactants, boronic ester 1 (0.049 M) and iodoarene 2 (0.033 M), and product 3 (0.033 M) in THF/MeOH (50/50 volume ratio) showed minimal differences to the naked eye (Fig. 2a). o prepare the training dataset, we first performed FTIR measurements on compounds 1, 2, and 3. Each compound was measured five times, and one measurement was randomly selected for use in linear combination, in order to prevent the model from learning noise patterns. Based on our previous study on flow coupling reactions36, we assumed that using 1.5 equivalents of compound 1 would be optimal for the reaction. Because the reaction system is well-characterized and known to involve few significant side products—particularly as 2 exhibits nearly ideal mass balance—we considered that the reaction mixture could be reasonably represented by a combination of the spectra of 1, 2, and 3. The training dataset was constructed as shown in Fig. 2b. In this dataset, y denotes the virtual percent yield of 3 (cyield), and X represents a one-dimensional data vector corresponding to a simulated spectrum as a linear combination of the FTIR spectra of compounds, reflecting a reaction that would produce 3 at cyield% yield. To account the decomposition of compound 1, a random number “r” was introduced to represent the decomposition rate. By varying cyield and r from 0 to 100 in integer steps, we generated 10,000 (X, y) datasets.

a FTIR spectra of the compounds and solvent. b Preparation of training data. c Model evaluation, where the x-axis represents the experimentally determined percent yield (cyield) of the prepared solution, and the y-axis indicates the predicted percent yield from its FTIR. d Inline FTIR and yield prediction of actual reactions. Gas chromatography (GC) yields were determined using an internal standard.
To evaluate the model, test solutions were prepared by dissolving compounds 1, 2, and 3 in arbitrary proportions, similar to the combinations used in Fig. 2b (see Supplementary Information section 2.2 and Table S1 for detail). FTIR measurements were then conducted to determine whether the model could predict the concentration of compound 3 (cyield) based on the spectral data. However, the initial attempt resulted in complete failure (Fig. 2c, left); the model predicted a cyield of approximately 100 for all spectra. We found that applying spectral differentiation significantly improved the prediction accuracy (Fig. 2c, middle). Furthermore, when we limited the spectral analysis to the fingerprint region (699–1692 cm−1) and applied differentiation, the model was able to predict cyield with high accuracy (Fig. 2c, right). These results indicate that restricting the spectral range can enhance the performance of the prediction model, and that using a larger number of data points is not necessarily beneficial. We note that while more complex spectral combination methods could be considered (e.g., weighted or nonlinear combinations), we deliberately selected simple linear combinations to prioritize methodological simplicity and ease of implementation, while still achieving high predictive accuracy. We applied the model to predict the yields of the SMC reaction under various conditions. According to our previous study34, the reaction was carried out in a column reactor packed with silica-supported palladium(0). A solution of 2 (0.033 M), containing 1 (1.5 equivalents) KOH (2 equivalents), was flowed through the column at 70 °C. By varying the flow rate, we generated multiple reaction conditions to evaluate the model’s prediction accuracy (Fig. 2d). This demonstrates that our linear combination approach allows the construction of a model using only the spectra of substrates and products, yet provides accurate yield prediction—a task challenging to achieve through manual interpretation. Importantly, the rapid yield prediction enabled by our model allows seamless integration into real-time, closed-loop optimization frameworks. Because the model requires only inline FTIR measurements and can be trained with synthetic spectra derived from pure compounds, it eliminates the need for extensive experimental screening. This makes it especially well-suited for Bayesian optimization, where each new experimental condition is selected based on predicted performance. The immediacy and accuracy of our predictive framework offer a practical advantage over conventional methods, enabling fast, data-efficient reaction optimization. This efficient analytical framework can accelerate the analysis stage of R&D.
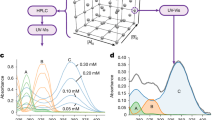
Having established the inline analysis model, we turned our attention to the development of a fully automated reaction optimization system. A schematic overview of the setup is shown in Fig. 3a. A programmable logic controller (PLC) was connected to the pump and the water bath heater to control the flow rate and temperature, respectively. To monitor system stability, the PLC was also connected to a flowmeter, pressure gauge (labeled “gauge” in Fig. 3a), and thermometer. Finally, the PLC was connected to a laptop computer, which controlled and monitored the entire system, including the FTIR unit. A process flowchart is shown in Fig. 3b. Initially, the reaction was carried out under randomly selected conditions from a total of 30 possibilities (five flow rates and six temperatures), defined as the “Initial” state in the flow chart. This initial state was repeated a predefined number of times, typically once or twice. After collecting the initial dataset, Bayesian optimization (BO)37,38,39,40,41 was applied to suggest the next candidate condition. The reaction was carried out automatically under the suggested conditions, and the resulting yield was analyzed. This iterative process continued until BO suggested a previously tested condition42.

a Schematic of setup. b Process flowchart. “Initial” means to collect initial data set. c Results of optimization trials. d Assessment of the accuracy of the prediction model.
In the first optimization trial, the initial conditions (6.0 mL/min and 50 °C, and 4.0 mL/min and 80 °C) were selected randomly. After six reaction runs, BO suggested conditions that had already been tested (2.0 mL/min, 80 °C). The system thus concluded that 2.0 mL/min and 80 °C were the optimal reaction conditions, yielding the highest predicted yield (Fig. 3c, 1st trial). Notably, this entire process required only 70 min to reach optimization. In the second trial, using a different set of initial conditions, the system again identified the same optimal parameters (Fig. 3c, 2nd trial). Although three more reactions were needed in the second trial, the total experimental time remained short at approximately 2 h. In our previous study36, we found that increasing the concentration fourfold under high-yielding conditions did not result in decreased yield. Therefore, it is likely that the optimal conditions identified here could be applied at higher, more productive concentrations, thereby improving throughput without compromising efficiency. These results highlight the rapidity and reproducibility of the developed automated optimization system.
We assessed the accuracy of the prediction model by comparing the predicted yields with those determined by gas chromatography (GC) using an internal standard for the reaction solutions obtained during the optimization process. To further validate the model, we conducted 63 additional reactions under varied conditions and similarly compared the predicted yields with experimentally measured values. As shown in Fig. 3d, the results demonstrated excellent agreement, with a high coefficient of determination (R² = 0.96). These findings confirm the robustness and general applicability of the model across a broader range of conditions.
Finally, we applied the prediction model for real-time analysis, demonstrating its utility as an example of process analytical technology (PAT)43, as described above and shown in Fig. 3a. Our optimization system allows for continuous monitoring of reaction parameters, with thermometer, flowmeter, and pressure gauge connected to the PLC. These instruments monitor the temperature, flow rate, and pressure in real time, while reaction yields are recorded every minute using inline FTIR analysis. Figure 4 shows a log of these measurements. The red line indicates the predicted yield based on FTIR data, demonstrating that the model can function effectively as a PAT system without further refinement. At some points, we collected the reaction solution, and confirmed the predicted yield is in good agreement with the yield determined by GC with the internal standard. Notably, the monitoring system was able to detect subtle differences in stabilized reaction conditions that were not evident from physical parameters alone, such as slight variations in temperature.

Red bald line: yields predicted by the model, black dash line: pressure measured by the pressure gage (Pres), blue dotted line: temperature measured by the thermometer (Temp), gray line: flow rate measured by the flowmeter (Rate).
Conclusion
In conclusion, we have developed a concise method for preparing FTIR data for machine learning, where individual FTIR spectra of the reactants and products are linearly combined to simulate the spectrum of a reaction mixture. With appropriate data preprocessing, we successfully constructed an accurate model for yield prediction. This was achieved with limited experimental input, as the model was trained on mimicked spectra generated from only the FTIR data of pure reactants and products, reducing the need for extensive reaction sampling. This framework enables real-time prediction of reaction yield within an integrated system, allowing us to achieve fully automated optimization of the Suzuki–Miyaura cross-coupling reaction. While this study employed a grid of reaction conditions, future implementations could incorporate dynamic flow techniques to further enhance experimental efficiency44. Furthermore, we demonstrated that the prediction model can function as a practical example of process analytical technology (PAT), capable of detecting subtle differences in reaction conditions that are not evident from physical parameters alone. We believe that the proposed system represents a meaningful step toward industrial automation. As often envisioned in science fiction45,46, it is becoming increasingly plausible that future industries may be operated entirely by robotic systems, without the need for human intervention.
Methods
Development of prediction model
Phenylboronic acid pinacol ester (1, 230 mg, 1.13 mmol) was dissolved in THF/MeOH (23 mL, 1:1 volume ratio) to be a 0.0049 M solution. The solution of 4-iodobenzonitrile (2, 0.033 M) and 4-cyanobiphenyl (3, 0.033 M) were similarly prepared. Each solution was pumped with a syringe pump at a flow rate of 1.0 mL/min and then in-line measurement was carried out. Five spectra were recorded in each solution considering noise patterns. Raw spectra were differentiated using the numpy.gradient function provided by the NumPy library. Only a spectral range from 1506 to 699 cm−1 (434 datapoints) was used for neural network (NN) training and prediction.
Training data was created with spectra of each compound (1, 2, and 3) according to the following equation;
where cyield and r are the yield and decomposed rate (integers from 0 to 100), SpecLC means the spectrum of the generated, Spec1, Spec2, Spec3 mean the spectra of 1, 2, and 3 respectively. In order to avoid learning noise pattern, one spectrum was randomly chosen for each linear combination. In this study, the number of training data was set to 10,000 throughout.
Automated optimization and reaction monitoring
Overall system control including file management and data logging, among other things, was written in C#.NET. Machine learning tasks (Bayesian optimization and prediction by NN model) were conducted using Python and the script was executed in C#.NET. The software for ReactIR (iC IR) was controlled through a mouse simulation. The pumps, sensors, heater and chiller were connected to PLC (Mitsubishi Electronic, MELSEC iQ-F series), and PLC was controlled via SoftGOT 2000 (Mitsubishi Electronic). The pump motor’s rotation speed was controlled through a voltage control (0–10 V). Values of pressure gauges and flowmeters were red through current measurements (4–20 mA). Thermocouples were connected to a dedicated unit of PLC, and the temperature was recorded. these values were logged every second. Power supply to a heater and a chiller pump were switched on and off by relays. According to our previous work39, the script was written in Python with GPyOpt library.
Process flow is as follows. At first, the search space in Bayesian optimization was determined, and a process was begun. In this work, a search space contained temperature (30, 40, 50, 60, 70, 80 °C, discrete) and flow rate (2, 3, 4, 5, 6 mL/min, discrete). The initial reaction condition was selected randomly, and the temperature and the flow rate were adjusted to the selected condition. After those values achieved the target ones, the system started stabilizing phase. The stabilizing phase lasted 480/F seconds (F means the flow rate). After stabilization, the system measured IR, and if necessary, sample solution was collected for GC analysis. After measurement, the flow rate was changed to 1.0 mL/min (stand-by mode) in order to reduce the waste of the reagents. During the stand-by mode, yield was predicted from the spectrum by using NN model, and the next candidate condition was selected randomly (as continuing Initial), or was suggested by the Bayesian optimization algorithm. If the suggested condition had not already selected in previous iterations, the next iteration began. If the suggested condition had already selected, the process completed
As a stock solution, 1 (9.356 g, 45.8 mmol), 2 (6.998 g, 30.6 mmol), potassium hydroxide (3.421 g, 61 mmol), n-tridecane (2.363 g, an internal standard) were dissolved in THF/MeOH (926 mL, 1:1 volume ratio). The stock solution was pumped into the tube reactor containing the polymer-supported palladiums in the water bath with heater and cooling jacket. In order to cool reaction mixture, another water bath was placed between tube reactor and spectrometer. In optimization process, first two iterations were carried out to obtain initial data and Bayesian optimization was executed from the third iteration onwards.
For the real-time yield monitoring, IR spectrum was measured every minute (128 scans). Moreover, additional waiting times of 240 and 120 seconds were taken before the spectrum measurement and after the sampling step, respectively.
Data availability
All data are included in the Supplementary Information.
Code availability
The code used in this study consists of simple data processing scripts that do not constitute a central component of the conclusions. Therefore, the code has not been deposited in a DOI-minting repository, but it is available from the corresponding author upon reasonable request.
References
Plante, J., Palmacci, E. R. & Seeberger, P. H. Automated solid-phase synthesis of oligosaccharides. Science 291, 1523–1527 (2001).
Adamo, A. et al. On-demand continuous-flow production of pharmaceuticals in a compact, reconfigurable system. Science 352, 61–67 (2016).
Chan, N., Cunningham, M. F. & Hutchinson, R. A. Continuous controlled radical polymerization of methyl acrylate with copper wire in a CSTR. Polym. Chem. 3, 486–497 (2012).
Soheilmoghaddam, F., Rumble, M. & Cooper-White, J. High-throughput routes to biomaterials discovery. Chem. Rev. 121, 10792–10864 (2021).
Workman, J. Jr., Lavine, B., Chrisman, R. & Koch, M. Process analytical chemistry. Anal. Chem. 83, 4557–4578 (2021).
Wood, B., Girard, K. P., Polster, C. S. & Croker, D. M. Progress to date in the design and operation of continuous crystallization processes for pharmaceutical applications. Org. Process. Res. Dev. 23, 122–144 (2019).
Hu, C. et al. Continuous reactive crystallization of an API in PFR-CSTR cascade with in-line PATs. React. Chem. Eng. 5, 1950–1962 (2020).
Fitzpatrick, D. E., Battilocchio, C. & Ley, S. V. Enabling technologies for the future of chemical synthesis. ACS Cent. Sci. 2, 131–138 (2016).
Cambié, D., Bottecchia, C., Straathof, N. J. W., Hessel, V. & Noël, T. Applications of continuous-flow photochemistry in organic synthesis, material science, and water treatment. Chem. Rev. 116, 10276–10341 (2016).
Plutschack, M. B., Pieber, B., Gilmore, K. & Seeberger, P. H. The Hitchhiker’s guide to flow chemistry. Chem. Rev. 117, 11796–11893 (2017).
Rial-Rodríguez, E. et al. Angew. Chem. Int. Ed. 63, e202412045 (2024).
Fukuyama, T., Fujita, Y., Rashid, M. A. & Ryu, I. Flow update for a cossy photocyclization. Org. Lett. 18, 5444–5446 (2016).
Ashikari, Y., Kawaguchi, T., Mandai, K., Aizawa, Y. & Nagaki, A. A synthetic approach to dimetalated arenes using flow microreactors and the switchable application to chemoselective cross-coupling reactions. J. Am. Chem. Soc. 142, 17039–17047 (2020).
Muta, K., Okamoto, K., Nakayama, H., Wada, S. & Nagaki, A. Defluorinative functionalization approach led by difluoromethyl anion chemistry. Nat. Commun. 16, 416 (2025).
Petersen, T. P., Becker, M. R. & Knochel, P. Continuous flow magnesiation of functionalized heterocycles and acrylates with TMPMgCl⋅LiCl. Angew. Chem. Int. Ed. 53, 7933–7937 (2014).
Usutani, H., Yamamoto, K. & Hashimoto, K. Process intensification of a napabucasin manufacturing method utilizing microflow chemistry. ACS Omega 8, 10373–10382 (2023).
Webb, D. & Jamison, T. F. Diisobutylaluminum hydride reductions revitalized: a fast, robust, and selective continuous flow system for aldehyde synthesis. Org. Lett. 14, 568–571 (2012).
Jiang, Y., Kurogi, T. & Yorimitsu, H. Reductive stereo- and regiocontrolled boryllithiation and borylsodiation of arylacetylenes using flow microreactors. Nat. Synth. 3, 192–201 (2024).
Elsherbini, M., Huynh, F., Dunbabin, A., Allemann, R. K. & Wirth, T. Selective hydroboration–oxidation of terminal alkenes under flow conditions. Chem. Eur. J. 26, 11423–11425 (2020).
Saito, Y., Nishizawa, K., Laroche, B., Ishitani, H. & Kobayashi, S. Continuous-flow synthesis of (R)-tamsulosin utilizing sequential heterogeneous catalysis. Angew. Chem. Int. Ed. 61, e202115643 (2022).
Leclercq, E. et al. Electrolyte-free electrochemical C–H trifluoromethylation of 2-pyridones under batch and flow conditions. Green. Chem. 24, 7388–7394 (2022).
Shamoto, O. et al. Peptide cyclization by the use of acylammonium species. Angew. Chem. Int. Ed. 62, e202300647 (2023).
Govindan, K., Chen, N.-Q., Venkatachalam, G., Leung, T.-F. & Lin, W.-Y. Batch vs continuous-flow method to synthesize N-(3-acylamidopropyl)lactams through N−C bond cleavage in amides with amidines. Adv. Synth. Catal. 367, e202401179 (2025).
Bédard, A.-C. et al. Reconfigurable system for automated optimization of diverse chemical reactions. Science 361, 1220–1225 (2018).
Mallia, C. J. et al. Development and manufacture of a curtius rearrangement using continuous flow towards the large-scale manufacture of AZD7648. Org. Process Res. Dev. 26, 3312–3322 (2022).
Ashikari, Y. et al. Flowmicro in-line analysis-driven design of reactions mediated by unstable intermediates: flash monitoring approach. Chem. Eur. J. 30, e202303774 (2024).
Trojanowicz, M. Flow chemistry vs. flow analysis. Talanta 146, 621–640 (2016).
Blower, A. L., Bakalis, S. & Watson, N. J. A review of in-line and on-line measurement techniques to monitor industrial mixing processes. Chem. Eng. Res. Des. 153, 463–495 (2020).
Rodriguez-Zubiri, M. & Felpin, F.-X. Analytical tools integrated in continuous-flow reactors: which one for what?. Org. Process Res. Dev. 26, 1766–1793 (2022).
Carter, C. F. et al. ReactIR flow cell: a new analytical tool for continuous flow chemical processing. Org. Process Res. Dev. 14, 393–404 (2010).
Lange, H. et al. A breakthrough method for the accurate addition of reagents in multi-step segmented flow processing. Chem. Sci. 2, 765–769 (2010).
Hafner, A. & Ley, S. V. Generation of reactive ketenes under flow conditions through zinc-mediated dehalogenation. Synlett 26, 1470–1474 (2015).
Brodmann, T., Koos, P., Metzger, A., Knochel, P. & Ley, S. V. Continuous Preparation of arylmagnesium reagents in flow with inline IR monitoring. Org. Process Res. Dev. 16, 1102–1113 (2012). Rough reaction monitoring based on the fingerprint area has been achieved. For see.
Sagmeister, P., Hiersegger, R., Williams, J. D., Kappe, C. O. & Kowarik, S. Artificial neural networks and data fusion enable concentration predictions for inline process analytics. Digit. Discov. 1, 405–412 (2022).
Malig, T. C., Kumar, A. & Kurita, K. L. Online and in situ monitoring of the exchange, transmetalation, and cross-coupling of a Negishi reaction. Org. Process Res. Dev. 26, 1514–1519 (2022). IR tracking for cross-coupling reactions with higher concentrations has been reported. See.
Ashikari, Y. et al. Flow grams-per-hour production enabled by hierarchical bimodal porous silica gel supported palladium column reactor having low pressure drop. Catal. Today 388–389, 231–236 (2022).
Mockus, J. Bayesian Approach to Global Optimization (Springer, 2011).
Sugisawa, N. et al. Rapid and mild one-flow synthetic approach to unsymmetrical sulfamides guided by bayesian optimization. Chem. Methods 1, 484–490 (2021).
Sato, E. et al. Application of an electrochemical microflow reactor for cyanosilylation: machine learning-assisted exploration of suitable reaction conditions for semi-large-scale synthesis. J. Org. Chem. 86, 16035–16044 (2021).
Naito, Y. et al. Bayesian optimization with constraint on passed charge for multiparameter screening of electrochemical reductive carboxylation in a flow microreactor. Chem. Commun. 58, 3893–3896 (2022).
Ashikari, Y., Mandai, K., Yao, Y., Tsuchihashi, Y. & Nagaki, A. Electrocatalytic reduction of (hetero)aryl halides in a proton-exchange membrane reactor and its application for deuteration. ChemElectroChem 10, e202300315 (2023).
Ashikari, Y. et al. Investigation of parameter control for electrocatalytic semihydrogenation in a proton-exchange membrane reactor utilizing bayesian optimization. Front. Chem. Eng. 3, 819752 (2022).
Chew, W. & Sharratt, P. Trends in process analytical technology. Anal. Methods 2, 1412–1438 (2010).
Martinuzzi, S. et al. Dynamic experiments in flow accelerate reaction network definition in a complex hydrogenation using catalytic static mixers. React. Chem. Eng. 9, 132–138 (2024). Kappe and Williams have demonstrated the potential of dynamic flow methods for high-throughput experimentation with minimal material usage. See.
Dick, P. K. Do Androids Dream of Electric Sheep? (Doubleday and Company, 1968).
Yoshiura, Y. Time of EVE (Studio RIKKA, 2008).
Acknowledgements
This work was supported by New Energy and Industrial Technology Development Organization (NEDO, JPNP19004). This work was also supported by JSPS KAKENHI (Grant Numbers; JP20K15276, JP20KK0121, JP21H01936, JP21H01706, JP23K21114, and JP24K08421), Japan Agency for Medical Research and Development (AMED, JP21ak0101156), Core Research for Evolutional Science and Technology (CREST, JPMJCR18R1), NEDO (JNPN14004), the Japan Keirin Autorace Foundation, the Ogasawara Foundation for the Promotion of Science & Engineering, and Hokkaido Startup Future Creation development by mutual support networks. The authors wish to express our gratitude to Messrs. Yoichi YAMASAKI and Yuki HARA (Mettler-Toledo International Inc.) for their valuable advice about in-line FTIR measurement.
Author information
Authors and Affiliations
Contributions
Y.A. designed the reaction and wrote the manuscript, T.T. carried out model construction, auto optimization and reaction monitoring. K.T. and Y.Y. carried out reactions. A.N. conceived of the project and supervised. All authors reviewed the manuscript. Y.A. and T.T. have contributed equally.
Corresponding author
Ethics declarations
Competing interests
Y.A., T.T., and N.A. are inventors on a Japanese patent application (“Method, device, and program for yield prediction, and method for compound synthesis”, Japanese Patent Application No. 2024-080773) related to the optimization method described in this manuscript. The application is currently under review. All other authors declare no competing interests.
Peer review
Peer review information
Communications Chemistry thanks Sooyeon Moon and the other, anonymous, reviewers for their contribution to the peer review of this work.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Ashikari, Y., Tamaki, T., Tomite, K. et al. Real-time inline-IR-analysis via linear-combination strategy and machine learning for automated reaction optimization. Commun Chem 8, 287 (2025). https://doi.org/10.1038/s42004-025-01676-y
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s42004-025-01676-y
