
Abstract
Background
Ischemic cardiomyopathy and resultant heart failure (HF) is a significant cause of morbidity and mortality worldwide. Downregulation of cardiac bridging integrator 1 (cBIN1), a membrane scaffolding protein responsible for organizing t-tubules and organizing the calcium handing apparatus, occurs in progressive HF. Therefore, gene therapy upregulating cBIN1 production may rescue failing muscle and clinical HF.
Methods
Adult mongrel dogs underwent ligation of the left anterior descending artery and developed progressive dilated cardiomyopathy and chronic HF. When left ventricular ejection fraction (LVEF) dropped below 40%, the animals received a one-time series of endocardial injections of either of low dose gene therapy composed of either adeno-associated virus serotype 9 packaged cBIN1 (AAV9-cBIN1, n = 6) or AAV9-GFP (green fluorescent protein, n = 4). Animals were followed up to 7 weeks after therapy delivery with laboratory, echocardiography, and endocardial mapping assessment.
Results
Post injection of the negative control, animals develop progressive symptomatic HF requiring early termination of all but one animal prior to the end of the study. In contrast, the AAV9-cBIN1-treated group reveals a significant improvement in LV function, with a noticeable improvement in LVEF (29 ± 3% vs. 42 ± 2%, p = 0.0095) and global longitudinal strain (−7.1 ± 0.9% vs. −12.5 ± 1.6%, p = 0.0095). Compared to the control animals, the AAV9-cBIN1-treated group displays improved T-tubule morphology, left ventricular chamber size, plasma biomarkers, and endocardial voltage, and survives the study period.
Conclusions
Chronic HF from ischemic cardiomyopathy can be successfully treated with low dose AAV9-cBIN1 gene therapy. This study indicates that myocardial specific therapy can dramatically reverse HF progression.
Plain Language Summary
Blocked heart arteries are known to damage heart muscle by impairing the ability of the heart to pump blood, resulting in progressive muscle damage and eventual heart failure. Medical interventions that recover failing heart muscle are needed to prevent hospitalization or death. Here, we developed a gene therapy to replace an important heart muscle protein, cardiac bridging integrator 1 (cBIN1), and tested it in a canine model of heart failure induced by blocked heart arteries. These findings show improved cardiac muscle function and survival rate following cBIN1 gene therapy, indicating the potential of this gene therapy to reduce the syndrome of heart failure.
Similar content being viewed by others
Introduction
The morbidity, mortality, and economic toll on the over six million Americans with HF is enormous and growing. For instance, the cost of HF in the USA was estimated at $43.6 billion in 2020 and is estimated to increase to $70 billion by 20301. Over the past decade, the annual number of HF hospitalizations has almost doubled with approximately 50% of patients being re-hospitalized within 6 months of discharge2. The traditional pharmacologic therapeutic options for chronic HF have systemic effects which mainly interfere with neurohormonal stress pathways3. However, the origin of chronic HF is failing myocardium and low cardiac output. Limitations of current therapeutic approaches have led to the pursuit of heart muscle specific therapies to recover failing myocardium. The choice of proper target gene and the availability of efficient gene carriers are requisite for a successful therapeutic approach4. Gene therapies, by which the DNA of critical and beneficial proteins are introduced to failing myocardium, are being explored as a new generation of therapeutic to limit the progression of myocardial decline and, ideally, reverse HF4,5. The pathophysiological hallmark of a failing heart is abnormal calcium handling6,7, and the loss of t-tubule structure and membrane microdomains is a common pathophysiology of acquired HF8.
cBIN1, a membrane sculpting protein that forms membrane microdomains within t-tubule invaginations, organizes the cardiomyocyte calcium handling apparatus responsible for cardiac contraction and relaxation9,10. cBIN1 is transcriptionally reduced in failing hearts, impairing t-tubule structure and intracellular organization of the calcium handling machinery9,11,12 which, in turn, negatively affects calcium transients and cardiac muscle function. and, in mouse models of HF11,12, cBIN1 gene therapy positively remodels cardiomyocyte t-tubule microdomains and can rescue left ventricular function. Further, loss of cBIN1 occurs in acquired human heart failure13,14,15 and its restoration by cBIN1 gene therapy in mouse models improves failing myocardium and HF outcomes11,12,16.
Mouse models of heart failure and mouse cardiomyocyte architecture have significant differences relative to their counterparts in larger animals and humans. To evaluate the potential of cBIN1 gene therapy to address human heart failure, we utilized a recently developed canine large animal model of dilated cardiomyopathy (DCM) of ischemic origin17. In this model, experimentally induced DCM is achieved with a thoracotomy and surgical ligation of the Left Anterior Descending (LAD) and its collateral coronary arteries17, resulting in a large anterior myocardial infarction. Post-coronary ligation, there is an acute decline in function followed by a brief stabilization and marginal recovery, and then progressive DCM with ventricular dilation occurring over a course of ten to sixteen weeks. During this time there is weekly assessment of both left ventricular function by echocardiography and clinical biomarkers by venous phlebotomy. Here we show that cBIN1 applied as an intramyocardial gene therapy in a large animal model of HF from DCM of ischemic origin reverses decline in left ventricular function, improves HF associated biomarkers, recovers low voltage endocardial zones, and improves mortality. We find that muscle damaged from chronic ischemic cardiomyopathy can be recovered by improving the intracellular architecture of failing cardiomyocytes.
Methods
All studies follow the guidelines from the National Institutes of Health Guide for the Care and Use of Laboratory Animals18 and the standards of United States regulatory agencies. The study protocol is approved by the Institutional Animal Care and Use Committee (IACUC) of The University of Utah.
Study design
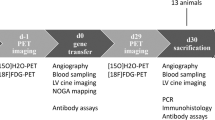
The objective of this study is to evaluate the therapeutic potential of AAV9-cBIN1 gene therapy in restoring the cardiac function of chronic DCM of ischemic origin. Twelve adult mongrel dogs (Canis lupus, Oak Hill Genetics, Ewing, IL, USA) age: 13 ± 3 months old and weighing 29 ± 3 kg (range: 25–33 kg with eight males) are housed according to guidelines with care by institutional veterinarians. Dogs are group housed with the exception of during recovery following procedures and in the case of animals showing aggression, in which cases they are housed in the same room but in separate cages to prevent injury. Environment stimulation is provided. Dogs are randomized into 3 groups as: AAV9-GFP-treated group, n = 4 (12 ± 1 months old, 30 ± 3 kg with four males); AAV9-cBIN1-treated group, n = 6 (13 ± 3 months old, 29 ± 3 kg with three males); and untreated heart failure control group, n = 2 (15 ± 1 months old, 25–31 kg with one male). We use a canine model of induced ischemic cardiomyopathy in which a thoracotomy is performed and the LAD and collateral coronary arteries are surgically ligated17. Post-coronary ligation, cardiac contractility (LVEF) and N-terminal-prohormone B-type natriuretic peptide (NT-proBNP) are evaluated biweekly to evaluate HF progression. As shown in Fig. 1a, in the first-month post-ligation, LVEF rapidly declines, followed by partial recovery in function as surviving animals transition from acute ischemic HF to chronic ischemic HF. Beyond week 1, systolic function recovers over 2–3 weeks and then slowly declines as the LV dilates and cardiac function worsens as indicated by cardiac biomarkers and echocardiogram measurements. The slower secondary decline is consistent with cardiac remodeling and the development of chronic HF. Over a course of 10–16 weeks post-ligation, progressive HF with reduced EF occurs. When LVEF falls below 40%, therapy is randomly initiated with injection with AAV9-cBIN1, AAV9-GFP (green fluorescent protein) or no-treatment (untreated control). Our approach to coronary ligation results in 91% survival 48 hours post ligation, and, of surviving animals a 100% success in development of chronic ischemic HF17. At the time of chronic ischemic HF, a NOGA XP+ cardiac mapping system (Biosense Webster, Diamond Bar, CA, a Johnson and Johnson Company) is used to deliver either AAV9-cBIN1 (n = 6) or AAV9-GFP (n = 4) to the endocardium. Animals are then followed for 5–8 weeks post-injection. Two animals develop chronic ischemic dilated cardiomyopathy, and do not receive any treatment. These animals are sacrificed at time zero and served as reference un-treated heart failure (UT-HF) animals. Two LV tissue samples per animal are collected away from the infarction at the time of study termination.

a Left ventricular ejection fraction (LVEF) from ligation through heart failure development, therapy, and for the post-therapy period. Time 0 refers to the day of injection for the AAV9-GFP (n = 4) and AAV9-cBIN1 (n = 6) treated cohorts. b Absolute LVEF from time of therapy to post-therapy week 5. c Left ventricular global longitudinal strain (GLS) from time of therapy to post-therapy week 5. All data were recorded in awake animals. In (a), two-way ANOVA mixed model was used to determine the statistical significance over time between two groups at day 14 (p = 0.2218), day 21 (p = 0.0327), day 28 (p = 0.0007) and day 35 (p = 0.0001). In (b and c), Mann–Whitney test is used to compute the statistical comparison between two groups (AAV9-GFP-treated group vs. AAV9-cBIN1-treated group) at post-therapy week 5 (LVEF: p = 0.0095 and GLS: p = 0.0095). In (b and c), Two-tailed paired t test is used to statistically compare the mean of the paired data between two different time points (pre-therapy vs. post-therapy week 5) within same group (for AAV9-GFP-treated group, LVEF: p = 0.0014 and GLS: p = 0.0228, and for AAV9-cBIN1-treated group, LVEF: p = 0.0005 and GLS: p = 0.0137). Data presented in (b and c) are shown as ‘mean’ and error bars are shown as ‘standard deviation’.
AAV9 viral packaging
Adeno-associated virus vector (AAV) is used because it is a promising gene therapy vehicle and exhibits the highest cardiac tropism, as previously described19. AAV9-GFP-V5 is used as the negative control virus because it does not induce cardiomyocyte toxicity and has been successfully used as a negative control virus in numerous gene therapy studies with animal models of cardiovascular diseases20, and in our previously reported mouse studies11. In this large animal preclinical study, four dogs receive a dose of 5 ×1012 vector genome (vg) of AAV9 transducing GFP, and another six dogs receive a dose of 5 ×1012 vg of AAV9 transducing cBIN1. After tittering, viruses are aliquoted and immediately frozen and placed at −80 °C for long-term storage. Five to ten minutes before the injection procedure, the aliquot is diluted in PBS to make a 5-mL injection solution, which is then administered at 20 injection sites with 0.25-mL and 2.5 × 1011 vg injected at each site.
Electroanatomical mapping and transendocardial injections
NOGA XP + 3D left ventricular electroanatomical mapping allows accurate determination of cardiac function and identification of sites of injury. Electroanatomical mapping and endocardial gene delivery using NOGA catheter systems is performed as described previously21. When LVEF falls below 40%, the therapeutic procedure is scheduled. For logistical reasons, the endocardial mapping and injection may occur a few weeks post falling below the 40% threshold. For all animals, time Day 0 in Fig. 1a corresponds to the exact day of therapeutic injection (in which LVEF was between LVEF 35% and 40%). Briefly, dogs are maintained on general anesthesia and are appropriately prepped and draped in a manner to allow sterile surgical access to the inguinal area. Femoral access is achieved by percutaneous access. A NogaStar catheter (Biosense Webster) is advanced into the left ventricle to first create a three-dimensional electroanatomical endocardial map. After mapping is completed, a MyoStar injection catheter (Biosense Webster) is primed with 0.15 mL of AAV9-cBIN1 or AAV9-GFP to fill the dead space before the start of the injection procedure. The needle is set to protrude from the end of the catheter by 4–6 mm. We perform a total of twenty transendocardial injections (0.25 mL per injection), with half in the border zone and half distributed throughout the remainder of the LV endocardium. For each injection, catheter is position perpendicular to the wall of the left ventricle wall, and the presence of a premature ventricular contraction upon extending the needle into the myocardium ensures proper delivery into the myocardium.
Echocardiography recordings and analysis
Left ventricular (LV) function is assessed by echocardiography (Acuson SC2000, Siemens, Munich, Germany) in awake dogs before the baseline coronary ligation procedure and throughout HF development. Data are acquired weekly (pre-therapy day to post-therapy week 8) to evaluate both systolic and diastolic function over time. Images are acquired in 4-chamber apical, parasternal long-axis, and mid papillary muscle short-axis views. LVEF is evaluated from a 4-chamber apical view. Left end-diastolic and end-systolic volumes are evaluated from the 4-chamber apical view. LV myocardial strain analysis is performed to assess the LV global longitudinal strain (GLS) using echocardiography (Acuson SC2000, Siemens, Munich, Germany). GLS is measured longitudinally in awake dogs at pre-therapy and at post-therapy week 5 (Fig. 1b). GLS data is acquired and analyzed as follows: (1) One cardiac cycle is defined from R wave to R wave, and the end‐diastolic frame is selected by the closure of the mitral aortic valve. (2) Reference points are placed at the mitral annulus, and the subsequent points are taken at the endocardial wall within the LV chamber. (3) LV GLS values are quantified using the Siemens analysis tools on the SC2000 and reported as absolute numbers. By convention, GLS is expressed as a negative number, and more negative LV GLS values represent better systolic contractility. GLS data are quantified from a 4-chamber apical view from at least three consecutive heart beats.
Sample processing and cBIN1 score determination
cBIN1 Score (CS) is an inverse index of plasma cBIN1 and is a marker of myocardial remodeling in HF patients14. Whole venous blood is drawn into EDTA tubes, stored immediately at 4 °C for less than 4 h, and then processed to plasma and flash frozen for storage at −80 °C prior to use, as previously described22. The concentration of cBIN1 is determined using a cBIN1 specific sandwich-ELISA assay provided by Sarcotein Diagnostics (Tampa, FL), as previously described23. Findings are reported using CS, the natural log of the inverse of cBIN1 plasma concentration which increases as cBIN1 decreases.
Histopathology data analysis
Tissue samples are collected from LV muscle away from the infarction at the time of study termination, and are assessed for t-tubule membrane cross-sectional area, interstitial fibrosis and inflammation. For t-tubule imaging, transmission electron microscopy (TEM) is demonstrated on the LV tissue samples (N) from following groups: Un-treated heart failure (UT-HF) group (N = 4 tissue samples, 2 animals, 2 samples each), AAV9-GFP-treated group (N = 6 tissue samples, 3 animals, 2 samples each) and AAV9-cBIN1-treated group (N = 6 tissue samples, 3 animals, 2 samples each). The number of t-tubules analyzed in these tissues are 75, 83 and 102 in UT-HF group, AAV9-GFP-treated group and AAV9-cBIN1-treated group, respectively. The samples are dissected into 1 mm pieces and immediately fixed with 2.5% glutaraledehyde, 1% paraformaldehyde, 0.1 M Cacodylate buffer, pH 7.4. Following overnight fixation, the specimens are rinsed two times in buffer and are post-fixed in 2% Osmium tetroxide for one hour at room temperature. The specimens are then rinsed in dH2O and incubated in saturated uranyl acetate for one hour at room temperature, then dehydrated in a graded ethanol series (50, 70, 2 × 95 and 4 × 100%), then three times in pure acetone, then infiltrated and embedded in epoxy resin using Embed 812 (cat # 14121, Electron Microscopy Sciences). The polymerization is performed at 60 °C for 48 h. Ultrathin sections are obtained at 70 nm using an ultratome Leica UC 6 (Leica) and counter stained with uranyl acetate for 20 min for membrane contrast followed by staining with lead citrate for 5 min. Grids are examined at an accelerating voltage of 120 kV in a JEOL-1400 plus (JEOL, Japan) transmission electron microscope equipped with CCD Gatan camera. For fibrosis and inflammation assessment, a detailed methodology is included in ‘Supplementary Methods’ of the supplementary information file. NT-proBNP Assay NT-proBNP values are obtained from the plasma of AAV9-GFP-treated group and AAV9-cBIN1-treated group pre- to post-therapy at different time points, and are sent to IDEXX BioAnalytics (West Sacramento, CA) to perform the NT-proBNP assay using electrochemiluminescence.
Statistics and reproducibility
Mann–Whitney test is used to compute unpaired data between two groups (AAV9-GFP-treated group vs. AAV9-cBIN1-treated group) at same time point (pre-therapy or post-therapy end-point). Two-tailed paired t test is used to statistically compare the mean of the paired data at two different time points (pre-therapy vs. post-therapy end point) for the same group (AAV9-GFP-treated group or AAV9-cBIN1-treated group). A two-way mixed analysis of variance (ANOVA) model with Bonferroni post hoc tests were used to examine the time effect (pre-therapy to post-therapy), and interaction between time and group effect on LVEF. Post hoc tests with Bonferroni correction are conducted to check for differences between two groups at different post-injection time-points (post-injection day 7, 14, 21, 28 and 35). For t-tubules imaging data, statistical comparison is included for only two groups (AAV9-cBIN1-treated group and AAV9-GFP-treated group). Details for statistics to support survival analysis, and fibrosis and inflammation assessment between AAV9-GFP-treated group and AAV9-cBIN1-treated group are included in subsections: ‘Survival Analysis’ and ‘Fibrosis and Inflammation Assessment’, respectively in the ‘Supplementary Methods’ section of the supplementary information file. All analyses are conducted in GraphPad PRISM 10.0. A p-value of <0.05 is considered statistically significant.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Results
In the canine model of DCM of ischemic origin (n = 10), therapy was applied when left ventricular ejection fraction (LVEF) falls from a baseline of 56 ± 4% to less than 40% and an increase in pre-load is confirmed with a rise in NT-proBNP to greater than 900 pmol/L from the baseline measure of 288 ± 55 pmol/L. In this large animal model, we utilized catheter-based endocardial injection. At the time of therapy, the animal is put under general anesthesia and a NogaStar Mapping catheter (Johnson & Johnson) is introduced percutaneously via the femoral artery to perform electroanatomical mapping of the left ventricular endocardium with the NOGA XP+ mapping system (Johnson & Johnson). Post-mapping, a Myostar catheter (Johnson & Johnson) is used to perform 20 endocardial injections of adeno-associated virus isotype 9 (AAV9) packaged with the mouse (mus musculus, mm) transgene with a C-terminal V5 tag mm-Bin1 + 13 + 17-V5. The presence of exons 13 and 17 is the cardiac t-tubule isoform of Bin1 (referred to as cBIN1) and the mouse isoform has 95% sequence homology to human hs-BIN1 + 13 + 17 gene10. Our Bin1 + 13 + 17-V5 plasmid is driven by the cytomegalovirus (CMV) promoter and the full therapy is termed here as AAV9-cBIN1. The mapping guided injections are uniformly distributed throughout the left ventricular endocardium without special consideration to the infarction scar so that most of the therapy is applied to non-infarcted yet failing muscle. Each injection is 2.5 × 1011 vg for a total of 5 × 1012 vg per animal (about 1.6 × 1011 vg/kg per 30 kg animal). Biologically inactive AAV9-GFP is injected in the negative control group. The animals continued to be monitored with weekly echocardiography and phlebotomy post therapy.
As seen in Fig. 1, AAV9-GFP injection did not affect the slope of progressive myocardial decline post infarction. All of the four animals treated with AAV9-GFP (red lines) developed severe HF signs and required veterinary mandated euthanasia 5–7 weeks post-injection (veterinary staff were blinded to treatment type). The animals were terminated due to, at day 34 (anorexia, emesis, and weight loss), day 36 (hypokinesis, anorexia), day 39 (anorexia, severe dyspnea with pulmonary edema), and day 49 (anorexia, and non-sustained ventricular tachycardia episodes). In contrast, all animals treated with AAV9-cBIN1 survived through at least day 48, and none had significant HF failure symptoms or negative medical reports. All the AAV9-cBIN1 treated animals experienced an improvement in LVEF at Day 48 which was the pre-determined time of sacrifice. None of these animals exhibited heart failure symptoms, and improved survival (p = 0.0011) - refer to subsection: ‘Survival Analysis’ in the ‘Supplementary Methods’ and as shown in Supplementary Fig. 1. We observed a trend in several of the animals of a small decline in LVEF from about day 35 (Fig. 1a). There was still an overall improvement in LVEF at day 48 compared to the time of injection at day 0, and future studies will help us confirm whether there is further decline in function or more simply a plateau in peak function is observed in these animals with significant permanent scar burden. The survival advantage of AAV9-cBIN1-treated animals is driven by not just a stabilization but a dramatic functional improvement of cBIN1-treated hearts. As seen in Fig. 1a there is improvement in LVEF beginning week-three post-injection. By post injection week-five, the LVEF of AAV9-GFP-treated animals decreased from 37 ± 1% to 29 ± 3% (Fig. 1b, p = 0.0014) and left ventricular global longitudinal strain (GLS) decreased from −9.8 ± 0.4 to −7.1 ± 0.9% (Fig. 1c, p = 0.0228), both of which are consistent with progressive heart failure. In contrast, in the AAV9-cBIN1-treated group, the LVEF increased from 37 ± 2% to 42 ± 2% (Fig. 1b, p = 0.0005), and GLS improved from −9.5 ± 1.5 to −12.5 ± 1.6% (Fig. 1c, p = 0.0137).
The improvement in left ventricular function in cBIN1 treated animals was accompanied by surprising reverse remodeling of the left ventricle. As seen in Fig. 2, in the AAV9-GFP-treated animals, from injection day 0 to five weeks post-injection, the change in left ventricular end diastolic and end systolic volumes increased by 18% and 10%, respectively (absolute change, relative increase in volumes was 11 ± 5 mL and 11 ± 4 mL, respectively). Whereas, in the AAV9-cBIN1-treated hearts, the change in left ventricular end diastolic and end systolic volumes decreased by 4% and 6%, respectively (relative reductions were 5 ± 4 mL and 4 ± 3 mL for end-diastolic and end-systolic volumes, respectively). As shown in Fig. 2a, b, between the two treatment groups (AAV9-GFP vs. AAV9-cBIN1), the change in LVEDV and the change in LVESV from pre-therapy to five weeks post-therapy is significant (p = 0.0095 and p = 0.0095, respectively). At time Day 0 (before therapy), the LVEDV is not significantly different (85 ± 19 mL vs. 81 ± 16 mL, p = 0.8670), but by five weeks following injection, LVEDV differs significantly between the two groups (106 ± 17 vs. 75 ± 12 mL, p = 0.0441). Similarly, at time Day 0 (before therapy), LVESV is not different (61 ± 9 mL vs. 58 ± 9 mL, p = 0.6094), but by five weeks post-therapy, LVESV differs significantly between two groups (70 ± 8 vs. 56 ± 5 mL, p = 0.0381). These data indicate that cBIN1 therapy-induced improvement of ventricular function is accompanied by reversal of the pathological remodeling and hemodynamics of heart failure (even at five weeks pos-therapy).

a Absolute change in LV end-diastolic volume from treatment to week 5. b Absolute change in LV end-systolic volume from treatment to week 5. c Absolute change in cBIN1 score (CS) from treatment to week 5. d Absolute change in N-terminal-prohormone B-type natriuretic peptide (NT-proBNP) from treatment to week 5. For a (p = 0.0095), b (p = 0.0095), c (p = 0.0095) and d (p = 0.0381), Mann–Whitney U tests are used to compute statistical significance between AAV9-GFP (n = 4) and AAV9-cBIN1 (n = 6) treated cohorts. ‘Δ’ refers to absolute change in parameter from time of therapy to post-therapy week 5. Data presented in (a–d) are shown as ‘mean’ and error bars are shown as ‘standard deviation’.
Blood based biomarker assays support the improvement in cardiac muscle function and hemodynamics. CS is independent of volume status reflecting the pathologic remodeling of myocardial muscle, and CS greater than 1 indicates failing heart muscle13. Between the two treatment groups (AAV9-GFP vs. AAV9-cBIN1), the change in CS from pre-therapy to five weeks post-therapy is significant (p = 0.0095), as shown in Fig. 2c. At time Day 0 (before therapy), CS between the groups is not different (1.19 ± 0.40 vs. 1.21 ± 0.20, p = 0.3816), but by five weeks post-therapy, CS increases (worsens) in the AAV9-GFP-treated group but decreases (improves) in the AAV9-cBIN1-treated group (1.59 ± 0.31 vs. 1.08 ± 0.28, p = 0.0412). The improved (lowering of) CS reflects an increase in plasma cBIN1 concentration which reflects an increase in cardiac content of cBIN116. Improved CS (Fig. 2c) indicates recovery of t-tubule microdomains facilitating organization of the calcium handling machinery for efficient excitation-contraction coupling and normalized calcium transients23, leading to improved systolic and diastolic function (Figs. 1a and 2b, respectively). Similarly, NT-proBNP, a marker of intracardiac pressure and hypervolemia24,25, predictably changed in this study. Between the two treatment groups (AAV9-GFP vs. AAV9-cBIN1), the change in NT-proBNP from pre-therapy to five weeks post-therapy is significant (p = 0.0381), as seen in Fig. 2d. At time Day 0 (before therapy), NT-proBNP was not meaningfully different (1084 ± 616 pmol/L vs. 1292 ± 317 pmol/L, p = 0.4968) and by five weeks post-therapy, the NT-proBNP of the AAV9-cBIN1-treated group decreased (improved) significantly (1007 ± 374 pmol/L, p = 0.0473), and NT-proBNP of the AAV9-GFP-treated group increased (worsened) (1451 ± 769 pmol/L, p = 0.0812), although did not reach statistical significance.
The lowering of CS reflects an increase in plasma cBIN1 concentration which reflects an increase in cardiac content of cBIN116. To confirm recovery of t-tubule membrane, transmission electron microscopy (TEM) was used to investigate t-tubule membrane morphology. Tissue samples were collected from the left ventricular free wall away from the infarction after termination. T-tubule membrane cross-sectional area of untreated heart failure, AAV9-GFP-treated, and AAV9-cBIN1-treated failing hearts were 0.053 um2, 0.050 ± 0.026 um2, and 0.031 ± 0.014 um2, respectively, as shown in Fig. 3. TT membrane area is reduced (positively remodeled) with AAV9-cBIN1 compared to AAV9-GFP (p = 0.0001). Increased t-tubule diameter is pathognomonic for failing heart muscle8 and narrowing of t-tubule diameters is consistent with the cBIN1 induced improvement in LVEF and mortality. As shown in Supplementary Fig. 2, the degree of fibrosis (left panel) and interstitial inflammation (right panel) were quantified. Fibrosis is significantly different (p = 0.0312) whereas inflammation is not (p = 0.3125).

a TT membrane (black arrows) captured using transmission electron microscopy - left panel. High resolution representative image of TT membrane - right panel. b Quantified area for TTs of tissues collected from untreated heart failure control group (n = 75), AAV9-GFP-treated group (n = 83), and AAV9-cBIN1-treated group (n = 102). UT-HF refers to heart failure group that did not receive treatment. Mann–Whitney U test is used to compute statistical significance between AAV9-GFP-treated group and AAV9-cBIN1-treated group (p = 0.0001). Data presented in (b) are shown as ‘mean’ and error bars are shown as ‘standard deviation’.
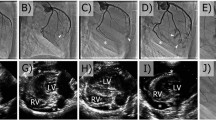
An interesting aspect of chronic ischemic cardiomyopathy is that myocardium can remain injured by both hemodynamic stress as well as limited blood flow to persistently ischemic regions26. Given that cBIN1 aids myocardial remodeling and function, our large animal model provides us with the ability to test for recovery of failing ischemic myocardium in the absence of revascularization. NOGA mapping allows for measurement of endocardial bipolar voltage. The effectiveness of catheter-based delivery of cBIN1 gene therapy in preserving bipolar voltage in a canine model of DCM was explored by comparing time of therapy to post-therapy (time of sacrifice) endocardial voltage maps (Fig. 4a, black dots in the voltage maps indicate the injection points). The AAV9-GFP-treated cohort demonstrated an increase (worsening) in area of low endocardial potentials, with the area of low voltage (<1.5 mV) increasing from 19.5 ± 3.4% to 28.7 ± 9.8% (p = 0.0023) of the LV endocardium. Thus, in the control cohort, myocardium became sicker. However, in the AAV9-cBIN1-treated hearts, there is recovery of endocardial potential, and the low voltage area (<1.5 mV) decreased from 18.9 ± 10.9% to 13.4 ± 9.1% (p = 0.0386) of the LV endocardium. As seen in Fig. 4b, compared to AAV9-GFP-treated group, AAV9-cBIN1-treated group indicates positive remodeling of endocardial voltage by reducing the size of low voltage areas (p = 0.0095). This recovery of endocardial voltage occurred in the absence of revascularization, indicating that lessening hemodynamics stress by improving cardiomyocyte efficiency and overall cardiac function aids in the recovery of ischemic myocardium.

a Time of treatment (left panel) and post-therapy (right panel) voltage maps for the animal treated with AAV9-GFP (top panel) and with AAV9-cBIN1 (bottom panel). b Change in low voltage area from pre- to post-therapy for AAV9-GFP (n = 4) and AAV9-cBIN1 (n = 6). Mann–Whitney U test is used to compute statistical significance between two groups (p = 0.0095). ‘Δ’ refers to the absolute percentage change in the bipolar voltage area (<1.5 mV) from time of treatment to post-therapy. Data presented in (b) are shown as ‘mean’ and error bars are shown as ‘standard deviation’.
Discussion
Here, we have found that AAV9-cBIN1 gene therapy not only halts deterioration of DCM of ischemic origin but that it can improve failing myocardium after a one-time therapeutic intracardiac application. Figure 1a indicates that the LVEF of the AAV9-GFP treated group continues to decline following injection of AAV9-GFP. The trajectory of the AAV9-cBIN1 treated group declines at a similar rate to the AAV9-GFP group until injection and then LVEF increases within 1-2 weeks following injection. These data indicate that the injection of AAV9-GFP does not substantially alter the progression of ischemic HF. Additionally, data from Masson Trichrome and H&E-stained tissues (Supplementary Fig. 2), and TEM (Fig. 3) confirm there is no change in inflammation between AAV9-cBIN1 and the controls (untreated HF and AAV9-GFP), and there is a significant decrease in fibrosis in the AAV9-cBIN1-treated group compared to AAV9-GFP-treated group. Together, these data provide compelling evidence that neither the injection procedure nor the introduction of AAV9-GFP substantially change the progression of ischemic HF, but that AAV9-cBIN1 therapy benefits cardiomyocyte organization resulting in tissue level, organ level, and hemodynamic benefit in chronic ischemic HF. Overall, the improvement is evidenced by 1) improved functional LVEF and global longitudinal strain measures, 2) positive remodeling of the left ventricle, 3) improved biomarker measures of cardiac preload and CS score, 4) normalization of t-tubule morphology, 5) reduced low voltage area on endocardial electroanatomical mapping of the LV endocardium, and 6) animal survival. Together, these measures indicate that cBIN1 gene therapy has potential to address the underlying disease substrate of t-tubule remodeling in the myocardium of ischemic cardiomyopathy, improving outcomes in patients with chronic HF due to DCM of ischemic origin.
In recent years, several gene therapy solutions have been explored for failing myocardium and chronic HF4,27. In the CUPID trial, SERCA2a therapy28 reached advanced phase II clinical testing29,30, yet the results were not encouraging. There are multiple potential reasons for lack of efficacy by SERCA2a gene therapy, included among them is the possibility that, increasing by exogenous means, a single important calcium related protein may not be the most effective approach to recover failing myocardium. Subcellular architecture of cardiomyocytes should be considered when picking a target for gene therapy. Use of cBIN1 as a target represents a shift in the approach to target selection. cBIN1 is not a protein critical to beat-to-beat calcium regulation such as SERCA2a. Instead, cBIN1 is a membrane sculpting protein that organizes SERCA2a as well as other critical calcium proteins such as the L-type calcium current and ryanodine receptors11,12. For instance, cBIN1 gene therapy not only increases total myocardium content of SERCA2a12 but, perhaps more importantly, cBIN1 therapy organized SERCA2a and other calcium handling proteins to the T-tubule for increased excitation-contraction efficiency and diastolic calcium removal12,31,32. With cBIN1 treatment, pathologically enlarged t-tubule cross sectional area improves (Fig. 3), further indicating the normalization of the t-tubule membrane structures within cardiomyocytes10 allowing retention of the critical proteins involved in EC coupling. In a separate study of trafficking related proteins, improving trafficking and proper destination of membrane bound proteins such as gap junctions also increases the overall level of gap junction protein available16,22 suggesting that trafficking pathways may be an attractive target in general to improve both intracellular organization and target protein content in cardiomyocytes.
Downregulation of endogenous cardiomyocyte cBIN1 during ischemic stress31, which is also measurable at the plasma level13,14, correlates with decreased cardiac inotropy15, decreased lusitropy12, and increased arrhythmia10. Reduction of cBIN1 has been observed in multiple animals models and humans with failing myocardium9,10,11,12,15,31,33,34,35. Derived from the inverse of plasma cBIN1 level, CS rises with worsening HF13, providing a blood available quantification of myocyte health. In this study, the increase of CS with worsening HF reverses with recovery of myocardial function (Fig. 2), suggesting that venous plasma-based CS can be used to track progression of HF as well as assess for myocardial recovery.
Intramyocardial injection of cBIN1 gene therapy can halt and reverse expansion of peri-infarction low voltage areas in a preclinical model of chronic ischemic HF. In Fig. 4a, the red zone denotes the endocardial voltage ≤1.5 mV referring the area including damaged tissue and scar. With AAV9-GFP as a control therapy, as shown in Fig. 4a – top-right, there was an expansion of peri-infarction low voltage areas compared to pre-therapy low voltage area. The lower panel in Fig. 4a suggests that non-infarcted yet failing and border zone tissues benefit significantly from cBIN1 treatment. While it is not expected that cBIN1 therapy can recover permanent scar, cBIN1 gene therapy positively remodels the left ventricle, decreasing wall stress. By decreasing demand, the ischemic tissue is allowed to recover (Fig. 4) and overall fibrosis is reduced (Supplementary Fig. 2).
The low therapeutic dose of cBIN1 may offer a paradigm for treating common acquired disease. Most current gene therapy targets attempt to outcompete a mutant gene by exogenous introduction of a healthy gene. Therefore, a high dose is needed for successful competitive inhibition and dominance of the exogenous gene. Our target, cBIN1, organizes the proteins of the calcium handling apparatus and thus is upstream of a potent second messenger system (the cardiac calcium transient). Our approach restores levels of endogenous cBIN1 downregulated by disease rather than competing with an existing poorly functioning mutant protein. Potency of cBIN1 therapy is understandable by being upstream of calcium signaling and in the absence of a competing protein. At the organ level, cBIN1 therapy does not have to be expressed in every cardiomyocyte to be effective. Once overall hemodynamics start to improve, stress signaling is reduced, and endogenous cBIN1 increases. In terms of precedent, other therapies adopting the approach of bolstering downregulated genes are having success in the lower therapeutic range. Sardocor’s SERCA2a (clinical trial NCT06061549) and AskBio’s protein phosphatase 1 inhibitor (clinical trial NCT04179643) are examples of this approach. In our study, we utilized a transendocardial delivery technique for direct intramyocardial injection of AAV9-cBIN1. While other routes for gene therapy exist such as intracoronary delivery, intrapericardial delivery, and direct epicardial surface application36, our positive results (Figs. 1–4) suggest payload retention occurs with intramyocardial injection. The low dose of AAV9-cBIN1 therapy utilized (1.6 × 1011 vg/kg) is sufficient for cardiac rescue. This dose compares favorability with previously reported dose in clinical trials37, suggesting off-target effects of AAV9 can be minimized. However, our finding that AAV9-GFP administration does not alter the progression of dilated cardiomyopathy is consistent with a similar report of no difference between AAV9-GFP and PBS treated animals in a minipig model of tachycardia induced cardiomyopathy38. As discussed above, given that virus induced inflammation is a known potential complication of gene therapy, additional studies with untreated animals (i.e. without AAV9-GFP as control) could help establish the best negative control in this growing field.
Our post-cBIN1 treatment follow-up timeline to monitor reverse LV remodeling and associated clinical HF symptoms was limited to seven weeks. Longer follow-up, post injection, is needed to validate an exit from neurohormonal stresses and HF hemodynamics, confirming sustained efficacy of AAV9-mediated cBIN1 therapy. Additionally, further investigation of systemic and off-target effects of AAV9-cBIN1 is necessary. In addition, it is known that humans are commonly exposed to AAVs and may have neutralizing antibodies19. Our study did not address the impact of neutralizing antibodies upon AAV9-cBIN1 treatment, and further investigation is required to address this consideration.
Gene therapy-based interventions to restore functional membrane microdomains at cardiac t-tubules can benefit diseased myocardium of DCM hearts of ischemic origin, improving cardiac function and improving mortality. Targeting intracardiomyocyte trafficking molecules such as cBIN1 presents a low dose opportunity to utilize gene therapy to treat a major form of acquired chronic HF in humans.
References
Heidenreich, P. A. et al. Forecasting the impact of heart failure in the united states a policy statement from the american heart association. Circ. Heart Fail. 6, 606–619 (2013).
Virani, S. S. et al. Heart Disease and Stroke Statistics - 2021 Update: A Report From the American Heart Association. Circulation 143, e254–e743 (2021).
von Lueder, T. G., Kotecha, D., Atar, D. & Hopper, I. Neurohormonal Blockade in Heart Failure. Card. Fail Rev. 03, 19 (2017).
Kieserman, J. M., Myers, V. D., Dubey, P., Cheung, J. Y. & Feldman, A. M. Current Landscape of Heart Failure Gene Therapy. J. Am. Heart Assoc. 8, e012239 (2019).
Sahoo, S., Kariya, T. & Ishikawa, K. Targeted delivery of therapeutic agents to the heart. Nat. Rev. Cardiol. 18, 389–399 (2021).
Gómez, A. M., Guatimosim, S., Dilly, K. W., Vassort, G. & Lederer, W. j. Heart failure after myocardial infarction altered excitation-contraction coupling. Circulation 104, 688–693 (2001).
Litwin, S. E., Zhang, D. & Bridge, J. H. B. Dyssynchronous Ca2+ sparks in myocytes from infarcted hearts. Circ. Res. 87, 1040–1047 (2000).
Setterberg, I. E., Le, C., Frisk, M., Li, J. & Louch, W. E. The Physiology and Pathophysiology of T-Tubules in the Heart. Front. Physiol. 12, 718404 (2021).
Hong, T. T. et al. BIN1 localizes the L-type calcium channel to cardiac T-tubules. PLoS Biol. 8, e1000312 (2010).
Hong, T. et al. Cardiac BIN1 folds T-tubule membrane, controlling ion flux and limiting arrhythmia. Nat. Med. 20, 624–632 (2014).
Li, J., Agvanian, S., Zhou, K., Shaw, R. M. & Hong, T. T. Exogenous Cardiac Bridging Integrator 1 Benefits Mouse Hearts With Pre-existing Pressure Overload-Induced Heart Failure. Front. Physiol. 11, 708 (2020).
Liu, Y. et al. In Mice Subjected to Chronic Stress, Exogenous cBIN1 Preserves Calcium-Handling Machinery and Cardiac Function. JACC Basic Transl. Sci. 5, 561–578 (2020).
Shaw, R. M. et al. Association of a Novel Diagnostic Biomarker, the Plasma Cardiac Bridging Integrator 1 Score, with Heart Failure with Preserved Ejection Fraction and Cardiovascular Hospitalization. JAMA Cardiol. 3, 1206–1210 (2018).
Hitzeman, T. C. et al. cBIN1 Score (CS) Identifies Ambulatory HFrEF Patients and Predicts Cardiovascular Events. Front. Physiol. 11, 503 (2020).
Hong, T. T. et al. BIN1 is reduced and Cav1.2 trafficking is impaired in human failing cardiomyocytes. Heart Rhythm. 9, 812–820 (2012).
Li, J. et al. Cardiac gene therapy rescues diabetic cardiomyopathy and lowers blood glucose. JCI Insight 8, e166713 (2023).
Khan, M. S. et al. A canine model of chronic ischemic heart failure. Am. J. Physiol. Heart Circulatory Physiol. 324, H751–H761 (2023).
National Research Council. Guide for the care and use of laboratory animals - Committee for the Update of the Guide for the Care and Use of Laboratory Animals, Institute for Laboratory. Anim. Res. Guide Care Use Lab. Anim. 327, 220 (2011).
Burdett, T. & Nuseibeh, S. Changing trends in the development of AAV-based gene therapies: a meta-analysis of past and present therapies. Gene Ther. 30, 323–335 (2023).
Denegri, M. et al. Single delivery of an adeno-associated viral construct to transfer the CASQ2 gene to knock-in mice affected by catecholaminergic polymorphic ventricular tachycardia is able to cure the disease from birth to advanced age. Circulation 129, 2673–2681 (2014).
Siimes, S., Järveläinen, N., Korpela, H. & Ylä-Herttuala, S. Endocardial Gene Delivery Using NOGA Catheter System. Methods Mol. Biol. 2573, 179–187 (2022).
Xiao, S. et al. Auxiliary trafficking subunit GJA1-20k protects connexin-43 from degradation and limits ventricular arrhythmias. J. Clin. Investig. 130, 4858–4870 (2020).
Li, J., Richmond, B. & Hong, T. Cardiac t-tubule cbin1-microdomain, a diagnostic marker and therapeutic target of heart failure. Int. J. Mol. Sci. 22, 1–17 (2021).
Hussain, A. et al. Association of NT-ProBNP, Blood Pressure, and Cardiovascular Events: The ARIC Study. J. Am. Coll. Cardiol. 77, 559–571 (2021).
Borgarelli, M. et al. The predictive value of clinical, radiographic, echocardiographic variables and cardiac biomarkers for assessing risk of the onset of heart failure or cardiac death in dogs with preclinical myxomatous mitral valve disease enrolled in the DELAY study. J. Vet. Cardiol. 36, 77–88 (2021).
Gálvez-Santisteban, M. et al. Hemodynamic-mediated endocardial signaling controls in vivo myocardial reprogramming. Elife 8, e44816 (2019).
Gabisonia, K. & Recchia, F. A. Gene Therapy for Heart Failure: New Perspectives. Curr. Heart Fail Rep. 15, 340–349 (2018).
Kawase, Y. et al. Reversal of Cardiac Dysfunction After Long-Term Expression of SERCA2a by Gene Transfer in a Pre-Clinical Model of Heart Failure. J. Am. Coll. Cardiol. 51, 1112–1119 (2008).
Jessup, M. et al. Calcium upregulation by percutaneous administration of gene therapy in cardiac disease (CUPID): A phase 2 trial of intracoronary gene therapy of sarcoplasmic reticulum Ca2+-ATPase in patients with advanced heart failure. Circulation 124, 304–313 (2011).
Greenberg, B. et al. Design of a Phase 2b Trial of Intracoronary Administration of AAV1/SERCA2a in Patients with Advanced Heart Failure. The CUPID 2 Trial (Calcium Up-Regulation by Percutaneous Administration of Gene Therapy in Cardiac Disease Phase 2b). JACC Heart Fail. 2, 84–92 (2014).
Fu, Y. et al. Isoproterenol Promotes Rapid Ryanodine Receptor Movement to Bridging Integrator 1 (BIN1)-Organized Dyads. Circulation 133, 388–397 (2016).
Perdreau-Dahl, H. et al. BIN1, Myotubularin, and Dynamin-2 Coordinate T-Tubule Growth in Cardiomyocytes. Circ. Res. 132, e188–e205 (2023).
Hong, T. T. et al. Plasma BIN1 correlates with heart failure and predicts arrhythmia in patients with arrhythmogenic right ventricular cardiomyopathy. Heart Rhythm. 9, 961–967 (2012).
Xu, B. et al. The ESCRT-III pathway facilitates cardiomyocyte release of cBIN1-containing microparticles. PLoS Biol. 15, e2002354 (2017).
Caldwell, J. L. et al. Dependence of cardiac transverse tubules on the BAR domain protein amphiphysin II (BIN-1). Circ. Res. 115, 986–996 (2014).
Shazly, T., Smith, A., Uline, M. J. & Spinale, F. G. Therapeutic payload delivery to the myocardium: Evolving strategies and obstacles. JTCVS Open 10, 185–194 (2022).
Maurya, S., Sarangi, P. & Jayandharan, G. R. Safety of Adeno-associated virus-based vector-mediated gene therapy—impact of vector dose. Cancer Gene Ther. 29, 1305–1306 (2022).
Li, J. et al. Cardiac bridging integrator 1 gene therapy rescues chronic non-ischemic heart failure in minipigs. NPJ Regen. Med. 9, 36 (2024).
Acknowledgements
The research has been supported through the National Institutes of Health grants R01HL171686 (RMS/DJD), R01HL152691 (RMS), R01HL159983 (RMS/TH), R01HL170196 (RMS/TH), R01HL128752 (DJD), 23CDA1057448 (MSK), R21HL156039 (DJD) and through research grants provided by the Nora Eccles Treadwell Foundation. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health or the Nora Eccles Treadwell Foundation.
Author information
Authors and Affiliations
Contributions
M.S.K. – design of the work, data acquisition, analysis, interpretation of data, developed software for data analysis, writing and revision of the manuscript, D.S. –design of the work, data acquisition, J.L. – design of the work, data acquisition, Y.I. – design of the work, acquisition and interpretation of the data, E.O. – data acquisition, M.S.R.C. – data acquisition, A.M.H. – data acquisition, P.B. – data collection and interpretation, J.H. – data collection, A.A. – data analysis and interpretation, M.P.R. – data analysis and interpretation, J.A.P. – data analysis and interpretation, C.H.S. – design of the work, R.R. – design of the work and data interpretation, T.H. – design of study, interpretation of data, D.J.D. – conception and design of the work, acquisition, analysis and interpretation of data, drafting and revision of manuscript, R.M.S. – conception and design of the work, analysis and interpretation of data, drafted and revised the manuscript. All authors approve of the submitted manuscript and revisions and confirm the accuracy and integrity of the presented work.
Corresponding authors
Ethics declarations
Competing interests
R.M.S. and T.T.H. have founder’s equity in TikkunLev Therapeutics which is developing gene therapy solutions for non-ischemic cardiomyopathy. All other authors declare no competing interests.
Peer review
Peer review information
Communications Medicine thanks the anonymous reviewers for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Khan, M.S., Smego, D., Li, J. et al. AAV9-cBIN1 gene therapy rescues chronic heart failure due to ischemic cardiomyopathy in a canine model. Commun Med 5, 93 (2025). https://doi.org/10.1038/s43856-025-00787-w
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s43856-025-00787-w
