
Abstract
Atmospheric amines, derivatives of ammonia, play a unique yet not fully understood role in air quality, climate and public health. Sub-5 parts per trillion Volume (pptV, <10-12 in volume) mixing ratios of amines facilitate the physical and/or chemical transformation of aerosols in the atmosphere, enhancing aerosol formation and growth rates, aerosol hygroscopicity, and the activation of cloud condensation nuclei. This serves as the initial step for cloud droplet formation and, consequently, influences cloud properties and the hydrological cycle. Ambient observations demonstrate more than a thousand-fold particle formation rates in the presence of amines as compared to ammonia. Yet, the challenges related to detecting minute levels of amines, the paucity of ambient amine measurements, and the limited process-based understanding of airborne aerosol production have resulted in amines being underrepresented in global climate models. Therefore, advanced techniques with extremely low detection limits and highly spatially and temporally resolved ambient amine measurements globally in diverse environments are essential.
Similar content being viewed by others

Introduction
The interaction between the Earth’s ecosystem and the atmosphere is a non-linear and dynamic process that has been evolving differently in recent decades1. This is due to changes in atmospheric factors, including levels of greenhouse and reactive gases, water vapor, and the uneven distribution of aerosol concentrations and sizes over time and space. Additionally, variations in atmospheric dynamics, the state of the oceans, and the cryosphere contribute to these changes. Therefore, the pressing issue of climate change – a major challenge of the 21st century, is driven by the need for a thorough process-level understanding of the diverse environmental factors that add to its intricacy. Despite advancements in regional to global climate models incorporated with process-based parameterizations based on controlled laboratory experiments, ambient measurements and satellite observations, the process-level understanding of the climate system remains elusive. One such complex driver is the interaction between aerosols and clouds, which remains the largest source of uncertainty in the climate system due to its dynamic and non-linear relationships2. This is mainly due to limitations in model assimilation and validation, exacerbated by the uneven geographical distribution and spatial heterogeneity of measurement networks, asynchronous monitoring, and inconsistencies in data collection methods of various atmospheric variables. This emphasizes the need for advanced climate prediction models with fewer uncertainties to reduce climate risk to the Earth’s ecosystem. Therefore, it is paramount to improve our current process-level understanding – specifically, how aerosols form, grow, activate, and interact with clouds and radiation, thereby affecting air quality, the hydrological cycle, climate, and human health.
Models estimate that up to 90% of aerosols are airborne via the condensation of low-volatility vapors, such as sulfuric acid (SA)3. However, the formation of initial molecular clusters in the planetary boundary layer would not be possible without binding agents like ammonia (NH3) and amines4,5. These agents promote the growth of molecular clusters by preventing their evaporation, making them essential for aerosol formation. One exception is a study that showed ion-induced nucleation of pure biogenic volatile organic compounds (VOCs) without SA in a chamber experiment under atmospheric conditions6. Atmospheric amines are ubiquitous organic bases that are emitted into the atmosphere from a wide range of natural sources, such as marine organisms, vegetation and forest fires, and anthropogenic sources, such as biomass and fossil-fuel burning, animal husbandry, waste incineration, sewage treatment and residential cooking7,8,9. Gas-phase concentrations of amines are typically reported to be 10 to 1000 times lower compared to NH3 concentrations10,11, but have a thousand-fold higher enhancement factor for particle nucleation rates than NH3 at low concentrations4,12. In laboratory settings, dimethylamine (DMA) has been shown to replace NH3 in molecular clusters with SA, making it an interesting target compound for ambient particle formation studies at the molecular level. It should be noted that near emission sources, amine mixing ratios can reach as high as hundreds of parts per billion in Volume (ppbV), as one study reported butylamine (BA) in the range from 27.3 to 187.7 ppbV in areas near livestock operations13. But, reports on amine speciation and ambient concentrations at the level relevant to new particle formation (NPF) are rare and discrepant14,15.
Ge et al.7,16 provided the first and the most comprehensive fundamental assessment of amines sources, fluxes, dynamics, and health effects. Gas-phase amines in the atmosphere primarily follow two reaction pathways: gas-phase reactions with oxidants and participation in heterogeneous reactions with acidic or carbonyl compounds, subsequently partitioning into nanoparticles. Lee and Wexler17 delineated the gas-phase reaction rates of amines with hydroxyl (OH) radical, ozone (O3), nitrate (NO3) radical, chlorine (Cl) atoms, and photolysis, as well as the products from these reactions. Previous studies also synthesized laboratory experiments of the multiphase chemistry of amines, including acid-base neutralization, carbonyl-amine interaction and particle-phase oxidation18, and measurements of reduced nitrogen-containing compounds in both the gas and particle phases19. Shen et al.8 recently summarized a decade of progress in understanding emission sources, detection methods, and oxidation reaction mechanisms to elucidate their direct or indirect effects on air quality and climate.
To obtain a holistic view of the role of amines in atmospheric chemistry, particle formation, air quality and climate, multiple laboratory experiments, quantum chemical calculations, and field observations have been conducted. These studies show that amines contribute to atmospheric particle formation and growth4,5,20,21,22,23,24,25, the formation of organic brown carbon (BrC)26,27, cloud water droplets28,29, aqueous fog and rain droplets30, and plays a crucial role in the biogeochemical nitrogen cycle31. Low molecular weight amines, like trimethylamine (TMA), not only have higher basicity than NH3 but also account for 14–35% of the concentrations of NH3 in aerosol particles32, yet amines are underrepresented in global climate models. The major uncertainties in the global climate model arise from limitations in measurement technology and model inaccuracies, which must be better constrained to accurately quantify the role of amines in air quality and climate33,34,35,36. Additionally, many amines pose risks to public health due to their toxicity and allergenic effects, with some capable of forming carcinogenic compounds, such as nitrosamines when they react with nitrites7,17. As carbon capture and storage (CCS) technologies increasingly rely on organic solvents such as monoethanolamine (MEA) or methyldiethanolamine (MDEA) to reduce carbon emissions from fossil-based energy and industry sources, ambient concentrations of amines will likely rise in the future37. While the Intergovernmental Panel for Climate Change (IPCC) highlights that accelerating CCS technologies could help the world to surpass climate tipping points2, the potential impacts of rising amines concentrations on the environment and climate are underrecognized in the IPCC report. Field observations and modelling proxies reveal decreasing emissions of sulfur dioxide (SO2, precursor of SA and the main sink of amines via acid-base neutralisation), condensation sink, particle formation rates, and NPF frequencies38, as a result, we should expect a longer atmospheric lifetime of amines and NH3, allowing their further transport to remote areas, including marine and polar areas. Furthermore, there is a risk of drinking water contamination by nitrosamines and nitramines39. Hence, it becomes increasingly important to monitor the ambient concentration of amines40 and study their gas-phase oxidation products to assess environmental impacts and health risks associated with amines from CCS facilities. Consequently, scientific research on atmospheric amines is rapidly growing globally, encompassing field observations, theoretical and numerical calculations, laboratory experiments, and advanced state-of-the-art techniques for detecting minute levels of amines in the air.
Why do we need accurate monitoring of atmospheric amines?
Prior to the 1980s, research on atmospheric amines primarily focused on regions with high emissions, such as livestock farms and waste treatment facilities, motivated by the potential carcinogenic risks of their derivatives, especially nitrosamines41. In addition to the potential negative effects of amines on human health, there has been colossal interest in amines and their derivatives in aerosols, their impact on air quality and climate. Laboratory experiments and field studies have both demonstrated the substantial contribution of amines to initial cluster formation. While NH3 effectively stabilizes clusters, amines (e.g., DMA, the strongest stabilizer among amine molecules22) can multiply the particle formation rate by up to 1000 times compared to NH34,20,42,43. Further, studies have demonstrated that the combined presence of amines and NH3 can lead to more efficient particle formation with SA and water compared to cases where NH3 is absent44. Since multiple particle formation pathways can be active simultaneously in the atmosphere, identifying dominant cluster formation pathways from field measurements is challenging. However, both atmospheric field measurements and laboratory experiments have highlighted the importance of SA, organic compounds and amines for NPF5,45,46,47,48,49. Previous studies have identified amines as an important organic component in sub-micron aerosols28,50,51,52,53,54,55, either via acid-base reactions with gas-phase inorganic acid or acidic aerosols56 or via condensation products following oxidation reactions with OH, O3, and NO357,58,59, with yields from 8 to 15% from oxidation of amines60. But, the exact contribution of amines, along with acids (e.g., SA, iodic acid), other bases (e.g., NH3) and organics (e.g., highly oxygenated organic molecules, HOMs), to the growth of sub-3 nm clusters in diverse environments, is still elusive61,62,63,64.
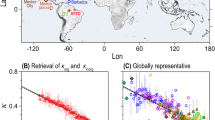
Figure 1a shows the geographic distribution of locations where ambient concentrations of gas-phase and/or particle-phase amines have been reported in the literature. This highlights the global scarcity of such observations, especially in the Southern Hemisphere, Southeast Asia and the Arctic, as well as discrepancies in reported amine concentrations in both the gas- and particle-phase. Although over 150 amines have been identified in the atmosphere17, only about 10 of them have been widely reported. The reported concentrations of gas-phase amines exhibit variability of over ten thousand-fold (ranging from <0.1 to >105 pptV) across different seasons, sources, and detection methods (Table 1). Particle-phase amine concentrations also show large variability, ranging from 0.02 to 425.9 ng m-3 (Table 2). The emission of amines from a particular source also shows a thousand-fold variability in both the gas- and particle-phase concentrations. For example, gas-phase amines from agriculture activities range from <0.5 to >100 pptV65,66,67,68. Moreover, the different techniques employed exhibit large variability in the measured levels of gas-phase and particle-phase amines. For example, gas-phase DMA concentrations ranged from <0.15 to >80 pptV in Hyytiälä boreal coniferous forest, Finland during spring or summer15,69,70,71, while particle-phase DMA concentrations in the particulate matter (particles with aerodynamic diameter of 2.5 µm or less, PM2.5) ranged from 2.4 to 280 ng m-3 in coastal Qingdao, China during winter72,73. Figure 1b, c depict the variability in different gas-phase and particle-phase amines based on reported ambient measurements (Tables 1 and 2). Livestock operations show the highest levels of different gas-phase amines, whereas other sources exhibited erratic behaviour in amines, encompassing data points across the entire range. Nevertheless, the accurate detection of amines, especially in the gas-phase, is lacking for several reasons; (i) current mass spectrometers cannot separate individual amine species and can only detect sums such as C2 or C4 amines due to their identical elemental composition, (ii) measurements are affected by contamination and memory effects, which increases detection limit, and insufficient resolution in mass spectrometers, resulting uncertainty in peak identification. The lack of analytical standards and zero samples exacerbates these issues, and (iii) there are discrepancies between laboratory/theoretical and ambient observations, such as the unobserved replacement of NH3 in clusters by DMA under ambient conditions5,74,75. Thus, the critical knowledge gap in amine measurement technology concerns whether the reported amine mixing ratios are subject to low or high bias from instrumental detection issues or by the natural variability stemming from varying source strengths. This indicates that the development of global gridded emission inventories for amines is currently not feasible. Notably, the oversimplified regional gridded inventories of amine emissions have been shown to hinder the simulation accuracy of atmospheric numerical models76. Although minute quantities of acidic compounds dominate aerosol numbers, resolving the role of amines in particle formation and growth has implications for aerosol-cloud-radiation-climate interaction research. Previous laboratory and atmospheric studies have observed high NPF rates involving precursor vapors (usually SA) stabilized by amines24,43,61, and these clusters subsequently grow to large sizes by condensing vapors which are typically low-volatile organic and inorganic compounds49,77,78. Several studies showed the vital contribution of NPF to cloud condensation nuclei (CCN) concentrations, thus affecting the cloud properties, especially in pristine areas3,79,80,81,82. However, amines influence CCN activity by either enhancing particle hygroscopicity through the formation of aminium salts32 or suppressing it through photochemical reactions with OH radicals83. Tang et al.83 demonstrated that secondary organic aerosols (SOA) formed from the reactions of TMA and BA with OH radicals consist of organic material with low hygroscopicity, characterized by a single hygroscopicity parameter (κ≤0.25). Although BA is less abundant in the atmosphere compared to TMA, it has been detected at high concentrations near dairy facilities, reaching concentration levels up to 187 ppbV13. This indicates that CCN activity for amines depends on the reaction pathways of specific amines and their magnitude, indicating climate impacts associated with amines are complex. Therefore, the accurate detection of a variety of amines and their chemical pathways are crucial for improving our process-level understanding of the climate system which could open new opportunities for Earth system modelling.

a Geographical distribution of locations where ambient observations of gas-phase amines (open circles), particle-phase amines (filled circles), or both (open left half and filled right half circles) are reported. The colour of the symbol indicates the type of environment (see legend). b Box-whisker plot of gas-phase concentrations of widely reported amines (Table 1). c Same as (b) but for particle-phase amines (Table 2). The black open square indicates the mean, the horizontal black line indicates the median, the bottom and top of the box represent the 25th and 75th percentiles, and the bottom and top of the whisker represent the 5th and 95th percentiles. The cross symbols indicate all individual data points, with the colour referring to the type of environment. ΣA refers to the sum of particle-phase amines. Details of amine measurement technology, location and time periods are presented in Tables 1 and 2. MMA Monomethylamine, DMA Dimethylamine, TMA Trimethylamine, EA Ethylamine, DEA Diethylamine, TEA Triethylamine, ΣA total mass of aliphatic amines.
Amines impact airborne aerosol production
Airborne aerosol production is a non-linear dynamic process involving multiple components in the formation of aerosols, with amines facilitating the fastest base-neutralisation mechanism, thereby decreasing the evaporation of nucleated clusters4. Field studies on atmospheric acids have become more prevalent with the development of mass spectrometric methods that have low detection limits (sub-pptV, <10-12 in volume) and high time and mass resolution. However, observations of base compounds (e.g., amines) in the field are almost solely carried out by estimating their abundance in acidic molecular clusters (e.g., SA)24,66,84,85. Direct measurements of amines at sub-5 pptV concentrations are difficult to reach, even in ultra-clean chamber facilities14. Further, simultaneous real-time measurements of gas-phase amines and their particulate counterparts in the marine and polar remote atmospheres remain challenging (Fig. 1), although this is not the case in the continental atmosphere54.
Amines are emitted into the atmosphere from diverse natural and anthropogenic sources in both continental and marine environments7. Anthropogenic activities predominantly contribute to amine emissions, although approximately 30% of amines in the atmosphere are believed to originate from the decomposition of organic matter in the oceans4,7 that can be transported to remote continental locations. Since amines from continental and marine sources can be transported regionally across atmospheric environments, the reported acid-amine neutralisation mechanisms can be broadly categories into continental and coastal/marine atmospheres.
Continental atmosphere
Amines in continental atmospheres come from both natural (vegetation and forest fires) and anthropogenic sources (biomass/fossil-fuel burning, animal husbandry, waste incineration, sewage treatment, and residential cooking). Once in the air, amines can rapidly react with SA to form stable salt particles through acid-base reactions. Whether amines form aminium salts depends not only on temperature but also on the specific amine involved, its concentration, and the type of acidic species present. In addition, the presence of NH3 competes with amines for acidic molecules56. The formation rate of nanometre-sized particles reaches saturation with amine mixing ratios as low as 5 pptV in atmospherically relevant SA concentrations4,86. The CLOUD chamber experiment showed that the addition of only 5 pptV DMA enhances the nucleation rate of SA by more than six orders of magnitude at 10 pptV NH3, 278 K and 38% relative humidity, but the addition of further DMA up to 140 pptV produces a negligible increase in nucleation rate4. Other studies showed that for SA concentrations <107 molecules cm-3 and DMA > ~ 10 pptV, nucleation proceeds at or near the kinetic limit, indicating that collision between SA molecules and clusters associated with DMA minimizes evaporation compared to NH35,20. The formation of the SA-base cluster and their subsequent stabilization by the presence of amines (e.g., DMA) and high NH3 concentrations at lower temperatures have been shown to drive NPF in polluted environments49,87,88,89. Studies in Chinese megacities showed that neutral SA-water-DMA nucleation was the dominant pathway for the formation of sub-3nm particles24,89. The molecular level ternary nucleation mechanism involving SA, DMA and TMA showed that TMA can accelerate the SA-DMA-based NPF by 50-100%, with its contribution up to 43% to the particle formation rate, in Beijing90. In contrast, measurements at the Finnish Antarctic research station (Aboa), approximately 130 km inland from the Southern Ocean coast, indicated that ion-induced SA-NH3 nucleation drives particle formation91 indicating an insufficient source of amines at the site.
Terrestrial vegetation is a dominant source of VOC emissions to the atmosphere92. Several studies have shown that organic compounds derived from various precursor VOCs are involved in particle formation with SA62,93,94,95,96 and that they are a dominant source of SOA77. According to a theoretical study by Zhao et al.97, 2-methylglyceric acid (MGA), an isoprene derivative, can efficiently form heterodimers with SA and MSA, thereby contributing to NPF. However, it remains unclear whether adding a base molecule (such as NH3 or amines) to this binary system would enhance or inhibit the formation of stable clusters. Kulmala et al.98 showed that the growth rate of particles in Beijing did not correlate with organic compounds and SA concentrations summed, indicating that either multiphase chemical reactions assist the growth rate or higher volatility compounds condense onto nanoparticles. On the contrary, there exists only one study showing pure biogenic nucleation from HOMs in the nucleating cluster without base molecules (NH3 or amine) at a high-altitude free-tropospheric site (3580 m a.m.s.l.) in Jungfraujoch, Switzerland85. Nevertheless, organic compounds are important for nanoparticle growth and hence for the survival of newly formed particles in the atmosphere47,78. It is well recognized that atmospheric SA concentrations are often insufficient to explain the observed growth of nanoparticles99,100 and organic compounds are believed to play an important role in the growth of nucleation mode particles101,102. It is plausible that the particle-phase reactions between amines and organic acids, along with a synergistic effect involving carbonyls, may contribute to nanoparticle growth. Organic molecules containing carbonyl functional groups, such as glyoxal and methyl glyoxal, are formed through the oxidation of both biogenic103,104 and anthropogenic105,106 compounds. These organic molecules can react with primary and secondary amines, such as monomethylamine (MMA) and DMA to form imine and enamine compounds, as well as polymerized products107,108, suggesting particle-phase reactions between amines and organic acids. Furthermore, Chen et al.109 reported significant enhancements in diethylamine (DEA) in particles, attributed to increased aerosol water content and aerosol acidity in an urban area of Chongqing, China, during winter.
At present, no single organic compound has conclusively demonstrated strong neutral nucleation potential110, particularly in clustering with base molecules (amine or NH3), except for HOMs containing an amino group that can contribute to methanesulfonic acid (MSA)-based NPF due to their high hydrogen bonding capacity111. However, the effect of organics on the MSA-amine system is not well understood in the continental atmosphere. Such nitrogen-containing organics suggest the involvement of amines in cluster formation and warrant thorough investigations in future studies. The specific contributions of amines, NH3, and organic compounds to the growth of clusters larger than 3 nm diameter remain unclear in continental environments due to the lack of suitable methods for measuring trace amounts of these bases.
Coastal and marine atmospheres
Amines in marine areas are released through various mechanisms, such as direct emissions from phytoplankton, excretion or decomposition of marine organisms50,112,113, bubble bursting at the air-sea interface114, or biological degradation of quaternary nitrogen osmolytes (R4N+, where R is an alkyl substituent on the N atom)115. Direct observations of both gas- and particle-phase amines in remote marine and polar atmospheres are extremely scarce (Fig. 1), and mostly reported from coastal environments which are also affected by anthropogenic emissions of various gaseous precursors from coastal continental areas, including amines. The sea ice microbiota and plankton in the marginal ice zones and adjacent open ocean of the Weddell Sea in the Antarctic have been reported to be important sources of volatile sulfur and alkylamines116, along with NH3 from penguin and seabird colonies in coastal Antarctica117,118. Measurements at the Spanish research station on the south coast of Livingston Island in the South Shetland Islands revealed abundant SA-amine peaks during NPF events63. Observations in the eastern Pacific Ocean indicated that an increase in gas-phase amines and organosulfur compounds has a profound impact on both particulate chemical composition and cloud properties119. Previous studies have shown that MSA, approximately 10% to 100% of that of SA over marine regions, contributes to particle formation and growth in coastal and oceanic regions120,121. While MSA, SA, and NH3/amines coexist in the marine atmosphere, how these compounds interact to form particles is not well understood. Given the exceedingly low binary nucleation efficiency of MSA-H2O under typical atmospheric conditions122,123, MSA’s contribution to NPF is predominately determined by the enhancing effect of other species, particularly atmospheric bases such as NH3 and amines124,125. Laboratory experiments have shown that the reactions of MSA and DMA can produce new particles, even in the absence of SA21,126, and MSA can contribute to the particle growth121,127. As a result, methanesulfonate clusters grow more rapidly in marine air with the addition of a DMA molecule compared to an NH3 molecule. Previous studies showed that amines, such as DMA, displace NH3 with near-unit reaction efficiency on the surfaces of ammonium methanesulfonate clusters128. This indicates that dimethylaminium methanesulfonate salts are preferred over ammonium methanesulfonate salts in small clusters. Such an exchange of ammonium in sub-micron aerosols by aminium salt is expected to occur within a few hours129. Laboratory experiments and ab initio calculations further demonstrated that the MSA-amine intermediate contributes to new particles for TMA due to its highly hygroscopic nature, whereas for MMA and DMA, water is required124. Nucleation experiments, however, provide evidence that MSA suppresses the SA-TMA pathway due to the steric hindrance of the MSA and TMA, while it enhances the SA-MMA pathway via the formation of the SA-MSA heterodimer130. Quantum chemical calculations and cluster kinetic modelling further highlight that the MSA-DEA system has a relatively stronger nucleation potential in marine environments than MSA-DMA and SA-MMA systems131. The hydrogen-bonding capacity of substituted MSA-amine species may contribute to increased water uptake, with an amine-specific effect on particle hygroscopicity and growth.
Oxalic acid, formed via oxidation of glyoxal, a highly prevalent dicarboxylic acid originating from both natural marine and anthropogenic continental sources, has been identified in tropospheric aerosols132,133, which was observed to have a stronger binding affinity with MMA than with NH3126,134,135 and TMA136. The addition of oxalic acid to the MSA-MMA system reasonably enhances NPF, likely due to increased hydrogen bonding capacity or promoting proton transfer. However, it does not affect the MSA-MMA-H2O system because water at atmospherically relevant concentrations overwhelms the contribution of the much smaller concentrations of organics126. It is important to note that both the basicity and hydrogen-bonding capacity are key factors in determining the enhancing effect of a base molecule on MSA-induced NPF125,137,138,139. Nevertheless, field measurements have reported a strong correlation between the methane sulfonate ion and particle number concentrations7,140, as well as between particle growth and MSA concentrations141. Several studies have observed the presence of amine, NH3, and MSA in sub-micron particles in coastal environments51,119,142,143, but observations of these compounds in ultrafine particles (diameter less than 100 nm) or nanoparticles (diameter less than 25 nm) are practically non-existent.
While SA, MSA, oxalic acid, and nitric acid all require an additional vapor (NH3 or amines) to form particles, organic compounds such as HOMs6 and iodine144,145,146 can form particles on their own under specific atmospheric conditions. The potential of iodine compounds to form new particles has been shown in laboratory experiments144,147,148 and field studies149,150,151,152,153. Among iodine compounds, iodic acid (IA, HIO3) has been identified as the key driver of NPF in the coastal, open ocean, and ice-covered polar regions146,151,154,155. However, atmospheric observations of IA cannot be correlated with predicted particle formation rates153. Huang et al.156 demonstrated that heterogeneous reactions between higher iodine oxides and condensing alcohols or carbonyls from the oxidation of marine VOCs lead to the formation of low-volatility oxidized organics for newly formed ultrafine particles. These organic acids can then react with basic molecules, such as amines, accelerating early particle growth to form highly hygroscopic salts. Semicontrolled seawater-air enclosure measurements in the Bay of Calvi (Corsica, France) by Sellegri et al.157 showed that iodine-containing species are the major precursors for new cluster formation and subsequent growth to larger diameters, which was mainly driven by Chlorophyll a (Chl a) related organic precursors. The high correlation between particle growth rate (1–10 nm) with TMA and the presence of amines in particles larger than 70 nm diameter and Chl a, plausibly suggests iodine-amines clustering. Using a quantum chemical approach and Atmospheric Cluster Dynamics Code (ACDC), Ning et al.155 showed that DMA can structurally stabilize IA through hydrogen and halogen bonds, and the clustering process is energy barrier-less. Their study further showed that DMA can enhance the formation rate of IA clusters by five orders of magnitude, demonstrating its much greater efficiency in promoting IA cluster formation compared to NH3. The IA-DMA clustering pathway exhibits greater stability and energy favourability compared to both the pure-IA and IA-NH3 systems. Consequently, these stable IA-DMA clusters could serve as abundant seeds for the marine NPF. Several precursors for IA have been proposed, including hydrated iodine atoms153,158, hydrated IO radicals158, iodine dioxide (OIO) radicals159, and larger iodine oxides (I2O3, I2O4 and I2O5)158,160,161. However, these mechanisms remain speculative and lack experimental confirmation, leaving atmospheric IA observations unexplained. Nevertheless, global iodine emissions have tripled over the past 70 years and are projected to rise further as sea ice thins162. The potential increase of iodic-induced CCN concentrations in the Arctic region could increase longwave radiative forcing from clouds and provide a positive feedback mechanism that would further accelerate the loss of sea ice. However, the potential impact of other nucleation precursors on IA-DMA and their molecular mechanism remains unclear and non-verified with field observations.
Amines in models: Efforts and knowledge gaps in governing processes
The development of process-based numerical models, involving many different acid and base molecules, organics, water, and ions, is crucial for integrating intricate and non-linear airborne aerosol production processes in regional and global climate models to simulate and predict their impact on climate163. However, modelling global amines remains a challenge due to the scarcity of ambient measurements (Fig. 1). Quantum chemical calculations suggest that alkylamines efficiently stabilize SA clusters164, and these clusters are further grown to larger diameters by abundant concentrations of VOCs77. A process model based on cluster kinetics and quantum chemistry has demonstrated that the collision of SA and amine (e.g., DMA) clusters is the dominant mechanism to trigger NPF events in Beijing165. Although amines can enhance aerosol formation rates by up to a thousand-fold compared to NH34,44, model simulations suggest that nearly all present-day atmospheric SA nucleation involves NH3 or biogenic volatile organic compounds47,166. Primary emissions and airborne secondary production are difficult to disentangle as these are intricately coupled, and therefore, current global climate models fail to produce realistic particle formation and growth rates.
Myriokefalitakis et al.35 used a three-dimensional chemical transport model to estimate SOA formation, focusing exclusively on the potential contribution of the oceanic amine sources, assuming that amines account for one-tenth of the ocean-derived NH3. Yu and Luo34, on the other hand, modelled the global distributions of gas-phase amines by adjusting emission ratios for particular amines (such as MMA, DMA and TMA) to investigate the primary mechanism regulating amine concentrations (including emission, transport, oxidation, deposition, and aerosol uptake), but validation of simulated concentrations of amines with observations was hindered by the limited availability of ambient measurement data33,34. The global aerosol-climate model ECHAM-HAMMOZ, which incorporates amine-enhanced NPF parameterization and a kinetic nucleation parameterization, showed that amine-enhanced NPF is limited to areas near the source regions of amines due to their short gas-phase residence time in the atmosphere. However, the simulated gas-phase concentrations of amines were mostly underestimated compared to ambient observations33. Their study further showed that kinetic nucleation, which depends solely on SA concentration, produces particles more uniformly globally due to the long-range transport of SO2. On the contrary, the baseline scenario simulation, using a coupled zero-dimensional cluster-aerosol dynamic and technology-based emission projection model, showed that a 10–20% increase in amine concentrations and a doubling of SO2 results in more than a 100% increase in particle formation rate across NPF events, outcompeting increase in condensation sink167. SO2 concentrations are declining in Europe, the USA, and China, due to an economic slowdown and government efforts to restrain anthropogenic emissions168,169, which could also alter the SA-amines pathways near and away from the SO2 sources. Julin et al.170 used the chemical transport model PMCAMx-UF, including the NH3-DMA pathway as well as the condensation of organic compounds onto particles to simulate particle number concentrations, and demonstrated that a decrease in NH3 and amines concentrations reduce total particle number concentrations by 10–50% across Europe, however, the amine species were treated as a surrogate compound representing MMA, DMA, and TMA, with the assumption that the amine emissions are equal to the NH3 emissions scaled by a factor of 0.0057. Subsequently, Mao et al.171 and Li et al.172 derived amine-to-NH3 mass emission ratios specific to the source to distinguish between different amines, including sources such as agriculture, residential, transportation, chemical industry and other industries, while they exclude other prominent sources such as livestock operations and marine in their simulation. Machine learning model constructed to study enhancing potential of amines showed that the nucleation rate of DEA, mainly emitted from ethanol gasoline vehicles, with SA is 3–7 times higher than that of DMA, which was thought to be key SA-driver nucleation173. A quantitative structure-activity relationship (QSAR) model constructed for 63 precursors of IA-containing dimer clusters demonstrated that DEA exhibits the highest potential to enhance IA-induced nucleation at a mere sub-pptV level (0.1 pptV), with nucleation rates comparable to the IA-iodous system175.
Furthermore, Zhang et al.76 developed an emission inventory for amines using multi-source data sets from marine biological emissions that account for air-sea exchange fluxes. Their findings revealed that marine regions can serve as either a sink or a source for amines, depending upon ambient conditions such as the atmospheric oxidation capacity50,52. The transfer of NH3 and MMA together with DMS, across the air-sea interface to the atmosphere is suggested to play an important role in the regulation of aerosol pH, cloud water and rainfall174,176. None of the above processes is currently incorporated into global climate models, and the gridded emission inventories predominately rely on a fixed amine-to-NH3 ratio method33,34,35 and lack highly spatially and temporally resolved amine emissions, except for the source-dependent amine-to-NH3 ratio established by Mao et al.171 and subsequently used in Energy Exascale Earth System Model (E3SM) version-1 incorporated with different NPF mechanisms177 and a three-dimensional Weather Research and Forecasting model coupled with Chemistry (WRF-Chem) regional model178. More recently, quantum chemical calculations, atmospheric cluster dynamic simulations, and WRF-Chem simulations showed that IA has great potential for participation in the SA–DMA nucleation process not only in marine NPF but also in continental NPF179, but this has not yet been validated with ambient measurements.
Recent computational and theoretical studies indicate that amines undergo autoxidation in ambient conditions, resulting in previously unidentified reaction products23,180. For instance, the autoxidation of TMA generates a completely novel type of nitrogen-containing compound, hydroperoxyl amides. Furthermore, atmospheric autoxidation pathways for the production of highly oxidised organic molecules involving peroxy radicals have been identified to oxidise amines at a rate that competes with bimolecular reactions with oxidants, such as oxides of nitrogen23. However, unimolecular autoxidation reactions of VOCs, and especially amines that compete with bimolecular reactions in atmospheric conditions are under-estimated23. Our understanding of the oxidation of various classes of amines and the chemical pathways leading to the formation of oxidized compounds from bases in the atmosphere is still extremely limited. While strong amines, even at low concentrations and when undetected in the smallest clusters, can be crucial for particle formation, the contribution of different amines and whether they compete among themselves or with other bases have yet to be quantified. Until now, climate models have not yet factored in amines, their chemical properties, or their effects on particle formation and growth, which is the major missing link in the Earth’s climate.
Atmospheric amines: Implications to climate and public health
Amines play a key role in aerosol formation and growth through acid-base neutralization4,5,22,24,28, potentially leading to sizes capable of acting as CCN, thus contributing to aerosol cooling effect via cloud microphysical processes. However, they can also act as precursors to climate-warming agents like methane and brown carbon, which may offset the cooling effect. Furthermore, CCS facilities utilize amine scrubbing technology to remove carbon from the air to combat climate change. However, CCS facilities release carcinogenic amines into the atmosphere, raising critical risk to public health.
Amines, such as TMA, serve as precursors for methane production over marine regions. Under oxygen-deprived conditions, microbial conversion of TMA originating from the degradation of quaternary amine precursors can account for up to 90% of methane emissions from salt marsh sediment or slurries181,182. Hence, amines found in marine ecosystems183 from surface seawaters to deep sediments50 have critical importance to global warming over oceans via the release of methane through methanogenesis in marine/coastal sediments184. Several studies report the formation of light-absorbing BrC185 via aqueous-phase reactions involving glyoxal, methylglyoxal, and formaldehyde26,186, as well as the oxidation of ethylamine (EA) mediated by nitrate (\({{{\rm{NO}}}}_{3}^{-}\)) photolysis187. Although BrC produced from the particle-phase reaction of methylglyoxal with MMA can contribute to atmospheric warming188, the BrC formation via aldehyde-amine-ammonium sulfate browning reactions is about ten times lower than that generated from wood burning, which accounts for <10% of light absorption in the atmosphere26. These findings suggest that specific reaction pathways involving particular amines may have differential effects on the climate.
Limiting the rise in global temperatures to less than 2 °C relative to pre-industrial times requires reductions in human-made greenhouse gas emissions, mainly carbon gases2. Several technologies for capturing and separating carbon dioxide (CO2) have been applied189, such as chemical absorption via amine scrubbing, separation by adsorption190,191, membrane separation192, and calcium chemical looping193. However, these technologies have several drawbacks, notably the degradation of the amines occurring at relatively low temperatures, particularly in the presence of oxygen in the inlet flue gas stream194. In the case of CCS facilities, 2-aminoethanol (MEA) is used as an absorption solvent in post-combustion capture195,196, leading to the release of 80 tons of MEA into the atmosphere for every 1 million tons of CO2 removed annually39,197. A modelling framework demonstrated that realistic emission of amines from a typical post-combustion CO2 capture results in the sum of carcinogenic amines (nitrosamines and nitramines) of 0.6–10 pg m-3 and 0.04–0.25 ngvL-1 in the ground-level air and drinking water, respectively, which are below the current safety guideline by National Institute of Public Health (NIPH) for human health36, while the deposition of MEA into small lakes in the Norwegian west coast could exceed toxicity limits for aquatic organisms39. This suggests that if the technology were widely deployed, a substantial amount of MEA would be released into the atmosphere from post-combustion CO2 capture units which could have implications to both climate and public health.
Summary
Despite vigorous research over the last more than three decades, a holistic understanding of the formation mechanisms of airborne nanometre-sized aerosols and their growth to CCN, toxicity and overall effect on the climate remain elusive. This is because the understanding of aerosol precursors is built on measurements of strictly selected groups of acidic or highly oxygenated organic compounds due to technological limitations. In laboratory conditions, amines are known to enhance aerosol formation rates by over a thousand-fold compared to ammonia, yet their roles in ambient aerosol production and interactions with other bases remain poorly quantified. Only a few pptV mixing ratios of DMA are needed to saturate the formation rate in atmospherically relevant SA concentrations. Mixing ratios below 1 pptV of DMA can enhance particle formation rates by stabilizing the clusters and minimizing evaporation compared to ammonia. This process seems to be valid for polluted areas, where neutral SA-DMA (-water) nucleation is found to produce sub-3 nm particles efficiently. Observations from the boreal forest or other continental areas do not seem to support a major role for DMA in SA stabilizing base compound but ammonia is observed to take the leading role. The exact contributions of DMA, ammonia and HOMs to the growth of sub-3 nm clusters are yet to be elucidated in most environments. The challenge to achieve this information is a major one; the current DMA mixing ratios measured from the same field site in Finland vary from ppqV to sub-ppbV levels, pointing toward colossal measurement errors in some or all used methods. Due to the technological challenges and lacking methodologies, only a minority of atmospheric bases have been quantified, and their properties and reactions are not understood well.
Further, ambient measurements of atmospheric amines are almost solely conducted in the Northern Hemisphere as shown in Fig. 1. However, there are limited data from the rest of the world, and no measurements exist from the continents of Africa, South America, Australia, or from the Arctic regions. Current studies are predominantly concentrated in urban and coastal areas, with minimal data from rural, agricultural, and polar environments, as well as regions with intensive livestock activities, which are believed to be a substantial source of amine emissions. Additionally, CCS technologies increasingly utilize specific amines to reduce carbon emissions from fossil-fuel-based energy and industrial sectors. However, detailed quantification of amine emissions from CCS facilities is still lacking, which limits our understanding of the broader environmental and climatic implications of amines, particularly within the context of the climate-air-health nexus.
Ambient base concentrations are only reported in polluted environments due to extremely low concentrations elsewhere in the atmosphere. Molecular clusters consisting of a few aerosol precursor molecules exist at mixing ratios approximately 1000 times lower (e.g., the maximum atmospheric concentrations of SA have been measured at <3 × 107 molecules cm−3, approximately 1.2 pptV), which makes their analysis extremely challenging. Current base molecule measurements suffer from contamination and memory effects that increase detection limits in field conditions, insufficient resolution mass spectrometers creating fatal uncertainty in peak identification and the lack of analytical standards and zero samples. This perspective article underscores the need for novel measurement technologies to measure sub-pptV concentrations of amines in the gas-phase as well as in nanoparticles. Furthermore, no single detection method is adequate to capture the full spectrum of amine emissions, highlighting the need for hybrid approaches that combine multiple techniques (chromatographic, spectroscopic, etc.) for comprehensive measurements.
Direct emissions from anthropogenic activities are often straightforward to mitigate through targeted interventions and regulatory measures, although airborne aerosol production involves the intricately mixed interactions between primary emission and secondary atmospheric processes, and usually are non-linear, thereby challenging to separate from primary emissions (Fig. 2). Airborne aerosol formation is the largest source of aerosol numbers in the global atmosphere, and therefore it is critical to understand the physiochemical mechanisms driving it to correctly represent this process in climate models, and thereby future climate predictions2. Very little information is available on the atmospheric bases involved in airborne aerosol production. This represents an important knowledge gap, as various bases, such as NH3 or amines, are known to stabilize negative ion clusters20,25,198,199,200, and results from ambient measurements are not sufficient to confirm or refute this information. The relevance of amines-driven NPF to the global CCN budget remains to be assessed due to the lack of measurements and accurate amines emission inventories. It is important to focus on reducing direct emissions of both aerosols and their precursor to effectively address their immediate impact on climate and air quality. This is because the climate and air quality represent two distinct, yet interrelated, entities. Any policy action aimed at mitigating one of these entities must consider the feedback from the other, as advancements in one entity may worsen conditions in the other. For example, the declining trend in SO2 emissions at the Station for Measuring Ecosystem-Atmosphere Relations (SMEAR-II, Hyytiälä, Finland), resulted in reduced SA concentrations, particle formation rates and NPF frequency38, and thereby lower aerosol numbers and improved air quality, which may dampen the cooling effect of aerosols. Therefore, ambient measurements integrated with laboratory experiments and theoretical studies aided with the development of the process-based models are required to improve the representation of amines (and other bases) in Earth system models to advance our understanding of the complex interactions of amines in atmospheric chemistry, ultimately addressing their climatic and environmental impacts more effectively.

Atmospheric amines, emitted from a wide range of natural and anthropogenic sources, facilitate the fastest base-neutralization mechanism, thereby reducing the evaporation of critical clusters and promoting the formation of new particles. These newly formed particles not only intricately mix with primary particles leading to haze formation but also have the potential to enhance aerosol hygroscopicity and the activation of cloud condensation nuclei. Therefore, the complex interplay and feedback mechanisms between air quality and climate are inadequately quantified, posing significant challenges to accurately constraining their combined impacts on air quality, climate, and human health.
Data availability
No new data were generated for this analysis.
References
Pielke R. A. et al. Interactions between the atmosphere and terrestrial ecosystems: influence on weather and climate. Glob. Change Biol. 4, 461-475 (1998).
IPCC. Climate Change 2023: Synthesis Report. Contribution of Working Groups I, II and III to the Sixth Assessment Report of the Intergovernmental Panel on Climate Change [Core Writing Team, H. Lee and J. Romero (eds.)]. IPCC, Geneva, Switzerland, pp. 35–115 (2023).
Gordon, H. et al. Causes and importance of new particle formation in the present-day and preindustrial atmospheres. J. Geophys. Res.: Atmos. 122, 8739–8760 (2017).
Almeida, J. et al. Molecular understanding of sulphuric acid–amine particle nucleation in the atmosphere. Nature 502, 359–363 (2013).
Kürten, A. et al. Neutral molecular cluster formation of sulfuric acid–dimethylamine observed in real time under atmospheric conditions. Proc. Natl. Acad. Sci. 111, 15019–15024 (2014).
Kirkby, J. et al. Ion-induced nucleation of pure biogenic particles. Nature 533, 521–526 (2016).
Ge, X., Wexler, A. S. & Clegg, S. L. Atmospheric amines – Part I. A review. Atmos. Environ. 45, 524–546 (2011).
Shen, X., Chen, J., Li, G. & An, T. A new advance in the pollution profile, transformation process, and contribution to aerosol formation and aging of atmospheric amines. Environ. Sci.: Atmos. 3, 444–473 (2023).
Zheng, J. et al. Measurement of atmospheric amines and ammonia using the high resolution time-of-flight chemical ionization mass spectrometry. Atmos. Environ. 102, 249–259 (2015).
Schade, G. W. & Crutzen, P. J. Emission of aliphatic amines from animal husbandry and their reactions: Potential source of N2O and HCN. J. Atmos. Chem. 22, 319–346 (1995).
Dawson, M. L. et al. Measurement of gas-phase ammonia and amines in air by collection onto an ion exchange resin and analysis by ion chromatography. Atmos. Meas. Tech. 7, 2733–2744 (2014).
Kirkby, J. et al. Role of sulphuric acid, ammonia and galactic cosmic rays in atmospheric aerosol nucleation. Nature 476, 429–433 (2011).
Rabaud, N. E., Ebeler, S. E., Ashbaugh, L. L. & Flocchini, R. G. Characterization and quantification of odorous and non-odorous volatile organic compounds near a commercial dairy in California. Atmos. Environ. 37, 933–940 (2003).
Simon, M. et al. Detection of dimethylamine in the low pptv range using nitrate chemical ionization atmospheric pressure interface time-of-flight (CI-APi-TOF) mass spectrometry. Atmos. Meas. Tech. 9, 2135–2145 (2016).
Sipilä, M. et al. Bisulfate–cluster based atmospheric pressure chemical ionization mass spectrometer for high-sensitivity (<100 ppqV) detection of atmospheric dimethyl amine: proof-of-concept and first ambient data from boreal forest. Atmos. Meas. Tech. 8, 4001–4011 (2015).
Ge, X., Wexler, A. S. & Clegg, S. L. Atmospheric amines – Part II. Thermodynamic properties and gas/particle partitioning. Atmos. Environ. 45, 561–577 (2011).
Lee, D. & Wexler, A. S. Atmospheric amines – Part III: Photochemistry and toxicity. Atmos. Environ. 71, 95–103 (2013).
Qiu, C. & Zhang, R. Multiphase chemistry of atmospheric amines. Phys. Chem. Chem. Phys. 15, 5738–5752 (2013).
Lee S.-H. Perspective on thE Recent Measurements Of Reduced Nitrogen Compounds In The Atmosphere. Front. Environ. Sci. 10, (2022).
Jen, C. N., McMurry, P. H. & Hanson, D. R. Stabilization of sulfuric acid dimers by ammonia, methylamine, dimethylamine, and trimethylamine. J. Geophys. Res.: Atmos. 119, 7502–7514 (2014).
Chen, H. et al. New particle formation and growth from methanesulfonic acid, trimethylamine and water. Phys. Chem. Chem. Phys. 17, 13699–13709 (2015).
Olenius, T. et al. New particle formation from sulfuric acid and amines: Comparison of monomethylamine, dimethylamine, and trimethylamine. J. Geophys. Res.: Atmos. 122, 7103–7118 (2017).
Møller, K. H., Berndt, T. & Kjaergaard, H. G. Atmospheric Autoxidation of Amines. Environ. Sci. Technol. 54, 11087–11099 (2020).
Yao L. et al. Atmospheric new particle formation from sulfuric acid and amines in a Chinese megacity. Science 361, 278–281 (2018).
Kurtén, T., Loukonen, V., Vehkamäki, H. & Kulmala, M. Amines are likely to enhance neutral and ion-induced sulfuric acid-water nucleation in the atmosphere more effectively than ammonia. Atmos. Chem. Phys. 8, 4095–4103 (2008).
Powelson, M. H., Espelien, B. M., Hawkins, L. N., Galloway, M. M. & De Haan, D. O. Brown Carbon Formation By Aqueous-phase Carbonyl Compound Reactions With Amines And Ammonium Sulfate. Environ. Sci. Technol. 48, 985–993 (2014).
De Haan, D. O. et al. Brown Carbon Production In Ammonium- Or Amine-containing Aerosol Particles By Reactive Uptake Of Methylglyoxal And Photolytic Cloud Cycling. Environ. Sci. Technol. 51, 7458–7466 (2017).
Youn, J. S., Crosbie, E., Maudlin, L. C., Wang, Z. & Sorooshian, A. Dimethylamine as a major alkyl amine species in particles and cloud water: Observations in semi-arid and coastal regions. Atmos. Environ. 122, 250–258 (2015).
Corral, A. F. et al. Dimethylamine in cloud water: a case study over the northwest Atlantic Ocean. Environ. Sci.: Atmos. 2, 1534–1550 (2022).
Zhang, Q. & Anastasio, C. Free and combined amino compounds in atmospheric fine particles (PM2.5) and fog waters from Northern California. Atmos. Environ. 37, 2247–2258 (2003).
Cape, J. N., Cornell, S. E., Jickells, T. D. & Nemitz, E. Organic nitrogen in the atmosphere — Where does it come from? A review of sources and methods. Atmos. Res. 102, 30–48 (2011).
Sorooshian, A. et al. Comprehensive airborne characterization of aerosol from a major bovine source. Atmos. Chem. Phys. 8, 5489–5520 (2008).
Bergman T., et al. Geographical and diurnal features of amine-enhanced boundary layer nucleation. JGR Atmos. 120, 9606–9624 (2015).
Yu, F. & Luo, G. Modeling of gaseous methylamines in the global atmosphere: impacts of oxidation and aerosol uptake. Atmos. Chem. Phys. 14, 12455–12464 (2014).
Myriokefalitakis, S. et al. Global Modeling of the Oceanic Source of Organic Aerosols. Adv. Meteorol. 2010, 939171 (2010).
Karl, M. et al. Uncertainties in assessing the environmental impact of amine emissions from a CO2 capture plant. Atmos. Chem. Phys. 14, 8533–8557 (2014).
Rao, A. B. & Rubin, E. S. A Technical, Economic, And Environmental Assessment Of Amine-based Co2 Capture Technology For Power Plant Greenhouse Gas Control. Environ. Sci. Technol. 36, 4467–4475 (2002).
Li, X. et al. Over 20 years of observations in the boreal forest reveal a decreasing trend of atmospheric new particle formation. Boreal Env. Res 29, 25–52 (2024).
Karl, M., Wright, R. F., Berglen, T. F. & Denby, B. Worst case scenario study to assess the environmental impact of amine emissions from a CO2 capture plant. Int. J. Greenh. Gas. Control 5, 439–447 (2011).
Nielsen, C. J., Herrmann, H. & Weller, C. Atmospheric chemistry and environmental impact of the use of amines in carbon capture and storage (CCS). Chem. Soc. Rev. 41, 6684–6704 (2012).
EPA. Scientific and technical assessment report on nitrosamines.). U.S. Environmental Protection Agency (EPA-600/6-77-001). (1976).
Kürten, A. et al. New particle formation in the sulfuric acid–dimethylamine–water system: reevaluation of CLOUD chamber measurements and comparison to an aerosol nucleation and growth model. Atmos. Chem. Phys. 18, 845–863 (2018).
Yu H., McGraw R., Lee S. H. Effects of amines on formation of sub-3 nm particles and their subsequent growth. Geophys. Res. Lett. 39, n/a-n/a (2012).
Glasoe W. A. et al. Sulfuric acid nucleation: An experimental study of the effect of seven bases. ADS 120, 1933–1950 (2015).
Kulmala, M, et al. Direct observations of atmospheric aerosol nucleation. Science 339, 943–946 (2013).
Dunne, E. M. et al. Global atmospheric particle formation from CERN CLOUD measurements. Science 354, 1119–1124 (2016).
Tröstl, J. et al. The role of low-volatility organic compounds in initial particle growth in the atmosphere. Nature 533, 527–531 (2016).
Cai, R. et al. Sulfuric acid–amine nucleation in urban Beijing. Atmos. Chem. Phys. 21, 2457–2468 (2021).
Yan C., et al. The Synergistic role of sulfuric acid, bases, and oxidized organics governing new-particle formation in Beijing. Geophys. Res. Lett. 48, e2020GL091944 (2021).
Gibb S. W., Mantoura R. F. C., Liss P. S. Ocean-atmosphere exchange and atmospheric speciation of ammonia and methylamines in the region of the NW Arabian Sea. Environ. Sci. Chem. 13, 161–178 (1999).
Facchini, M. C. et al. Important Source of Marine Secondary Organic Aerosol from Biogenic Amines. Environ. Sci. Technol. 42, 9116–9121 (2008).
van Pinxteren, M. et al. Aliphatic amines at the Cape Verde Atmospheric Observatory: Abundance, origins and sea-air fluxes. Atmos. Environ. 203, 183–195 (2019).
Tao, Y. et al. Effects of amines on particle growth observed in new particle formation events. J. Geophys. Res.: Atmos. 121, 324–335 (2016).
VandenBoer, T. C., Petroff, A., Markovic, M. Z. & Murphy, J. G. Size distribution of alkyl amines in continental particulate matter and their online detection in the gas and particle phase. Atmos. Chem. Phys. 11, 4319–4332 (2011).
Kanawade, V. P., Tripathi, S. N., Chakraborty, A. & Yu, H. Chemical Characterization of Sub-micron Aerosols during New Particle Formation in an Urban Atmosphere. Aerosol Air Qual. Res. 20, 1294–1305 (2020).
Murphy, S. M. et al. Secondary aerosol formation from atmospheric reactions of aliphatic amines. Atmos. Chem. Phys. 7, 2313–2337 (2007).
Malloy, Q. G. J., Warren, Li. Q., Cocker Iii, B., Erupe, D. R. & Silva, M. E. PJ. Secondary organic aerosol formation from primary aliphatic amines with NO3 radical. Atmos. Chem. Phys. 9, 2051–2060 (2009).
Erupe, M. E. et al. Determination of methylamines and trimethylamine-N-oxide in particulate matter by non-suppressed ion chromatography. J. Chromatogr. A 1217, 2070–2073 (2010).
Silva, P. J. et al. Trimethylamine as Precursor to Secondary Organic Aerosol Formation via Nitrate Radical Reaction in the Atmosphere. Environ. Sci. Technol. 42, 4689–4696 (2008).
Nielsen C. J. et al. Atmospheric Degradation of Amines (ADA) Summary Report: Photo-Oxidation of Methylamine, Dimethylamine and Trimethylamine CLIMIT. In: Project No. Norge: Norsk Institutt for Luftforskning) (2016).
Dada, L. et al. The synergistic role of sulfuric acid, ammonia and organics in particle formation over an agricultural land. Environ. Sci. Atmos. 3, 1195–1211 (2023).
Schobesberger, S. et al. Molecular understanding of atmospheric particle formation from sulfuric acid and large oxidized organic molecules. Proc. Natl. Acad. Sci. 110, 17223–17228 (2013).
Brean, J. et al. Open ocean and coastal new particle formation from sulfuric acid and amines around the Antarctic Peninsula. Nat. Geosci. 14, 383–388 (2021).
Jokinen, T. et al. Solar eclipse demonstrating the importance of photochemistry in new particle formation. Sci. Rep. 7, 45707 (2017).
VandenBoer, T. C. et al. Ion chromatographic separation and quantitation of alkyl methylamines and ethylamines in atmospheric gas and particulate matter using preconcentration and suppressed conductivity detection. J. Chromatogr. A 1252, 74–83 (2012).
Kürten, A. et al. Observation of new particle formation and measurement of sulfuric acid, ammonia, amines and highly oxidized organic molecules at a rural site in central Germany. Atmos. Chem. Phys. 16, 12793–12813 (2016).
Grönberg, L., Lövkvist, P. & Jönsson, J. Å. Measurement of aliphatic amines in ambient air and rainwater. Chemosphere 24, 1533–1540 (1992).
Freshour, N. A. et al. Amine permeation sources characterized with acid neutralization and sensitivities of an amine mass spectrometer. Atmos. Meas. Tech. 7, 3611–3621 (2014).
Sellegri, K., Hanke, M., Umann, B., Arnold, F. & Kulmala, M. Measurements of organic gases during aerosol formation events in the boreal forest atmosphere during QUEST. Atmos. Chem. Phys. 5, 373–384 (2005).
Hemmilä, M. et al. Amines in boreal forest air at SMEAR II station in Finland. Atmos. Chem. Phys. 18, 6367–6380 (2018).
Kieloaho, A.-J. et al. Gas-phase alkylamines in a boreal Scots pine forest air. Atmos. Environ. 80, 369–377 (2013).
Liu, Z. et al. Large contributions of anthropogenic sources to amines in fine particles at a coastal area in northern China in winter. Sci. Total Environ. 839, 156281 (2022).
Chen, D. et al. Mapping gaseous dimethylamine, trimethylamine, ammonia, and their particulate counterparts in marine atmospheres of China’s marginal seas – Part 1: Differentiating marine emission from continental transport. Atmos. Chem. Phys. 21, 16413–16425 (2021).
Quéléver, L. L. J. et al. Investigation of new particle formation mechanisms and aerosol processes at Marambio Station, Antarctic Peninsula. Atmos. Chem. Phys. 22, 8417–8437 (2022).
Kupiainen, O., Ortega, I. K., Kurtén, T. & Vehkamäki, H. Amine substitution into sulfuric acid – ammonia clusters. Atmos. Chem. Phys. 12, 3591–3599 (2012).
Zhang, Q. et al. Contribution of marine biological emissions to gaseous methylamines in the atmosphere: An emission inventory based on multi-source data sets. Sci. Total Environ. 898, 165285 (2023).
Ehn, M. et al. A large source of low-volatility secondary organic aerosol. Nature 506, 476–479 (2014).
Riipinen, I. et al. The contribution of organics to atmospheric nanoparticle growth. Nat. Geosci. 5, 453–458 (2012).
Merikanto, J., Spracklen, D. V., Mann, G. W., Pickering, S. J. & Carslaw, K. S. Impact of nucleation on global CCN. Atmos. Chem. Phys. 9, 8601–8616 (2009).
Wang, M. & Penner, J. E. Aerosol indirect forcing in a global model with particle nucleation. Atmos. Chem. Phys. 9, 239–260 (2009).
Yu, F. & Luo, G. Simulation of particle size distribution with a global aerosol model: contribution of nucleation to aerosol and CCN number concentrations. Atmos. Chem. Phys. 9, 7691–7710 (2009).
Sebastian M. et al. New Particle Formation and Growth to Climate-Relevant Aerosols at a Background Remote Site in the Western Himalaya. J. Geophys. Res.: Atmos. 126, (2021).
Tang, X. et al. Cloud condensation nuclei (CCN) activity of aliphatic amine secondary aerosol. Atmos. Chem. Phys. 14, 5959–5967 (2014).
Zhao, J. et al. Observation of neutral sulfuric acid-amine containing clusters in laboratory and ambient measurements. Atmos. Chem. Phys. 11, 10823–10836 (2011).
Bianchi, F. et al. New particle formation in the free troposphere: A question of chemistry and timing. Science 352, 1109–1112 (2016).
Lawler, M. J. et al. Unexpectedly acidic nanoparticles formed in dimethylamine–ammonia–sulfuric-acid nucleation experiments at CLOUD. Atmos. Chem. Phys. 16, 13601–13618 (2016).
Yao, L. et al. Detection of atmospheric gaseous amines and amides by a high-resolution time-of-flight chemical ionization mass spectrometer with protonated ethanol reagent ions. Atmos. Chem. Phys. 16, 14527–14543 (2016).
Brean, J. et al. Molecular insights into new particle formation in Barcelona, Spain. Atmos. Chem. Phys. 20, 10029–10045 (2020).
Xiao, M. et al. The driving factors of new particle formation and growth in the polluted boundary layer. Atmos. Chem. Phys. Discuss 2021, 1–28 (2021).
Cai, R. et al. Significant contributions of trimethylamine to sulfuric acid nucleation in polluted environments. npj Clim. Atmos. Sci. 6, 75 (2023).
Jokinen, T. et al. Ion-induced sulfuric acid–ammonia nucleation drives particle formation in coastal Antarctica. Sci. Adv. 4, eaat9744 (2018).
Goldstein, A. H. & Galbally, I. E. Known and unexplored organic constituents in the Earth’s atmosphere. Environ. Sci. Technol. 41, 1514–1521 (2007).
Kulmala, M. et al. Toward direct measurement of atmospheric nucleation. Science 318, 89–92 (2007).
Riccobono, F. et al. Oxidation products of biogenic emissions contribute to nucleation of atmospheric particles. Science 344, 717–721 (2014).
Metzger, A. et al. Evidence for the role of organics in aerosol particle formation under atmospheric conditions. Proc. Natl. Acad. Sci. 107, 6646–6651 (2010).
Jokinen, T. et al. Production of extremely low volatile organic compounds from biogenic emissions: Measured yields and atmospheric implications. Proc. Natl. Acad. Sci. 112, 7123–7128 (2015).
Zhao, F. et al. Enhancement of atmospheric nucleation by highly oxygenated organic molecules: a density functional theory study. J. Phys. Chem. A 123, 5367–5377 (2019).
Kulmala, M. et al. The contribution of new particle formation and subsequent growth to haze formation. Environ. Sci.: Atmos. 2, 352–361 (2022).
Erupe M. E. et al. Correlation of aerosol nucleation rate with sulfuric acid and ammonia in Kent Ohio: An Atmospheric Observation. J. Geophys. Res. 115, (2010).
Yli-Juuti, T., Tikkanen, O.-P., Manninen, H., Nieminen, T. & Kulmala, M. Analysis of sub-3 nm particle growth in connection with sulfuric acid in boreal forest. Boreal Environ. Res. 21, 287–298 (2016).
Zhang, R., Khalizov, A., Wang, L., Hu, M. & Xu, W. Nucleation and growth of nanoparticles in the atmosphere. Chem. Rev. 112, 1957–2011 (2012).
Riccobono, F. et al. Contribution of sulfuric acid and oxidized organic compounds to particle formation and growth. Atmos. Chem. Phys. 12, 9427–9439 (2012).
Lei, W. & Zhang, R. Theoretical study of hydroxyisoprene alkoxy radicals and their decomposition pathways. J. Phys. Chem. A 105, 3808–3815 (2001).
Zhang, D., Lei, W. & Zhang, R. Mechanism of OH formation from ozonolysis of isoprene: kinetics and product yields. Chem. Phys. Lett. 358, 171–179 (2002).
Lei, W., Zhang, R., Sean McGivern, W., Derecskei-Kovacs, A. & North, S. W. Theoretical study of isomeric branching in the isoprene–OH reaction: implications to final product yields in isoprene oxidation. Chem. Phys. Lett. 326, 109–114 (2000).
Zhao, J., Zhang, R., Misawa, K. & Shibuya, K. Experimental product study of the OH-initiated oxidation of m-xylene. J. Photochem. Photobiol. A: Chem. 176, 199–207 (2005).
De Haan D. O., Tolbert M. A., Jimenez J. L. Atmospheric condensed-phase reactions of glyoxal with methylamine. Geophys. Res. Lett. 36, (2009).
Trainic, M., Abo Riziq, A., Lavi, A. & Rudich, Y. Role of interfacial water in the heterogeneous uptake of glyoxal by mixed glycine and ammonium sulfate aerosols. J. Phys. Chem. A 116, 5948–5957 (2012).
Chen, Y. et al. Characterization of urban amine-containing particles in southwestern China: seasonal variation, source, and processing. Atmos. Chem. Phys. 19, 3245–3255 (2019).
Elm, J. et al. Quantum chemical modeling of organic enhanced atmospheric nucleation: A critical review. WIREs Comput. Mol. Sci. 13, e1662 (2023).
Duporté, G. et al. Chemical characterization of gas- and particle-phase products from the Ozonolysis of α-Pinene in the presence of Dimethylamine. Environ. Sci. Technol. 51, 5602–5610 (2017).
Steiner, M. & Hartmann, T. The occurence and distribution of volatile amines in marine algae. Planta 79, 113–121 (1968).
Sun, J., Mausz, M. A., Chen, Y. & Giovannoni, S. J. Microbial trimethylamine metabolism in marine environments. Environ. Microbiol 21, 513–520 (2019).
Gorzelska K., Galloway J. N. Amine nitrogen in the atmospheric environment over the North Atlantic Ocean. Glob. biochem. Cycles 4, 309–333 (1990).
Beale, R. & Airs, R. L. Quantification of glycine betaine, choline and trimethylamine N-oxide in seawater particulates: Minimisation of seawater associated ion suppression. Anal. Chim. Acta 938, 114–122 (2016).
Dall’Osto, M. et al. Simultaneous detection of alkylamines in the surface ocean and atmosphere of the antarctic sympagic environment. ACS Earth Space Chem. 3, 854–862 (2019).
Legrand, M, Ducroz, F, Wagenbach, D, Mulvaney, R & Hall, J Ammonium in coastal Antarctic aerosol and snow: Role of polar ocean and penguin emissions. J. Geophys. Res. Atmos. 103, 11043–11056 (1998).
Otero, X. L., De La Peña-Lastra, S., Pérez-Alberti, A., Ferreira, T. O. & Huerta-Diaz, M. A. Seabird colonies as important global drivers in the nitrogen and phosphorus cycles. Nat. Commun. 9, 246 (2018).
Sorooshian A. et al. On the link between ocean biota emissions, aerosol, and maritime clouds: Airborne, ground, and satellite measurements off the coast of California. Glob. Biochem. Cycles 23, (2009).
Eisele, F. L. & Tanner, D. J. Measurement of the gas phase concentration of H2SO4 and methane sulfonic acid and estimates of H2SO4 production and loss in the atmosphere. J. Geophys. Res. Atmos. 98, 9001–9010 (1993).
Berresheim H. et al. Gas-aerosol relationships of H2SO4, MSA, and OH: Observations in the coastal marine boundary layer at Mace Head, Ireland. J. Geophys. Res. Atmos. 107, PAR 5-1-PAR 5-12 (2002).
Kreidenweis, S. M., Flagan, R. C., Seinfeld, J. H. & Okuyama, K. Binary nucleation of methanesulfonic acid and water. J. Aerosol Sci. 20, 585–607 (1989).
Wyslouzil, B. E., Seinfeld, J. H., Flagan, R. C. & Okuyama, K. Binary nucleation in acid–water systems. II. Sulfuric acid–water and a comparison with methanesulfonic acid–water. J. Chem. Phys. 94, 6842–6850 (1991).
Dawson M. L. et al. Simplified mechanism for new particle formation from methanesulfonic acid, amines, and water via experiments and ab initio calculations. Proc. Natl Acad. Sci. USA 109, 18719-18724 (2012).
Chen, H., Varner, M. E., Gerber, R. B. & Finlayson-Pitts, B. J. Reactions of Methanesulfonic Acid with Amines and Ammonia as a Source of New Particles in Air. J. Phys. Chem. B 120, 1526–1536 (2016).
Arquero, K. D., Gerber, R. B. & Finlayson-Pitts, B. J. The Role of Oxalic Acid in New Particle Formation from Methanesulfonic Acid, Methylamine, and Water. Environ. Sci. Technol. 51, 2124–2130 (2017).
Kerminen, V.-M., Aurela, M., Hillamo, R. E. & Virkkula, A. Formation of particulate MSA: deductions from size distribution measurements in the Finnish Arctic. Tellus B: Chem. Phys. Meteorol. 49, 159–171 (1997).
Bzdek B. R., Ridge D. P., Johnston M. V. Reactivity of methanesulfonic acid salt clusters relevant to marine air. J. Geophys. Res.: Atmos. 116, (2011).
Lloyd, J. A., Heaton, K. J. & Johnston, M. V. Reactive uptake of Trimethylamine into Ammonium nitrate particles. J. Phys. Chem. A 113, 4840–4843 (2009).
Johnson, J. S. & Jen, C. N. Role of Methanesulfonic acid in sulfuric acid-amine and ammonia new particle formation. ACS Earth Space Chem. 7, 653–660 (2023).
Xu, C.-X. et al. Formation of atmospheric molecular clusters of methanesulfonic acid–Diethylamine complex and its atmospheric significance. Atmos. Environ. 226, 117404 (2020).
Kerminen, V.-M., Teinilä, K., Hillamo, R. & Mäkelä, T. Size-segregated chemistry of particulate dicarboxylic acids in the Arctic atmosphere. Atmos. Environ. 33, 2089–2100 (1999).
Rinaldi M. et al. Evidence of a natural marine source of oxalic acid and a possible link to glyoxal. J. Geophys. Res. Atmos. 116, (2011).
Guo, T., Li, K., Zhu, Y., Gao, H. & Yao, X. Concentration and size distribution of particulate oxalate in marine and coastal atmospheres – Implication for the increased importance of oxalate in nanometer atmospheric particles. Atmos. Environ. 142, 19–31 (2016).
Hong, Y. et al. Interaction of oxalic acid with methylamine and its atmospheric implications. RSC Adv. 8, 7225–7234 (2018).
Zhu, Y. et al. New particle formation in the marine atmosphere during seven cruise campaigns. Atmos. Chem. Phys. 19, 89–113 (2019).
Xie, H.-B. et al. Atmospheric fate of Monoethanolamine: Enhancing new particle formation of sulfuric acid as an important removal process. Environ. Sci. Technol. 51, 8422–8431 (2017).
Yang, Y., Waller, S. E., Kreinbihl, J. J. & Johnson, C. J. Direct link between structure and hydration in ammonium and aminium bisulfate clusters implicated in atmospheric new particle formation. J. Phys. Chem. Lett. 9, 5647–5652 (2018).
Waller, S. E. et al. The interplay between hydrogen bonding and coulombic forces in determining the structure of sulfuric acid-amine clusters. J. Phys. Chem. Lett. 9, 1216–1222 (2018).
Quinn P. K. et al. A 3-year record of simultaneously measured aerosol chemical and optical properties at Barrow, Alaska. J. Geophys. Res. 107, AAC 8-1-AAC 8-15 (2002).
Willis, M. D. et al. Growth of nucleation mode particles in the summertime Arctic: a case study. Atmos. Chem. Phys. 16, 7663–7679 (2016).
Müller, C. et al. Seasonal variation of aliphatic amines in marine sub-micrometer particles at the Cape Verde islands. Atmos. Chem. Phys. 9, 9587–9597 (2009).
Gaston, C. J., Pratt, K. A., Qin, X. & Prather, K. A. Real-time detection and mixing state of Methanesulfonate in single particles at an inland urban location during a phytoplankton bloom. Environ. Sci. Technol. 44, 1566–1572 (2010).
Hoffmann T., O’Dowd C. D. & Seinfeld J. H. Iodine oxide homogeneous nucleation: An explanation for coastal new particle production. Geophys. Res. Lett. 28, 1949–1952 (2001).
O’Dowd, C. D. et al. Marine aerosol formation from biogenic iodine emissions. Nature 417, 632–636 (2002).
Sipilä, M. et al. Molecular-scale evidence of aerosol particle formation via sequential addition of HIO3. Nature 537, 532–534 (2016).
Jimenez J. L. et al. New particle formation from photooxidation of diiodomethane (CH2I2). J. Geophys. Res. Atmos. 108, (2003).
Burkholder, J. B., Curtius, J., Ravishankara, A. R. & Lovejoy, E. R. Laboratory studies of the homogeneous nucleation of iodine oxides. Atmos. Chem. Phys. 4, 19–34 (2004).
McFiggans, G. et al. Direct evidence for coastal iodine particles from Laminaria macroalgae – linkage to emissions of molecular iodine. Atmos. Chem. Phys. 4, 701–713 (2004).
Allan, J. D. et al. Iodine observed in new particle formation events in the Arctic atmosphere during ACCACIA. Atmos. Chem. Phys. 15, 5599–5609 (2015).
Baccarini, A. et al. Frequent new particle formation over the high Arctic pack ice by enhanced iodine emissions. Nat. Commun. 11, 4924 (2020).
Wan, Y. et al. Probing key organic substances driving new particle growth initiated by iodine nucleation in coastal atmosphere. Atmos. Chem. Phys. 20, 9821–9835 (2020).
He, X. C. et al. Role of iodine oxoacids in atmospheric aerosol nucleation. Science 371, 589–595 (2021).
Beck, L. J. et al. Differing mechanisms of new particle formation at two Arctic sites. Geophys. Res. Lett. 48, e2020GL091334 (2021).
Ning, A. et al. The critical role of dimethylamine in the rapid formation of iodic acid particles in marine areas. npj Clim. Atmos. Sci. 5, 92 (2022).
Huang, R.-J. et al. Heterogeneous iodine-organic chemistry fast-tracks marine new particle formation. Proc. Natl. Acad. Sci. USA 119, e2201729119 (2022).
Sellegri K. et al. Evidence of atmospheric nanoparticle formation from emissions of marine microorganisms. Geophys. Res. Lett. 43, 6596–6603 (2016).
Gómez Martín, J. C. et al. A gas-to-particle conversion mechanism helps to explain atmospheric particle formation through clustering of iodine oxides. Nat. Commun. 11, 4521 (2020).
Plane, J. M. C., Joseph, D. M., Allan, B. J., Ashworth, S. H. & Francisco, J. S. An experimental and theoretical study of the reactions OIO + NO and OIO + OH. J. Phys. Chem. A 110, 93–100 (2006).
Khanniche, S., Louis, F., Cantrel, L. & Černušák, I. Computational study of the I2O5+H2O=2 HOIO2 gas-phase reaction. Chem. Phys. Lett. 662, 114–119 (2016).
Xia, D. et al. Formation mechanisms of Iodine–Ammonia clusters in polluted coastal areas unveiled by thermodynamics and kinetic simulations. Environ. Sci. Technol. 54, 9235–9242 (2020).
Cuevas, C. A. et al. Rapid increase in atmospheric iodine levels in the North Atlantic since the mid-20th century. Nat. Commun. 9, 1452 (2018).
Roldin, P. et al. The role of highly oxygenated organic molecules in the Boreal aerosol-cloud-climate system. Nat. Commun. 10, 4370 (2019).
Han, J. et al. Determinant factor for thermodynamic stability of sulfuric acid–amine complexes. J. Phys. Chem. A 124, 10246–10257 (2020).
Cai, R. et al. The missing base molecules in atmospheric acid–base nucleation. Natl. Sci. Rev. 9, nwac137 (2022).
Contini, D., Vecchi, R. & Viana, M. Carbonaceous aerosols in the atmosphere. Atmosphere 9, 181 (2018).
Brean, J., Rowell, A., Beddows, D. C. S., Shi, Z. & Harrison, R. M. Estimates of future new particle formation under different emission scenarios in Beijing. Environ. Sci. Technol. 57, 4741–4750 (2023).
Li, C. et al. India is overtaking china as the world’s largest emitter of anthropogenic sulfur dioxide. Sci. Rep. 7, 14304 (2017).
Krotkov, N. A. et al. Aura OMI observations of regional SO2 and NO2 pollution changes from 2005 to 2015. Atmos. Chem. Phys. 16, 4605–4629 (2016).
Julin, J. et al. Impacts of future European Emission Reductions On Aerosol Particle Number Concentrations Accounting For Effects Of Ammonia, Amines, And Organic Species. Environ. Sci. Technol. 52, 692–700 (2018).
Mao, J. et al. High-resolution modeling of gaseous methylamines over a polluted region in China: source-dependent emissions and implications of spatial variations. Atmos. Chem. Phys. 18, 7933–7950 (2018).
Li, Y. et al. A dynamic parameterization of sulfuric acid–dimethylamine nucleation and its application in three-dimensional modeling. Atmos. Chem. Phys. 23, 8789–8804 (2023).
Ma F. et al. Sulfuric acid-driven nucleation enhanced by amines from ethanol gasoline vehicle emission: machine learning model and mechanistic study. Environ. Sci. Technol., (2024).
Van Neste A., Duce R. A., & Lee C. Methylamines in the marine atmosphere. Geophys. Res. Lett. 14, 711–714 (1987).
Ma, F. et al. Enhancement of atmospheric nucleation precursors on iodic acid-induced nucleation: predictive model and mechanism. Environ. Sci. Technol. 57, 6944–6954 (2023).
Charlson, R. J., Lovelock, J. E., Andreae, M. O. & Warren, S. G. Oceanic phytoplankton, atmospheric sulphur, cloud albedo and climate. Nature 326, 655–661 (1987).
Zhao, B. et al. Global variability in atmospheric new particle formation mechanisms. Nature 631, 98–105 (2024).
Shrivastava M., et al. Anthropogenic Extremely Low Volatility Organics (ELVOCs) govern the growth of molecular clusters over the southern great plains during the springtime. JGR Atmos. 129, e2024JD041212 (2024).
Ning, A. et al. Overlooked significance of iodic acid in new particle formation in the continental atmosphere. Proc. Natl. Acad. Sci. USA 121, e2404595121 (2024).
Ma, F. et al. Autoxidation mechanism for atmospheric oxidation of tertiary amines: Implications for secondary organic aerosol formation. Chemosphere 273, 129207 (2021).
King, G. M., Klug, M. J. & Lovley, D. R. Metabolism of acetate, methanol, and methylated amines in intertidal sediments of lowes cove, maine. Appl. Environ. Microbiol. 45, 1848–1853 (1983).
Oremland, R. S., Marsh, L. M. & Polcin, S. Methane production and simultaneous sulphate reduction in anoxic, salt marsh sediments. Nature 296, 143–145 (1982).
Mausz, M. A. & Chen, Y. Microbiology and ecology of methylated amine metabolism in marine ecosystems. Curr. Issues Mol. Biol. 33, 133–148 (2019).
Hippe, H., Caspari, D., Fiebig, K. & Gottschalk, G. Utilization of trimethylamine and other N-methyl compounds for growth and methane formation by Methanosarcina barkeri. Proc. Natl. Acad. Sci. USA 76, 494–498 (1979).
Galloway, M. M. et al. Secondary Organic Aerosol Formation during Evaporation of Droplets Containing Atmospheric Aldehydes, Amines, and Ammonium Sulfate. Environ. Sci. Technol. 48, 14417–14425 (2014).
De Haan, D. O. et al. Formation of nitrogen-containing oligomers by methylglyoxal and amines in simulated evaporating cloud droplets. Environ. Sci. Technol. 45, 984–991 (2011).
Tian X., Chan V. Y. Chan C. K. Secondary Organic aerosol formation from aqueous ethylamine oxidation mediated by particulate nitrate photolysis. ACS ES&T Air, (2024).
Marrero-Ortiz, W. et al. Formation and optical properties of brown carbon from small α-Dicarbonyls and Amines. Environ. Sci. Technol. 53, 117–126 (2019).
MacDowell, N. et al. An overview of CO2 capture technologies. Energy Environ. Sci. 3, 1645–1669 (2010).
Webley, P. A. Adsorption technology for CO2 separation and capture: a perspective. Adsorption 20, 225–231 (2014).
Bahamon, D. & Vega, L. F. Systematic evaluation of materials for post-combustion CO2 capture in a Temperature Swing Adsorption process. Chem. Eng. J. 284, 438–447 (2016).
Luis, P. & Van der Bruggen, B. The role of membranes in post-combustion CO2 capture. Greenh. Gases: Sci. Technol. 3, 318–337 (2013).
Abanades, J. C., Anthony, E. J., Wang, J. & Oakey, J. E. Fluidized Bed Combustion Systems Integrating CO2 Capture with CaO. Environ. Sci. Technol. 39, 2861–2866 (2005).
Lourdes F. V. et al. How Molecular Modelling Tools Can Help in Mitigating Climate Change (2021).
Karl, M. et al. Modelling atmospheric oxidation of 2-aminoethanol (MEA) emitted from post-combustion capture using WRF-Chem. Sci. Total Environ. 527-528, 185–202 (2015).
Xie, H.-B., He, N., Song, Z., Chen, J. & Li, X. Theoretical investigation on the different reaction mechanisms of aqueous 2-Amino-2-methyl-1-propanol and Monoethanolamine with CO2. Ind. Eng. Chem. Res. 53, 3363–3372 (2014).
Veltman, K., Singh, B. & Hertwich, E. G. Human and environmental impact assessment of postcombustion CO2 capture focusing on emissions from amine-based scrubbing solvents to air. Environ. Sci. Technol. 44, 1496–1502 (2010).
Myllys, N. et al. Guanidine: A Highly Efficient Stabilizer in Atmospheric New-Particle Formation. J. Phys. Chem. A 122, 4717–4729 (2018).
Paasonen, P. et al. On the formation of sulphuric acid – amine clusters in varying atmospheric conditions and its influence on atmospheric new particle formation. Atmos. Chem. Phys. 12, 9113–9133 (2012).
Zollner, J. H. et al. Sulfuric acid nucleation: power dependencies, variation with relative humidity, and effect of bases. Atmos. Chem. Phys. 12, 4399–4411 (2012).
You, Y. et al. Atmospheric amines and ammonia measured with a chemical ionization mass spectrometer (CIMS). Atmos. Chem. Phys. 14, 12181–12194 (2014).
Wang, Y. et al. Detection of gaseous dimethylamine using vocus proton-transfer-reaction time-of-flight mass spectrometry. Atmos. Environ. 243, 117875 (2020).
Tzitzikalaki E., Kalivitis N., & Kanakidou M. Observations of gas-phase alkylamines at a coastal site in the East Mediterranean atmosphere. Atmosphere 12, 1454 (2021).
Yu H., & Lee S.-H. Chemical ionisation mass spectrometry for the measurement of atmospheric amines Environ. Chem. 9, 190-201 (2012).
Akyüz, M. Simultaneous determination of aliphatic and aromatic amines in ambient air and airborne particulate matters by gas chromatography-mass spectrometry. Atmos. Environ. 42, 3809–3819 (2008).
Tiszenkel, L., Flynn, J. H. & Lee, S. H. Measurement report: Urban ammonia and amines in Houston, Texas. Atmos. Chem. Phys. 24, 11351–11363 (2024).
Huang, G., Hou, J. & Zhou, X. A measurement method for atmospheric ammonia and primary amines based on aqueous sampling, OPA Derivatization and HPLC analysis. Environ. Sci. Technol. 43, 5851–5856 (2009).
Deng, C. et al. Seasonal characteristics of new particle formation and growth in urban Beijing. Environ. Sci. Technol. 54, 8547–8557 (2020).
Zhu, S. et al. Observation and source apportionment of atmospheric alkaline gases in urban Beijing. Environ. Sci. Technol. 56, 17545–17555 (2022).
Hanson, D. R., McMurry, P. H., Jiang, J., Tanner, D. & Huey, L. G. Ambient Pressure Proton Transfer Mass Spectrometry: Detection of Amines and Ammonia. Environ. Sci. Technol. 45, 8881–8888 (2011).
Hellén, H., Kieloaho, A. J. & Hakola, H. Gas-phase alkyl amines in urban air; comparison with a boreal forest site and importance for local atmospheric chemistry. Atmos. Environ. 94, 192–197 (2014).
Hu Q., et al. Concentration, size distribution, and formation of Trimethylaminium and Dimethylaminium Ions in atmospheric particles over marginal seas of China. J. Atmos. Sci. 72, 3487–3498 (2015).
Xie, H. et al. Concentration and size distribution of water-extracted dimethylaminium and trimethylaminium in atmospheric particles during nine campaigns - Implications for sources, phase states and formation pathways. Sci. Total Environ. 631-632, 130–141 (2018).
Yu, P. et al. Characteristics of dimethylaminium and trimethylaminium in atmospheric particles ranging from supermicron to nanometer sizes over eutrophic marginal seas of China and oligotrophic open oceans. Sci. Total Environ. 572, 813–824 (2016).
Violaki, K. & Mihalopoulos, N. Water-soluble organic nitrogen (WSON) in size-segregated atmospheric particles over the Eastern Mediterranean. Atmos. Environ. 44, 4339–4345 (2010).
Chen, D. et al. Competitive uptake of Dimethylamine and Trimethylamine against ammonia on acidic particles in marine atmospheres. Environ. Sci. Technol. 56, 5430–5439 (2022).
Huang, X., Kao, S.-J., Lin, J., Qin, X. & Deng, C. Development and validation of a HPLC/FLD method combined with online derivatization for the simple and simultaneous determination of trace amino acids and alkyl amines in continental and marine aerosols. PLOS ONE 13, e0206488 (2018).
Yang H., Xu J., Wu W.-S., Wan C. H. Yu J. Z. Chemical characterization of water-soluble organic aerosols at Jeju Island Collected During ACE-Asia. Environ. Chem. 1, 13–17 (2004).
Du, W. et al. Particulate amines in the background atmosphere of the Yangtze River Delta, China: Concentration, size distribution, and sources. Adv. Atmos. Sci. 38, 1128–1140 (2021).
Feng, X. et al. Outbreaks of Ethyl-Amines during Hazeepisodes in North China Plain: A potential source of amines from ethanol gasoline vehicle emission. Environ. Sci. Technol. Lett. 9, 306–311 (2022).
Suzuki, Y., Kawakami, M. & Akasaka, K. 1H NMR application for characterizing water-soluble organic compounds in urban atmospheric particles. Environ. Sci. Technol. 35, 2656–2664 (2001).
Yang, H. et al. The chemical composition of inorganic and carbonaceous materials in PM2.5 in Nanjing, China. Atmos. Environ. 39, 3735–3749 (2005).
Liu, F. et al. Concentration, size distribution and dry deposition of amines in atmospheric particles of urban Guangzhou, China. Atmos. Environ. 171, 279–288 (2017).
Ho, K. F. et al. Characteristics of water-soluble organic nitrogen in fine particulate matter in the continental area of China. Atmos. Environ. 106, 252–261 (2015).
Ho, K.-F. et al. Chemical composition and bioreactivity of PM2.5 during 2013 haze events in China. Atmos. Environ. 126, 162–170 (2016).
Calderón, S. M., Poor, N. D. & Campbell, S. W. Estimation of the particle and gas scavenging contributions to wet deposition of organic nitrogen. Atmos. Environ. 41, 4281–4290 (2007).
Zhou, S., Lin, J., Qin, X., Chen, Y. & Deng, C. Determination of atmospheric alkylamines by ion chromatography using 18-crown-6 as mobile phase additive. J. Chromatogr. A 1563, 154–161 (2018).
Cheng, G. et al. Characteristics and potential source areas of aliphatic amines in PM2.5 in Yangzhou, China. Atmos. Pollut. Res. 11, 296–302 (2020).
Place, B. K., Quilty, A. T., Di Lorenzo, R. A., Ziegler, S. E. & VandenBoer, T. C. Quantitation of 11 alkylamines in atmospheric samples: separating structural isomers by ion chromatography. Atmos. Meas. Tech. 10, 1061–1078 (2017).
Acknowledgements
We acknowledge the funding support by the European Union ERC-2022-STGERC-BAE-Project: 101076311. Views and opinions expressed are however those of the author(s) only and do not necessarily reflect those of the European Union or the European Research Council Executive Agency. Neither the European Union nor the granting authority can be held responsible for them. This project has also received funding from the European Union’s Horizon 2020 research and innovation programme under grant agreement No. 856612 and the Cyprus Government. The authors thank the anonymous reviewers for their valuable suggestions and comments.
Author information
Authors and Affiliations
Contributions
T.J. conceived the idea. V.P.K. wrote the first draft and revised it with critical inputs from T.J.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Communications Earth & Environment thanks the anonymous reviewers for their contribution to the peer review of this work. Primary Handling Editor: Alice Drinkwater. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Kanawade, V.P., Jokinen, T. Atmospheric amines are a crucial yet missing link in Earth’s climate via airborne aerosol production. Commun Earth Environ 6, 98 (2025). https://doi.org/10.1038/s43247-025-02063-0
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s43247-025-02063-0
This article is cited by
-
Diverse mixing states and atmospheric processes of urban amine-containing particles in the North China Plain
ENGINEERING Environment (2026)
-
Multiple oceanic sources of alkylamines in Southern Ocean atmospheres
npj Climate and Atmospheric Science (2025)
-
Estimating aqueous solubility and hygroscopicity of amino acid particles with COSMOtherm
Communications Chemistry (2025)
