
Abstract
The accurate prediction of reaction performance based on empirical knowledge paves the way to efficient molecule design. Compared with the human-summarized reaction knowledge of a focal dataset, the machine-learned quantitative structure–performance relationship of larger-scale datasets is more effective at accessing the entire chemical space. Here we report a multitask learning workflow combined with a mechanism-informed graph neural network to predict site selectivity for ruthenium-catalysed C–H functionalization of arenes. The multitask architecture enables the acquisition of related knowledge from the simultaneous learning tasks. The embedded reaction graph bridges the gap between previous mechanistic studies and reaction representation. Along with this mechanistic embedding, the developed multitask model demonstrates excellent interpolative and extrapolative ability on the reported dataset composed of 256 reactions, achieving an average site-selectivity prediction accuracy of 0.934 with a standard deviation of 0.007. The prediction scope ranges from simple to fused arenes and was even extended to heterocyclic indole derivatives in the additional out of sample tests containing 14 unseen instances. Furthermore, interpretation of the model promotes the development of a para-selective mechanistic model verified by density functional theory calculations.
Similar content being viewed by others
Main
Precise chemo-, stereo- and position-selectivity control is a continuous pursuit throughout the era of modern organic synthesis1,2. A fully selective reaction typically derives from rational design based on the understanding of the underlying reaction mechanism. Even with tools of quantum mechanics computations3,4,5 or theoretical frameworks6,7,8, the traditional reaction design is still often restricted to a specific reactant subclass. The advent of machine learning (ML) algorithms offers exciting opportunities for selectivity control of the reaction outcome with their accurate predictive capabilities9,10. Recent progress has flourished in the prediction of molecular properties11,12,13,14,15, reaction mechanism16,17, yield18,19,20,21, chemoselectivity22,23,24, site selectivity25,26,27,28,29,30,31 and stereoselectivity32,33,34,35,36,37 with the aid of data-driven techniques. This effective strategy originates from the processing of multidimensional data and the establishment of the resulting structure–performance relationship, which sets the stage for a comprehensive understanding of the overall chemical space as compared with traditional methods operating in a substantially reduced chemical space (Fig. 1a).

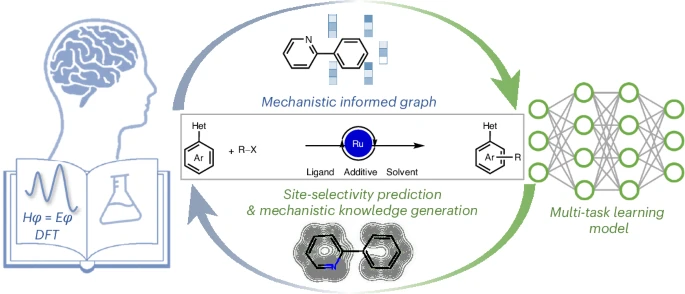
a, Two different paradigms for chemical space exploration. b, Ruthenium-catalysed C–H functionalization of arenes with synergistic effect of six reaction components on the site selectivity. c, This work: multitask learning with informed GNN for site-selectivity prediction and mechanistic insight acquisition. Het, heteroarenes.
Ruthenium-catalysed direct C–H activations have emerged as one of the most robust tools for the assembly of ortho-, meta- or para-substituted arenes38,39,40 with high position-selectivity control (Fig. 1b). The change of selectivity arises from the subtle changes in the substrates or reaction conditions, which, as yet, cannot be rationalized by a unified theoretical framework, contrasting with the available in-depth mechanistic studies, both experimentally and computationally41,42,43,44,45,46. This type of reaction features diverse data and complex structure–activity relationships and is highly suitable for using ML algorithms to access rational and predictive modelling47. Considering the synergistic effect of multiple reaction components on site selectivity, effective reaction representations and model architecture are indispensable to reach accurate predictions over the full domain of interest. The envisioned representation is expected to leverage previous mechanistic insights as an augmentation, as well as to provide a comprehensive description of the reaction entry including reaction conditions.
Among various machine-readable molecular representations, string (for example, Simplified Molecular Input Line Entry System (SMILES) and International Chemical Identifier), chemical table (MDL Molfile and so on) and topology-based representations (such as molecular fingerprints) encounter challenges in effectively encoding expert knowledge48,49. This impedes the capture of the structure–performance relationship and undermines the generalizability. A molecular graph, however, is an intuitive and concise way to represent molecules with featurized nodes (atoms) and edges (bonds)50. Harnessing graph representation, neural network algorithms have proven to be powerful in reaction performance prediction51. This automatic, learned molecular representation was further improved by the integration of general chemical knowledge and the use of a condensed graph52 and a three-dimensional (3D) graph53. The general chemical knowledge includes quantum mechanical descriptors and predicted physical property descriptors, with major contributions by Jensen28, Green28, Coley54,55, Luo56, Hong57 and Guan26, among others. Here, we surmised whether merging a GNN with mechanism-informed reaction graphs would enable a challenging site-selectivity prediction of ruthenium-catalysed C–H activations.
To this end, we used a multitask learning strategy combined with an informed GNN to predict the reaction site selectivity alongside two molecular property tasks simultaneously (Fig. 1c). The design of multitask architecture aims to benefit the site-selectivity classification with the aid of learning mechanism-related physical properties of the substrates. Likewise, the summarized information of previous mechanistic studies on ruthenium-catalysed C–H functionalization is embedded in the reaction graph, which enriches the reaction representation. The resulting model achieved excellent interpolative and extrapolative prediction accuracy even in additional experimental tests with unseen substrates. In addition to the outstanding prediction, further heuristic information extracted from the model, verified by density functional theory (DFT) calculations, reinforced our mechanistic understanding of the distinctive site selectivity in ruthenium-catalysed C–H functionalization. Our study highlights the feasibility and effectiveness of the multitask graph neural network (MT-GNN) algorithm, accelerating the design of molecular synthesis while unravelling the hidden origins of site selectivity.
Results
Building a comprehensive sample space is a key factor for a useful and generalized model47. To achieve the target site-selectivity prediction, we first manually collected the related reaction data (Fig. 2a). The ruthenium-catalysed C–H activation reaction data were collected from a wide range of publications, covering selectivity with different sites and chemical environments (Supplementary Table 1). Various arenes with distinct exogenous hetero-functionalities and scaffolds of electrophiles with a major distribution on halides were taken into consideration (Supplementary Figs. 1 and 2). The record of each transformation contains the details of arene, electrophile, catalyst, ligand, additive, solvent, reaction performance and source publication. In total, 256 individual reactions were included, whose distribution based on functionalization type is depicted in Fig. 2b along with the corresponding site selectivity. The collection of 95 arenes and 67 electrophiles, together with the reaction condition components, constitutes a vast chemical space for ruthenium-catalysed site-selective C–H functionalization. Further analyses of the database are provided in Supplementary Fig. 4.

a, Data collection for ruthenium-catalysed site-selective C–H functionalization. b, The distribution of functionalization types in the collected database. c, The workflow of the MT-GNN and the model architecture. Mes, 2,4,6‐trimethylphenyl.
Multitask learning aims to improve learning efficiency, predictive accuracy and robustness through the cooperation of related tasks58,59,60,61. We surmised that multitask architecture can benefit target site-selectivity predictions from the knowledge jointly learned in other tasks. In addition, we chose a two-dimensional (2D)—instead of 3D—GNN to avoid extra computational costs on the conformational searches for all reaction components. This option also considered that the ruthenium-catalysed arene functionalization basically occurred on a 2D planer with minor steric effect on the site selectivity. The operation of our designed MT-GNN begins with the construction of batches of graphs for arenes, electrophiles, catalyst, ligand, solvent and additive (Fig. 2c). The two substrate graphs whose node features contain prior mechanistic knowledge41 (condensed Fukui indices f0, f−, f+ and atomic charges Qc) are subgraphs of the original reaction graph. Through message passing, the six reaction component subgraphs are condensed into six virtual nodes, constituting a complete reaction graph with six nodes and 30 edge vectors. Although the model has already been informed with atom-level mechanistic information, the understanding for the key reaction components at the molecular level is still missing. As the subgraphs of reaction components are available, the independent learning of molecular properties by the model itself can complement the shortness of global perception. Therefore, we designed the multitask learning architecture, in which site-selectivity classification task and molecular properties regression tasks for arenes and electrophiles were processed in parallel. Considering the radical nature of ruthenium-catalysed C–H functionalization, and other electronic as well as steric effects presented in this reaction38,41, the molecular property prediction targets included electron affinity, lowest unoccupied molecular orbital energy, singly occupied molecular orbital energy, spin density and buried volumes62. As a result, three clusters consisting of the reaction graph, arene subgraph and electrophile subgraph are set as input, subsequently passing through the attention layer, max pooling, convolutional layer and linear layer. The separated loss functions of the three tasks are finally weighted and summed up to a shared loss function, which then back propagates and continues optimizing by minimizing the shared loss function. By leveraging multitask learning as well as embedded graphs, the model is expected to acquire molecular- and atomic-level understanding of the reaction, ultimately providing satisfying predictions.
The predictive ability of the MT-GNN model for site-selectivity tasks as well as molecular property tasks was evaluated and compared using the dataset of 256 reactions (Fig. 3a). MT-GNN with a mechanistic–embedded reaction graph achieved impressive results across all three tasks through tenfold cross-validation based on random splitting, with an average accuracy of 0.934 for site-selectivity classification after three repetitions and a s.d. of 0.007. The single-task GNN without a mechanistic–embedded reaction graph underperformed compared with our designed model for all three tasks. Likewise, another neural network model applying multitask architecture with molecular descriptors in RDKit63 and the same mechanistic features in MT-GNN showed limited performance, emphasizing the necessity of graph-based representation. Other tested models comprised random forest (RF), k-nearest neighbours (KNN) and support vector machine (SVM), which performed less effectively in classification task with RDKit descriptors and mechanistic features. For molecular property regression tasks of arenes and electrophiles, the MT-GNN presented Pearson correlation coefficients (R) of 0.864 and 0.830, respectively. By contrast, other tested models failed to surpass MT-GNN on the same two regression tasks simultaneously.

a, Prediction comparison of site-selectivity classification (accuracy) and molecular property regression (Pearson correlation coefficient) among various models (MT-GNN, single-task GNN, multitask neural network (NN), RF, KNN and SVM). b, Prediction comparison of site-selectivity accuracy under different data-splitting methods among various models (MT-GNN, single-task GNN, RF, on-the-fly QM-GNN and aGNN3D). The results are the average accuracy, with s.d., of three repetitions. The numbers in parentheses represent the proportion of each specific functionalization type in the collected database.
Focusing on the site-selectivity classification, two state of the art (SOTA) models for site-selectivity prediction, on-the-fly quantum mechanics-graph neural network (QM-GNN)28 and 3D atomistic graph neural network (aGNN3D)53, were selected to investigate the effectiveness of the MT-GNN (Fig. 3b). In the interpolation test of tenfold cross-validation (random 90/10), our model exhibited marginal improvement over the SOTA model on-the-fly QM-GNN (0.918 ± 0.008). Expanding the validation set with twofold cross-validation (random 50/50) resulted in a slight drop in performance for both MT-GNN (0.905 ± 0.005) and on-the-fly QM-GNN (0.878 ± 0.005). Nonetheless, through extrapolation tests, the model performances were differentiated. When specific functionalization types, such as alkylation and benzylation, were treated as extra tests, the MT-GNN model outperformed all other single-task models in most cases. In two reaction condition optimization tests (Supplementary Schemes 4 and 6), the MT-GNN (0.917 ± 0.042 and 0.733 ± 0.067) exhibited a clear advantage over other tested models, especially on-the-fly QM-GNN (0.127 ± 0.003 and 0.362 ± 0.002). This benefit results from the thorough reaction representation, modelling every reaction component as a graph, thus enhancing its sensitivity against variations in reaction conditions. Under various comparisons, the reliability and robustness of the MT-GNN model were confirmed, validating the design of multitask architecture in facilitating mutual assistance among different tasks and the incorporation of mechanistic information for ruthenium-catalysed C‒H functionalization.
To further challenge the extrapolative ability of the MT-GNN model, the boundary of the test was extended to a scenario of substrate scope exploration with an experimental test set. Fused aromatic rings are a group of compounds that exhibit different global as well as site-specific properties compared with mono-aromatic rings, but were thus far underrepresented in ruthenium-catalysed C–H activations. We performed a series of experiments using previously unseen substrates composed of fused aromatic rings (Fig. 4a). A wide variety of bromides were selected as electrophiles including primary, secondary and tertiary alkyl bromides, as well as aryl electrophiles. These alkyl and aryl bromides (2a–e) reacted with benzoquinoline 1a and naphthalene derivatives 1b–c, demonstrating a constant change in the site selectivity. The MT-GNN model achieved excellent predictions for all the experimental cases even those with noticeable changes in arenes. A key advantage of the MT-GNN model potentially lies in the automatically learned reaction representation, which compiles the knowledge from the molecular properties of two substrates as well as site-specific mechanistic embedding. To visualize the learned representation by the MT-GNN, the encoding of the training data and experimental data was extracted from the convolutional layer and reduced to a single dimension through principal component analysis (PCA). Features for training the RF model were directly used for its PCA visualization. The encoding space of the MT-GNN, RF and single-task GNN model along the single principal component is visualized in Fig. 4b, with the corresponding quantitative distance between data points in Fig. 4c. The MT-GNN model had the shortest average distance from the nearest training data point to each experimental data point compared with RF and single-task GNN. All the learned representations of experimental data were located within the distribution of training data, contributing to the excellent prediction accuracy of MT-GNN in this extrapolative task. In addition, the average distance between experimental data points in the MT-GNN model was relatively small, probably owing to the similarity of the experimental data involving fused ring substrates.

a, Experimental test data and prediction accuracy by MT-GNN, RF and single-task GNN. b, Visualization of the encoding space of MT-GNN, RF and single-task GNN using PCA. c, Quantitative distance between data points along the principal component. d, An additional extrapolative test containing N-substituted indoles as substrates by the MT-GNN model.
Inspired by the experimental test, the indole scaffold, which is also a type of fused aromatic ring, could potentially fall within the prediction limits. Therefore, an out-of-sample test was collected from the literature for ruthenium-catalysed C–H functionalization of N-substituted indoles64,65 (Fig. 4d). To our delight, indole derivatives proved to be another successful extrapolation of substrates by the MT-GNN model. When the versatility of reaction components was expanded in the aforementioned extrapolation tests, our model consistently delivered accurate prediction by leveraging the distilled structure–performance relationship from the original dataset, avoiding the necessity for building a new mechanistic framework.
The black-box nature of the GNN means it is not readily interpretable owing to the high degree of complexity55,66. Meanwhile, the internal logic of the black box model is a reservoir of meaningful physical insights to be excavated. In addition to the predictive ability of the MT-GNN model, translating the hidden patterns identified by the model into interpretable information also deserves to be explored as they unveil opportunities for advancing chemical understanding. To this end, node attention values were extracted from the attention layer of the MT-GNN architecture. When tuning the site selectivity, different attention value patterns were observed and the visualization of node attention values of arenes are shown in Fig. 5a. For the meta-selective reactions using activated primary alkyl bromide, the node attention of substrates with a biaryl skeleton emphasized the imine nitrogen, which might correspond to the crucial role of directing groups in C–H activation, forming a reactive six-membered ruthenacycle. By contrast, the node attention shifted to amine nitrogen for aniline derivatives when the siteselectivity is para. This changing of highlighted atoms probably suggests a new underlying para-selective mechanistic model with direct participation of the amine nitrogen, generating a four-membered ruthenacycle with N–H bond cleavage. To validate this hypothesis guided by the exploration of the attention layer, we carried out DFT studies.

a, Node attention values of arenes and different mechanistic models. b, DFT calculations on the competing radical attack pathways of ruthenacycles int2 and int3. c, Node attention values of phenol derivative and DFT calculations on the competing radical attack pathways (computational methods: MN15-D3(BJ)/6-311+G(d,p)-SDD-SMD(m-xylene)//B3LYP-D3(BJ)/6-31G(d)-LANL2DZ, thermal correction at 120 °C. The energies are reported in the unit of kcal mol−1.) Ad, adamantyl; DG, directing group.
Detailed DFT computations were performed to investigate the origins of the para-selectivity using an aniline derivative (Fig. 5b). The formation pathways of four-membered ruthenacycle int2 and six-membered ruthenacycle int3 from the substrate-coordinated intermediate int1 are shown in Supplementary Figs. 15 and 17. For these two plausible pathways, the rate-determining step is the radical attack, which determines the site selectivity. When the radical attack occurs at the para-position of the phenyl ring in int2 via the open-shell singlet transition state TS4, the resulting closed-shell intermediate int5 is a relatively stable imine species (Fig. 5, labelled in blue). Alternatively, the attack at the meta- or ortho-position forms highly unstable diradical intermediates int6 and int7, given the absence of stabilization by the nitrogen. On the other hand, radical attack at the para-position of the Ru–C bond in int3 through the open-shell singlet transition state TS8 is more favourable than other positions. This is attributed to the formation of a singlet ruthenium carbene intermediate int9, which results in the meta-selectivity (Fig. 5, labelled in red). However, the stabilization of radical by the amine nitrogen in the four-membered ruthenacycle pathway is stronger than that by the metal atom in the six-membered ruthenacycle pathway, which lowers the para-selective radical attack barrier. Thus, the effective radical stabilization of the nitrogen atom in the aniline skeleton, which is highlighted by the attention layer, results in para-selectivity.
The interpretation of the MT-GNN model inspired us to develop a new mechanistic model for the origins of para-selectivity with aniline derivatives, which was previously unclear. Replacing the nitrogen of aniline with oxygen gives rise to a phenol derivative, which is also encompassed within our database. Here, the top one node attention of 2-phenoxypyridine is located in the nitrogen of the pyridyl group instead of oxygen (Fig. 5c). Inferred from the aforementioned conclusion, the oxygen is unable to stabilize the para-selective radical pathway, therefore the active species is six-membered ruthenacycle, which undergoes the meta-selective pathway. Further DFT studies for 2-phenoxypyridine again validated the implication by the node attention. Radical attack at the para-position to the oxygen forms the unstable diradical intermediate int13, while attack at the para-position to the Ru–C bond is more favourable.
Discussion
Improving the efficiency and selectivity of chemical space exploration for molecular synthesis design is challenging, yet of prime importance to various applied areas such as crop protection and drug development. Rather than being solely promoted by chemical intuition or a specific theoretical framework, the introduction of machine learning allows access to thus far uncharted territory. Our MT-GNN model achieved effective synchronous learning at high accuracies over three related tasks derived from the ruthenium-catalysed site-selective C–H functionalization database through cross-validation. With a combination of mechanistic-informed reaction representation and multitask architecture, the MT-GNN model outperformed SOTA models under most of the data-splitting methods based on functionalization types. The extrapolative prediction of MT-GNN in the experimental test and indole derivative test further demonstrated the robustness of our strategy.
The interpretability of the MT-GNN model enabled by the attention layer facilitated the extraction of hidden information at the level of individual atoms. Although the node attention values are unable to directly predict the changes in selectivity or provide definitive mechanistic insights, they may offer preliminary indications that inspire the formulation of testable hypotheses regarding the origins of the distinctive siteselectivity. Alongside computational studies, a mechanistic rationale was proposed for the ruthenium-catalysed para-selective C–H functionalization. A four-membered ruthenacycle was identified as the reactive species in the para-selective pathway rather than six-membered ruthenacycle. The directing role and extra-radical stabilization provided by the amine nitrogen atom in the aniline skeleton was clarified, which lowered the open-shell para-selective radical attack process.
Our findings mirror the rationality of the MT-GNN architecture and mechanistic-informed reaction representation, providing a uniquely useful tool for the assembly-line synthesis of multisubstituted arenes. At the same time, the interpretability of the MT-GNN model notably accelerates the knowledge-generation process, thus completing the mechanistic feedback loop between human chemists and artificial intelligence.
Methods
Data collection
The ruthenium-catalysed C‒H functionalization database comprises 256 reactions from 19 representative publications. Each entry includes detailed information on the arene, electrophile, catalyst, ligand, additive, solvent, yield, site selectivity and source publication. Since potassium carbonate is added to every reaction, it is excluded from the database. Site selectivity for each reaction is determined by the major product. All six reaction components are recorded in SMILES format and stored in a CSV file in the GitHub repository67. To embed mechanistic information at the possible reactive sites of arenes and electrophiles, we also provided MDL Mol files for 95 arenes and 67 electrophiles.
Generation of reaction graphs
One complete reaction graph consists of six nodes and 30 edge vectors. The six nodes are the virtual nodes aggregated from the corresponding reaction component graphs using the built-in mean function in the Deep Graph Library (DGL) package, which averages the features of neighbouring nodes during message passing. The original graphs of reaction condition components, including catalyst, ligand, additive and solvent, are generated from the SMILES strings using DGL with default features of atoms and bonds. These default features for atoms encompass one-hot encoding of atom type, atom degree, number of implicit hydrogens, formal charge, number of radical electrons, hybridization, aromaticity and total hydrogens. The original graphs for arenes and electrophiles are generated from the Mol files retaining the information of atom number to facilitate the embedding of mechanistic features (Fukui indices f0, f−, f+ and atomic charges Qc) to the reactive sites. Besides the mechanistic features, the default DGL atom features are also used for the atoms in the original arene graphs and electrophiles graphs before message passing.
MT-GNN architecture
The MT-GNN was developed to handle three tasks in parallel, including two regression tasks and one classification task. The model architecture consisted of multiple layers starting from graph attention layers that utilize three attention heads. Following the attention layers, a max pooling operation aggregated the node features, which were then passed through two consecutive graph convolutional layers. The outputs of the max pool layer were collected to provide insights for the input dataset. A rectified linear unit activation function was used after each layer to introduce nonlinearity into the model. The model produced three outputs for each regression task and logits for the classification task through the final linear layers. The model utilized 250 hidden dimensions for both regression and classification tasks.
Training details
The model is trained using a mini-batch gradient descent approach with the Adam optimizer, initialized with a learning rate of 10−3. During each training epoch, the losses for the three tasks are computed, where mean squared error is used for the regression tasks, and cross-entropy loss is applied to the classification task. A total loss Ltotal is computed as a weighted sum of these individual losses, where the regression tasks contribute 40% each (L1 and L2), and the classification task contributes 20% (L3).
The model is evaluated on both training and validation datasets, with merits of accuracy for site-selectivity prediction and mean absolute error for molecular property regression to monitor the performance throughout the training process. The model is trained for a maximum of 200 epochs with a batch size of 30. When the model is trained through tenfold cross-validation, average values of the top one prediction accuracy for classification task of each fold is finally recorded.
General procedure for ruthenium-catalysed C‒H functionalization
A flame-dried Schlenk tube was added with substrate 1 (0.2 mmol), [RuCl2(p-cymene)]2 (5 mol%, 0.01 mmol), PPh3 (20 mol%, 0.04 mmol), MesCOOH (30 mol%, 0.06 mmol) and K2CO3 (0.4 mmol). The Schlenk tube was then sealed, purged and backfilled with N2 three times. Then, 1,4-dioxane (1.0 ml) and bromide 2 (0.6 mmol) were added via syringe, and the resulting mixture was stirred at 120 °C for 24 h. After cooling to ambient temperature, the mixture was diluted with ethyl acetate and filtered through a pad of Celite. Then, the solvent was removed in vacuo. The residue was purified by column chromatography on silica gel to afford the desired product 3.
Data availability
The collected ruthenium-catalysed C–H functionalization database and extrapolative test data are available via GitHub at https://github.com/xinranchen95/MT-GNN (ref. 67). ML details, experimental procedures, NMR spectra, DFT details, DFT-optimized structures and the ruthenium-catalysed C–H functionalization dataset are available in the Supplementary Information.
Code availability
Codes for mechanistic-informed reaction graph generation, MT-GNN, model training and extrapolative prediction are freely available via GitHub at https://github.com/xinranchen95/MT-GNN (ref. 67).
References
Trost, B. M. & Fleming, I. Comprehensive Organic Synthesis: Selectivity, Strategy, and Efficiency in Modern Organic Chemistry (Pergamon, 1991).
Gaich, T. & Winterfeldt, E. Directed Selectivity in Organic Synthesis: A Practical Guide (Wiley, 2014).
Ahn, S., Hong, M., Sundararajan, M., Ess, D. H. & Baik, M. H. Design and optimization of catalysts based on mechanistic insights derived from quantum chemical reaction modeling. Chem. Rev. 119, 6509–6560 (2019).
Poree, C. & Schoenebeck, F. A holy grail in chemistry: computational catalyst design: feasible or fiction? Acc. Chem. Res. 50, 605–608 (2017).
Houk, K. N. & Cheong, P. H. Computational prediction of small-molecule catalysts. Nature 455, 309–313 (2008).
Bickelhaupt, F. M. & Houk, K. N. Analyzing reaction rates with the distortion/interaction–activation strain model. Angew. Chem. Int. Ed. 56, 10070–10086 (2017).
Fernandez, I. & Bickelhaupt, F. M. The activation strain model and molecular orbital theory: understanding and designing chemical reactions. Chem. Soc. Rev. 43, 4953–4967 (2014).
Geerlings, P., De Proft, F. & Langenaeker, W. Conceptual density functional theory. Chem. Rev. 103, 1793–1873 (2003).
Oliveira, J. C. A. et al. When machine learning meets molecular synthesis. Trends Chem 4, 863–885 (2020).
Yang, L.-C., Zhu, L.-J., Zhang, S.-Q. & Hong, X. Machine learning prediction of structure–performance relationship in organic synthesis. Chin. J. Chem. 40, 2106–2117 (2020).
Nie, W., Liu, D., Li, S., Yu, H. & Fu, Y. Nucleophilicity prediction using graph neural networks. J. Chem. Inf. Model. 62, 4319–4328 (2022).
Fang, X. et al. Geometry-enhanced molecular representation learning for property prediction. Nat. Mach. Intell. 4, 127–134 (2022).
Wen, M., Blau, S. M., Spotte-Smith, E. W. C., Dwaraknath, S. & Persson, K. A. Bondnet: a graph neural network for the prediction of bond dissociation energies for charged molecules. Chem. Sci. 12, 1858–1868 (2020).
St. John, P. C., Guan, Y., Kim, Y., Kim, S. & Paton, R. S. Prediction of organic homolytic bond dissociation enthalpies at near chemical accuracy with sub-second computational cost. Nat. Commun. 11, 2328 (2020).
Roszak, R., Beker, W., Molga, K. & Grzybowski, B. A. Rapid and accurate prediction of pKa values of C–H acids using graph convolutional neural networks.J. Am. Chem. Soc. 141, 17142–17149 (2019).
Bures, J. & Larrosa, I. Organi creaction mechanism classification using machine learning. Nature 613, 689–695 (2023).
Jorner, K., Brinck, T., Norrby, P. O. & Buttar, D. Machine learning meets mechanistic modelling for accurate prediction of experimental activation energies. Chem. Sci. 12, 1163–1175 (2021).
Zuranski, A. M., Martinez Alvarado, J. I., Shields, B. J. & Doyle, A. G. Predicting reaction yields via supervised learning. Acc. Chem. Res. 54, 1856–1865 (2021).
Chen, Y. et al. Electro-descriptors for the performance prediction of electro-organic synthesis. Angew. Chem. Int. Ed. 60, 4199–4207 (2021).
Sandfort, F., Strieth-Kalthoff, F., Kühnemund, M., Beecks, C. & Glorius, F. A structure-based platform for predicting chemical reactivity. Chem 6, 1379–1390 (2020).
Ahneman, D. T., Estrada, J. G., Lin, S., Dreher, S. D. & Doyle, A. G. Predicting reaction performance in C–N cross-coupling using machine learning. Science 360, 186–190 (2018).
Maley, S. M. et al. Quantum-mechanical transition-state model combined with machine learning provides catalyst design features for selective Cr olefin oligomerization. Chem. Sci. 11, 9665–9674 (2020).
Coley, C. W., Barzilay, R., Jaakkola, T. S., Green, W. H. & Jensen, K. F. Prediction of organic reaction outcomes using machine learning. ACS Cent. Sci. 3, 434–443 (2017).
Wei, J. N., Duvenaud, D. & Aspuru-Guzik, A. Neural networks for the prediction of organic chemistry reactions. ACS Cent. Sci. 2, 725–732 (2016).
King-Smith, E. et al. Predictive minisci late stage functionalization with transfer learning. Nat. Commun. 15, 426 (2024).
Guan, Y., Lee, T., Wang, K., Yu, S. & McWilliams, J. C. SNAr regioselectivity predictions: machine learning triggering DFT reaction modeling through statistical threshold. J. Chem. Inf. Model. 63, 3751–3760 (2023).
Caldeweyher, E. et al. Hybrid machine learning approach to predict the site selectivity of iridium-catalyzed arene borylation. J. Am. Chem. Soc. 145, 17367–17376 (2023).
Guan, Y. et al. Regio-selectivity prediction with a machine-learned reaction representation and on-the-fly quantum mechanical descriptors. Chem. Sci. 12, 2198–2208 (2020).
Li, X., Zhang, S. Q., Xu, L. C. & Hong, X. Predicting regioselectivity in radical C–H functionalization of heterocycles through machine learning. Angew. Chem. Int. Ed. 59, 13253–13259 (2020).
Beker, W., Gajewska, E. P., Badowski, T. & Grzybowski, B. A. Prediction of major regio-, site-, and diastereoisomers in diels-alder reactions by using machine-learning: the importance of physically meaningful descriptors. Angew. Chem. Int. Ed. 58, 4515–4519 (2019).
Tomberg, A., Johansson, M. J. & Norrby, P. O. A predictive tool for electrophilic aromatic substitutions using machine learning. J. Org. Chem. 84, 4695–4703 (2019).
Zhang, Z. J. et al. Data-driven design of new chiral carboxylic acid for construction of indoles with C-central and C–N axial chirality via cobalt catalysis. Nat. Commun. 14, 3149 (2023).
Xu, L.-C. et al. Enantioselectivity prediction of pallada-electrocatalysed C–H activation using transition state knowledge in machine learning. Nat. Synth. 2, 321–330 (2023).
Gallarati, S. et al. Reaction- based machine learning representations for predicting the enantioselectivity of organocatalysts. Chem. Sci. 12, 6879–6889 (2021).
Singh, S. et al. A unified machine-learning protocol for asymmetric catalysis as a proof of concept demonstration using asymmetric hydrogenation. Proc. Natl Acad. Sci. USA 117, 1339–1345 (2020).
Zahrt, A. F. et al. Prediction of higher-selectivity catalysts by computer-driven workflow and machine learning. Science 363, eaau5631 (2019).
Reid, J. P. & Sigman, M. S. Holistic prediction of enantioselectivity in asymmetric catalysis. Nature 571, 343–348 (2019).
Dutta, U., Maiti, S., Bhattacharya, T. & Maiti, D. Arene diversification through distal C(sp2)–H functionalization. Science 372, eabd5992 (2021).
Korvorapun, K., Samanta, R. C., Rogge, T. & Ackermann, L. Remote C–H functionalizations by ruthenium catalysis. Synthesis 53, 2911–2946 (2021).
Leitch, J. A. & Frost, C. G. Ruthenium-catalysed sigma-activation for remote meta-selective C–H functionalisation. Chem. Soc. Rev. 46, 7145–7153 (2017).
Chen, X. et al. Close-shell reductive elimination versus open-shell radical coupling for site-selective ruthenium-catalyzed C–H activations by computation and experiments. Angew. Chem. Int. Ed. 62, e202302021 (2023).
Wang, X. G. et al. Three-component ruthenium-catalyzed direct meta-selective C–H activation of arenes: a new approach to the alkylarylation of alkenes. J. Am. Chem. Soc. 141, 13914–13922 (2019).
Korvorapun, K., Kuniyil, R. & Ackermann, L. Late-stage diversification by selectivity switch in meta-C–H activation: evidence for singlet stabilization. ACS Catal. 10, 435–440 (2019).
Simonetti, M., Cannas, D. M., Just-Baringo, X., Vitorica-Yrezabal, I. J. & Larrosa, I. Cyclometallated ruthenium catalyst enables late-stage directed arylation of pharmaceuticals. Nat. Chem. 10, 724–731 (2018).
Korvorapun, K. et al. Sequential meta-/ortho-C–H functionalizations by one-pot ruthenium(II/III) catalysis. ACS Catal. 8, 886–892 (2018).
Paterson, A. J. et al. Alpha-halo carbonyls enable meta selective primary, secondary and tertiary C–H alkylations by ruthenium catalysis. Org. Biomol. Chem. 15, 5993–6000 (2017).
Raghavan, P. et al. Dataset design for building models of chemical reactivity. ACS Cent. Sci. 9, 2196–2204 (2023).
Gallegos, L. C., Luchini, G., St. John, P. C., Kim, S. & Paton, R. S. Importance of engineered and learned molecular representations in predicting organic reactivity, selectivity, and chemical properties. Acc. Chem. Res. 54, 827–836 (2021).
David, L., Thakkar, A., Mercado, R. & Engkvist, O. Molecular representations in AI-driven drug discovery: a review and practical guide. J. Cheminform. 12, 56 (2020).
Wigh, D. S., Goodman, J. M. & Lapkin, A. A. A review of molecular representation in the age of machine learning. Wiley Interdiscip. Rev.: Comput. Mol. Sci. 12, e1603 (2022).
Ding, Y. et al. Exploring chemical reaction space with machine learning models: representation and feature perspective. J. Chem. Inf. Model. 64, 2955–2970 (2024).
Heid, E. & Green, W. H. Machine learning of reaction properties via learned representations of the condensed graph of reaction. J. Chem. Inf. Model. 62, 2101–2110 (2022).
Nippa, D. F. et al. Enabling late-stage drug diversification by high-throughput experimentation with geometric deep learning. Nat. Chem. 16, 239–248 (2024).
Goldman, S., Li, J. & Coley, C. W. Generating molecular fragmentation graphs with autoregressive neural networks. Anal. Chem. 96, 3419–3428 (2024).
Stuyver, T. & Coley, C. W. Quantum chemistry-augmented neural networks for reactivity prediction: performance, generalizability, and explainability. J. Chem. Phys. 156, 084104 (2022).
Zhang, B. et al. Chemistry-informed molecular graph as reaction descriptor for machine-learned retrosynthesis planning. Proc. Natl Acad. Sci. USA 119, e2212711119 (2022).
Li, S.-W., Xu, L.-C., Zhang, C., Zhang, S.-Q. & Hong, X. Reaction performance prediction with an extrapolative and interpretable graph model based on chemical knowledge. Nat. Commun. 14, 3569 (2023).
Taylor, C. J. et al. Accelerated chemical reaction optimization using multi-task learning. ACS Cent. Sci. 9, 957–968 (2023).
Lu, J. & Zhang, Y. Unified deep learning model for multitask reaction predictions with explanation. J. Chem. Inf. Model. 62, 1376–1387 (2022).
Biswas, S., Chung, Y., Ramirez, J., Wu, H. & Green, W. H. Predicting critical properties and acentric factors of fluids using multitask machine learning. J. Chem. Inf. Model. 63, 4574–4588 (2023).
Struble, T. J., Coley, C. W. & Jensen, K. F. Multitask prediction of site selectivity in aromatic C–H functionalization reactions. React. Chem. Eng. 5, 896–902 (2020).
Poater, A. et al. Thermodynamics of N-heterocyclic carbene dimerization: the balance of sterics and electronics. Organometallics 27, 2679–2681 (2008).
RDKit: open-source chemoinformatics and machine learning. Release_2022.09.4 (RDKit, 2022); http://www.rdkit.org
Leitch, J. A., McMullin, C. L., Mahon, M. F., Bhonoah, Y. & Frost, C. G. Remote C6-selective ruthenium-catalyzed C–Halkylation of indole derivatives via σ-activation. ACS Catal. 7, 2616–2623 (2017).
Simonetti, M. et al. Ruthenium-catalyzed C–Harylation of benzoic acids and indole carboxylic acids with aryl halides. Chem. Eur. J. 23, 549–553 (2017).
Esterhuizen, J. A., Goldsmith, B. R. & Linic, S. Interpretable machine learning for knowledge generation in heterogeneous catalysis. Nat. Catal. 5, 175–184 (2022).
Chen, X. et al. MT-GNN. GitHub https://github.com/xinranchen95/MT-GNN (2024).
Acknowledgements
We gratefully acknowledge the support from the ERC Advanced Grant (no. 101021358) and the DFG (Gottfried-Wilhelm-Leibniz-Preis and SPP2363) to L.A., National Natural Science Foundation of China (nos. 22122109 and 22271253), National Key R&D Program of China (no. 2022YFA1504301), Zhejiang Provincial Natural Science Foundation of China (no. LDQ23B020002), the Starry Night Science Fund of Zhejiang University Shanghai Institute for Advanced Study (no. SN-ZJU-SIAS-006), Beijing National Laboratory for Molecular Sciences (no. BNLMS202102), CAS Youth Interdisciplinary Team (no. JCTD-2021-11), Fundamental Research Funds for the Central Universities (nos. 226-2022-00140, 226-2022-00224, 226-2023-00115 and 226-2024-00003), the State Key Laboratory of Physical Chemistry of Solid Surfaces (no. 202210), the Leading Innovation Team grant from Department of Science and Technology of Zhejiang Province (no. 2022R01005), Open Research Fund of School of Chemistry and Chemical Engineering of Henan Normal University (no. 2024Z01 to X.H.) and the Alexander von Humboldt Foundation (fellowship to Z.-J.Z.).
Funding
Open access funding provided by Georg-August-Universität Göttingen.
Author information
Authors and Affiliations
Contributions
L.A., X.H. and X.C. designed the overall project. X.C. designed and implemented the MT-GNN model and algorithm. X.C. and Z.-J.Z. designed the experimental test. Z.-J.Z. performed experiments and analysed the data. All the authors participated in the discussion and preparation of the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Synthesis thanks Kenneth Atz, Robert Pollice and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. Primary Handling Editor: Peter Seavill, in collaboration with the Nature Synthesis team.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Supplementary Information
ML and experimental details, Supplementary Figs. 1–20, Supplementary Tables 1–9 and Supplementary Schemes 1–9.
Supplementary Data 1
Dataset of ruthenium-catalysed C‒H functionalization.
Supplementary Data 2
DFT-optimized structures.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Chen, X., Zhang, ZJ., Hong, X. et al. Integrating a multitask graph neural network with DFT calculations for site-selectivity prediction of arenes and mechanistic knowledge generation. Nat. Synth 4, 877–887 (2025). https://doi.org/10.1038/s44160-025-00770-2
Received:
Accepted:
Published:
Version of record:
Issue date:
DOI: https://doi.org/10.1038/s44160-025-00770-2
