
Abstract
Although genetic variant effects often interact nonadditively, strategies to uncover epistasis remain in their infancy. Here we develop low-signal signed iterative random forests to elucidate the complex genetic architecture of cardiac hypertrophy, using deep learning-derived left ventricular mass estimates from 29,661 UK Biobank cardiac magnetic resonance images. We report epistatic variants near CCDC141, IGF1R, TTN and TNKS, identifying loci deemed insignificant in genome-wide association studies. Functional genomic and integrative enrichment analyses reveal that genes mapped from these loci share biological process gene ontologies and myogenic regulatory factors. Transcriptomic network analyses using 313 human hearts demonstrate strong co-expression correlations among these genes in healthy hearts, with significantly reduced connectivity in failing hearts. To assess causality, RNA silencing in human induced pluripotent stem cell-derived cardiomyocytes, combined with novel microfluidic single-cell morphology analysis, confirms that cardiomyocyte hypertrophy is nonadditively modifiable by interactions between CCDC141, TTN and IGF1R. Our results expand the scope of cardiac genetic regulation to epistasis.
Similar content being viewed by others


Main
Heart disease is closely tied to the structure of the heart1. Heart failure, a syndrome characterized by increased pressure within, or decreased output from, the heart is influenced by structural features including atrial and ventricular chamber size and wall thickness2,3. Left ventricular hypertrophy—increased thickness of the left ventricle (LV)—can be the result of Mendelian genetic diseases such as hypertrophic cardiomyopathy (HCM)4 but is also a complex phenotypic trait influenced by multiple factors, genetic and environmental. Progressive LV hypertrophy carries substantial independent risk for incident heart failure, atrial arrhythmia and sudden death5,6, highlighting the need to understand genetic determinants of cardiac phenotype.
Recent discoveries leveraging cardiac magnetic resonance imaging (MRI) in the UK Biobank (UKBB) have revealed that cardiac structure is in part determined by complex genetics7,8,9. Common genetic variants, many located near genetic loci associated with dilated cardiomyopathy and heart failure, have been found to influence LV size and systolic function7. Furthermore, specific genetic variants that influence LV trabeculation have been shown to impact systolic function and overall risk of cardiomyopathy8. However, these variants remain inadequate to explain the total heritable disease risk10. Indeed, common genetic variants rarely act independently and additively as modeled by most genome-wide association studies (GWAS)11. There is growing biological and clinical evidence to support a disease risk model in which multiple genes interact nonadditively with each other through epistasis12,13. While some computational studies estimated a minor average epistatic component compared with the additive component within the total genetic variance, these epistatic variance estimates exhibit a large trait-to-trait variation14. In addition, it is important to distinguish between the concepts of statistical epistasis, estimated through variance components and influenced by allele frequencies, and biological epistasis (for example, gene actions), which is independent of allele frequencies15. A recent study has shown that common genetic variation influences susceptibility to and expressivity of HCM9. This raises the possibility that common epistatic interactions drive cardiac phenotype, holding critical potential for uncovering disease mechanisms and developing potential therapeutic strategies.
Several computational and experimental challenges need to be resolved to allow robust identification of epistasis. First, the combinatorial nature of possibly high-order interactions makes an exhaustive search computationally intractable. To reduce the computational burden and ensure stable discoveries, we developed an approach based on signed iterative random forests (siRF)16,17 to uncover higher-order (not limited to pairwise) nonlinear interactions in a computationally tractable manner. Second, many previously reported epistatic relationships were not replicated18. To achieve more trustworthy results, we adhered to a new framework for veridical data science19, centered around the principles of predictability, computability and stability (PCS) and the need for transparent documentation of decisions made in data analysis pipelines. A third challenge is the generally small effect size of common genetic variants10, which impedes both the data-driven discovery and functional validation of epistatic interactions. In human biobanks, recent advances in deep-learning-enabled phenotyping20 using cardiac magnetic resonance images have led to more refined phenotypes at larger scales. At the cellular level, high-throughput microfluidic technologies21,22 have been integrated with artificial intelligence-based image analysis of single-cell morphology23 and human induced pluripotent stem cell-derived cardiomyocytes24, opening up new possibilities for rapid, label-free detection of the phenotypic consequences of genetic perturbations.
Results
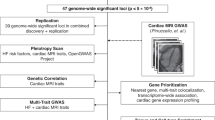
In contrast to many studies12,14,18 that have investigated the statistical significance or causality of epistasis solely from observational data, we tackle the aforementioned challenges and conceptual gap between statistical epistasis and biological epistasis15 via a multistage approach. This approach begins with a data-driven prioritization of promising statistical epistasis followed by extensive functional interpretations and experimental validations to reliably assess the biological epistasis consistency. More specifically, our methodology includes four major stages: derivation of LV mass estimates (green boxes, Fig. 1), computational prioritization of epistatic drivers (orange boxes, Fig. 1), functional interpretation of the hypothesized epistatic genetic loci (purple boxes, Fig. 1) and experimental confirmation of epistasis through perturbation (blue boxes, Fig. 1).

The study workflow includes four major stages: (1) derivation of LV mass from cardiac MRIs (green boxes), (2) computational prioritization of epistatic drivers (orange), (3) functional interpretation of the hypothesized epistatic genetic loci (purple) and (4) experimental confirmation of epistasis in cardiac tissues and cells (blue). LVM, left ventricular mass; LVMi, LVM indexed by body surface area; PLINK26 and BOLT-LMM27, two different GWAS software packages; ANNOVAR74, a software for functional annotation of genetic variants; CADD36 scores the deleteriousness of variants; RegulomeDB35, a database that scores functional regulatory variants; ChromHMM34, a multivariate Hidden Markov Model for chromatin state annotation; PheWAS, phenome-wide association study; D40, cultured for 40 days post-differentiation from iPSCs.
Deep learning of cardiac imaging quantifies LV hypertrophy
We accessed all cardiac magnetic resonance images from the UKBB substudy (44,503 people at the time of this analysis)25. We focused on the largest ancestry subset of 29,661 unrelated individuals (summary characteristics in Supplementary Table 1) and analyzed the most recent image per individual. We leveraged a recent deep learning model20 to quantify LV hypertrophy from these 29,661 multislice cine magnetic resonance images (Fig. 2a). A fully convolutional network had been previously trained for image segmentation and was evaluated on manual pixelwise annotations of images from 4,875 UKBB participants20. This fully convolutional network learns features across five different resolutions through sequential convolutional layers interspersed with nonlinearities, and has shown accurate performance compared with cardiac segmentation by human experts20. Using this segmentation model, we extracted areas of the LV chamber wall in each slice of the short-axis image at the end of diastole. Areas extracted from each image slice in the same image stack were then integrated to calculate the heart muscle volume, which was converted to LV mass and normalized by body surface area to obtain the LV mass index (LVMi; Extended Data Fig. 1). Details regarding this analysis can be found in Methods.

a, Lo-siRF took as input SNV data and cardiac MRI-derived LV mass indexed by body surface area (LVMi) from 29,661 UKBB participants. b, Two GWAS studies were applied using different software to reduce the dimensionality of SNVs. c, LVMi was binarized into high and low categories using three thresholds (shown as stacked boxes). d, For each threshold, an siRF was fitted using the GWAS-filtered SNVs to predict the binarized LVMi, achieving prediction accuracy better than or comparable to other methods, before interpreting the model fit. PRS, polygenic risk score; ML, machine learning; DL, deep learning. e, siRF aggregates SNVs into genetic loci using ANNOVAR74 and finally ranks loci and their interactions based on a stability-driven importance score averaged across the three binarization thresholds.
Low-signal signed iRF prioritizes epistatic genetic loci
We developed low-signal signed iterative random forests (lo-siRF; Fig. 2) to prioritize statistical epistatic interactions from the extracted LV mass and single-nucleotide variants (SNVs) from UKBB. Given the inherent low signal-to-noise ratio and aforementioned challenges, lo-siRF aims to recommend reliable candidate interactions for experimental validation rather than directly assessing claims of statistical significance from data. This prioritization pipeline is guided by the PCS framework19 and builds upon siRF16,17, a computationally tractable algorithm to extract predictive and stable nonlinear higher-order interactions that frequently co-occur along decision paths in a random forest. More specifically, lo-siRF proceeds through four steps:
-
(1)
Dimension reduction (Fig. 2b): we combined the results of two initial GWAS, implemented via PLINK26 and BOLT-LMM27 (Extended Data Fig. 2 and Supplementary Data 1) to reduce the interaction search space from 15 million imputed variants down to 1,405 variants (Supplementary Data 2). Details can be found in the Methods section ‘Lo-siRF step 1: dimension reduction of variants via GWAS’.
-
(2)
Binarization (Fig. 2c): we partitioned the LVMi measurements into high, middle and low categories using three different partitioning schemes (Supplementary Table 2). The partitioning enabled us to transform the original low-signal regression problem for a continuous trait into a relatively easier binary classification task for predicting individuals with high-versus-low LV mass (omitting the middle category). This transformation is necessary to obtain a sufficient prediction signal, ensuring that the model indeed captures pertinent information about reality (Supplementary Table 3). Further justification and details on the partitioning can be found in the Methods section ‘Lo-siRF step 2: binarization of the left ventricular mass phenotype’.
-
(3)
Prediction (Fig. 2d): we trained an siRF using the 1,405 GWAS-filtered SNVs to predict the binarized LVMi measurements. The learnt model yields, on average, the highest (balanced) classification accuracy (55%), area under the receiver operator characteristic (0.58) and area under the precision–recall curve (0.57) compared with common machine learning prediction algorithms (Supplementary Table 4). Details about the model and prediction check can be found in the Methods section ‘Lo-siRF step 3: prediction’.
-
(4)
Prioritization (Fig. 2e): we developed a stability-driven feature importance score (Extended Data Fig. 3), which leveraged the fitted siRF and a permutation test, to aggregate SNVs into genetic loci and prioritize interactions between genetic loci. This importance score provides the necessary new interpretable machine learning ingredient to complete the lo-siRF discovery pipeline. Details can be found in the Methods section ‘Lo-siRF step 4: prioritization’.
Additional discussion of the philosophy and modeling decisions driving lo-siRF can be found in Supplementary Note 1, an interactive HTML webpage hosted at https://yu-group.github.io/epistasis-cardiac-hypertrophy/. The webpage also provides a comparison of lo-siRF with alternative epistasis detection methods (implementation details in Supplementary Note 2), including an exhaustive regression-based pairwise interaction search28, MAPIT29, and a locus-level variant of MAPIT using gene set enrichment analysis30, demonstrating the challenges and limitations of existing methods for analyzing low-signal, complex phenotypes.
Lo-siRF identified six genetic risk loci that showed stable and reliable associations with LV mass (Table 1). Because these loci are located either within a gene body or in between two genes (Fig. 3a), for convenience, we denote these loci by their nearest genes. Notably, out of the six loci, three (TTN, CCDC141 and IGF1R) were prioritized by lo-siRF as epistatic loci. These loci not only interact with other loci, but also marginally affect LV mass. The other three lo-siRF-prioritized loci are LOC157273;TNKS, MIR588;RSPO3 and LSP1. The LOC157273;TNKS locus is located within the intergenic region between genes LOC157273 and TNKS (the semicolon indicates intergenic region). This locus was prioritized by lo-siRF to be hypostatic (that is, effects are deemed stable by lo-siRF only when interacting with the CCDC141 locus). Interestingly, all three identified epistatic interactions involved the CCDC141 locus (Fig. 3a, green links in circle 1). Furthermore, while the MIR588;RSPO3 and LSP1 loci lacked evidence for epistasis by lo-siRF, they were each identified to be marginally associated with LV mass. The specific prioritization order of these loci can be found in Supplementary Table 5, and details regarding the direction or sign of the interactions can be found in Supplementary Note 1. In total, lo-siRF identified 283 SNVs located within the 6 loci (Supplementary Data 3 and Extended Data Fig. 4). Of the 283 SNVs, 90% have previously been shown to harbor multiple distinct cardiac function associations31 in phenome-wide analyses (for example, pulse rate; Supplementary Data 3), suggesting a strong likelihood that these lo-siRF-prioritized loci contribute to determining cardiac structure and function.

a, Circos plot illustrating genetic risk loci identified by lo-siRF (green, circle 2) and regions after clumping FUMA-extracted SNVs in LD (r2 > 0.6) with lo-siRF-prioritized SNVs (black, circle 3). Circle 1 shows the top 300 epistatic SNV–SNV pairs ranked by occurrence frequency in lo-siRF (green), SNV–gene linkages (FDR < 0.5) from GTEx37 v8 cis-eQTL data of heart and skeletal muscle tissues (purple), and 3D chromatin interactions33 from Hi-C data of LV tissue (GSE87112). Circle 5 and 6 show occurrence frequencies and the number of partner SNVs in epistasis (normalized by the maximum value per locus) identified by lo-siRF, respectively. Circle 7 displays the ChromHMM34 core-15 chromatin state for the LV, right ventricle (RV), right atrium (RA) and fetal heart (Fetal), using the standard color scheme (bottom right)34. Circle 8 presents a GWAS Manhattan plot (points, two-sided P < 0.05 from PLINK26), in which SNVs are color coded by their maximum r2 value relative to the 283 lo-siRF-prioritized SNVs. The outer heatmap layer represents LD-linked SNVs (r2 > 0.6) extracted from the FUMA reference panel, which lack GWAS P values. SNVs not in LD (r2 ≤ 0.6) with any of the 283 lo-siRF-prioritized SNVs appear in gray. The dashed line indicates GWAS P = 5 × 10−8. Circle 9 shows genes functionally mapped by FUMA. b, Pie charts depicting ANNOVAR enrichment results for the six lo-siRF loci (circle 2 in Fig. 3a and Table 1). The slice arc length represents the proportion of SNVs with a specific functional annotation. The slice radius indicates log2(E + 1), where E is the enrichment score computed as (proportion of SNVs with an annotation for a given locus)/(proportion of SNVs with an annotation relative to all available SNVs in the FUMA reference panel). The dashed circle indicates E = 1 (no enrichment). Asterisks denote two-sided Fisher’s exact test P values for the enrichment of each annotation. **P < 1 × 10−5; *P < 0.002. Details in Supplementary Data 4 and 5.
Considering the correlations between LV hypertrophy and hypertension32, we evaluated whether these identified variants affect LV mass by regulating blood pressure. Specifically, we repeated the lo-siRF analysis using only the subset of UKBB individuals without hypertension (details in Methods). All previously highlighted loci and interactions maintained priority in this non-hypertensive subset, except for the MIR588;RSPO3 locus (Table 1), which was not stably prioritized across all three binarization thresholding schemes. In addition, none of the lo-siRF-prioritized variants showed a strong marginal association with hypertension, failing to meet the genome-wide (P < 5 × 10−8) and even the suggestive (P < 1 × 10−5) significance level. However, the MIR588;RSPO3 locus with lead SNV rs2022479 gave the smallest P value of 5 × 10−5, which may suggest a possible pleiotropic effect of MIR588;RSPO3 on both LV hypertrophy and blood pressure. In brief, while we cannot completely rule out pleiotropy, the highly stable prioritization of all three epistatic interactions in both analyses with and without hypertensive individuals suggests that the identified epistases on LV mass is not solely driven by blood pressure (additional discussion in Supplementary Note 1).
Loci associated with LV mass show regulatory enrichment
We performed functional mapping and annotation (FUMA)33 for the 283 lo-siRF-prioritized SNVs (Fig. 1, purple, and Fig. 3). For linkage disequilibrium (LD), we used a default threshold of r2 = 0.6 and chose the UKBB release 2b reference panel created for British and European participants to match the population group used for lo-siRF prioritization. FUMA identified 572 additional candidate SNVs (Supplementary Data 4) in strong LD (r2 > 0.6) with any of the 283 lo-siRF-prioritized SNVs, including 492 SNVs from the input GWAS associations (points in Fig. 3a, circle 8) and 80 non-GWAS-tagged SNVs extracted from the selected reference panel (heatmap tracks in Fig. 3a, circle 8). We then assigned these 572 FUMA-extracted candidate SNVs to a lo-siRF-prioritized locus (Table 1) based on the corresponding lo-siRF-prioritized SNV (out of the 283 SNVs), which has the maximum r2 value with the candidate SNV.
The two loci contributing to the top-ranked epistatic interaction by lo-siRF, the CCDC141 and IGF1R loci (Table 1), both showed a significant enrichment of intronic variants relative to the background reference panel (Fig. 3b and Supplementary Data 5). Over 88% of the SNVs in or in LD with these two loci were mapped to actively transcribed chromatin states (TxWk) or enhancer states (Enh) in LVs based on the ChromHMM core 15-state model34 (Fig. 3a, circle 7). More than 47% and 76% of the identified SNVs in or in LD with the CCDC141 and IGF1R loci, respectively, showed the highest RegulomeDB33,35 categorical score (ranked within category 1 from the 7 main categories). The Combined Annotation-Dependent Depletion (CADD) score36 was used to judge the deleteriousness of prioritized variants (Supplementary Data 4). As expected, GTEx37 data revealed that 82% of SNVs in or in LD with the IGF1R locus are expression quantitative trait loci (eQTL) for the gene IGF1R. By contrast, of the SNVs in or in LD with the CCDC141 locus, only 14% are eQTLs for gene CCDC141 and 22% are splicing quantitative trait loci (sQTL) for gene FKBP7. Furthermore, high-throughput chromosome conformation capture (Hi-C) data indicated that all SNVs identified in or in LD with the IGF1R locus are in three-dimensional (3D) chromatin interaction with gene SYNM while more than 54% of SNVs identified in or in LD with the CCDC141 locus are in 3D chromatin interaction with gene TTN. These known 3D chromatin interactions could suggest a possibility of higher-order interactions between more than two genes.
The CCDC141 and TTN loci exhibit genomic proximity (Fig. 3a). Their interaction, however, does not appear to stem from this proximity. Indeed, the CCDC141 and TTN genes have been individually associated with LV mass38,39. Due to this proximity, previous studies40,41 have assumed CCDC141 as a secondary gene that affects LV mass through the TTN gene expression. However, we found low LD (r2 < 0.6) between any two of the 283 lo-siRF-prioritized SNVs, suggesting that the identified CCDC141–TTN interaction is unlikely driven by nonrandom LD associations between SNVs in these two loci. In addition, we compared all the epistasis-contributing SNVs that were aggregated to the TTN locus, including both lo-siRF-prioritized SNVs and their LD-linked variants, with the complementary set of TTN-annotated SNVs in lo-siRF. We found that the TTN locus showed a significant depletion of SNVs located close to (<10 kb) the gene CCDC141 (P = 2.38 × 10−9, two-sided Fisher’s exact test). Similarly, the CCDC141 locus showed a significantly decreased enrichment of SNVs that are close to gene TTN (P = 0.02, two-sided Fisher’s exact test). These results suggest that although the CCDC141 and TTN loci are located close to each other in the genome, the prioritized epistatic SNVs are located farther apart relative to randomly selected SNVs from the two loci.
In contrast to the CCDC141 and IGF1R loci, the TTN locus showed a significant enrichment of exonic variants and intronic variants that are transcribed into noncoding RNA (ncRNA_intronic, Fig. 3b). Of those exonic variants, 62% are nonsynonymous. This differential enrichment of exonic variants for the TTN locus may suggest a potential epistatic contribution to the structural alterations in the titin protein. Over 90% of SNVs in or in LD with the TTN locus were mapped to actively transcribed states (Tx, TxWk)34 in LVs (Fig. 3a, circle 7). Interestingly, these SNVs were associated with a quiescent chromatin state (Quies) in the right atrium, indicating that the epistatic effects of the TTN locus may be specific to ventricular tissues. Nearly half of SNVs in or in LD with the TTN locus are eQTLs for the gene FKBP7. In addition, 83% of these SNVs are sQTLs for gene FKBP7 or TTN, suggesting a regulatory effect of the TTN locus on the expression and splicing of gene FKBP7. Moreover, the TTN locus was suggested to impact genes PDE11A, RBM45, PRKRA and PJVK through 3D chromatin interactions.
The hypostatic locus LOC157273;TNKS showed a significant enrichment of variants within noncoding RNA regions of exons and introns (Fig. 3b). Over 95% of identified SNVs in or in LD with this locus were mapped to inactive chromatin states (ReprPCWk, Quies)34 in LVs (Fig. 3a, circle 7). This suggests that in the absence of an epistatic partner, the LOC157273;TNKS locus is epigenetically quiescent or repressed by polycomb group proteins. In addition, of all the SNVs in or in LD with this locus, 66% are eQTLs for MFHAS1 or CLDN23 and 22% are in 3D chromatin interaction with gene TNKS.
Functional annotations for the other two lo-siRF-prioritized loci that were marginally associated with LV mass can be found in Supplementary Data 4 and 5.
Epistatic loci functionally map to 20 protein-coding genes
Three strategies, positional, eQTL and chromatin interaction, mapped the six LV hypertrophy risk loci to 20 protein-coding genes (Fig. 4a). Genes prioritized by eQTL and chromatin interaction mapping are not necessarily located in the corresponding risk locus, but they are linked to SNVs within or in LD with the locus (Fig. 3a). Among the 20 genes, CCDC141 and IGF1R were prioritized by all the three mapping strategies (Fig. 4a), suggesting that these two genes are very likely involved in determining LV mass. Interestingly, none of the SNVs mapped to IGF1R were statistically significant in our GWAS studies using BOLT-LMM and PLINK (Extended Data Fig. 2 and Supplementary Data 1). Set-based association tests42 also failed to identify the IGF1R locus (details in Methods and Supplementary Note 1). This reveals the potential of lo-siRF to identify risk loci that may be overlooked by GWAS. Based on the expression data from GTEx v8, TTN, TNNT3 and SYNM are upregulated while CLDN23 and MFHAS1 are downregulated in both heart and muscle tissues (Fig. 4b). By contrast, CCDC141 is upregulated specifically in heart tissues whereas RSPO3 is downregulated in heart but upregulated in muscle tissues (Fig. 4b).

a, UpSet plot showing the number of lo-siRF-prioritized SNVs (dark blue) and their LD-linked (r2 > 0.6) SNVs (light blue, circle 8 in Fig. 3a) functionally mapped to 20 protein-coding genes via positional, eQTL and/or chromatin interaction (CI) mapping in FUMA33. CCDC141 and IGF1R (highlighted red) are prioritized by all three mapping strategies (details in Supplementary Data 4). b, Heatmap of averaged expression (GTEx, 50% winsorization, log2(TPM + 1)) across different tissues for these functionally mapped genes. c, Co-association network built from an enrichment analysis integrating multiple annotated gene set libraries for GO and pathway terms from Enrichr43. Nodes represent genes (green) functionally linked from lo-siRF-prioritized epistatic and hypostatic loci (Table 1) and top enriched GO and pathway terms. Edge width indicates the co-association strength between enriched terms and genes ranked by two-sided P values from an exhaustive permutation of the co-association score for all possible gene–gene combinations in the network (details in Methods and Supplementary Data 6). d, Comparison between the top 10 TFs enriched from genes prioritized (top) and deprioritized (bottom) by lo-siRF. Prioritized genes are functionally linked to lo-siRF-prioritized SNVs (a), while deprioritized genes are genes linked to SNVs that failed to pass the lo-siRF prioritization thresholds. Enrichment was performed against nine TF-annotated gene set libraries from ChEA344 and Enrichr43, ranked by the number of significantly (P < 0.05, two-sided Fisher’s exact test) overlapped libraries (numbers in the ‘nLibraries’ column) and the mean scaled rank across all libraries containing that TF (colored boxes in the ‘nLibraries’ column). The balloon plot shows the lowest two-sided Fisher’s exact test P values (horizontal axis) and the proportion of overlapped genes (balloon size) between the input gene set and the corresponding TF-annotated gene set. DE, differential expression. e, Heatmap showing the TF co-association strength of gene–gene combinations among lo-siRF-prioritized genes relative to randomly selected gene pairs in the co-association network. Further details in Extended Data Fig. 5 and Supplementary Data 7.
Epistatic genes show strong network co-associations
We performed gene ontology (GO) and pathway enrichment analysis on the 20 genes mapped from lo-siRF loci. We adopted previously established approaches43,44 and integrated enrichment results across libraries from multiple sources to establish a GO and pathway co-association network (Fig. 4c). To evaluate the correlation strength between any two genes in the network, we calculated a co-association score for every possible gene–gene combination (n = 72,771) from both genes prioritized and deprioritized by lo-siRF. Lo-siRF-prioritized genes are the 20 genes functionally mapped from the 283 lo-siRF-prioritized SNVs and their LD-linked SNVs (Fig. 4a). Lo-siRF-deprioritized genes are those functionally mapped from the SNVs that failed to pass the lo-siRF prioritization threshold. Compared with random gene pairs in the network, ten genes that were functionally mapped from the lo-siRF-prioritized epistatic and hypostatic loci showed significant co-associations with multiple GOs and pathways (Fig. 4c and Supplementary Data 6). Consistent with our hypothesized epistasis (Table 1), gene CCDC141 showed a significant co-association to SYNM (functionally linked to the IGF1R locus) and PDE11A and PLEKHA3 (both functionally linked to the TTN locus) through the GO term of hyperactivity (excessive movement), which has been linked to increased risk of cardiac disease45. Beyond that, TTN, IGF1R and SYNM are co-associated with kinase activity and cardiac-structure-related GO terms, indicating that these genes may jointly affect cardiac structure by regulating the process of kinase activity.
Epistatic genes share myogenic regulatory factors
We next performed an integrative enrichment analysis to assess transcriptional regulation of genes prioritized and deprioritized by lo-siRF. Due to assay-specific limitations and biases, we integrated the enrichment results across nine distinct gene set libraries43,44 (Fig. 4d and Supplementary Data 7). We found that the lo-siRF-prioritized epistatic genes shared important myogenic regulatory factors, such as MYOD1, MYF6 and MYOG (Fig. 4d, top). These myogenic regulatory factors coordinate to regulate muscle development and differentiation. By contrast, transcription factors (TFs) enriched from lo-siRF-deprioritized genes display a less coordinated regulatory pattern (Fig. 4d, bottom). These analyses enriched TFs based on their associations to given sets of individual genes rather than co-association to gene pairs43,44. To further evaluate the correlation strength between any two genes that share TFs, we calculated a TF co-association score for all the 72,771 possible gene–gene combinations (Methods). Compared with random gene pairs,16 gene–gene combinations from the lo-siRF-prioritized genes showed a significant co-association (empirical P < 0.05; Fig. 4e). These co-associations were found in gene–gene combinations from both intra- and inter-lo-siRF-prioritized loci (Fig. 4e). In particular, pairwise combinations among TTN, TNNT3, CCDC141 and SYNM share a common splicing regulator, RBM20 (Extended Data Fig. 5). RBM20 has been reported to regulate the alternative splicing of genes important for cardiac sarcomere organization46. This suggests that the splicing patterns of these four genes are likely to be co-regulated by RBM20, which is consistent with the exhibited enrichment of sQTLs by the CCDC141, TTN and LSP1 lo-siRF loci (Supplementary Data 4).
Human heart transcriptomic correlations reflect epistasis
We proceeded to the fourth stage for experimental confirmation (Fig. 1, blue) and evaluated how the identified epistases contribute to the progression of heart failure (Fig. 5). We used a series of weighted gene co-expression networks derived from human cardiac transcriptomic data from 177 failing hearts isolated at the time of heart transplant and 136 non-failing hearts collected from cardiac transplant donors whose organs were not able to be placed47 (Fig. 5a). We compared the molecular connectivity of genes identified by lo-siRF as statistical epistatic interactors. We defined connectivity as the edge weights between two genes normalized to the distribution of all network edge weights, and compared this with the connectivity of all other available gene–gene combinations in the network. This revealed strong co-expression correlations between CCDC141 and genes functionally linked to the IGF1R locus (SYNM and LYSMD4) and TTN locus (TTN and FKBP7) in the healthy control network (Fig. 5b). By contrast, most of these gene pairs (except for CCDC141–TTN) no longer exhibit a strong connectivity in the heart failure network (Fig. 5c). All of these connectivities showed a significant decrease (indicated by the negative connectivity difference score and P < 0.05 in Fig. 5d) in the differential network, suggesting a declined co-expression correlation between these gene pairs relative to random gene pairs during the progression of failing hearts. This difference is potentially related to the rewired gene modular assignments between the control and heart failure networks47 (Fig. 5e and Extended Data Fig. 6). For instance, CCDC141, SYNM, TTN and TNNT3 are co-associated with the electron transport chain/metabolism module in the control network. In the failing hearts, SYNM and TTN rewire to the muscle contraction/cardiac remodeling module, whereas CCDC141 and TNNT3 remain associated with the metabolism module (Fig. 5e). In addition, other genes functionally linked to IGF1R and TTN lo-siRF loci are co-associated with the membrane transport or unfolded protein response module in healthy hearts and rewire to the muscle contraction/cardiac remodeling or cell surface/immune/metabolism module in failing hearts.

a, Gene co-expression networks for control (blue) and heart failure (red) conditions were established using weighted gene co-expression network analysis (WGCNA) on transcriptomic data from 313 non-failing and failing human heart tissues47. b,c, The connectivity between lo-siRF-prioritized genes was assessed against the full connectivity distributions of all possible gene–gene combinations in the control (b) and heart failure (c) networks. CCDC141 showed a significant connectivity to SYNM and LYSMD4 (IGF1R lo-siRF locus) and TTN and FKBP7 (TTN lo-siRF locus) in the control network. The color scales indicate two-sided empirical P values. d, Comparing between the control and heart failure networks indicated a significant connectivity decrease of these gene pairs during heart failure progression. e, A Sanky plot showing the rewired gene modular assignments for the lo-siRF loci-associated genes (middle column) in the control versus heart failure networks. Module names47 in the control (left column) and heart failure (right column) networks were derived from KEGG and Reactome associations of genes within each module.
Perturbation confirms epistasis in cardiomyocyte hypertrophy
We interrogated epistatic associations in a genetic model of cardiac hypertrophy (Fig. 1, blue): induced pluripotent stem cell cardiomyocytes derived from patients with and without HCM caused by the cardiac myosin heavy chain (MYH7) p.R403Q variant24 (Fig. 6a). Cardiac myosin heavy chain 7 is a key component of the cardiac sarcomere, and the most common cause of HCM24. The patient presented with typical symptoms, and echocardiography revealed severe LV hypertrophy and a small LV cavity24. At the cellular level, cardiomyocytes show an elevated mean cell size and non-Gaussian size distribution with a long tail relative to the unaffected control (Fig. 7a).

a, hiPSC-CMs, with and without HCM (MYH7-R403Q mutation), were transfected with scramble siRNA or siRNAs specifically targeting single (CCDC141, IGF1R, TTN) or combined (CCDC141–IGF1R, CCDC141–TTN) loci prioritized by lo-siRF. RISC, RNA-induced silencing complex. b, Gene-silenced cardiomyocytes were bifurcated into large and small cells using a spiral microfluidic device (Extended Data Fig. 7) for high-resolution single-cell imaging. c, Image analysis workflow. Single-cell images were analyzed using a customized MATLAB program to extract cell size and shape features through a multistep process.

a, Violin plots showing total cell diameter distributions for unaffected (blue, n = 36,327) and MYH7-R403Q variant (red, n = 46,542) cardiomyocytes from three independent cell batches (P = 2.1 × 10−36, two-sided Mann–Whitney U test). In all panels, box plots show the interquartile range (IQR) with the median line, and whiskers extend to 1.5× IQR. Overlaid points represent median diameters from individual batches. b, Gene-silencing efficiency measured by RT-qPCR in unaffected (blue) and MYH7-R403Q variant (red) cells from at least n = 3 cell batches (exact n numbers provided in Source Data). The bars, overlaid points and error bars show mean values, individual biological replicates and standard deviations, respectively. c–h, Gene-silencing effects on single-cell morphology analyzed for n = 3 independent cell batches (n = 4 for unaffected cells silencing CCDC141), with over 33,000 cells measured per condition per batch. c, Percentage change in median cell diameter for each gene-silencing condition relative to scramble controls (that is, relative size difference). The squares represent mean relative size differences across batches, with overlaid points indicating individual batches. The violins display mean relative size differences from 1,000 bootstrap samples, with error bars indicating standard deviations. *P < 0.05; **P < 6 × 10−5 (exact P values in Source Data), two-sided Mann–Whitney U test. d, Violin plots showing nonadditive interaction effects (\({\hat{\beta }}_{{\boldsymbol{12}}}\)) for unaffected (blue) and MYH7-R403Q variant (red) cells compared with marginal effects (\({\hat{\beta }}_{{\boldsymbol{1}}}\) and \({\hat{\beta }}_{{\boldsymbol{2}}}\), gray), estimated via a quantile regression model across 10,000 bootstrap samples (details in Methods). e, Scatterplots showing gene-silencing effects on cellular texture (‘norm. peak no.’, x-axis) and boundary (roundness error, y-axis) features. The markers represent mean values across cell batches, with error bars indicating standard deviations from 1,000 bootstrap samples. Bootstrapped values are visualized as density contours (scattered dots represent a density level lower than 0.2). f, Cell boundary waviness and texture irregularity were measured by roundness error (top) and normalized peak number (bottom), respectively. g,h, Representative single-cell images illustrating boundary irregularity (g) and textural variation (h). Norm. peak no., normalized peak number. Scale bars, 10 μm. Statistical details in Supplementary Data 8 and Source Data.
To determine whether CCDC141 can act both independently and in epistatic interactions with other genes to attenuate the pathologic cellular hypertrophy caused by MYH7-R403Q, we silenced genes CCDC141, IGF1R, TTN and gene pairs CCDC141–IGF1R and CCDC141–TTN using small interfering RNAs (siRNAs) in both diseased and healthy cardiomyocytes and compared them with cells transfected with scramble siRNAs (control) (Figs. 6a and 7b). Phenotypic consequences of these perturbations on cellular morphology were then evaluated in high throughput using a spiral inertial microfluidic device (Fig. 6b) in combination with automated single-cell image analysis (Fig. 6c). The microfluidic device adopted the Dean flow focusing principle22 (details in Extended Data Fig. 7 and Methods) to mitigate the nonuniform cell focusing48, thereby enhancing the imaging resolution49 affected by the large variations in cardiomyocyte diameter (Fig. 7a).
We first assessed the knockdown effects of the CCDC141–IGF1R interaction on cardiomyocyte size (Fig. 7c). Bootstrapped hypothesis tests were performed, for which the P values are capped below by P < 1 × 10−4 (Supplementary Data 8). Silencing IGF1R alone reduces the median cell size by 5.3% ± 0.3% (P < 1 × 10−4) in diseased cells compared with scrambled control and 6.6% ± 0.3% (P < 1 × 10−4) in healthy cells. Silencing CCDC141 alone also decreases median cell size by 3.2% ± 0.3% (P < 1 × 10−4) in diseased cells, but had no impact on healthy cells. Digenic silencing of CCDC141 and IGF1R reveals a synergistic effect on attenuating pathologic cell hypertrophy in diseased cells, resulting in an 8.5% ± 0.2% (P < 1 × 10−4) decrease in the median cell size. This is consistent in healthy cells, in which silencing CCDC141 alone fails to affect cell size, but digenic silencing of CCDC141 and IGF1R decreases the median cell size by 9.3% ± 0.3% (P < 1 × 10−4). Moreover, according to our estimated quantile regression analysis (details in Methods), this interaction effect appears to be nonadditive for both healthy and diseased cells (\({\hat{\beta }}_{{\boldsymbol{12}}}\) < 0, Fig. 7d; P < 1 × 10−4 for nonadditivity, Supplementary Data 8), consistent with an epistatic mechanism. These findings serve to confirm the strongest epistatic association identified by lo-siRF (Table 1).
We found a comparable nonadditive effect for the CCDC141–TTN interaction. Digenic silencing of CCDC141–TTN leads to a pronounced reduction in median cell size (by 5.8% ± 0.3% for healthy cells and 3.3% ± 0.3% for diseased cells, P < 1 × 10−4) relative to monogenic silencing (Fig. 7c). This interaction appears to be nonadditive for both healthy and diseased cells (P values in Supplementary Data 8), yet demonstrating opposite epistatic directions in these two cell states (Fig. 7d). In addition, CCDC141 and TTN show distinctive independent roles in repressing cardiomyocyte hypertrophy. In healthy cells, monogenic silencing of TTN leads to a larger cell size reduction compared with the case of silencing CCDC141. By contrast, diseased cells display a larger size reduction in response to monogenic silencing of CCDC141.
Furthermore, both CCDC141–IGF1R and CCDC141–TTN interactions show a stronger effect on rescuing larger cardiomyocytes over smaller ones in both cell lines (Extended Data Figs. 8 and 9). By contrast, monogenic silencing does not exhibit such a nonuniform effect on reshaping the cell size distribution, which reinforces the hypothesized nonadditivity of these two epistatic interactions (details in Supplementary Data 8 and Supplementary Note 4).
Recent studies have shown that cellular morphological features, such as cell boundary and textural irregularities, are informative readouts of cytoskeletal structure, which is highly associated with disease state in HCM23,50. We analyzed relative changes in cell shape and texture (Fig. 7e) by measuring the counts of peak intensities normalized to the total number of pixels enclosed by the cell boundary (Fig. 7f). Cells with a high normalized peak number display a ruffled texture, which manifests in unevenly distributed two-dimensional (2D) intensities (Fig. 7h). Our analysis shows that silencing both CCDC141 and IGF1R (circles in Fig. 7e, left) yields a larger increase in intensity peak number than silencing IGF1R alone (triangles in Fig. 7e, left) for both cell lines, exhibiting a synergistic epistasis between CCDC141 and IGF1R (P < 1 × 10−4 for nonadditivity). We also analyzed cell roundness error, a measure of how far radii measured on the cell outline deviate from a perfect circle (Fig. 7f). This parameter increases with an increasing cell boundary waviness or elongation (Fig. 7g). We show that the silencing of CCDC141 and IGF1R synergistically interact to increase the roundness error of diseased cardiomyocytes (P < 1 × 10−4 for nonadditivity; Fig. 7e, left). In addition, CCDC141 and TTN display antagonistic epistasis and synergistic epistasis in their impact on roundness error for healthy and diseased cells (P < 1 × 10−4 for nonadditivity; Fig. 7e, right), respectively.
Discussion
While computational models12,13 have supported epistatic contributions to human complex traits and disease risk, examples in the literature are rare, with even fewer experimentally confirmed. Here we developed a veridical machine learning19 approach to identify epistatic associations with cardiac hypertrophy derived from a deep learning model that estimates LV mass from cardiac imaging of almost 30,000 individuals in the UKBB. We report novel epistatic effects on LV mass of common genetic variants associated with CCDC141, TTN and IGF1R. We used established tools to functionally link risk loci to genes and then confirmed gene-level co-associations through network analyses, leveraging shared TFs and pathways enriched against multiple annotated gene set libraries and co-expression networks we built using transcriptomic data from over 300 healthy and diseased human hearts. Finally, using a cellular disease model incorporating monogenic and digenic silencing of individual genes, we assessed phenotypic changes in cardiomyocyte size and morphology using a novel microfluidic system, confirming the nonadditive nature of the interactions.
Our approach advances epistasis discovery in several key ways. First, unlike studies relying on linear-based models51,52, we leverage a more realistic, nonlinear tree-based model that mirrors the thresholding (or switch-like) behavior commonly observed in biomolecular interactions53. Second, in contrast to other tree-based approaches that evaluate interactions on a variant-by-variant basis54,55, our novel stability-driven importance score consolidates individual variants into loci for the assessment of feature importance, allowing for more reliable extraction of epistatic interactions from weak association signals. This is particularly valuable for evaluating noncoding variants and resembles ideas from marginal association mapping with sets of SNVs42,56. Moreover, instead of exhaustively searching all possible interactions, siRF internally employs a computationally efficient algorithm, which automatically narrows the search space of interactions to only those that stably appear in the forest and thus achieves a scalability much higher than that of existing tree-based approaches54. This allows lo-siRF to handle larger datasets without the need for LD pruning before the interaction search, which may inadvertently eliminate important epistatic variants, given that epistasis between loci in strong LD has been evidenced by a recent study57. Furthermore, our computational prioritization is rigorously validated through multiple functional network analyses and robust experimental confirmation.
Our results add to a small literature on epistasis in cardiovascular disease. Two recent studies have found epistasis influencing the risk of coronary artery disease12,13. One study13 identified epistasis between ANRIL and TMEM106B in coronary artery tissues. Although their method predicted functionally interpretable interactions between risk loci of interest, they relied heavily on previous knowledge and careful selection of the causal gene pairs13, making the approach challenging to scale. The other study12 used population-scale data and performed epistasis scans from regions around 56 known risk loci. This study identified epistasis between variants in cis at the LPA locus without experimental confirmation. By contrast, our approach allows discovery of not only cis-epistasis but also long-range interactions between interchromosomal loci (for example, CCDC141 and IGF1R) and is supported by gene perturbation experiments. More importantly, both studies searched for interactions around known risk loci identified by genome-wide association, which can be far away from the possible epistatic or hypostatic loci that are statistically insignificant in linear univariate association studies. In addition, both studies relied on a logistic regression model, which imposes restrictive assumptions that can be avoided using a nonlinear machine learning approach as in lo-siRF.
Our study has limitations. Given our primary interest in biological epistasis rather than statistical epistasis15, we tailored lo-siRF to conservatively prioritize reliable targets for experimental validation as opposed to finding all possible epistatic drivers. Lo-siRF should ideally be used as a first-stage hypothesis-generation tool within a broader scientific discovery pipeline. To assess significance of the lo-siRF-prioritized targets, we rely on and encourage follow-up investigations such as the high-throughput gene-silencing experiments conducted here. We focused this analysis on a single ancestry to enhance the likelihood of finding reliable interactions from weak association signals. These findings cannot be automatically applied to others. It was not feasible to conduct a formal genetic replication study because the UKBB is the only large-scale population cohort with integrated cardiac magnetic resonance images and genetic data. However, to help reduce the possibility of overfitting and increase generalizability, lo-siRF used numerous stability analyses (Supplementary Note 1) in addition to a proper training–validation–test data split. Beyond these computational checks, we also present functional supporting evidence and experimental validation. Our computational prioritization via lo-siRF currently groups SNVs based on genomic proximity, without accounting for their functional interdependencies, but this could be addressed by integrating functional annotation into the lo-siRF pipeline. Lo-siRF also relies on a GWAS to reduce the number of SNVs to a computationally manageable size, but this could be improved with more sophisticated epistasis detection algorithms such as MAPIT29. Lastly, lo-siRF is not as scalable as linear-based methods, although it is more scalable than alternative tree-based methods for epistasis detection. It also should be noted that although this study did not identify stable higher-order (beyond pairwise) interactions owing to the weak association signal between SNVs and LV mass, the method retains the capability to detect such interactions for broader phenotypes and complex traits without incurring additional computational cost.
In summary, our study adds to the discovery toolkit for the genomic architecture of complex traits and expands the scope of genetic regulation of cardiac structure to epistasis.
Methods
Study participants
The use of human participants (IRB-4237) and human-derived induced pluripotent stem cells (SCRO-568) in this study has been approved by the Stanford Research Compliance Office. The UKBB received ethical approval from the North West—Haydock Research Ethics Committee (21/NW/0157).
The UKBB is a biomedical database with detailed phenotypic and genetic data from over 500,000 UK individuals aged 40–69 at recruitment58. In this study, we focused on the largest ancestry subset (that is, the White British population) of 29,661 unrelated individuals who have both genetic and cardiac MRI data (Supplementary Table 1). More specifically, we considered UKBB individuals who self-reported as White British and have similar genotypic backgrounds based on principal components analysis58. To ensure unrelatedness, we identified and excluded third-degree relatives or closer via kinship estimation, retaining one individual per related group58,59. This yielded a cohort of 337,535 unrelated White British individuals, of which 29,661 have both genetic and cardiac MRI data. We randomly split these data (29,661) into training (15,000), validation (5,000) and test (9,661) sets.
Genotyping and quality control
For the 29,661 individuals in the study cohort, we leveraged genotype data from approximately 15 million imputed autosomal SNVs. These variants were imputed from 805,426 directly assayed SNVs (obtained by the UKBB from one of two similar Affymetrix arrays) using the Haplotype Reference Consortium and UK10K reference panels58. Imputed variants were subject to several quality-control filters, including outlier-based filtration on effects due to batch, plate, sex, array and discordance across control replicates. We excluded variants owing to extreme heterozygosity, missingness, minor allele frequency (<10−4), Hardy–Weinberg equilibrium (<10−10) and poor imputation quality (<0.9)58,59.
Quantification of LV hypertrophy
We retrieved cardiac MRI images from 44,503 UKBB participants and followed a method previously described20. Briefly, a fully convolutional network20 was trained on 4,875 participants with 93,500 pixelwise segmentations of UKBB short-axis cardiac MRI multislice images generated manually with quality control checks for interoperator consistency60. The cardiac MRI image resolution was 1.8 × 1.8 mm2, with a slice thickness of 8.0 mm and a 2.0-mm gap, typically consisting of 10 slices. Each slice was converted to an image and cropped to a 192 × 192 square, and measurements were 0–1 normalized. The network architecture used multiple convolutional layers to learn image features across five resolution scales. Each scale involved two or three convolutions with kernel size 3 × 3 and stride 1 or 2 (2 appearing every 2 or 3 layers), followed by batch normalization and ReLU transformation. Feature maps from the five scales were upsampled back to the original resolution, combined into a multi-scale feature map and processed through three additional convolutional layers with kernel size 1 × 1, followed by a softmax function to predict the segmentation label for each pixel. Notably, the pixelwise annotations used for training and evaluation were hand segmented and validated by a human expert. The model showed strong concordance with the human-generated gold standard in UKBB datasets20. We thus applied this trained deep learning model to our dataset of 44,503 cardiac MRIs, obtaining segmentation of the LV cavity and myocardium from each short-axis frame. After quality control checks20, 44,219 segmentations were retained. The LV myocardium volume was calculated by integrating segmented areas over slices, converted to LV mass using a standard density estimate of 1.05 g ml−1 (ref. 61) and then normalized by an estimate of body surface area using the Du Bois formula62 to obtain LVMi. From the 44,219 segmentations, we focused our analysis on LVMi measurements for 29,661 unrelated White British individuals, using data from their most recent imaging visit if multiple imaging visits were recorded.
Lo-siRF step 1: dimension reduction of variants via GWAS
As the first step in lo-siRF, we performed a GWAS on rank-based inverse normal-transformed LVMi from the training dataset using PLINK26 and BOLT-LMM27. Similar to typical screening in fine mapping63 and other tree-based epistasis detection methods64,65, this step reduces over 15 million SNVs to a more computationally feasible size (Fig. 2b). As BOLT-LMM and PLINK use different statistical models, we used both to mitigate model dependence due to this arbitrary choice. Specifically, we performed two GWAS runs: one using a linear regression model implemented via ‘glm’ in PLINK66 and another with BOLT-LMM27, a fast Bayesian-based linear mixed model. Each GWAS was adjusted for the first five principal components of ancestry, sex, age, height and body weight. SNVs were ranked by P value for each GWAS run separately, and the top 1,000 from each run were combined, yielding 1,405 GWAS-filtered SNVs for downstream lo-siRF analysis. PLINK and BOLT-LMM results are provided in Supplementary Data 1, and the 1,405 GWAS-filtered SNVs are listed in Supplementary Data 2. These 1,405 SNVs strictly contain the SNVs that passed the genome-wide significance threshold (P = 5 × 10−8).
Lo-siRF step 2: binarization of the LV mass phenotype
Next, we partitioned the raw (continuous) LVMi phenotype into low, middle and high groups before fitting the siRF (Fig. 2c). Specifically, for a given threshold x, we binned individuals within the top and bottom x% of LVMi values into high and low LVMi classes, respectively, while omitting individuals in the middle quantile range. Given the sex-specific variation of LVMi (Supplementary Note 1), this partitioning was performed separately for males and females, with cutoffs per sex and binarization threshold detailed in Supplementary Table 2. This binarization step simplifies the original low-signal regression problem, predicting continuous LVMi, into a relatively easier binary classification problem, distinguishing individuals with very high versus very low LVMi. The decision to use this approach was driven by the observation that regression models yielded validation R2 values below 0 (Supplementary Table 3), suggesting poor relevance to reality. The PCS framework for veridical data science19 advocates that a model should fit the data well, as measured by prediction accuracy, before trusting any of its interpretations. Because the threshold choice is arbitrary, we ran the remainder of the lo-siRF pipeline using three binarization thresholds (15%, 20% and 25%) to balance prediction signal improvement and data lost. Results stable across all binarization thresholds were aggregated (Methods, ‘Lo-siRF step 4.4: ranking genetic loci and interactions between loci’).
Lo-siRF step 3: prediction
Lo-siRF step 3.1: fitting siRF on the binarized LVMi phenotype
For each binarization threshold, we trained an siRF model17 using the 1,405 GWAS-filtered SNVs to predict the binarized LVMi phenotype and identify candidate interactions (Fig. 2d). siRF iteratively grows a sequence of feature-weighted random forests, re-weighting features in each iteration proportional to their feature importance from the previous iteration to stabilize the decision paths. If the stabilized forest provides reasonable prediction performance (Methods, ‘Lo-siRF step 3.2: prediction check’), siRF leverages random intersection trees (RIT)67 to identify sets of features that frequently co-occur along decision paths. These co-occurring features are more likely to interact and are outputted as candidate interactions by siRF. siRF is well-suited for prioritizing epistatic interactions because (1) it efficiently searches for nonlinear higher-order interactions at a computational cost similar to a traditional random forest and (2) its decision tree thresholding mimics the switch-like behavior observed in biomolecular interactions53. Improved upon its predecessor, iterative random forests16, siRF also tracks the sign of features17. In brief, a signed feature X– (or X+) indicates a decision rule of X < t (or X > t) for some threshold t. In SNV data, SNV+ typically represents a heterozygous or homozygous mutation, while SNV– represents no mutation at the locus. We trained siRF using the iRF2.0 R package with the following hyperparameters: number of iterations = 3, number of trees = 500, number of bootstrap replicates = 50, depth of RIT = 3, number of RIT = 500, number of children in RIT = 5 and minimum node size in RIT = 1. Hyperparameter tuning was not performed, as siRF is robust to different choices of hyperparameters17. We trained siRF using 10,000 randomly sampled training samples (from 15,000 total), with the remaining 5,000 training samples reserved for selecting genetic loci for the permutation test (Methods, ‘Lo-siRF step 4.3: permutation test for difference in LSI scores’).
Lo-siRF step 3.2: prediction check
Per the PCS framework for veridical data science19, we assessed the validation prediction accuracy of siRF (Fig. 2d) to ensure it captures biologically relevant phenotypic signals rather than simply noise before interpreting the model. We compared siRF with several machine learning prediction methods: L1-regularized (LASSO)68 and L2-regularized (ridge)69 logistic regression, random forests70, support vector machines71, a multilayer perceptron72 (fully connected feedforward neural network with one hidden layer and ReLU activations) and AutoGluon TabularPredictor73 (an auto machine learning framework that ensembles multiple models, including neural networks, LightGBM, boosted trees, random forests and k-nearest neighbors, by stacking them in multiple layers). Implementation and hyperparameters (tuned via 5-fold cross-validation) are detailed in Supplementary Table 6. We also compared siRF with a basic polygenic risk score, constructed using PLINK with lead SNVs from the LVMi PLINK GWAS identified by FUMA at the suggestive significance threshold of 1 × 10−5 (Supplementary Data 1). Logistic regression was performed using this polygenic risk score as a predictor for binarized LVMi. Prediction performance for each of these methods was assessed using classification accuracy, area under the receiver operator curve (AUROC) and area under the precision–recall curve (AUPRC). Although the prediction power of siRF was modest (~55% balanced classification accuracy, ~0.58 AUROC, ~0.57 AUPRC), it consistently outperformed random guessing (that is, >50% balanced classification accuracy and >0.5 AUROC/AUPRC, which is not guaranteed given the high phenotypic diversity of LVMi) and exceeded all other prediction methods across almost all binarization thresholds and evaluation metrics (Supplementary Table 4). We thus deemed that the siRF fit for LVMi passed the prediction check.
Lo-siRF step 4: prioritization
To interpret the siRF fit, we developed a novel stability-driven importance score to prioritize genetic loci and their interactions for follow-up experimental validation (Fig. 2e). Our proposed importance score aggregates weak, unstable variant-level importances into stronger, more stable locus-level importances by (1) assigning each variant to a genetic locus, (2) evaluating the local (or per-individual) importance of each locus or locus–locus interaction in the siRF fit via a stability-driven measure and (3) conducting a permutation test to summarize the importance of the locus or interaction across all individuals. We detail each step next.
Lo-siRF step 4.1: aggregation of SNVs into loci
We aggregated SNVs into genetic loci based on genomic proximity, using ANNOVAR74 and hg19 RefSeq Gene annotations. Each SNV was assigned to a single locus, with a default 1 kb maximum distance from gene boundaries. In lo-siRF, a genetic locus is a non-overlapping group of SNVs, and a signed genetic locus consists of signed SNVs (that is, Locus+ consists of SNV1+, …, SNVp+, while Locus‾ consists of SNV1‾, …, SNVp‾).
Lo-siRF step 4.2: local stability importance score
Next, we measured the importance of a genetic locus or locus–locus interaction based on their stability, defined as frequency of occurrence within the siRF fit (that is, how often SNVs from a locus or interaction were split upon in the fitted forest). As raw occurrence frequencies are inherently biased toward larger loci containing more SNVs, we developed a local (or per-individual) stability importance (LSI) score, which quantifies the importance of a signed locus or interaction for predicting each individual’s response. Let G = {g1, …, gK} denote a signed order-K interaction involving the signed loci g1, …, gK, and let \({v}_{1}^{(j)},\ldots ,{v}_{{p}_{j}}^{(j)}\) denote the signed SNVs within locus gj. Given a forest T, a signed interaction G and an individual i, the LSI score is defined as:
where ∣T∣ is the number of trees in the forest T and DT(G,i) is the number of decision paths in the forest T satisfying two criteria: (1) individual i appears in its terminal node and (2) for each j = 1, …, K, there exists an \(l\in \{1,\ldots ,{p}_{j}\}\) such that \({v}_{l}^{(j)}\) was used in a decision split along the path (Extended Data Fig. 3a). Thus, LSIT(G,i) is the proportion of trees in the forest T where at least one signed variant from each signed locus in G was used to predict individual i. A high LSI score indicates that the interaction G was frequently used to predict the response of individual i and is an important interaction for individual i. As a single locus can be viewed as an order-1 interaction, the LSI score also applies to individual loci for assessing their marginal importance.
Lo-siRF step 4.3: permutation test for difference in LSI scores
After computing LSI scores for all individuals, we performed a two-sample permutation test (Extended Data Fig. 3a) to assess whether the LSI scores for a given signed locus or interaction, G, differ between individuals with high and low LVMi (conditional on the rest of the fitted forest). This permutation test tests the null hypothesis L = H versus the alternative hypothesis L ≠ H, where L and H are the LSI distributions for low and high LVMi individuals, respectively. A small nominal P value indicates that G can differentiate between high- and low-LVMi individuals, suggesting an important risk locus or interaction for LVMi in the fitted siRF. We performed this permutation test using 10,000 permutations, the difference in means as the test statistic and the 5,000 validation samples. To enhance the reliability of our findings, we followed the PCS framework and conservatively tested a subset of genetic loci and interactions that passed predictive and stability checks. Namely, we tested:
-
(1)
The 25 genetic loci with the highest average LSI scores across the 5,000 validation samples, which were set aside from the 15,000 training samples and not used in fitting siRF (Methods, ‘Lo-siRF step 3.1: fitting siRF on the binarized LVMi phenotype’)
-
(2)
The signed interactions between loci that were stably identified across 50 siRF bootstrap replicates. RIT searches within siRF were performed at the locus level (using the ‘varnames.grp’ argument when running siRF in R). We defined an interaction as ‘stable’ if it passed the following siRF stability criteria (Supplementary Table 7):
-
Stability score > 0.5 (interaction appears frequently in siRF)
-
Stability score for mean increase in precision > 0 (interaction is predictive)
-
Stability score for feature selection dependence > 0 (filters out additive interactions)16,17
Given the challenges of analyzing low-signal data, these nominal permutation P values were used primarily to rank candidate loci and interactions rather than as formal tests of statistical significance, which relies heavily on untestable model assumptions that often do not hold in practice.
Lo-siRF step 4.4: ranking genetic loci and interactions between loci
As a final stability check, we recommended only those signed loci and interactions that underwent the permutation test and consistently yielded a nominal P < 0.1 across all three binarization runs. These stably identified signed loci and interactions were ranked by their mean nominal P value, averaged across the three binarization thresholds (Supplementary Table 5). To prioritize candidates for experimental validation, if both the positive and negative versions of a signed locus (or interaction) appeared, the final ranking was based on the smaller of the two mean nominal P values (Table 1). Nominal P values for all permutation tests, including those for loci and interactions that were unstable across binarization thresholds, are provided in Supplementary Note 1.
Lo-siRF: PCS documentation, additional stability analyses and simulations
We acknowledge that many human judgment calls were inevitably made throughout our veridical machine learning pipeline and that alternative choices could have been made (for example, different dimension reduction techniques, binarization procedures and prediction models). We provide extensive documentation, discussion and justification for these decisions in Supplementary Note 1. We also performed stability analyses19 to ensure that our findings are stable and robust to these human judgment calls. We lastly conducted a simulation study using the simChef R package75, showing that lo-siRF produces calibrated P values under a null response model and investigated the performance of lo-siRF under both a marginal effect and interaction effect simulation model. Supplementary Note 1 is an HTML document, which can be downloaded and shown in a browser or found at https://yu-group.github.io/epistasis-cardiac-hypertrophy/.
Non-hypertensive cohort analysis
We defined hypertensive individuals as anyone with self-reported hypertension, a doctor-diagnosed high blood pressure or any ICD-10 billing code diagnosis in I10–I16. Out of the 29,661 UKBB participants in the original lo-siRF analysis, 7,371 had hypertension, leaving 22,290 for the non-hypertensive analysis. Using the same 1,405 GWAS-filtered SNVs as in the original lo-siRF analysis, we repeated steps 2–4 of lo-siRF exclusively in the non-hypertension cohort. In addition, we assessed the marginal effect of each of the 1,405 GWAS-filtered SNVs on hypertension by fitting logistic regression models that regressed hypertension status (binary) on each SNV while adjusting for the first five principal components of ancestry, sex, age, height and body weight. A detailed discussion of the non-hypertension analysis results can be found in Supplementary Note 1.
Functional interpretation of lo-siRF-prioritized variants
Functional interpretation step 1: extraction of candidate SNVs and LD structures
We used the SNP2GENE function in FUMA (v1.5.4)33 to incorporate LD structure and prioritize candidate genes. Taking GWAS summary statistics from PLINK26 and BOLT-LMM27 as an input, we submitted the 283 lo-siRF-prioritized SNVs into SNP2GENE as predefined SNVs. This allows SNP2GENE to define LD blocks for each SNV and include both the input 283 SNVs and those in LD with them for further annotations. Using the default r2 threshold (0.6), FUMA defined all 283 SNVs as independent significant SNVs, as any two of them are in LD with each other at r2 < 0.6. To match the population used in lo-siRF prioritization, we selected the UKBB release 2b reference panel for British and European participants to compute r2 and minor allele frequencies. FUMA identified 572 candidate SNVs in strong LD (r2 < 0.6) with the 283 independent significant SNVs, extracted from both the input GWAS (P < 0.05) and the reference panel. Each candidate SNV was then assigned to one of the six lo-siRF-identified loci (Table 1) based on the independent significant SNV (from the 283 SNVs), with which it had the highest r2 value. A combination of the 283 independent significant SNVs and the 572 candidate SNVs (details in Supplementary Data 4) was defined as the ‘lo-siRF-prioritized SNV set’ and used to generate the lo-siRF-prioritized gene list (Fig. 4a) for enrichment analysis (Fig. 4c–e). For comparison, we also uploaded all 1,405 GWAS-filtered SNVs (Supplementary Data 2) as the predefined SNVs in a separate SNP2GENE job. Using the same approach and parameter settings, FUMA identified 929 independent significant SNVs within the input set and 5,771 candidate SNVs in LD with the 929 SNVs. The combined set of 929 independent significant SNVs and 5,771 candidate SNVs was defined as the ‘reference SNV set’. Unlike the lo-siRF-prioritized SNV set, this reference SNV set was derived purely from GWAS prioritization, without considering epistatic effects.
Functional interpretation step 2: ANNOVAR enrichment test
We performed an ANNOVAR enrichment test on the lo-siRF-prioritized SNV set against the selected reference panel in FUMA (see previous step). SNP2GENE generated unique ANNOVAR74 annotations for all identified SNVs. The enrichment score for a given annotation in each lo-siRF-prioritized locus (Fig. 3b) was computed as the proportion of SNVs with that annotation in the locus, divided by the proportion of SNVs with the same annotation in the reference panel. To compute the enrichment P value for the ith ANNOVAR annotation in the jth lo-siRF-prioritized locus, we performed a two-sided Fisher’s exact test on the 2-by-2 contingency table containing (Supplementary Data 5):
-
nj(i) = the number of SNVs with the ith annotation in the jth lo-siRF-prioritized locus
-
∑tnj(t) − nj(i), with ∑tnj(t) being the summation of nj(i) for all available annotations in the jth lo-siRF-prioritized locus
-
N(i) − nj(i), with N(i) being the number of SNVs with the ith annotation in the reference panel
-
∑tN(t) − ∑tnj(t) − 𝑛𝑗(𝑖), where ∑tN(t) is the summation of N(i) for all available annotations in the reference panel
Functional interpretation step 3: functional annotations
In addition to ANNOVAR annotations, FUMA annotated all identified SNVs for potential regulatory functions (core-15 chromatin state prediction and RegulomeDB score) and deleterious effects (CADD score). The core-15 chromatin state was annotated to all SNVs of interest by ChromHMM45 derived from five chromatin markers (H3K4me3, H3K4me1, H3K36me3, H3K27me3 and H3K9me3) for four relevant tissue and cell types, including LV (E095), right ventricle (E105), right atrium (E104) and fetal heart (E083) (Fig. 3a, circle 7). Data and a description of the core-15 chromatin state model can be found at https://egg2.wustl.edu/roadmap/web_portal/chr_state_learning.html. RegulomeDB33,35 annotations guide the interpretation of regulatory variants through a seven-level categorical score, with category 1 (including six subcategories ranging from 1a to 1f) indicating the strongest functional evidence. Because the RegulomeDB database (v1.1) used in FUMA has not been updated, we queried all SNVs identified by lo-siRF and FUMA in the RegulomeDB database v2.2 (https://regulomedb.org/regulome-search). Deleteriousness annotations were obtained from the CADD database (v1.4)36 by matching the chromosome, position, reference and alternative alleles of all SNVs. High CADD scores indicate the highly deleterious effects of a given variant, with a threshold score of 12.37 (ref. 36). We also retrieved eQTL and sQTL data for all 283 independent significant SNVs and 572 candidate SNVs from GTEx v8 (ref. 37). All functional annotation results are summarized in Supplementary Data 4.
Functional interpretation step 4: functional gene mapping
We performed three functional gene mapping strategies in SNP2GENE—positional, eQTL and 3D chromatin interaction mapping—using the lo-siRF-prioritized and reference SNV set (Methods, ‘Functional interpretation step 1: extraction of candidate SNVs and LD structures’). For positional mapping33,35, a default 10-kb maximum distance was used between SNVs and genes. For eQTL mapping, we used cis-eQTL data of heart LV, atrial appendage and muscle skeletal tissues from GTEx v8 (ref. 37), with significant SNV–gene pairs (false discovery rate (FDR) < 0.05, P < 1 × 10−3). For 3D chromatin interaction mapping, Hi-C data of LV tissue (GSE87112) with a default FDR < 1 × 10−6 threshold was used. A default promoter region window was defined as 250 bp upstream and 500 bp downstream of the transcription start site (TSS)33,35. Using these strategies, we mapped the lo-siRF-prioritized SNV set to 20 HGNC-recognizable protein-coding genes (Fig. 4a). Each gene was functionally linked to a specific lo-siRF-prioritized locus (Table 1), to which the highest proportion of SNVs mapped to that gene was assigned. A Circos plot (Fig. 3a) was created by TBtools76 to visualize the lo-siRF-prioritized epistatic interactions, eQTL SNV-to-gene connections and 3D chromatin interactions, as well as LD structures and the 20 prioritized genes. These 20 genes were submitted to GENE2FUNC in FUMA, yielding GTEx gene expression data for 19 genes across multiple tissue types (Fig. 4b). In a separate SNP2GENE job, we mapped the reference SNV set to 382 HGNC-approved genes using the same approach, and used both gene sets for enrichment analysis.
GO and pathway enrichment analysis
To assess differential GO and pathway co-association among lo-siRF-prioritized genes versus their counterparts deprioritized by lo-siRF, we performed an integrative GO and pathway enrichment analysis followed by an exhaustive permutation of co-association scores for all possible gene–gene combinations within the aforementioned 382 HGNC-approved genes (Methods, ‘Functional interpretation step 4: functional gene mapping’).
To improve GO and pathway prioritization, we adopted the concept from Enrichr-KG77 and ChEA3 (ref. 44) to assess enrichment results across libraries and domains of knowledge as an integrated network of genes and their annotations. We queried the 382 HGNC-approved genes against various previous-knowledge gene set libraries in Enrichr43 (https://maayanlab.cloud/Enrichr/), including GO biological process78,79, GO molecular function78,79, MGI Mammalian Phenotypes80, Reactome pathways81 and Kyoto Encyclopedia of Genes and Genomes (KEGG) pathways82. This allowed us to search for a union of enriched GO and pathway terms and their correspondingly annotated gene sets, from which we built a co-association network, in which nodes are either the enriched terms or genes. To measure the degree of co-association to specific GO or pathway terms for two given genes, we computed a co-association score for each of the 72,771 possible gene pairs (from the 382 queried genes). The co-association score was calculated as:
where N(A⋂B) denotes the number of GO or pathway terms significantly enriched for both gene A and gene B in a gene pair, and N(A⋃B) is the number of GO or pathway terms enriched for either gene A or gene B. For cases in which N(A⋃B) = 0, R = 0. Note that the 20 genes mapped from lo-siRF-prioritized SNVs form a subset of the 382 HGNC-approved genes. We compared the co-association scores R for the 20 lo-siRF-prioritized genes relative to the full distribution of R from an exhaustive permutation of all possible gene pairs within the 382 HGNC-approved genes, ranking gene pairs by two-sided empirical P values (Fig. 4c). Further details are in Supplementary Data 6.
TF enrichment analysis
Owing to the limitations and biases of various specific assays, we performed an integrative TF enrichment analysis against the following nine annotated gene set libraries in ChEA3 (ref. 44) and Enrichr43 from distinct sources:
-
1.
Putative TF target gene sets determined by ChIP-seq experiments from ENCODE83
-
2.
Putative TF target gene sets determined by ChIP-seq experiments from ReMap84
-
3.
Putative TF target gene sets determined by ChIP-seq experiments from individual publications44
-
4.
TF co-expression with other genes based on RNA-seq data from GTEx37
-
5.
TF co-expression with other genes based on RNA-seq data from ARCHS4 (ref. 85)
-
6.
Single TF perturbations followed by gene differential expression43
-
7.
Putative target gene sets determined by scanning position weight matrices (PWMs) from JASPAR86 and TRANSFAC87 at promoter regions of all human genes
-
8.
Gene sets predicted by transcriptional regulatory relationships unraveled by sentence-based text-mining (TRRUST)88
-
9.
Top co-occurring genes with TFs in a large number of Enrichr queries44
Of the mentioned nine gene set libraries, libraries 1, 3 and 5 were assembled by combining gene set libraries downloaded from both ChEA3 (ref. 44) and Enrichr43. Libraries 2, 4 and 9 were downloaded from ChEA3 (ref. 44). Libraries 6, 7 and 8 were downloaded from Enrichr43. Following the ChEA3 (ref. 44) integration method, when multiple gene sets were annotated to the same TF, the gene set with the lowest two-sided Fisher’s exact test (FET) P value was used for each library. As mentioned previously, we have mapped lo-siRF-prioritized SNVs and all GWAS-filtered SNVs to two gene sets: a lo-siRF-prioritized set (20 HGNC-recognizable genes) and a reference set (382 HGNC-recognizable genes). The 362 genes in the reference set complementary to the 20 lo-siRF-prioritized genes served as the lo-siRF-deprioritized gene set. We then performed a separate enrichment analysis on the lo-siRF-prioritized (20 genes) and lo-siRF-deprioritized (362 genes) sets across the 9 gene set libraries. TF enrichment significance was ranked for each library by FET P values, with ties assigned the same integer rank. The integer rank of each TF was scaled by dividing by the maximum rank within its respective library. We then integrated the nine sets of TF rankings and reordered the TFs by two sequential criteria: (1) the number of libraries showing significant enrichment (FET P < 0.05) and (2) the mean scaled rank across all libraries containing that TF. This approach prioritized distinct sets of TFs for the lo-siRF-prioritized genes (Fig. 4d, top) and lo-siRF-deprioritized genes (Fig. 4d, bottom).
To assess TF co-association43,44 between lo-siRF-prioritized genes, we computed TF co-association scores using equation (2), where N(A⋂B) and N(A⋃B) denote the number of enriched TF terms instead of GO and pathway terms (Methods, ‘GO and pathway enrichment analysis’). TF co-associations between lo-siRF-prioritized genes were ranked by empirical P values (Fig. 4e) from an exhaustive permutation of all 72,771 possible gene pairs from the 382 genes. Further details are in Supplementary Data 7.
Disease-state-specific gene co-expression network analysis
To evaluate gene connectivity and its role in myocardial transition from healthy to failing states, we analyzed the topological structure of gene co-expression networks in human heart tissues (Fig. 5). To construct gene co-expression networks, cardiac tissue samples from 177 failing hearts and 136 donor, non-failing (control) hearts were collected from operating rooms and remote locations for RNA expression measurements. We performed weighted gene co-expression network analysis (WGCNA) on the covariate-corrected RNA microarray data for the control and heart failure networks separately47 (Fig. 5a). Data for these networks are available at https://doi.org/10.5281/zenodo.2600420. To evaluate the degree of connectivity between correlating genes in each of the networks, we compared edge weights between lo-siRF-prioritized genes relative to the distribution of all possible gene pairs (Fig. 5b,c). We also evaluated the difference of edge weights (Z-score normalized) between the control and heart failure networks to understand how these gene–gene connectivities change between non-failing and failing hearts (Fig. 5d). Two-tailed empirical P values represent the proportion of absolute difference in edge weights of all gene pairs that exceed the absolute difference score for gene pairs of interest. In addition, we compared the structure of modules derived from dendrograms on the WGCNA control and heart failure networks (Extended Data Fig. 6). Modules were labeled according to Reactome enrichment analysis of genes within each module. Full gene module descriptions and Benjamini–Hochberg-adjusted enrichment P values are available in Supplementary Data 5 and 6 of a previous study47.
Induced pluripotent stem cell cardiomyocyte differentiation
The studied patient-specific human induced pluripotent stem cells (hiPSCs) were derived from a 45-year-old female proband with a heterozygous MYH7-R403Q mutation. Derivation and maintenance of hiPSC lines were performed following a previous study24. Briefly, hiPSCs were maintained in mTeSR (StemCell Technologies) and split at a low density (1:12) onto fresh 1:200 matrigel-coated 12-well plates. Following the split, cells were left in mTeSR medium supplemented with 1 μM thiazovivin. The hiPSCs were maintained in mTeSR until cells reached 90% confluency, which began on day 0 of the cardiomyocyte differentiation protocol. Cardiomyocytes were differentiated from hiPSCs using small-molecule inhibitors. For days 0–5, cells were given RPMI 1640 medium + l-glutamine and B27 − insulin. On days 0 and 1, the medium was supplemented with 6 μM of the GSK3β inhibitor CHIR99021. On days 2 and 3, the medium was supplemented with 5 μM of the Wnt inhibitor IWR-1. The medium was switched to RPMI 1640 medium + l-glutamine and B27 + insulin on days 6–8. On days 9–12, cells were maintained in RPMI 1640 medium + l-glutamine − glucose, B27 + insulin, and sodium lactate. On day 13, cells were detached using Accutase for 7–10 min at 37 °C and resuspended in neutralizing RPMI 1640 medium + l-glutamine and B27 + insulin. This mixture was centrifuged for 5 min at 1,000 rpm (103g). The cell pellet was resuspended in 1 μM thiazovivin-supplemented RPMI 1640 medium + l-glutamine and B27 + insulin. For the rest of the protocol (days 14–40), cells were exposed to RPMI 1640 medium + l-glutamine − glucose, B27 + insulin, and sodium lactate. Medium changes occurred every other day on days 14–19 and every 3 days for days 20–40. On day 40, cardiomyocytes reached maturity.
RNA silencing in induced pluripotent stem cell-derived cardiomyocytes
Mature hiPSC-derived cardiomyocytes (hiPSC-CMs) were transfected with Silencer Select siRNAs (Thermo Fisher Scientific) using TransIT-TKO Transfection reagent (Mirus Bio). Cells were incubated for 48 h with 75 nM siRNA treatments. Four wells of cells were transfected with each of the six siRNAs: scramble, CCDC141 (ID s49797), IGF1R (ID s223918), TTN (ID s14484), CCDC141 and IGF1R, and CCDC141 and TTN. After 2 days, hiPSC-CMs were collected for RNA extraction.
RT-qPCR analysis for siRNA gene silencing efficiency
Following cell morphology measurement, all cells for each condition were centrifuged for 5 min at 1,000 rpm (103g). Cell pellets were frozen at −80 °C before RNA extraction. RNA was extracted using Trizol reagent for reverse transcription quantitative polymerase chain reaction (RT-qPCR) to confirm that gene knockdown occurred. Reverse transcription of RNA was done using a High-Capacity cDNA Reverse Transcription Kit (Thermo Fisher Scientific). qPCR of the single-stranded cDNA was performed using TaqMan Fast Advanced MM (Thermo Fisher Scientific) with the following annealing temperatures: 95 °C for 20 sec and 40 cycles of 95 °C for 1 sec and 60 °C for 20 sec. qPCR of the silenced genes was performed using TaqMan Gene Expression Assays, including CCDC141 (Hs00892642_m1), IGF1R (Hs00609566_m1) and TTN (Hs00399225_m1). For gene-silencing efficiency analysis, gene RPLP0 (Hs00420895_gH) was used as a reference gene. Data were analyzed using the delta–delta Ct method.
Cell sample preparation for cell morphology measurement
Following siRNA treatments, cells were detached for microfluidic single-cell imaging using a mixture of 5 parts Accutase and 1 part TrypLE, treated for 6 min at 37 °C. Cells were then added to the neutralizing RPMI 1640 medium + l-glutamine and B27 + insulin. These mixtures were centrifuged for 5 min at 1,000 rpm (103g). For each gene-silencing condition, four wells of cells were resuspended in 4 ml of MEM medium, which is composed of minimum essential medium (MEM, Hank’s Balanced Salt Solution balanced), 10% fetal bovine serum and 1% Pen Strep (Gibco). Cells were filtered with 100-μm strainers (Corning) before introducing them into the microfluidic device.
Microfluidic inertial focusing device
We adopted the ‘Dean flow focusing’ concept from a previous study22 and developed a spiral inertial microfluidics system to focus randomly suspended cells into single streams based on cell size for high-resolution, high-throughput single-cell imaging. The microfluidic device (Extended Data Fig. 7) contains a five-loop spiral microchannel with an expanding radius (3.3–7.05 mm) and a cross section with a slanted ceiling (dimensions and fabrication details in Supplementary Note 3). This geometry induces strong Dean vortices in the outer half of the channel cross section, optimizing size-based separation and cell focusing. Two inlets at the spiral center are used to introduce cell suspensions and sheath flow of fresh medium, while the top and bottom outlets enable high-throughput imaging of hypertrophic and non-hypertrophic cardiomyocytes (Extended Data Fig. 9).
High-throughput single-cell imaging
Before each experiment, microchannels were flushed with 3 ml of the MEM medium. Prepared cell samples and fresh MEM medium were loaded into 3-ml syringes, which were connected to the corresponding microchannel inlets using Tygon PVC tubing (McMaster). Both cells and the fresh MEM medium were infused into the microchannel using a Pico Plus Elite syringe pump (Harvard Apparatus) at 1.2 ml min−1. Microscope image sequences of cells focused to the top and bottom observation channels were captured using a VEO 710S high-speed camera (Phantom) with a sampling rate of 700 fps and a 5-μs light exposure.
Image analysis for cell feature extraction
For each gene-silencing condition in each biological replicate, 21,000 images were processed to extract cell morphology features. To analyze cell size and shape changes induced by gene silencing, we developed a MATLAB-based image analysis pipeline with three major steps: image preparation, feature extraction and post-processing (Fig. 6c). In step 1, image sequences were background corrected by subtracting an automatically generated background image, in which each pixel’s value was computed as the mode intensity value of that pixel location across the entire image sequence. After illumination correction, step 2 detects cell edges using local maxima of the bright-field intensity gradient, following which the program closes edge gaps, removes border-adjacent cells, filters small features (noise) and fills holes to generate binary images with centroid positions for each cell. A double-counting filter was used to exclude repeated measurements of stuck cells collected in the same location and cell size range using a Gaussian kernel density method (bandwidth = 0.09). Binary images passing the double-counting filter were used to create cell outline coordinates (X, Y), which leads to various morphological features, including cell area (2D integration of the cell outline), diameter (\(2\sqrt{{\rm{area}}/\pi }\)), solidity (cell area/convex hull area), roundness error (s.d./mean of radii from the centroid), circularity (4π × area/perimeter2) and intensity spatial relationship enclosed within the cell boundary. The 2D intensity distributions within cell outlines were used to derive peak locations and count peak numbers, which are measures of intensity spatial relationship and a gating parameter to remove clumped cells. In the post-processing step, data were filtered using three gating thresholds. To remove large clumps, the peak-solidity filter removes data outside of the polygonal region defined by {(0.9, 0), (0.9, 3.2), (0.934, 8.26), (1, 28), (1, 0)} in the (solidity, peak number) space. Then, the roundness filter removes cells with weird shapes by excluding data with a roundness error higher than 0.3 or a circularity lower than 0.6. Finally, the small-size filter removes cell debris whose major diameter is lower than 15 μm (12 μm) or minor diameter is lower than 12 μm (10 μm) for images photographed at the top (bottom) outlet microchannels. All these filters were manually validated through visual inspection.
Statistical assessment of gene-silencing effects on single-cell morphology
We conducted various statistical analyses to investigate how and where cell size distributions differ between the gene- or gene-pair-silenced groups and their respective scrambled control groups. First, we compared differences in median cell size (that is, diameter in μm) using both a two-sided Mann–Whitney U test (Fig. 7c) and a bootstrap quantile test at the 0.5 quantile level. In accordance with the PCS framework, we used two different tests to ensure that our findings are robust to this arbitrary modeling choice. Median cell size difference was of greater interest than mean size difference owing to the heavy right skewness of cell size distributions. In addition, we compared differences in upper quantiles (0.6, 0.7, 0.8 and 0.9 quantile levels) of size distributions between gene- or gene-pair-silenced cells and their scrambled controls using a bootstrap quantile test (Extended Data Fig. 8). Identifying cell size differences at these upper quantiles, which highlight larger, hypertrophic cells, is particularly relevant for understanding the pathologic phenotype of cardiac hypertrophy and its clinical implications. All tests were performed separately for each experimental batch, and the most conservative P value (maximum across batches) was reported. Similar analyses were conducted for assessing differences in morphological features (that is, cell roundness error and normalized peak number).
Statistical assessment of nonadditivity in gene-silencing effects
We next assessed whether gene pairs (CCDC141–TTN and CCDC141–IGF1R) affect cell size additively or nonadditively (that is, through epistasis; Fig. 7d). To assess this (non-)additivity for a given gene pair, say gene 1 and gene 2, we fit the following quantile regression:
where yi is the diameter (μm) of cell i; xi1 and xi2 indicate whether gene 1 and gene 2 were silenced in cell i, respectively; \({\mu }_{{g}_{i}}\) accounts for batch effects (with gi denoting the batch identifier for cell i); and ϵi is the random error or noise term for cell i. This regression was fitted using scrambled control cells (xi1 = xi2 = 0), gene 1-silenced cells (xi1 = 1; xi2 = 0), gene 2-silenced cells (xi1 = 0; xi2 = 1) and double-gene-silenced cells (xi1 = xi2 = 1). Under this regression model, we tested the null hypothesis β12 = 0 versus the alternative hypothesis β12 ≠ 0 via a percentile bootstrap t-test and a traditional t-test (Supplementary Data 8) for varying quantile levels (0.5, 0.6, 0.7, 0.8, 0.9). A small P value suggests nonadditive epistatic interaction. Again, two different tests were performed to ensure robustness against modeling assumptions. To further bolster the robustness of our conclusions, we repeated this assessment of epistasis using a rank-based inverse normal-transformed cell diameter as the response y in an ordinary linear regression model, yielding similar P values and directions of nonadditive interaction effects (Supplementary Data 8). As gene-silencing efficiency varied across conditions (for example, silencing CCDC141 and TTN versus silencing only CCDC141 versus silencing only TTN), we restricted the analysis to experimental batches with high gene-silencing efficiencies (>60%) to minimize spurious epistatic signals. See Supplementary Note 5 (available on the website https://yu-group.github.io/epistasis-cardiac-hypertrophy/simulations_efficiency) for further discussion and simulations. Similar analyses were conducted to assess differences in morphological features (that is, cell roundness error and normalized peak number).
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
All genotype and cardiac MRI data used as input to the lo-siRF pipeline are available from the UKBB (https://www.ukbiobank.ac.uk/). This work was conducted under UKBB application 22282. Data for the gene co-expression networks from 313 explanted human hearts are available via Zenodo at https://doi.org/10.5281/zenodo.2600420. The RNA-seq data used to establish the healthy and heart failure gene co-expression networks are available in the Gene Expression Omnibus under accession number GSE57338. Other databases used in this study include RegulomeDB (v2.2), CADD (v1.4), GTEx (v8), Enrichr (last updated version: 8 June 2023) and ChIP-X Enrichment Analysis Version 3 (ChEA3). All other data have been provided in Supplementary Information.
Code availability
All code for running the custom lo-siRF analysis and analyzing the experimental results can be found via GitHub at https://github.com/Yu-Group/epistasis-cardiac-hypertrophy. This custom lo-siRF analysis was conducted using R v3.6.1, Python v3.6.1 and iRF2.0 (https://github.com/karlkumbier/iRF2.0). The LVMi derivation from cardiac MRI images was computed using the ukbb_cardiac package (https://github.com/baiwenjia/ukbb_cardiac) and Python v3.9. PLINK v1.90b5.3 (https://www.cog-genomics.org/plink/) and BOLT-LMM v2.3.4 (https://alkesgroup.broadinstitute.org/BOLT-LMM/BOLT-LMM_manual.html) were used to perform the GWAS dimension reduction. ANNOVAR (downloaded version: 8 June 2020; https://annovar.openbioinformatics.org/en/latest/) was used to map each SNV to a genetic locus within lo-siRF. Comparisons with existing methods were performed using the SKAT R package (v2.2.5), mvMAPIT R package (v2.0.3), GSEA v4.3.3 (https://www.gsea-msigdb.org/gsea/index.jsp) and MAGMA (v1.6). Functional annotation and gene mapping were performed using FUMA v1.5.4 (https://fuma.ctglab.nl/), ChromHMM v1.10 core 15-state (https://egg2.wustl.edu/roadmap/web_portal/chr_state_learning.html), RegulomeDB v2.2 (https://regulomedb.org/regulome-search/), CADD v1.4 (https://cadd.gs.washington.edu/) and GTEx v8 (https://www.gtexportal.org/home/). The Circos plot was made using TBtools (v2.210). Gene set enrichment analyses for GOs, pathways and TFs were performed using Enrichr (last updated version: 8 June 2023; https://maayanlab.cloud/Enrichr/), ChEA3 (https://maayanlab.cloud/chea3/) and MATLAB R2019a. Healthy and heart failure network analyses were performed previously in R v3.5.2. Cell boundary extraction and morphology analysis from single-cell images was performed using MATLAB R2019a.
References
Weldy, C. S. & Ashley, E. A. Towards precision medicine in heart failure. Nat. Rev. Cardiol. 18, 745–762 (2021).
Bastos, M. B. et al. Invasive left ventricle pressure–volume analysis: overview and practical clinical implications. Eur. Heart J. 41, 1286–1297 (2019).
Burkhoff, D., Mirsky, I. & Suga, H. Assessment of systolic and diastolic ventricular properties via pressure–volume analysis: a guide for clinical, translational, and basic researchers. Am. J. Physiol. Heart Circ. Physiol. 289, H501–H512 (2005).
Marian, A. J. & Braunwald, E. Hypertrophic cardiomyopathy: genetics, pathogenesis, clinical manifestations, diagnosis, and therapy. Circ. Res. 121, 749–770 (2017).
Haider, A. W., Larson, M. G., Benjamin, E. J. & Levy, D. Increased left ventricular mass and hypertrophy are associated with increased risk for sudden death. J. Am. Coll. Cardiol. 32, 1454–1459 (1998).
Kawel-Boehm, N. et al. Left ventricular mass at MRI and long-term risk of cardiovascular events: the Multi-Ethnic Study of Atherosclerosis (MESA). Radiology 293, 107–114 (2019).
Pirruccello, J. P. et al. Analysis of cardiac magnetic resonance imaging in 36,000 individuals yields genetic insights into dilated cardiomyopathy. Nat. Commun. 11, 2254 (2020).
Meyer, H. V. et al. Genetic and functional insights into the fractal structure of the heart. Nature 584, 589–594 (2020).
Harper, A. R. et al. Common genetic variants and modifiable risk factors underpin hypertrophic cardiomyopathy susceptibility and expressivity. Nat. Genet. 53, 135–142 (2021).
O’Sullivan, J. W. et al. Polygenic risk scores for cardiovascular disease: a scientific statement from the American Heart Association. Circulation 146, e93–e118 (2022).
Guindo-Martínez, M. et al. The impact of non-additive genetic associations on age-related complex diseases. Nat. Commun. 12, 2436 (2021).
Zeng, L. et al. Cis-epistasis at the LPA locus and risk of cardiovascular diseases. Cardiovasc. Res. 118, 1088–1102 (2022).
Li, Y. et al. Statistical and functional studies identify epistasis of cardiovascular risk genomic variants from genome‐wide association studies. J. Am. Heart Assoc. 9, e014146 (2020).
Hivert, V. et al. Estimation of non-additive genetic variance in human complex traits from a large sample of unrelated individuals. Am. J. Hum. Genet. 108, 786–798 (2021).
Mackay, T. F. & Moore, J. H. Why epistasis is important for tackling complex human disease genetics. Genome Med. 6, 124 (2014).
Basu, S., Kumbier, K., Brown, J. B. & Yu, B. Iterative random forests to discover predictive and stable high-order interactions. Proc. Natl Acad. Sci. USA 115, 1943–1948 (2018).
Kumbier, K. et al. Signed iterative random forests to identify enhancer-associated transcription factor binding. Preprint at https://arxiv.org/abs/1810.07287 (2023).
Murk, W., Bracken, M. B. & DeWan, A. T. Confronting the missing epistasis problem: on the reproducibility of gene–gene interactions. Hum. Genet. 134, 837–849 (2015).
Yu, B. & Kumbier, K. Veridical data science. Proc. Natl Acad. Sci. USA 117, 3920–3929 (2020).
Bai, W. et al. Automated cardiovascular magnetic resonance image analysis with fully convolutional networks. J. Cardiovasc. Magn. Reson. 20, 65 (2018).
Wang, Q. et al. Microfluidic dielectrophoresis illuminates the relationship between microbial cell envelope polarizability and electrochemical activity. Sci. Adv. 5, eaat5664 (2019).
Guan, G. et al. Spiral microchannel with rectangular and trapezoidal cross-sections for size based particle separation. Sci. Rep. 3, 1475 (2013).
Wu, P.-H. et al. Single-cell morphology encodes metastatic potential. Sci. Adv. 6, eaaw6938 (2020).
Dainis, A. et al. Silencing of MYH7 ameliorates disease phenotypes in human iPSC-cardiomyocytes. Physiol. Genomics 52, 293–303 (2020).
Littlejohns, T. J. et al. The UK Biobank imaging enhancement of 100,000 participants: rationale, data collection, management and future directions. Nat. Commun. 11, 2624 (2020).
Purcell, S. et al. PLINK: a tool set for whole-genome association and population-based linkage analyses. Am. J. Hum. Genet. 81, 559–575 (2007).
Loh, P.-R. et al. Efficient Bayesian mixed-model analysis increases association power in large cohorts. Nat. Genet. 47, 284–290 (2015).
Zhu, S. & Fang, G. MatrixEpistasis: ultrafast, exhaustive epistasis scan for quantitative traits with covariate adjustment. Bioinformatics 34, 2341–2348 (2018).
Crawford, L., Zeng, P., Mukherjee, S. & Zhou, X. Detecting epistasis with the marginal epistasis test in genetic mapping studies of quantitative traits. PLoS Genet. 13, e1006869 (2017).
Subramanian, A. et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl Acad. Sci. USA 102, 15545–15550 (2005).
McInnes, G. et al. Global Biobank Engine: enabling genotype–phenotype browsing for biobank summary statistics. Bioinformatics 35, 2495–2497 (2019).
Yildiz, M. et al. Left ventricular hypertrophy and hypertension. Prog. Cardiovasc. Dis. 63, 10–21 (2020).
Watanabe, K., Taskesen, E., van Bochoven, A. & Posthuma, D. Functional mapping and annotation of genetic associations with FUMA. Nat. Commun. 8, 1826 (2017).
Roadmap Epigenomics Consortium. et al. Integrative analysis of 111 reference human epigenomes. Nature 518, 317–330 (2015).
Boyle, A. P. et al. Annotation of functional variation in personal genomes using RegulomeDB. Genome Res. 22, 1790–1797 (2012).
Kircher, M. et al. A general framework for estimating the relative pathogenicity of human genetic variants. Nat. Genet. 46, 310–315 (2014).
GTEx Consortium. The Genotype-Tissue Expression (GTEx) project. Nat. Genet. 45, 580–585 (2013).
Khurshid, S. et al. Clinical and genetic associations of deep learning-derived cardiac magnetic resonance-based left ventricular mass. Nat. Commun. 14, 1558 (2023).
Aung, N. et al. Genome-wide analysis of left ventricular image-derived phenotypes identifies fourteen loci associated with cardiac morphogenesis and heart failure development. Circulation 140, 1318–1330 (2019).
Verweij, N., van de Vegte, Y. J. & van der Harst, P. Genetic study links components of the autonomous nervous system to heart-rate profile during exercise. Nat. Commun. 9, 898 (2018).
Thorolfsdottir, R. B. et al. Genetic insight into sick sinus syndrome. Eur. Heart J. 42, 1959–1971 (2021).
Lee, S. et al. Optimal unified approach for rare-variant association testing with application to small-sample case-control whole-exome sequencing studies. Am. J. Hum. Genet. 91, 224–237 (2012).
Kuleshov, M. V. et al. Enrichr: a comprehensive gene set enrichment analysis web server 2016 update. Nucleic Acids Res. 44, W90–W97 (2016).
Keenan, A. B. et al. ChEA3: transcription factor enrichment analysis by orthogonal omics integration. Nucleic Acids Res. 47, W212–W224 (2019).
Li, L. et al. Attention-deficit/hyperactivity disorder as a risk factor for cardiovascular diseases: a nationwide population-based cohort study. World Psychiatry 21, 452–459 (2022).
Parikh, V. N. et al. Regional variation in RBM20 causes a highly penetrant arrhythmogenic cardiomyopathy. Circ. Heart Fail. 12, e005371 (2019).
Cordero, P. et al. Pathologic gene network rewiring implicates PPP1R3A as a central regulator in pressure overload heart failure. Nat. Commun. 10, 2760 (2019).
Hood, K., Kahkeshani, S., Di Carlo, D. & Roper, M. Direct measurement of particle inertial migration in rectangular microchannels. Lab Chip 16, 2840–2850 (2016).
Stavrakis, S., Holzner, G., Choo, J. & deMello, A. High-throughput microfluidic imaging flow cytometry. Curr. Opin. Biotechnol. 55, 36–43 (2019).
Alizadeh, E., Xu, W., Castle, J., Foss, J. & Prasad, A. TISMorph: a tool to quantify texture, irregularity and spreading of single cells. PLoS ONE 14, e0217346 (2019).
Wan, X. et al. BOOST: a fast approach to detecting gene–gene interactions in genome-wide case-control studies. Am. J. Hum. Genet. 87, 325–340 (2010).
Ueki, M. & Cordell, H. J. Improved statistics for genome-wide interaction analysis. PLoS Genet. 8, e1002625 (2012).
Nelson, D. L., Lehninger, A. L. & Cox, M. M. Lehninger Principles of Biochemistry (Macmillan, 2008).
Adams, S. M. et al. Genome wide epistasis study of on-statin cardiovascular events with iterative feature reduction and selection. J. Pers. Med. 10, 212 (2020).
Hornung, R. & Boulesteix, A.-L. Interaction forests: identifying and exploiting interpretable quantitative and qualitative interaction effects. Comput. Stat. Data Anal. 171, 107460 (2022).
Demetci, P. et al. Multi-scale inference of genetic trait architecture using biologically annotated neural networks. PLoS Genet. 17, e1009754 (2021).
Singhal, P. et al. Evidence of epistasis in regions of long-range linkage disequilibrium across five complex diseases in the UK Biobank and eMERGE datasets. Am. J. Hum. Genet. 110, 575–591 (2023).
Bycroft, C. et al. The UK Biobank resource with deep phenotyping and genomic data. Nature 562, 203–209 (2018).
Morgan, M. D. et al. Genome-wide study of hair colour in UK Biobank explains most of the SNP heritability. Nat. Commun. 9, 5271 (2018).
Petersen, S. E. et al. Reference ranges for cardiac structure and function using cardiovascular magnetic resonance (CMR) in Caucasians from the UK Biobank population cohort. J. Cardiovasc. Magn. Reson. 19, 18 (2017).
Grothues, F. et al. Comparison of interstudy reproducibility of cardiovascular magnetic resonance with two-dimensional echocardiography in normal subjects and in patients with heart failure or left ventricular hypertrophy. Am. J. Cardiol. 90, 29–34 (2002).
Du Bois, D. & Du Bois, E. F. Clinical calorimetry: tenth paper a formula to estimate the approximate surface area if height and weight be known. JAMA Intern. Med. 17, 863–871 (1916).
Schaid, D. J., Chen, W. & Larson, N. B. From genome-wide associations to candidate causal variants by statistical fine-mapping. Nat. Rev. Genet. 19, 491–504 (2018).
Yoshida, M. & Koike, A. SNPInterForest: a new method for detecting epistatic interactions. BMC Bioinformatics 12, 469 (2011).
Saha, S., Perrin, L., Röder, L., Brun, C. & Spinelli, L. Epi-MEIF: detecting higher order epistatic interactions for complex traits using mixed effect conditional inference forests. Nucleic Acids Res. 50, e114 (2022).
Chang, C. C. et al. Second-generation PLINK: rising to the challenge of larger and richer datasets. Gigascience 4, 7 (2015).
Shah, R. D. & Meinshausen, N. Random intersection trees. J. Mach. Learn. Res. 15, 629–654 (2014).
Tibshirani, R. Regression shrinkage and selection via the lasso. J. R. Stat. Soc. Ser. B 58, 267–288 (1996).
Hoerl, A. E. & Kennard, R. W. Ridge regression: biased estimation for nonorthogonal problems. Technometrics 12, 55–67 (1970).
Breiman, L. Random forests. Mach. Learn. 45, 5–32 (2001).
Cortes, C. & Vapnik, V. Support-vector networks. Mach. Learn. 20, 273–297 (1995).
Pedregosa, F. et al. Scikit-learn: machine learning in Python. J. Mach. Learn. Res. 12, 2825–2830 (2011).
Erickson, N. et al. AutoGluon-tabular: robust and accurate AutoML for structured data. Preprint at https://arxiv.org/abs/2003.06505 (2020).
Wang, K., Li, M. & Hakonarson, H. ANNOVAR: functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res. 38, e164 (2010).
Duncan, J., Tang, T., Elliott, C. F., Boileau, P. & Yu, B. simChef: high-quality data science simulations in R. J. Open Source Softw. 9, 6156 (2024).
Chen, C. et al. TBtools: an integrative toolkit developed for interactive analyses of big biological data. Mol. Plant 13, 1194–1202 (2020).
Evangelista, J. E. et al. Enrichr-KG: bridging enrichment analysis across multiple libraries. Nucleic Acids Res. 51, W168–W179 (2023).
Ashburner, M. et al. Gene Ontology: tool for the unification of biology. The Gene Ontology Consortium. Nat. Genet. 25, 25–29 (2000).
Gene Ontology Consortium. et al. The Gene Ontology knowledgebase in 2023. Genetics 224, iyad031 (2023).
Smith, C. L. & Eppig, J. T. The mammalian phenotype ontology: enabling robust annotation and comparative analysis. Wiley Interdiscip. Rev. Syst. Biol. Med. 1, 390–399 (2009).
Gillespie, M. et al. The reactome pathway knowledgebase 2022. Nucleic Acids Res. 50, D687–D692 (2022).
Kanehisa, M. & Goto, S. KEGG: Kyoto Encyclopedia of Genes and Genomes. Nucleic Acids Res. 28, 27–30 (2000).
Davis, C. A. et al. The Encyclopedia of DNA Elements (ENCODE): data portal update. Nucleic Acids Res. 46, D794–D801 (2018).
Chèneby, J., Gheorghe, M., Artufel, M., Mathelier, A. & Ballester, B. ReMap 2018: an updated atlas of regulatory regions from an integrative analysis of DNA-binding ChIP-seq experiments. Nucleic Acids Res. 46, D267–D275 (2018).
Lachmann, A. et al. Massive mining of publicly available RNA-seq data from human and mouse. Nat. Commun. 9, 1366 (2018).
Portales-Casamar, E. et al. JASPAR 2010: the greatly expanded open-access database of transcription factor binding profiles. Nucleic Acids Res. 38, D105–D110 (2010).
Matys, V. et al. TRANSFAC and its module TRANSCompel: transcriptional gene regulation in eukaryotes. Nucleic Acids Res. 34, D108–D110 (2006).
Han, H. et al. TRRUST v2: an expanded reference database of human and mouse transcriptional regulatory interactions. Nucleic Acids Res. 46, D380–D386 (2018).
Acknowledgements
We would like to acknowledge D. Amar (Stanford University), S. Upadhyayula (University of California, Berkeley), H. Huang (University of California, Berkeley), Z. Obermeyer (University of California, Berkeley) and L. Crawford (Microsoft Research; Brown University) for their critical comments and discussions on this work; E. Enriquez (Stanford Nanofabrication Facility) for the technical support of microfluidic device fabrication; and A. Shcherbina (Stanford University) and M. Rivas (Stanford University) for their technical help with the UKBB data. We would also like to thank the editor for their invaluable feedback and discussions, which have greatly improved this work. This work was supported by the Chan Zuckerberg Biohub—San Francisco through the Intercampus Research Awards (2019–2022) (R.A., J.R.P., J.B.B., A.J.B., E.A.A., B.Y.); the National Institutes of Health through grant numbers 1R01HL144843 (E.A.A.), R01GM152718 (B.Y.), K08HL143185 (V.N.P.), R01HL105993 (W.H.W.T., K.B.M., T.P.C., E.A.A.), and F32HL160067, K08HL167699 and L30HL159413 (C.S.W.); the American Heart Association through grant numbers 23POST1023278 (Q.W.) and 23CDA1042900 (C.S.W.); the National Science Foundation through grants DMS-1613002 and IIS 1741340 (B.Y.); the Graduate Research Fellowship Program DGE-2146752 (T.M.T.); and a Weill Neurohub grant (B.Y.).
Author information
Authors and Affiliations
Contributions
Q.W., T.M.T., C.S.W., J.W.H., A.J.B., R.A., J.B.B., J.R.P., V.N.P., B.Y. and E.A.A. conceived and designed the research. J.W.H. performed LVMi extraction from cardiac MRI images. T.M.T., A.A., X.L., M.B. and K.K. performed exploratory data investigations leading to the development of lo-siRF. T.M.T., A.A. and B.Y. developed the lo-siRF pipeline. T.M.T. performed the lo-siRF analysis. Q.W. and T.M.T. performed the FUMA SNP2GENE process using lo-siRF-prioritized SNVs and GWAS-filtered SNVs. Q.W. evaluated functional annotation results from FUMA, performed the ANNOVAR enrichment test for each lo-siRF loci and functional gene mapping. Q.W. performed integrative biological enrichment analyses and evaluated the co-associations between lo-siRF hypothesized gene–gene interactions and the enriched GOs, pathways and TFs. E.T.C., C.S.M., W.H.W.T., K.B.M., T.P.C., V.N.P. and E.A.A. contributed to the data collection and construction of WGCNA healthy and heart failure co-expression networks. Q.W. and E.T.C. evaluated connectivity differences of epistatic genes hypothesized by lo-siRF between cardiac co-expression networks of failing and non-failing hearts. Q.W. designed and created microfluidic devices; M.Y., S.C.S. and V.N.P. created gene-silenced hiPSC-CM lines; Q.W. and M.Y. performed the microfluidic single-cell imaging experiments. Q.W., O.R. and A.M.K. performed single-cell image analysis and morphological feature extraction. Q.W., T.M.T., A.M.K., O.R., C.S.W., V.N.P., J.W.H., B.Y. and E.A.A. interpreted the results of the experiments; Q.W., T.M.T. and A.M.K. prepared the figures; Q.W., T.M.T., C.S.W. and J.W.H. drafted the paper. All authors contributed to the editing and revising of the paper.
Corresponding authors
Ethics declarations
Competing interests
E.A.A. is a founder of Personalis, Deepcell, Svexa, Candela, Saturnus Bio and Parameter Health; an advisor to SequenceBio, Foresite Labs, Pacific Biosciences and Versant Ventures; a non-executive director for AstraZeneca and Svexa; and a stockholder in Pacific Biosciences and AstraZeneca; and has received in-kind collaborative support from Illumina, Pacific Biosciences, Oxford Nanopore, Cache and Cellsonics. V.N.P. is a Scientific Advisory Board (SAB) member for and receives research support from BioMarin, Inc., a SAB member for Lexeo Therapeutics and a consultant for Constantiam Biosciences and viz.ai. C.S.W. is a consultant for AiRNA Bio and Avidity Biosciences. While these companies may have interests in genomics or cardiovascular health, they had no direct role in the design, data presentation, analysis or interpretation of this study. The other authors declare no competing interest.
Peer review
Peer review information
Nature Cardiovascular Research thanks Declan O’Regan and the other, anonymous, reviewer(s) for their contribution to the peer review of this work.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Extended data
Extended Data Fig. 1 Distribution of LVM and LVMi measurements for 29,661 UK Biobank participants.
Left ventricular mass (LVM, a) and LVM indexed to body surface area (LVMi, b) measurements were extracted from cardiac magnetic resonance imaging for 29,661 unrelated White British individuals via deep learning20. c, A high Pearson correlation of 0.92 was observed between these LVM and LVMi measurements.
Extended Data Fig. 2 LVMi GWAS using BOLT-LMM and PLINK.
Two-sided GWAS p-values from BOLT-LMM (a) and PLINK (b) identified LVMi-associated SNVs, of which the top lead SNV rs3045696 showed the highest significance in both analyses. Other labeled lead SNVs were significant in only one of the two methods. The red dashed line indicates the genome-wide significance threshold (p < 5E-8, two-sided). Notably, rs3045696 and rs67172995 were also stably prioritized by lo-siRF as epistasis interactor variants. Further details are provided in Supplementary Data 1.
Extended Data Fig. 3 Differences in local stability scores between high and low LV mass highlight the importance of the lo-siRF-prioritized interactions between genetic loci.
a, Schematic of local stability importance score computation. Given a locus (light blue transcript), the local stability importance score for an individual is defined as the proportion of trees for which at least one SNV (shaded black region) in the locus is used in the individual’s decision path. This computation (top) was performed for each individual (denoted by the stacked boxes). Then, a two-sided permutation test was conducted to assess the difference in these local stability importance scores between the low and high LVMi individuals (bottom). b, Differences in the distribution of local stability importance scores suggest that the identified interactions between genetic loci are important for differentiating individuals with high (dark gray) and low (orange) LVMi in the siRF fit. This result, evaluated on the validation data, is stable across the three binarization thresholds and is quantified by a nominal two-sided p-value from a permutation test given in the top right corner of each subplot. Given the use of 10,000 permutations, p-values cannot be reported with greater precision than p = 1E-4.
Extended Data Fig. 4 Top SNVs from lo-siRF-prioritized loci and interactions between loci.
The most important SNVs and SNV-SNV pairs, as measured by their proportion of occurrence in the siRF fit, are annotated for the top lo-siRF-prioritized interactions between loci in a and top genetic loci in b. The y-axis shows the proportion of decision paths in siRF, for which the SNV or SNV-SNV pair occurs, averaged across all three binarization thresholds. In each of the interactions between genetic loci, rs7591091 in the CCDC141 locus appears most frequently, suggesting a key role in cardiac hypertrophy.
Extended Data Fig. 5 Genes mapped from epistatic loci share transcription factors and splicing regulators.
Gene pairs exhibiting strong co-association with transcription factors (TFs) and RNA-binding regulators (p < 0.05, two-sided Fisher’s exact test, Fig. 4e) are displayed. Horizontal bars indicate the number of shared TFs or RNA-binding regulators (bottom axis), with the top-ranked TF or regulator, determined by the lowest two-sided enrichment p-value (dots, top axis), labeled on each bar. Bars are color-coded by the corresponding gene set library. Detailed information can be found in Supplementary Data 7.
Extended Data Fig. 6 Dendrograms from WGCNA network analyses.
Dendrograms from WGCNA control (a) and heart failure (b) networks show distinctive gene module structures and modular assignments for CCDC141, IGF1R, and TTN.
Extended Data Fig. 7 Spiral-shaped inertial microfluidic channel for cell focusing and imaging.
a, Schematic of an inertial microfluidic cell focusing device. Cell suspensions and fresh medium were introduced into the microfluidic device through the cell and sheath flow inlets, respectively, using a syringe pump and flowed down the 5-loop spiral microchannel with the same flow rate (1.2 mL/min). Inserted microscopy images show that randomly dispersed cells were separated by size and bifurcated into the top (large cells) or bottom (small cells) outlets. Scale bar, 10 μm. Outlet channels are connected to straight observation channels where flowing cells were further focused in the channel height direction and imaged using a high-speed camera for morphological feature extraction (Fig. 6c). b, Schematic of the cell focusing principle. The spiral microchannel has a cross-section with a slanted ceiling, resulting in different depths at the inner and outer side of the microchannel. This geometry induces strong Dean vortices (counter rotating vortices in the plane perpendicular to the main flow direction) in the outer half of the microchannel cross-section. The interplay between drag forces (FD) induced by Dean vortices and lift forces (FL) due to shear gradient and the channel wall drives cell transverse migration towards equilibrium positions where the net force is zero. As a result, large cells in a heterogeneous population progressively migrate closer to the inner channel wall, while smaller cells move towards the outer channel wall. Details about microchannel dimensions can be found in Methods.
Extended Data Fig. 8 Epistatic genes non-uniformly reshape cardiomyocyte size distributions.
a, Heatmap showing relative differences in cell sizes at various quantile levels between gene-silenced and scramble control conditions for unaffected and MYH7-R403Q variant cardiomyocytes. Larger quantiles correspond to larger cells in the cell size distribution. Dark red indicates strong size reduction at the specified quantile level in gene-silenced cells relative to scramble controls. The corresponding statistical differences (b) were evaluated by the maximum (two-sided) p-values across n = 3 (n = 4 for unaffected cells silencing CCDC141) independent cell batches using a bootstrap quantile test with 10,000 bootstrapped samples (exact p values per batch are available in Source Data). c, QQ-plots of averaged cell size quantiles across cell batches compare gene-silenced cells against scramble controls in unaffected (top row) and MYH7-R403Q variant (bottom row) cardiomyocytes, revealing a clear size-dependent effect of silencing CCDC141-IGF1R on correcting cardiomyocyte hypertrophy.
Extended Data Fig. 9 Effects of lo-siRF-prioritized genes and gene-gene interactions on hypertrophic and non-hypertrophic cell morphology.
Relative differences in median cell size (a), normalized peak number (b), and roundness error (c) were analyzed separately for cells size-sorted into the top (hypertrophic cells, green) and bottom (non-hypertrophic cells, orange) microchannel outlets (Extended Data Fig. 7), as well as for all cells combined (blue). For both unaffected (left) and MYH7-R403Q variant (right) cardiomyocytes, the gene-silencing induced relative differences in each morphological feature (a-c) were quantified as \(\left({m}_{s}-{m}_{c}\right)/{m}_{c}\times 100 \%\), where \({m}_{s}\) and \({m}_{c}\) denote median values in gene-silencing and scrambled control conditions, respectively. Squares represent mean relative differences across n = 3 independent cell batches (n = 4 for unaffected cells silencing CCDC141), with overlaid points indicating medians of individual replicates. Violins display mean relative differences from 1,000 bootstrap samples, with error bars indicating standard deviations. Asterisks indicate significant differences compared to scramble controls, based on the maximum p-values of two-sided Mann-Whitney U test across all cell batches (*p < 0.05, **p < 1E-3, ***p < 1E-4). Exact p-values and corresponding cell counts per condition per batch are provided in Source Data.
Supplementary information
Supplementary Information
Supplementary Note 1: a description of the HTML (webpage style) PCS documentation for lo-siRF. Supplementary Note 2: methodological details for the implementation of alternative epistasis detection methods as a comparison to lo-siRF. Supplementary Note 3: descriptions of the microfluidic device geometry, dimensions and fabrication. Supplementary Note 4: discussion of the nonadditive gene-silencing effects of CCDC141, IGF1R and TTN on cardiomyocyte size distribution. Supplementary Note 5: a description of the HTML (webpage style) documentation for the simulations used to examine how various gene-silencing efficiencies impact the significance testing for epistasis.
Supplementary Tables
Supplementary Tables 1–7.
Supplementary Data 1
GWAS results for left ventricular mass (LVM). A summary of the LVM and LVMi GWAS results using PLINK and BOLT-LMM. This includes a list of the LVM and LVMi risk loci, lead SNVs, independent significant SNVs and positionally mapped genes from FUMA GWAS.
Supplementary Data 2
List of GWAS-filtered SNVs used in the lo-siRF pipeline. A list of the 1,405 SNVs that passed the GWAS dimension reduction step in the lo-siRF pipeline. This table includes the rsID, chromosome, hg19 position, allelic information, corresponding genetic loci in lo-siRF and inferred (exonic) function.
Supplementary Data 3
The lo-siRF-prioritized SNVs and SNV–SNV epistatic interactions. A summary of the 283 lo-siRF-prioritized SNVs that show stable association with LVMi, which are aggregated into six left ventricular hypertrophy risk loci (Table 1) according to ANNOVAR gene annotation in lo-siRF. The spreadsheet also includes separate tabs showing the prioritized SNV–SNV pairs for each of the three locus-to-locus interactions (CCDC141–IGF1R, CCDC141–TTN and CCDC141–LOC157273;TNKS) stably identified by all the three LVMi binarization thresholds in lo-siRF.
Supplementary Data 4
Functional annotations for lo-siRF-prioritized SNVs and their LD-linked SNVs extracted by FUMA, along with the functionally mapped genes. The spreadsheet includes a list of the 283 lo-siRF-prioritized SNVs and the FUMA-extracted candidate SNVs in LD with any of the 283 lo-siRF-prioritized SNVs, as well as their corresponding functional annotations from ANNOVAR, ChromHMM core 15 chromatin state, CADD score, RegulomeDB score, eQTL and sQTL information from GTEx, and GWAS statistics. The spreadsheet also includes a list of protein-coding genes functionally linked from the 283 lo-siRF-prioritized SNVs and their LD-linked SNVs by positional, eQTL and chromatin interaction mapping in FUMA.
Supplementary Data 5
ANNOVAR enrichment analysis results. ANNOVAR enrichment of functional consequences of all SNVs (both the lo-siRF-prioritized SNVs and their LD-linked SNVs extracted by FUMA, details in Supplementary Data 4) associated with each of the six lo-siRF-prioritized risk loci for left ventricular hypertrophy.
Supplementary Data 6
GO and pathway co-association network. Co-association network between genes that are functionally linked to the lo-siRF-prioritized epistatic and hypostatic loci (Supplementary Data 4) and their shared GO or pathway terms. GOs and pathways are enriched from multiple annotated gene set libraries from Enrichr (Fig. 4c).
Supplementary Data 7
Transcription factor co-association network. The spreadsheet includes two lists of transcription factors and RNA-binding regulators enriched from lo-siRF-prioritized and lo-siRF-deprioritized genes (Fig. 4d), respectively. It also includes a summary of co-associations between lo-siRF-prioritized genes and their shared regulatory factors (Fig. 4e). Transcription factors and RNA-binding regulators in the co-association network are enriched from nine distinct annotated gene set libraries from Enrichr and ChEA3.
Supplementary Data 8
Single cardiomyocyte morphology assessment and statistical analysis. A summary of the epistatic relationships in cardiomyocyte morphology confirmed by gene perturbation experiments (Fig. 7). The spreadsheet includes an evaluation of epistatic effects on cell size (at various quantile levels), cell textural irregularity and cell boundary irregularity, supported by multiple statistical analyses and assessment of nonadditivity for the investigated gene–gene interactions.
Supplementary Webpage 1
An HTML webpage providing a comprehensive PCS documentation for lo-siRF. Please unzip the file and open it in a web browser to view the content. Descriptions of the webpage are available in Supplementary Note 1.
Supplementary Webpage 2
An HTML webpage summarizing the simulation studies we used to examine the effect of varying gene-silencing efficiencies on epistasis testing. Descriptions of the webpage are provided in Supplementary Note 5.
Source data
Source Data Fig. 3
Source data and statistical results for Fig. 3a,b.
Source Data Fig. 4
Source data and statistical results for Fig. 4a–e.
Source Data Fig. 5
Source data and statistical results for Fig. 5b–e.
Source Data Fig. 7
Source data and statistical results for Fig. 7a–e.
Source Data Extended Data Fig. 1
Source data for Extended Data Fig. 1a–c.
Source Data Extended Data Fig. 2
Source data for Extended Data Fig. 2a,b.
Source Data Extended Data Fig. 3
Source data for Extended Data Fig. 3b.
Source Data Extended Data Fig. 4
Source data for Extended Data Fig. 4a,b.
Source Data Extended Data Fig. 6
Source data for Extended Data Fig. 6a,b.
Source Data Extended Data Fig. 8
Source data and statistical results for Extended Data Fig. 8a–c.
Source Data Extended Data Fig. 9
Source data and statistical results for Extended Data Fig. 9a–c.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Wang, Q., Tang, T.M., Youlton, M. et al. Epistasis regulates genetic control of cardiac hypertrophy. Nat Cardiovasc Res 4, 740–760 (2025). https://doi.org/10.1038/s44161-025-00656-8
Received:
Accepted:
Published:
Version of record:
Issue date:
DOI: https://doi.org/10.1038/s44161-025-00656-8
This article is cited by
-
CCDC141 is a Connectin/Titin and Nesprin-1 binding protein that adapts cardiomyocytes to mechanical stress
Communications Biology (2025)
-
Epistasis in cardiac traits
Nature Cardiovascular Research (2025)
