
Abstract
After decades of neglect and a decline in antibiotic research and development, we are now finally witnessing the advent of new funding programs dedicated to new therapies. In addition to traditional new chemical entities that directly kill or arrest the growth of bacteria, alternative approaches are being identified and advanced towards proof-of-concept trials in the clinic. We briefly review the current pipeline of conventional new antibiotics and highlight in more depth promising alternatives, including potentiators of antibiotic action, bacteriophage, lysins and microbiome modulation. More innovative approaches, such as adaptive and innate immune modulators, CRISPR-Cas and diagnostic guided ‘theranostics’ are discussed and contrasted. Such exploratory therapies may require the development of alternative regulatory and clinical development pathways, but represent a potential circuit breaker from the current ‘arms race’ between bacteria and traditional antibiotics.
Similar content being viewed by others


Why is antibiotic discovery so difficult?
Developing a new drug is hard. At the discovery stage, the odds of a molecule making it to market are less than 1 in 2001. Even this number is misleading, as it is based on a starting point of a ‘declared candidate’, which may be the result of years of discovery effort and thousands to millions of compounds tested. Finding and developing a new antibiotic is even harder. Most medicines are designed to bind to and then modulate the function of a human target, which is usually evolutionary conserved and similar in most people. In contrast, antibiotics have to hit moving targets, as bacteria can evolve and adapt so quickly under selective pressure that drug resistance can appear even during a clinical trial, and within patients2. Even if one bacterium survives an antibiotic treatment, an hourly reproduction rate means within one day it can produce over 224 = > 16,000,000 offspring. This potential for rapid resistance development creates an additional challenge for antibiotic development that is unique compared to other drug classes. Cultural innovations can lead to new threats, such as the advent of air-conditioning units and cooling towers providing stagnant water that led to a rise in infections of Legionella pneumophila3, while the healthcare system itself can drive and amplify antimicrobial resistance (AMR)4. Bacteria don’t need passports to cross national boundaries and are sexually promiscuous, moving genetic material rapidly between strains, species and geographies. We are thus in a classic Red Queen situation5; having to run just to keep up with the fast-evolving superbug threat. Currently we are losing this race, with the impact of bacterial infections on mortality and morbidity continuing to rise as the number of effective drugs diminishes.
How to get to the clinic with a new antibiotic?
If one is fortunate enough to discover a new drug candidate for a common cancer, heart disease or inflammation, there are hundreds of biotech and pharma companies willing and able to take this discovery through development to the market. In the antibiotic space, there are few options left, and those still in the space have limited resources and bandwidth to take on new external projects. The capitalistic model that has overtaken most pharmaceutical companies (many originally family-founded with more altruistic goals) and now drives science innovation based on return on investment does not work anymore for antibiotics. Large pharmaceutical companies have abandoned antibiotic research, not just because it is extremely challenging, but because it cannot be justified economically. The direct net present value of an antibiotic is close to zero. The societal value of an antibiotic is no doubt far greater, particularly given current6,7 and future8 projections for the global burden of AMR, but whilst the value of antivirals and vaccines has been quantified for viral pandemics9, the fully encapsulated societal value of antibiotics is less well studied10,11.
Very few medicines can be said to cure a disease. Antibiotics are still ‘miracle drugs’ that support and enable modern medicine. They can protect organ transplant patients and those undergoing many types of cancer therapy who become immunocompromised. Routine hip and knee replacements still rely on antibiotics to prevent infections during surgery. Given this value to human health and society, why is there so little funding for antibiotics? Back in 2012, analysis of funds distributed by the US National Institute for Allergy and Infectious Diseases (NIAID) showed that ~$1,500 was spent per MRSA (Golden Staph) related death and $750 was spent per C. difficile-related death, compared to ~$75,000 per HIV-related death12. However, antibiotics cost just as much to develop as other drugs, with a mean estimate of $1.3b for systemic anti-infectives matching the overall mean for all drug classes (despite a Phase 1 to approval success rate of 25% that is better than the 14% average)13. Post-approval costs for a new antibiotic have been estimated to add an additional $240-622m of expenses over 5 years14. A recent commentary15 highlighted estimates that a new antibiotic needs at least $300m in annual revenue to be sustainable16, yet most companies make between $15m and $50m in US sales per year17. A 2021 study calculated the average sales of new antibiotics during their first 8 years on the market, finding an average revenue of $240m in total per antibiotic, with the US market accounting for 84% of those sales18. The cost of clinical trials is another barrier, with thousands of patients generally needed for the Phase 3 trials in order to meet the non-inferiority comparisons to existing therapy that new antibiotics need to achieve19,20. This means multiple sites are needed for enrolment. Attempts to run trials against resistant infections are even more costly: Achaogen attempted to evaluate the efficacy and safety of plazomicin compared against infections caused by CRE. The trial had to be stopped prematurely because only 39 out of 2000 screened patients were successfully enrolled21,22, with an estimated cost of $1 million per recruited patient23.
Large Pharma continues to exit from antibiotic R&D
In response to this calamitous situation, we have produced dozens of government-sponsored recommendations, think tank reports, white papers, declarations and commentaries purporting to address the threat of drug-resistant infections to human health and modern medicine. The Center for Global Development published an analysis of the economics of antibiotic resistance in 202424, along with a 2023 working group report on how to improve the antimicrobial market25, with another 2024 report “Forecasting the Fallout from AMR” generated by the same center along with World Organisation for Animal Health and the World Bank26. More than 80 companies from around the world signed a declaration at the World Economic Forum in Davos in 2016 calling for new economic models to support incentives for antibiotic R&D27. AstraZeneca, GlaxoSmithKline, Johnson & Johnson, Merck and Pfizer were some of the large pharma signing the declaration, which argued that the economic value assigned to antibiotics does not reflect the benefits they bring to society. New economic models and new ideas are desperately needed now to be put into action.
These companies are right, so what did they do next? Pfizer essentially exited antibiotic pre-clinical research in 2011, when announcing it would move its antibiotic research to China28. AstraZeneca spun out its antibiotic assets into Entasis Therapeutics in 2015, but this meant an antibiotic unit with 175 staff was reduced to a new company with only 2129. Entasis raised $US75m in an IPO in Sept 2018 at $15 a share30, but it had the biggest drop for a biotech IPO in 16 years on debut, falling 29%31. In 2022 Entasis was acquired by holding company Innoviva at a $2.20 share price32. Merck fired Cubist’s R&D team after acquiring the developer of daptomycin in 2015 (though daptomycin was originally discovered at Eli Lilly and Company as LY 146032)33, then shortly thereafter let go many of its own internal team, and in 2018 licensed most of its preclinical assets to a new startup, Prokaryotics. In 2017 The Medicines Company exited the field, selling their antibiotic programs to Melinta, but then in Nov 2018 Melinta eliminated most of its R&D efforts soon after receiving European approval to market Vabomere34, and it filed for Chapter 11 bankruptcy in Dec 2019. In 2018 Allergan35, Sanofi and Novartis36 pulled out, with Sanofi transferring its infectious diseases Research & Development (R&D) unit to Evotec37. This left Roche, its Genentech subsidiary and GSK as the only remaining major pharma with significant research in the field. However, GSK initiated a strategic review of its cephalosporin antibiotic program, and in 2018 announced a focus on investment in oncology and immuno-inflammation38.
A 2024 report by the AMR Industry Alliance outlined the ‘brain drain’ of antimicrobial researchers, estimating that there are only approximately 3,000 AMR researchers currently active in the world39. Almost all antibiotic research is now conducted by small biotech companies or academics: a study of 213 systemic antibacterial drugs having active clinical development under a US IND during the past 40-year shows that antibiotic INDs filed by large companies have declined from over 75% of the total in the 1980s to under 20% in the 2010’s40. Successfully achieving a New Drug Approval (NDA) is normally the ‘pot of gold’ for small biotechs, but perversely this is not the case in the antibiotic field. Achaogen received approval for Plazomicin in June 2018, but filed for Chapter 11 bankruptcy in April 2019. An analysis of Achaogen’s inability to fund commercialisation was published in 202441. Tetraphase had eravacycline approved in Aug 2018, but was acquired by La Jolla Pharmaceuticals in July 2020 for $US43m upfront, a dramatic fall from its market cap peak of $1.9b in July 201542. The most recent casualties are Destiny Pharma, appointing administrators in 2024 after 27 years of R&D43. Today Evotec, a contract research organisation, is arguably the major industry player in the space with a bolt-on acquisition of IP rights and 100 employees from Sanofi’s portfolio transferred to them in June 2018. In 2022 Evotec combined with Boehringer Ingelheim and bioMérieux to form Aurobac Therapeutics SAS, a joint venture to create antimicrobials along with diagnostics44. Another bright spot is Shionogi, a mid-tier pharmaceutical company recognised in a 2020 Antimicrobial Resistance Benchmark survey report45 as having the highest annual ratio of R&D investment for anti-bacterial and anti-fungal agents of any of the companies surveyed. In 2024 Shionogi acquired Qpex, an antibiotic-focused biotech, which launched in 2018 with the preclinical-stage anti-infective assets of The Medicines Company46. Shionogi will establish a discovery lab in San Diego focused on antimicrobial and pandemic preparedness research47. The collective loss of expertise in the specialized field of antimicrobial research exacerbates the difficulties in discovering new antibiotics. An analysis of reviewer comments on 91 funding applications for antibacterial drug discovery projects submitted to two major global funders identified faults that included “scientific and technical shortcomings, unclear potential societal impact, and insufficient capability and expertise of the project team regarding the R&D process”48. “Innovations” in antibiotic discovery are often re-badged traditional approaches with new technology, with the loss of expertise meaning similarities in approaches are not recognised.
The abandonment of antibiotic research has continued despite the increasing attention on push and pull incentives and does not augur well for investment in antibiotic research in the near future. Indeed, the Novo Repair Fund compared private financing investment in oncology-focused companies vs those focused on antibiotics & antifungals from 2018 to 2023, with 42-fold more money raised ($US 50b vs $US1.2b) and 15-fold more deals done (1695 vs 102)49. The median time from IND submission to marketing approval appears to be lengthening: from 6.0 and 5.9 years for INDs filed in the 1980s and 1990s to over 9 years expected for INDs filed in the 2000s40, a timeline inconsistent with the rapid returns generally expected by venture capital investors. However, former analyst and current Spero Therapeutics CFO Joel Sendek has compared the current situation to that of oncology drugs in 1997, when one company was attempting to launch a novel product in a genericized market that most big pharmaceutical companies already abandoned. At that time Joel put an above-consensus pricing on a stock that most analysts were pessimistic about—Genentech and their first oncology product, Rituxan50. The antibiotic market is further constrained due to a lack of access to both old and new antibiotics, which varies between countries and can be caused by short or long-term shortages, deregistrations, or lack of registration51, with a 2024 WHO report on policy and regulatory interventions to address antibiotic shortages in low and middle-income countries52. Despite barriers to access, antibiotic consumption increased 16% from 2016 to 2023, and is projected to increase by 52% from 2023 levels by 203053.
How to fix a broken market?
The lack of commercial viability to develop new antibiotics has led to a variety of suggestions on how to encourage more research. A working group from the Innovative Medicines Initiative that examined new economic models for antibiotic development (see below) proposed large market entry rewards, on the order of $1 billion per antibiotic globally, as ‘pull’ incentives. Such large sums of money obviously require a daunting level of international cooperation, but the argument has been made that if tens to hundreds of billions of dollars can be organised to fund international initiatives such as the International Space Station54 or Large Hadron Collider, and maintain the CHF$1.1b annual budget for CERN55, it is not unachievable. Similarly, the Duke-Margolis Center for Health Policy proposed a Priority Antimicrobial Value and Entry (PAVE) Award, which combines a more limited initial market entry reward with subsequent ‘value-based’ payments for effective antibiotics56. Other proposed pull incentives include proposed US legislation for an add-on Medicare payment for certain antibiotics (DISARM; S. 3787, Developing an Innovative Strategy for Antimicrobial Resistant Microorganisms, a bill introduced by Senators Orrin Hatch and Bob Casey in the US 115th Congress, Dec 2018 and reintroduced in the 116th Congress in 2019)57,58, and a transferred exclusivity concept (REVAMP, RE-valuing AntiMicrobial Products, a bipartisan bill introduced by Rep John Shimkus, R-Ind., and Rep Tony Cardenas, D-Calif in July 2018 where an antibiotic approval gives 12 months extended market exclusivity that can be transferred to another, more lucrative, product)59. An updated version of the The Pioneering Antimicrobial Subscriptions to End Upsurging Resistance (PASTEUR) Act was re-introduced to the US Senate in Sept 2020, with a proposed $US11b fund to reward new ‘Critical Need Antibiotics’ with a subscription contract (valued at $750m to $3b) that prepays for all US federal use of the drug60,61. In 2018 former FDA Commissioner Scott Gottlieb proposed a model in which acute-care institutions would pay a fixed licensing fee in return for access to a certain number of annual doses of a new antibiotic62. In 2019 a new rule was implemented that provided improved reimbursement for newer antibiotics administered in US hospitals63.
The UK announced a 5-year national action plan in 2019, with one goal to investigate a new payment model that is based on the value of the medicine to the health system, de-linking payments based on volumes of antibiotics sold64,65,66, and this was implemented with a trial to pay drug companies for antibiotics using a subscription-style model67, with the first two treatments selected for evaluation in Dec 202068. The success of the program led to full implementation in Aug 2024, with the NHS tendering contracts with an estimated value of almost £1.9b over 16 years for new antimicrobial medicines69. The USA launched its own updated National Action Plan for Combatting Antimicrobial Resistant Bacteria in Oct 2020, with one of the five goals including a sub-objective to “promote sustainability of the commercial market for new antibiotic products”, with several specific initiatives listed70. More radical solutions have also been raised, with economist Lord Jim O’Neil, head of a British global review of antimicrobial resistance in 2016, suggesting a “play or pay” levy on all drug sales for pharmaceutical companies that do not have antibiotic development programs, or a nationalisation of antibiotic R&D71. The potential for non-profit enterprises or public benefit corporations with low-profit expectations to undertake antibiotic development has also been explored72. While funding is critical, we do need to be clear that money alone will not solve the problem if all we do is more of the same: scientific advances that develop truly inventive approaches are essential.
The fightback begins!
The past decade has at last seen new initiatives arising from the public sector. In the US, these include the Generating Antibiotic Incentives Now (GAIN) act that extends the patent and commercial exclusivity for a new antibiotic and the Limited Population Pathway for Antibacterial and Antifungal Drugs (LPAD) that allows for smaller, more efficient clinical trials. A range of organisations have been created to advance antibiotic discovery. The European Innovative Medicines Initiative (IMI) founded the New Drugs for Bad Bugs (ND4BB) program73, with the first project in 2013, although the bulk of this funding was allocated to academic consortia carrying out basic research and clinical trials. Over €650m funded the TRANSLOCATION project (examining antibiotic penetration and efflux from Gram-negative bacteria), ENABLE (establishing an antibiotic development platform), five projects focused on clinical trials (COMBACTE-CARE, COMBACTE-MAGNET, COMBACTE-NET, COMBACTE-CDI, iABC) and DRIVE AB, which examined new economic models for antibiotic development. DRIVE AB released its final report in Jan 2018, highlighting four incentives to stimulate the antibiotic pipeline while ensuring access to critical antibiotics. The incentives include traditional ‘push’ incentives, such as nonrepayable R&D grants, but also recommend substantial market entry rewards ($1 billion per antibiotic globally) as ‘pull’ incentives. The other suggestions are ‘pipeline coordinators’ to track and stimulate the antibiotic pipeline, and a long-term continuity model to ensure continued supply of generic antibiotics. The Combating Antibiotic Resistant Bacteria Accelerator (CARB-X), is an initiative to rescue and accelerate underfunded or abandoned assets in pre-clinical or early clinical development from industry and (since 2018) academia, co-funding stages from Hit-to-Lead into Phase 174,75,76. Since its inception in 2016 the US-based CARB-X has invested more than $US490m in 104 projects from 13 countries (as of Dec 11, 2024), with funding contributed by BARDA (Biomedical Advanced Research and Development Authority), Wellcome Trust, BMGF (Bill and Melinda Gates Foundation), GAMRIF (The Global AMR Innovation Fund), UK AID, NIAID (National Institute of Allergy and Infectious Diseases), ASPR (Administration for Strategic Preparedness and Response), Novo Nordisk Foundation, and the German and Canadian governments. CARB-X solicits applications in different funding rounds that have focused both on direct acting small molecule antibiotics (new classes or targets) as well as ‘non-traditional’ approaches such as vaccine and biotherapeutics; diagnostics are also in scope. A similar initiative, PACE (Pathways to Antimicrobial Clinical Efficacy), was launched in 2023 with £30m in funds from LifeArc, Catapult and Innovate UK.
BARDA is an office in the U.S. Department of Health and Human Services that is responsible for countermeasures against biological chemical, nuclear and radiological threats. It acts as an interface between the U.S. Government and the biomedical industry, and is responsible for stockpiling key medicines, including antibiotics. BARDA works with partners such as the NIH, DoD, CDC, industry, and academia to develop and advance innovative solutions for medical countermeasures. supporting drugs and diagnostics from research through advanced development via funding, technical assistance and core services. A key driver of this effort’s focus on emerging infectious diseases is through the founding of CARB-X. BARDA founded the Broad Spectrum Antimicrobial Program in 2010, focused on developing new antibacterial drugs and diagnostics via public-private partnerships with industry. As of April 2023, BARDA77 had 27 active therapeutic and diagnostic candidates under development in the antimicrobial portfolio (in addition to CARB-X projects), including five out of the 15 antibacterial therapeutic products in active Phase 3 clinical studies.
The USA National Institute of Allergy and Infectious Diseases (NIAID) also has a range of funding programs to address antimicrobial resistance78 with a report in 2019 outlining the institute’s portfolio of basic, translation and clinical research79. These services include a range of preclinical in vitro and animal model screening tools to support Investigational New Drug applications. In Europe, the WHO and the Drug for Neglected Diseases Initiative (DNDi) launched the Global Antibiotics Research and Development Partnership (GARDP) in 201680. Now an independent not-for-profit Swiss foundation, GARDP is focused on neonatal sepsis, paediatric antibiotics, sexually transmitted infections and the development of new antibiotics along with curation and retention of antibiotic development knowledge, with a REVIVE initiative to connect and support the AMR research community. The US-based PEW Trust funded a similar initiative, the Shared Platform for Antibiotic Research and Knowledge (SPARK)81, in attempt to capture some of the learnings from successes and failures of the many antibiotic developers who have been let go or moved to other fields as their parent companies exit the field. CO-ADD, the Community for Open Antimicrobial Drug Discovery, is an initiative targeted at earlier stage antibiotic discovery82,83,84,85,86. It is designed to prime the antibiotic pipeline by helping academic chemists identify potential new antibiotics and provide sufficient antimicrobial characterisation to enable them to enter programs such as CARB-X and IMI-ENABLE. SPARK was transferred to CO-ADD in 2021. Both the Wellcome Trust87 and the WHO88 are highly invested in advancing antibiotic research.
On the for-profit side, Novo Holdings launched the REPAIR (Replenishing and Enabling the Pipeline for Anti-Infective Resistance) Impact Fund of $US165m in 2018 to invest in about 20 early-stage companies developing therapies targeting resistant microorganisms89. The portfolio includes 11 companies, as of Dec 2024. In 2020, the WHO, European Investment Bank (EIB), Wellcome Trust and more than 20 biopharmaceutical companies led by the International Federation of Pharmaceutical Manufacturers & Associations (IFPMA) launched the AMR Action Fund90, with over $US1b pledged to bring 2–4 new treatments to patients by 203091,92. Investments have supported 10 companies as of Dec 2024. Overall, the CARB-X, REPAIR Fund and AMR Action Fund reflect a potential injection of over $1 billion into antibiotic development between 2016 and 2024. The BEAM Alliance (Biotech companies from Europe innovating in Anti-Microbial resistance research), established in June 2015, is an advocacy platform for a network of over 60 European SMEs involved in antimicrobial research, collectively working on over 140 R&D projects. Similarly, the AMR Industry Alliance is a private sector coalition set up as an industry response to the 2016 UN call for action. Comprising over 100 biotech and pharmaceutical companies and associations, the organisation facilitates collaborations between the public and private sectors. Combined with funds from CARB-X, this means a potential injection of over $1 billion into antibiotic development between 2014 and 2021.
These combined initiatives may be having a beneficial effect. As illustrated in Fig. 1, analyses of the antibacterial clinical pipeline from 2011-2024 by several different groups93,94,95,96,97,98,99,100,101,102,103,104) indicate a trend towards replenishment of Phase 1 studies, though overall the number of compounds in clinical development is vastly lower than other indications such as oncology. The number of ‘nontraditional’ antibacterial therapies is even more sparse. Preclinical pipeline analyses have also been published99,100,101,105,106.

Top: Evolution of the clinical pipeline of antibiotics since 2011, derived from assessments by Butler et al.93,94,95,96,97, the WHO (2018)98 and 202399,100,101), the Pew Trust (2019102,103), and The Biotechnology Innovation Organization (BIO) (Oct 2021104) compared to non-traditional antimicrobials in 2015 (Czaplewski et al.)107 2019 (Pew Trust)251 and 202399. Bottom: The number of antibiotics approved each year over the past 25 years (see SI for detailed list). Where indicated by the reference, divided into antibiotic spectrum of activity. Note that summaries complied by different entities have varying inclusion/exclusion criteria (e.g. TB drugs, topicals, virulence agents) so the totals are not directly comparable.
How to discover new antimicrobial therapies?
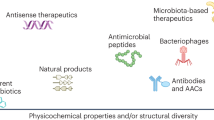
Focusing on the development of antibiotics alone will not be able to address the threat of antimicrobial resistance. Expansion of proven alternative strategies, such as prevention through vaccines, or modernisation of historically marginal approaches such as phage therapy or probiotics, are all attracting substantial interest107,108. This does not mean we will not need antibiotics in the future, but helps to alleviate the pressure driving their overuse and provides options when they are ineffective. The therapeutic programs funded by CARB-X encapsulate these different strategies, including antibiotics, alternative therapeutics, prophylactics (vaccines), at stages from Hit-to-Lead through to Phase 1. New diagnostics are also funded. These projects, as summarised in Fig. 2, include eight programs based on existing classes of antibiotics, 26 on new classes of small-molecule antibiotics (including 15 acting on new targets), nine antimicrobial peptides and proteins (some of which overlap with new classes and/or targets), 23 ‘non-traditional’ approaches (ten potentiators/anti-virulence factors, six phage/lysin-derived, four microbiome and three immune modulation), 18 vaccines and antibodies, and 16 diagnostics (as of May 13 2024)109. Of these programs, 14 have ‘graduated’ and 44 programs are no longer funded in the pipeline for undisclosed reasons. In the following sections we will use examples from the CARB-X pipeline to illustrate the variety of antibacterial therapies under development.


Programs that have ‘graduated’ indicated with graduation cap. Programs no longer funded are greyed out. Where structure of lead compound or compound series is known, it is included. Current, or most advanced stage of development, indicated.
Traditional approaches to antibiotic discovery: improve existing antibiotics
A well validated approach that has been most successful in generating new antibiotics over the past five decades is to generate improved versions of existing classes of antibiotics, designed to have greater activity, overcome resistance, and/or have reduced side effects. This is why we have multiple classes of different antibiotics: e.g. fourth generation fluoroquinolones, third generation tetracyclines and fifth generation cephalosporins. The potential for this strategy to continue to combat rising resistance was recently reviewed110. Programs funded by BARDA’s BSAP portfolio fall into this approach, with Vabomere (approved Aug 2017) a combination of the β-lactam antibiotic meropenem and the β-lactamase inhibitor vaborbactam, Plazomicin (approved June 2018) an aminoglycoside, Eravacycline (Tetraphase, FDA + EMA approved Aug 2018) a tetracycline, Solithromycin (New Drug Application rejected Dec 2016)) a macrolide, and BAL30072 an intravenous monosulfactam antibiotic. Other antibiotics approved since 2017 include 3 fluoroquinolones (Lascufloxacin, Kyorin Pharmaceutical, PDMA 2019; Levonadifloxacon, Wockhardt, DCSCO 2020; Delafoxacin (Melinta Therapeutics, FDA 2017), two more tetracyclines (omadacycline, Gurnet Point Capital/Novo Holdings, FDA 2018) and an oxazolidinone (contezolid, MicuRx, NMPA 2021). CARB-X has funded several projects that ‘improve’ existing antibiotics109. For example, ‘graduated’ sulopenem is a thiopenem lactam antibiotic that has activity against Enterobacteriaceae with resistance to third generation cephalosporins due to encoding ESBLs or AmpC-type β-lactamases, with MIC90 of <0.25 µg/mL against a range of resistant organisms, compared to ceftriaxone MIC90 of 1 to >32 µg/mL111. Bugworks’ BWC09777 (supported by CARB-X since 2017 with additional potential funding from GARDP announced in 2023 to conduct Phase 2 and 3 studies112), is an oxazolidinone scaffold which targets bacterial DNA gyrase and topoisomerase, but at a site distinct from fluoroquinolones113 (and a completely different target than the oxazolidinone antibotics such as linezolid, which bind to the 23S portion of the 50S ribosomal subunit). BWC0977 has broad-spectrum activity against many “global priority” pathogens, including carbapenem-resistant Enterobacteriaceae, carbapenem-resistant Pseudomonas aeruginosa, carbapenem-resistant Acinetobacter baumannii and methicillin-resistant Staphylococcus aureus.
As highlighted in the WHO analysis of antibacterial agents in preclinical and clinical development in 202399, the clinical pipeline is still dominated by ‘traditional’ antibacterial agents (57 of 97): excluding those targeting M. tuberculosis/C. difficile/H. pylori, 15 of the 32 remaining were β-lactam or β-lactam/β-lactamase inhibitor (β-lactam/BLI) combinations, and 10/13 of the traditional agents approved since July 2017 belong to existing antibiotic classes for which resistance mechanisms are well known. The potential liability with this approach is that bacteria are already well-versed in developing resistance to these classes, so resistance is either already present, or may quickly evolve, as has been seen progressively with penicillins, cephalosporins, then carbapenems. Evaluation of the propensity to develop resistance is critical during antibiotic development: a 2025 study explored in vitro emergence of resistance to 13 recent antibiotics over 60 days, compared with ‘in-use’ antibiotics, against multiple bacterial species114. The newer antibiotic candidates showed similar susceptibility to resistance development as the existing antibiotics with overlapping resistance mechanisms.
Modifications of existing antibiotics can lead to compounds acting on new targets. The veterinary antibiotic florfenicol was used as the base scaffold for computer-assisted drug design based on the peptidyl transferase centre (PTC) of the bacterial 50S ribosome subunit as the target. The most promising compound, with in vitro activity against a range of pathogen including E. coli, S. aureus, E. faecalis, S. typhi, P. multocida, and H. parasuis, and in vivo efficacy against MRSA infections. However, investigations into its bactericidal mechanism found inhibition of ornithine carbamoyl transferase (arcB), an enzyme involved in arginine degradation metabolism biosynthesis, leading to disruption of the bacterial cell membrane115.
Traditional approaches: novel antibiotics acting on existing targets
There is a very limited set of clinically validated antibiotic targets: cell wall components (e.g. β-lactams and glycopeptides inhibiting steps of peptidoglycan synthesis), the 50S (macrolides, oxazolidinone) or 30S (aminoglycosides, tetracyclines) ribosomes of the protein translational machinery, and DNA gyrase (fluoroquinolones)/ RNA polymerase (rifamycins) component of transcription. Novel chemical classes that inhibit validated targets, but bind to a different target site, provide an opportunity to develop antibiotics that escape existing resistance mutations. Alternatively, new chemotypes might possess superior PK-PD or safety margins compared to current therapies. Examples within the CARB-X portfolio include Idorsia and Redx, who were funded to advance new chemotypes targeting topoisomerase/gyrase enzymes, Bioversys, Curza and Melinta for inhibitors that bind to unique sites of the ribosome, Mutabilis, Entasis and VenatoRx for compounds targeting penicillin-binding protein with non-β-lactam structures, and the University of Queensland for a class of lipopeptides similar to the polymyxins, which also interact with Lipid A, yet are effective against polymyxin-resistant bacteria with mutated Lipid A116.
Traditional approaches: novel antibiotics acting on novel targets
Completely new antibiotic chemical classes acting on novel bacterial targets are the holy grail of antibiotic discovery: something that people want and are looking for but that is extremely difficult to find or get. The hope is that the combination of new chemical structures blocking previously unutilised pathways would avoid existing resistance mechanisms, and potentially take longer to develop new ones. The advent of genomics and target-based drug discovery in the 1980’s-90’s gave hope that essential targets unique to bacteria could quickly be identified and leveraged through rational drug design, but, as summarised by reviews of research programs at GlaxoSmithKline117 and AstraZeneca118, the approach was largely unsuccessful. Potent and selective inhibitors could be identified against isolated targets, but when tested in phenotypic assays they generally lost activity due to the many mechanisms bacteria have developed to block entry to, or pump out undesirable compounds. The obstacle of lack of cellular accumulation (poor permeability, efflux) is also accompanied by dangers or rapid development of resistance via single-step mutational changes – particularly for protein targets where a single amino acid change could lead to resistance. An exemplary warning story is provided by GlaxoSmithKline’s GSK2251052, a novel boron-based benzoxaborole small-molecule candidate acquired from Anacor in 2010 that targets the bacterial enzyme leucyl tRNA synthetase119. In 2012 GSK halted a Phase 2b trial for the treatment of complicated urinary tract infections (cUTI) due to the development of resistance in patients during treatment120, with ≥32-fold increases in the GSK2251052 MIC of the infecting pathogen being detected in 3 of 14 patients being treated121. Whole genome sequencing confirmed that resistance was due to specific mutations, selected on the first day of therapy, in the LeuRS editing domain. A single T247I change was the most commonly encountered mutation, leading to high resistance. A simple in vitro frequency of spontaneous resistance assay in both standard ATCC25922 E. coli and patient isolates clearly identified high rates of resistance (1 X 10-7 to 4 X 10-7); it is unclear whether this assay was not conducted or just ignored during the preclinical development121. The challenge of rapid resistance arising from new drugs targeting novel proteins could potentially be addressed by taking a page from the therapeutic approaches used to treat TB. New single-targeted agents are never used alone, but in combination with multiple other antibiotics, where either resistance to one antibiotic leads to greater susceptibility to another, or the accumulation of multiple resistance leads to bacterial fitness costs.
Despite these setbacks, research has continued into new targets, with one of the most promising being LpxC122,123. This enzyme, UDP-3-O-(R-3-hydroxymyristoyl)-N-acetylglucosamine deacetylase, is a Zn2+ metalloenzyme essential for the synthesis of lipid A, a component of the outer membrane of Gram-negative bacteria. The enzyme was first identified by Christian H.R. Raetz at Merck in 1993124. LpxC inhibitors were actually discovered before the enzyme’s identity was known, via screening for LPS synthesis inhibitors in the mid-1980s122. Researchers at the University of Washington and at Chiron developed a prototypical inhibitor structure, consisting of a Thr-hydroxamic acid that complexes to the active-site zinc, N-acylated with rigid hydrophobic groups (two aromatic rings separated by one or two triple bonds) that occupy a tunnel where the fatty acyl chain of the substrate normally resides. The first inhibitor that entered human testing, Achaogen’s ACHN-975, replaced the Thr with a β-amino-Val residue, and modified the aryl-bisalkyne substituent. ACHN-975 completed a Phase 1 single-ascending-dose study (NCT01597947) in 2012, but this revealed a peak plasma concentration (Cmax)-driven dose-limiting toxicity (DLT) of transient hypotension without tachycardia125. Work continued on a series selective for P. aeruginosa, producing compounds with MIC90 of 2 µg/mL against 250 resistant clinical respiratory isolates, but this program was also discontinued in Nov 2017 due to unexpected cardiovascular toxicity126. In Sept 2018 Achaogen announced that they would non-exclusively transfer compounds, scientific data and assays to Forge Therapeutics (who have a CARB-X funded LpxC program) and share their data via SPARK. While almost all reported LpxC inhibitors rely on a chelating hydroxamic acid ‘warhead’, Forge Therapeutics has identified alternative motifs that retain good potency127,128. Other companies working on LpxC inhibitors, with patents filed between 2010 and 2016129, include Merck and Co (Schering Corporation), AstraZeneca, Pfizer, Actelion, Novartis, and Viamet Pharmaceuticals, along with Duke University and University of California. CARB-X has also funded LpxC inhibitor development by Recida Therapeutics, while Cubist Pharmaceutical (acquired by Merck)130, Kyorin Pharmaceutical Co131, Vernalis/Taisho Pharmaceutical132 and Entasis133 have recently published in this area. Other members of the lipid A pathway are also targets, such as LpxH, with a pyridinyl sulfonyl piperazine scaffold inhibitor active against Enterobacterales such as E. coli and K. pneumoniae reported in 2024134. Another promising new target is FabI, an NADH-dependent enoyl reductase from the type II bacterial fatty acid biosynthesis pathway (FAS-II)135, which is generally conserved across pathogens but distinct from the corresponding system found in mammals. In contrast to LpxC, compounds have advanced further into human testing, with Phase 2 trials of a benzyl pyridinone CG400549 by CrystalGenomics Inc and a benzofuran napthyridine Afabicin (Debio 1450) by Debiopharm International. CARB-X has funded two earlier-stage Debiopharm compounds, DEBIO1453 for Neisseria gonorrhoeae, and DEBIO1464 for A. baumannii.
Aminoacyl tRNA synthetases are part of the protein synthesis pathway and can be inhibited at multiple sites136. Oxford Drug Design was CARB-X funded for DaaRSi small-molecule aaRS inhibitors with activity against Gram-negative ESKAPE pathogens. Recent publications include MRS-2541, a novel inhibitor of methionyl-tRNA synthetase with selective activity against gram-positive bacteria137.
Additional new targets, along with an overview of their targets, were summarised in a 2023 review138. The advances in new molecules acting on new targets are reflected by several recent high impact publications demonstrating in vivo efficacy.
Traditional approaches: antibiotic potentiators
Potentiators or antibiotic adjuvants work in combination with existing antibiotics to either increase their activity or overcome resistance mechanisms. This is not a new idea – β-lactamase inhibitors (BLIs) have been used on the clinic for many years to prevent the rapid hydrolysis of β-lactam (BL) antibiotics. CARB-X has had a number of potentiators in its portfolio – including improved BL/BLI combinations. A non-BLI potentiator, SPR741, was under development by Spero Therapeutics. A CARB-X ‘graduate’, SPR741is a polymyxin analogue with limited antimicrobial activity on its own. Instead, it sensitises bacteria to certain classes of antibiotics, synergistically increasing their effectiveness. Originally developed by Northern Antibiotics decades earlier as NAB741139, this molecule replaces the polymyxin N-terminal fatty acid tripeptide linear component with a truncated acetyl-Thr-DSer sequence, removing two of the polymyxin’s five positive charges and the lipophilic tail. While not able to directly kill bacteria, it sufficiently perturbs the membrane so that the activity of hydrophobic antibiotics (e.g. macrolides or rifampicin) or large amphiphilic antibiotics (e.g. vancomycin) can be potentiated by 10- to 2000-fold140,141,142. The reduced positive charge is purported to ameliorate the nephrotoxicity associated with the polymyxins and a Phase 1 trial in 2018 did report good safety. In vitro potentiation has been demonstrated for a range of lactam antibiotics, including ceftazidime, piperacillin-tazobactam, mecillinam and temocillin, with other studies showing good synergy with azithromycin c143 or minocycline142 against Enterobacteriaceae, and with rifampin against extensively drug-resistant A. baumannii144. However, development was discontinued in 2020145. The University of Queensland was also funded by CARB-X to investigate whether their octapeptin analogues could act in a similar manner against polymyxin-resistant bacteria146.
Anti-virulence strategies are similar to potentiators, in that they do not directly kill bacteria, but help subdue the virulent characteristics of pathogenic bacteria147. They will most likely still require co-administration with a conventional antibiotic to gain clinical acceptance. A range of approaches are possible, such as developing inhibitors that block FimH, a protein involved in the adhesion of E. coli to the bladder wall during urinary tract infections148. GlaxoSmithKline is a CARB-X graduate for a Phase 1 trial for GSK3882347, a small molecule FimH antagonist co-developed with Fimbrion Therapeutics. As of Dec 2024, a Phase 1b study (NCT05138822) was recruiting to evaluate a double-blind, double-dummy, nitrofurantoin-controlled study designed to evaluate microbiological response at the test of cure (ToC) visit along with safety, tolerability and pharmacokinetic (PK) response. The Helmholtz Institute has an active CARB-X project on inhibitors of S. aureus α-hemolysin, an important virulence factor that contributes to the capacity of the bacteria to infect the host. Several antibodies against S. aureus toxins have progressed into clinical trials149: unfortunately a Phase II trial of suvratoxumab, an alpha-toxin human monoclonal antibody150, did not significantly lower the incidence of S. aureus pneumonia151,152.
Microbiotix, a 2020 recipient of CARB-X funding, is developing an inhibitor of the type III secretion system (T3SS) of Pseudomonas aeruginosa. This complex delivers toxins into macrophages and neutrophils, disabling the host immune response. Inhibition can weaken infection, but does not directly affect bacterial growth153. Other anti-virulence approaches target the toxins produced by bacteria, or the components required to construct biofilms – several of these are vaccines, discussed later in this article.
Alternate antibacterial approaches: phage and lysins
Bacteriophages, discovered in 1915, are Nature’s defence against bacteria – viral ‘bacteria-eaters’154. They were first developed in the former Soviet Union and Eastern Europe at a time when antibiotics were prohibitively expensive and virtually inaccessible locally. One of the long-perceived liabilities of phage therapy, its species and even strain specificity, is now being recognised as a potential strength, with narrow spectrum agents favoured to preserve protective or beneficial microbiota. Rapid diagnostics potentially allow for the matching of a phage cocktail to specific microbial pathogens. The ability to engineer phage through synthetic biology, and the development of manufacturing capabilities suitable for regulatory approval have all contributed to a renaissance in bacteriophage therapy, though not without setbacks155. Resistance to phage therapy has been reported156,157, though at a fitness cost158. Interestingly, clinical isolates of A. baumannii that developed resistance to two bacteriophages contained loss-of-function mutations in genes responsible for capsule biosynthesis, meaning they lost their capsule and became resensitized to beta-lactam antibiotics159. This suggests the potential for combination therapy.
Ad-hoc reports of clinical use still predominate160, with, for example, high profile coverage for treating an often fatal disseminated Mycobacterium abscessus infection in a 15-year-old cystic fibrosis patient161,162. Researchers at the University of California, San Diego (UCSD) successfully employed phage as a last resort to treat a drug-resistant strain of Acinetobacter baumannii that had put a colleague into a coma163. UCSD subsequently launched a Center for Innovative Phage Applications and Therapeutics (IPATH). The company that supplied the bacteriophage cocktail used at UCSD, AmpliPhi Biosciences, developed natural phage cocktails for clinical trials, with AB-SA01 a cGMP-manufactured three-phage therapeutic164,165 targeting MDR S. aureus that completed Phase 1 topical studies (NCT02757755). In 2019 AmpliPhi merged with C3J Therapeutics, a company developing synthetic phage through a proprietary phage engineering platform, to form Armata Pharmaceuticals. Armata is now focusing on AP-SA02, another S. aureus-targeting cocktail that started a Phase 2a trial in 2023 (NCT05184764), looking at efficacy of intravenous AP-SA02 as an adjunct to the best available antibiotic therapy compared to the best available antibiotic therapy alone. AB-PA01 is an earlier stage four-phage cocktail for P. aeruginosa165, potentially for nebulised delivery to treat lung infections. Both AB-SA01 and AB-PAO1 were made available through FDA Expanded Access programs for life-threatening infections (NCT03395769, NCT03395743). AP-PA01, successfully treated an MDR P. aeruginosa infection in a cystic fibrosis patient166, with a second-generation product (AP-PA02) completing a Phase 1b/2 trial in 2023 (NCT04596319). A Phase 2 trial in non-cystic fibrosis bronchiectasis completed enrolment in July 2024 (NCT05616221). A new cocktail, AP-PA03, has been developed for the different physiology of acute pneumonia lung infections167. An Israeli company, BiomX, raised $32 m in Feb 2019 to advance the preclinical development of phage cocktail therapies targeting bacteria causing acne and inflammatory bowel disease168.
There appear to be both scientific and regulatory barriers preventing phage therapies from becoming mainstream. The ability to develop a phage cocktail which has widespread coverage against most strains of a pathogenic species, without development of resistance, is incredibly challenging, while the creation of bespoke cocktails tailored to individual patients is time, labour and money intensive. From a regulatory perspective, developing phage cocktails that meet GMP standards, including analytical methods to assess phage identity, titer and purity is a significant impediment169.
Engineered phage approaches are also under development170. A current active CARB-X project is targeting ventilator-associated pneumonia caused by Pseudomonas aeruginosa. Phico Therapeutics’ approach is to use engineered bacteriophages combined with antibacterial small acid-soluble spore proteins (SASPs): the phage delivers the SASP gene cassette, directing the bacteria to produced SASs, which bind bacterial DNA in a non-sequence-specific manner and inactivate it171. Phico also has constructs targeting K. pneumoniae (SASPject PT4), E. coli (SASPject PT5) and S. aureus (SASPject PT1.2). As discussed further below, engineered bacteriophage are also being used to deliver CRSIPR-Cas9 for site-specific DNA cleavage of crucial resistance or virulence domains of bacterial genomes.
A closely related approach relies on the active component of bacteriophages – the lysin enzyme produced by lytic phages that is responsible for bacterial cell lysis via cleavage of the structural peptidoglycan. As such, they can kill dormant cells and have been shown to eradicate biofilms, so could be useful for the treatment of recalcitrant infections such as endocarditis172. As a therapeutic, lysins have the advantage over phages in that they are well-defined proteins that can be produced under standard manufacturing conditions, and so are much more palatable from a regulatory perspective. Lysins traditionally have only Gram-positive activity, as the Gram-negative outer membrane prevents access to periplasmic peptidoglycan. However, an engineered lysin containing the lytic-domain of T4 lysozyme fused to domain that binds to an iron transporter receptor, FyuA, killed clinical E. coli isolates173,174. Natural lysins active against Gram-negative bacteria, with a membrane-disruptive cationic peptide domain, have also been identified175. Companies focused on the development of lysins include GanaGen Inc, an Indian company with P128, an ectolysin effective against 120 strains of S. aureus, representing more than 3,000 isolates, including MRSA, VRSA and other antibiotic-resistant strains in Phase 2 testing for nasal decolonisation and preclinical development for systemic applications. ContraFect Corporation received two CARB-X awards to advance its early stage phage-derived protein programs, with the first using lysins to target invasive infections caused by P. aeruginosa, and the second based on ‘amurins’ with, broad spectrum activity against Gram-negative ESKAPE pathogens, good biofilm clearance, and synergistic activity with current antibiotics. The amurins are distinct but related to the lysins, consisting of a family of novel antimicrobial peptides 40-50 amino acids in length that cause lysis by the inhibition of cell wall synthesis176. ContraFect’s lead lysin program, CF-301 (exebacase)177,178 has completed a Phase 2 trial for S. aureus bacteremia. The Dutch company Micreos has marketed a non-prescription skin-care product (Gladskin), which contains endolysins targeting S. aureus, for conditions such as acne and eczema and licensed its technology to L’Oréal in 2020. It raised €30 m in 2019 to fund the development of another S. aureus endolysin (XZ.700), enrolling its first patients in a Phase 1/2a trial for atopic dermatitis in Sept 2020. In Feb 2025 Basilea announced that it was not exercising an option to progress tonabacase, a S. aureus endolysin developed by iNtron Biotechnology (Korea), into clinical trials. A ‘stringent risk-return criteria’ was provided as the reason179.
Alternate approaches: microbiome modulation and supplementation
Manipulation of the microbiome has attracted increasing awareness as an alternative approach to preventing and treating infections. Probiotic therapy is proposed to maintain or rebuild a healthy population of the natural flora, allowing them to compete with pathogenic bacteria to prevent them from becoming established. It is perhaps not surprising that Clostridium difficile infections (CDI) have become the focus of initial attempts, as they are located in the gut and often appear to be induced by antibiotic treatment wiping out the normal microbiome180. Fecal transplant therapy has garnered significant attention in recent years for recalcitrant and recurrent CDI, where normal antibiotic therapies (vancomycin, metronidazole of fidaxomicin) have failed. Donor-transmitted infections are a concern181,182. Companies such as Seres Therapeutics, SciBac, Finch Therapeutics, Rebiotix, NuBiyota and Vedanta Biosciences are developing specialized cocktails of bacteria or spores with rationally defined composition. While the first Phase 2 trial (NCT02437487) of Seres Therapeutic’s lead compound SER-109, completed in July 2016, showed no improvement in the degree of CDI recurrence (44%) compared to placebo (53%), a Phase 3 trial was initiated (NCT03183128) and successfully met its primary endpoint in 2020183. A 2019 perspective summarises the pros and cons of probiotics, summarising studies treating C. difficile–associated diarrhoea, neonatal sepsis and acute respiratory infection184.
CARB-X has funded several microbiome programs, with Seres developing SER-155 as a preventative consortium of bacterial spores to reduce the risk of both graft vs. host disease, and bacterial infection for CRE/VRE in patients receiving stem cell or whole organ transplantation. Vedanta’s VE303 and SciBac’s SCB-102 focused on C. difficile. Vedanta has an active project with VE707 as a preventative infection control strategy via intestinal decolonization of CRE, ESBL, and VRE, to reduce these common hospital-acquired infections. Outside of CARB-X, a phase 1 randomized clinical trial demonstrated that healthy human skin commensal Staphylococcus hominis A9 (ShA9) could be used to treat S. aureus-colonised atopic dermatitis, leading to a significant decrease in S. aureus and some benefits in local eczema severity185.
Alternate approaches: co-opting our immune system
Most infections occur in immunocompetent individuals, although immunosuppression during cancer therapy or organ transplant can increase the risk of infection significantly. However, in early drug discovery, nearly all compounds are assayed in vivo using animals treated with the immunosuppressant cyclophosphamide to increase the bacterial load in an animal in a reliable manner and short time frame. This of course ablates the function of any putative drug candidate that is designed to co-opt innate or adaptive immunity to exert its anti-bacterial effect. There are relatively few labs or CROs that can run quantitative, reliable immunocompetent bacterial infection models. At the time of writing, non-clinical models have been developed for only a handful of clinically relevant microbial pathogens, viz. MRSA, E. coli, caecal puncture peritonitis and the fungi C. albicans and A. fumigatus. There is also a paucity of sites that are able to run human challenge models, where a non-virulent, drug-sensitive pathogen is given to healthy volunteers to derive an early indication of human PK/PD for a drug candidate. Several CROs offer malarial and viral human challenge protocols, however the limited number of clinical investigators working with microbes are principally geared up to test new vaccines, not drug PK/PD. We outline the differences between a traditional and a potential future alternative compound progression plan in Fig. 3.

The steps required to discover and develop a new antimicrobial agent are compared for traditional versus non-traditional approaches.
Alternate approaches: antibodies and adaptive immunity
There are two primary components of our immune system – adaptive (resulting in an antibody response) and innate (primarily leukocytic cell responses). Initial antibacterial strategies focused on adaptive immunity using antibodies186,187,188,189 against bacterial virulence factors or bacteria themselves. In the latter case antibody-drug conjugates can deliver a toxic payload190, a strategy often used successfully in oncology. Visterra has ligated macrocyclic antimicrobial peptides onto an E. coli targeted monoclonal antibody191. The adducts had no detectable haemolytic activity, could kill E. coli at nanomolar concentrations, and showed preferential killing for E. coli over Pseudomonas aeruginosa and Klebsiella pneumoniae, though only in vitro. A more advanced system targeting MRSA has been developed by Genentech/Symphogen, using an engineered IgG1 antibody linked to a rifamycin analogue antibiotic through a valine-citrulline linker192,193,194. The antibody–antibiotic conjugate DSTA4637S (also known as RG7861) is designed to target intracellular S. aureus, a reservoir inside cells such as phagocytes that can be difficult to kill by standard approaches. The ‘rifalogue’ antibiotic component was selected as it kills intracellular bacteria, non-replicating bacteria, and antibiotic-resistant persister cells. The antibody is designed to bind to the outside of S. aureus by attaching to wall-teichoic acid, but only release the attached antibiotic after entry of the opsonized bacterium into a mammalian cell, via cleavage of the peptide linker by the lysosomal enzyme cathepsin D. The antibody–antibiotic conjugate killed bacteria inside macrophages, prevented colonisation of infections in mice, and was more effective than vancomycin in a delayed therapy bacteraemia model. The antibody-drug conjugate successfully completed a phase I study (NCT02596399) in 2017195. Rather than direct killing of bacteria, antibodies can also be used to neutralise bacterial-produced toxins For example, Bezlotoxumab (approved Oct 2016), binds and neutralises toxin B from C. difficile infections. Medimmune has developed multifunctional bispecific antibodies that protect against Pseudomonas aeruginosa infections in mice, with MEDI3902 targeting three components, the serotype-independent type III secretion system (injectisome), the virulence factor PcrV and the persistence factor Psl exopolysaccharide196. As mentioned earlier, antibodies have been developed against the S. aureus α-hemolysin virulence factor149. Within the CARB-X portfolio, Bravos was funded to develop BB100, a monoclonal antibody with directly bactericidal activity against a specific strain of hyper-virulent, multi-drug resistant E. coli (ST131-025b), which is often associated with complicated urinary tract, bloodstream, and prostate infections. CARB-X graduate Clarametyx Biosciences developed CMTX-101, a humanised monoclinal antibody effective against ESKAPE and MRSA biofilms that acts by binding to DNABII binding proteins, which stabilize and maintain the integrity of bacterial biofilms197. Trellis Bioscience TRL1068 also targets this protein, with a successful Phase 1 trial focused on periprosthetic joint infection (NCT04763759) in 2023198,199,200. Lumen Bioscience’s LMN-GI-EEC-401 is an oral mAB cocktail to prevent diarrheal diseases caused by Campylobacter jejuni and enterotoxigenic E. coli. Cytokine modulation has been targeted in sepsis trials using clinically approved modulators of TNFα, IL-1β, IL-6, IL-7 and many others. Whilst these have shown promising results201 in secondary outcome measures such as lymphocyte, CD4+ and CD8+ T-cell counts, they have had limited impact on the key primary outcome measures of morbidity and mortality202. This is most likely due to the lack of appropriately staged biomarkers to guide timed precision therapy. Whilst master cytokines TNFα, IL-1β, are reliable markers of chronic inflammation, they are not strictly relevant in the context of acute hyperinflammation found in sepsis patients, as they are elevated only for a very short period of time in the initial phase of sepsis, when patients may not yet have been admitted to the ICU. IL-6 is elevated for a longer period of time and elevated levels of IL-6 are found in sepsis, but it is also observed in many conditions driven by sterile inflammation. Research continues on MCP-1, procalcitonin and IL-8, however, many challenges remain203.
Alternate approaches: innate immune modulation
Research in immuno-oncology and immuno-metabolism has led to the launch of blockbuster drugs, such as the PD1 mAb Pembrolizumab (Keytruda), that are disease modifying for patients. In stark contrast, the study of immuno-infection, or what may be termed ‘inflabiotics’, has been comparatively neglected. This is due to a number of factors: i) there is no incentive to take significant additional risks in a clinically unproven area when the fundamental economics for investment in R&D are already so dire, ii) investigational drug candidate trials of immunotherapies have been almost exclusively in the area of sepsis, which is an incredibly difficult clinical indication to work on with multiple pathologies, staging of disease and drivers of pathophysiology within a patient population in acute, critical danger of death, iii) early immuno-modulatory agents were ‘blunt’ tools that often had significant off-target effects deleterious to host survival, iv) the antibiotic R&D community is, in part, dominated by incumbents reluctant to depart from the in vitro MIC/MBC screening paradigm that has been used in the field for the last 80 years, v) there are knowledge and language barriers between many immunologists and microbiologists, in contrast to the more fertile exchange of ideas between immunologists and those working in oncology, cardiology, neurology, osteology, rheumatology and inflammation, where immunology has been embraced academically and commercially, and vi) there is no accepted or standardized alternate pathway to progress drug hits to leads to candidates; from a biologically relevant primary screen to robust immunocompetent non-clinical models through to human challenge Phase 0 / 1b trials and indication-centric Phase 2 proof-of-concept trials. On the bright side, we now have more precise pharmacological tools that can very selectively modulate immune pathways. Many of these target histone deacetylases (HDACs), complement pathway proteins and receptors, recombinant cytokines (e.g. IL-7), colony-stimulating factors and epigenetic bromodomain modulators. These have been systematically reviewed elsewhere and are beyond the scope of this review. Most clinical efforts to treat sepsis using this approach have focused on antibody- or small molecule-mediated modulation of complement or toll-like receptor activity204 (in particular TLR2, 3, 4, 5, 7 & 9), but have met with limited success205. Selected, smaller peptide ligands that exert both an antimicrobial activity and modulate innate immunity are highlighted in the next section.
Innate immune modulation: peptide ‘inflabiotics’
Antimicrobial peptides that also exert immunomodulatory effects have received a lot of attention in academia206. However, whilst there are thousands of articles describing peptide antibiotics with antimicrobial activity in vitro, there are very few examples demonstrating in vivo activity. This is principally because in animals, these peptides are synthesised and secreted locally by immune cells recruited to the site of infection. These peptides, which are much larger than tissue-penetrant small-molecule antibiotics, only need to diffuse small distances to exert their effect on bacteria and other immune cells at the infection site. Generating sufficient bioavailability and exposure following parenteral delivery is much more challenging. Defensins are cationic, disulphide cross-linked peptides that can act directly against Gram-positive and -negative bacteria with modest MICs in vitro, but which also modulate and coordinate innate immune responses with a much greater impact on bacterial burden and survival seen in vivo207. α-defensins are produced by neutrophils and are chemotactic for immature dendritic cells, T cells, and mast cells. The β-defensins are expressed in most leukocytes and epithelial cells, where they enhance bactericidal responses via enhanced phagocytosis, pro-inflammatory cytokine induction (particularly in gut and pulmonary epithelial cells), monocyte and immature dendritic cells chemotaxis, and binding to CCR6 receptors on dendritic cells, linking the innate immune system to adaptive immunity208.
Another canonical example is human cathelicidin LL37 and its murine ortholog mCRAMP, which possess weak activity against bacteria in vitro, but remarkable efficacy in animal models of bacterial infection and inflammatory diseases. These effects are thought to be mediated via induction of macrophage differentiation to the M1 phenotype, enhancement of IL1-β signalling via activation of the NLRP3 inflammasome, and concomitant down-regulation LPS-induced activation of TLR-4, and inflammatory cytokines such as IL-12, TNF-α, TNF-γ, IL-4 and IL-12 in leukocytic cells. Further protection may be mediated by its neutrophil and eosinophil chemotactic effects, which are mediated via binding to the formyl peptide receptor 2209. Indeed, the combination of low-grade bacterial infection and LL37-mediated immunomodulation has been implicated in atopic dermatitis, psoriasis, asthma, COPD, systemic lupus erythematosus, rheumatoid arthritis and even atherosclerosis, where defensins and LL-37 are thought to contribute disease progression210. More recently, lignin, a cyclic peptide antibiotic that inhibits S. aureus epithelial colonization in humans and rodents, was reported211 to act via increased expression and release of LL-37 and CXCL8/MIP-2 in human keratinocytes and mouse skin, resulting in the recruitment of monocytes and neutrophils via a TLR/MyD88-dependent mechanism. Bacterial elimination by lugdunin was further enhanced by synergistic application of LL-37 and dermcidin-derived peptides. These results have spurred researchers to develop improved, protease-resistant derivatives of progenitor cathelicidins and defensins, for example a synthetic LL-37 variant was reported to show efficacy in a murine catheter-associated biofilm model, reducing levels of TNFα, whilst boosting protective MCP-1, IL-17A and IL-10212.
A novel immunomodulatory & antimicrobial approach was described in 2021213,214, using a two-pronged approach where an inhibitor of the bacterial enzyme IspH, part of the isoprenoid-synthesis pathway, effected direct bacterial killing. However, inhibition of the enzyme also caused accumulation of the enzyme-substrate, (E)-4-hydroxy-3-methyl-but-2-enyl pyrophosphate (HMBPP), which was recognised by the human defence system to trigger an immune response. A different bacterial-targeting immune response tactic relies on the antibody-recruiting small molecules (ARMs) approach. This strategy, predominantly applied to cancer targets so far215,216, appends a bacterial-targeting ligand to an antibody-triggering stimulant, such as the trisaccharide alpha-Gal217. This elicits uptake and killing of the bacteria by human phagocytes. Within the CARB-X portfolio, Centauri Therapeutics is applying this approach, with ABX01 targeting drug-sensitive and MDR Gram-negative bacteria.
Innate immune modulation: inflammasome inhibitors
Inflammasomes are a family of proteins present in all leukocytes that act as sensors of pathogen- and danger-associated signals. They have evolved to sense bacterial, fungal, viral and parasite threats, however one member of the family, NLRP3, is also known to sense amyloids, crystals, metabolic dysbiosis and mitochondrial stress. Activation of NLRP3 drives caspase 1-dependent release of the pro-inflammatory cytokines IL-1β and IL-18, as well as gasdermin D-mediated pyroptotic cell death218. IL-1β, which downstream drives IL-6 and CRP, is a major pro-inflammatory cytokine that has been implicated in causing many inflammatory diseases. Blocking IL-1β with an antibody (canakinumab) significantly reduced atherothrombosis and non-fatal stroke, with striking effects on mortality due to non-small cell lung carcinoma (NSCLC) and other cancers219. However, there was an adverse two-fold higher incidence of fatal infection; similar to that seen with the IL-1 receptor antagonist Anakinra220. In contrast to IL-1β blockers, inhibition of NLRP3 is likely to be largely redundant during infection, exemplified by studies such of Aspergillus infection, where AIM2 can compensate for a lack of NLRP3221, and with Toxoplasma gondii, where NLRP1 can compensate for a lack of NLRP3222.
In 2015, MCC950 (CP-424,174) originally discovered at Pfizer, was identified as a potent, orally available and highly selective small molecule inhibitor of NLRP3223. It has since been used to show therapeutic effects in disease models involving a number of bacterial, viral, fungal and parasitic pathogens. For example, MCC950 improved survival and reduced (by 4-log fold) bacterial load in the lung in a murine model of S. aureus pneumonia. In contrast blockage of IL1-β with Anakinra or IL-18 with a mAb (alone or in combination) showed a lesser effect on mortality and no effect on bacterial load. It was posited that these effects were mediated via ablation of the effect of staphylococcal virulent alpha-toxin (AT), which the bacterium uses to evade detection and avoid phagocytosis. Other notable examples include the pharmacological application of MCC950 to treat infections associated with haemolytic enterotoxin BL (HBL) induced B. cereus224, Group A Streptococcus (GAS)225, uropathogenic E. coli (UPEC) urinary tract infection226, a cecal ligation and puncture (CLP) model of polymicrobial sepsis227,228, and S. suis, which can cause Streptococcal toxic shock-like syndrome229. Whilst these studies present promising early results in non-clinical models, caution is needed regarding the timing of the dosing regimen and translation to human disease. When MCC950 was used in a combined bacterial and viral pulmonary infection model (influenza plus S. aureus superinfection), there was a decrease in pulmonary bacterial load in treated animals, but this was not associated with improved survival230.
Alternate approaches: CRISPR-Cas
The clustered regularly interspaced short palindromic repeats (CRISPR) and CRISPR-associated (Cas) proteins in bacteria function to protect them from foreign invaders via adaptive and heritable immunity, with the spacer sequences storing genetic information from invading bacteriophages, plasmids and transposons so it can subsequently guide Cas proteins to destroy a recurrent attack. The system has been used widely in recent years for gene editing, including for basic research into understanding host-pathogen interactions. It is now being investigated for potential antibacterial therapeutic applications231,232,233, as summarised in recent reviews234,235,236,237,238, by removing genes encoding for virulence factors or drug resistance. In 2017 Locus Biosciences raised $US19m to help advance a CRISPR-Cas3 bacteriophage platform technology into Phase 1 testing targeting E. coli urinary tract infections or recurrent C. difficile infection. French biotech Eligo Biosciences, a spinout from Massachusetts Institute of Technology and Rockefeller University based at Institute Pasteur, raised €17 m for a bacteriophage-delivered system, using the capsid delivered from a bacteriophage virus with a CRISPR/Cas9 payload. In June 2020 CARB-X awarded funding to Eligo Bioscience for the development of EB004 to eliminate extended-spectrum β-lactamase (ESBL)-producing and CRE from the microbiome of organ transplant patients before their procedure in order to prevent the onset of infection239. In Jan 2021 Eligo signed an agreement with GlaxoSmithKline to adapt Eligo’s technology into a topical treatment for the bacteria that cause acne240. CARB-X-funded Danish microbiome technology company SNIPR Biome completed a successful phase 1 study of a CRISPR-based phage therapy targeting Escherichia coli in the gastrointestinal tract in 2023239. Their product, SNIPR001, contains four bacteriophages armed with CRISPR/Cas DNA editing technology containing sequences specific to E. coli, including antibiotic-resistant strains241.
Alternate approaches: vaccines
Vaccines have been remarkably effective at reducing pneumococcal (Streptococcus pneumoniae), meningococcal (Neisseria meningitidis) and Haemophilus influenzae type B infections, the two classes of bacteria where vaccines are available and widely used. Vaccines can indirectly reduce AMR by reducing antibiotic use, and because the immune response stimulated by vaccination targets only one type of bacteria, vaccines spare the commensal microbiome from the collateral damage caused by antibiotics. Resistance to vaccines is rare (e.g. diphtheria and pertussis vaccines in use for 70 years) and they are suggested to reduce AMR rates242,243, although a recent study indicated an increase in frequency of antibiotic resistance in serotypes not covered by the S. pneumoniae vaccines244. In contrast to antibiotic research, pharmaceutical companies are investing strongly in the development of new vaccines, driven by the ability to have blockbuster sales. In 2017, Pfizer’s Prevnar vaccine was its biggest product, generating $US5.6b. Multiple companies are attempting to develop vaccines against a range of organisms, including S. aureus, Group A Streptococcus, and C. difficile. A 2018 report245 from the Wellcome Trust and the Boston Consulting Group suggests that vaccine efforts should be focused on pathogens on the WHO list including M. tuberculosis, N. gonorrhoeae, P. aeruginosa, S. aureus, E. coli, non-typhoidal Salmonella and Shigella, with less emphasis on WHO-listed pathogens that have low incidence or alternative strategies, such as A. baumannii, Campylobacter, E. faecium, Enterobacteriaceae, H. pylori, K. pneumoniae and S. paratyphi. Ten vaccines were identified in clinical development against C. difficile, S. aureus, Group B Streptococcus and E. coli in 2017243, with another thirteen against M. tuberculosis, while the Pew Trust summary107 of non-traditional products for bacterial infections (Dec 2018) lists eight.
Vaccines are increasingly prominent within the CARB-X portfolio, with 11 active projects (out of 31 therapeutic/preventatives as of May 2024, vs only 1 of 53 formerly funded). These include GlyProVac GPV02 glycosylated protein maternal vaccine against E. coli in neonatal sepsis, GlaxoSmithKline for a Strep A vaccine and an invasive nontyphoidal salmonellosis (iNTS) typhoid conjugate vaccine (TCV) against Salmonella enterica serovars Enteritidis and Typhimurium, LimmaTech Biologics LBT-SA7 (formerly Integrated Biotherapeutics IBT-V02) multivalent vaccine against the three major classes of toxins produced by S. aureus as well as a separate project for a multivalent gonococcus vaccine, Idorsia VXN-319 K. pneumoniae vaccine, Intravacc Avacc 11 outer membrane vesicle vaccine against N. gonorrhoeae bacterial infections, Jenner Institute (University of Oxford) dmGC_0817560 Native Outer Membrane Vesicles (NOMV) vaccine against N. gonorrhoeae, Syntiron Alloy-EK maternal vaccine which targets iron receptor proteins for E. coli and K. pneumoniae neonatal infections, Vaxcyte (formerly SutroVax) VAX-A1 Streptococcus pyogenase carbohydrate conjugate vaccine, Vaxdyn KAPAVAX trivalent vaccine against A. baumannii, P. aeruginosa and K. pneumoniae. However, antibacterial vaccine development is not straightforward. The Phase 3 results of a vaccine targeting the toxoids A and B of C. difficile were reported in 2021, with no prevention of infection identified despite eliciting an immune response246,247. Attempts to develop a Group B streptococcal vaccine have yet to be successful, despite attempts since the 1980’s248. Drawbacks of the vaccination strategy include the difficulty in engineering a vaccine to cover the multiple serotypes of a bacterial species, and the increasing public resistance to employing vaccines, largely based on discredited links to autism. A recent report describes a novel vaccine approach, targeting antigens linked to the transmission of resistance determinants, rather than those directly involved in resistance mechanisms. Incompatibility group HI (IncHI) plasmids encode for surface-located bacterial immunoglobulin-like (Big) domains (Big proteins). Purified RSP protein protected mice from infection by antibiotic-resistant S. Typhimurium 249,250. Nanobodies targeting these proteins interfered with the conjugative transfer of IncHI plasmids.
Alternate approaches: diagnostic-guided therapy
Improved diagnostics are an underappreciated tool in the fight against antimicrobial resistance. While modern technology is able to rapidly identify bacterial species in a pathology laboratory, particularly with high throughput centralised MALDI-TOF analysis, these techniques generally rely on a time-consuming culture step, and are unable to determine full resistance profiles. An inexpensive point-of-care diagnostic that could rapidly distinguish bacterial from viral infections or sterile inflammation would have a dramatic impact on reducing unnecessary antibiotic prescription, and if resistance profiling was possible the most appropriate antibiotic (and least powerful needed to do the job) could be administered from the start of treatment. Importantly, from an antibiotic development perspective, advanced diagnostics would make possible both pathogen-specific therapies, and the ability to conduct clinical trials on MDR infections, as recruiting patients with infections caused by a specific pathogen or known MDR without prior exposure to other antibiotics becomes logistically and financially feasible. The CARB-X portfolio include 6 current, 5 former and 5 graduated diagnostic projects advancing a range of technologies to rapidly identify infections and their antibiotic susceptibility.
Conclusion
The discovery and development of antibiotics revolutionized medicine, transforming once deadly infections into curable diseases. Antibiotic prophylaxis and therapy allowed innovation in many other medical areas, from the implantation of prosthetics, transplantation of tissues and organs, to the use of aggressive immunosuppressive cancer chemotherapy. Sadly, the prospects for the field are dire. The antibiotic market failure continues to be the major driver of an accelerating exit of people and companies from antibiotic R&D. The recent entry of new push incentives such as CARB-X and GARDP is encouraging and can help to sustain small biotechs in the space, but they cannot fix the base economics of antibiotic development. In the face of these challenges, the field is begging to explore alternate, potentially more sustainable modes of clearing or reducing the pathogenicity of infections. These are at a very early stage and will require more informed use of companion diagnostics, biomarker-guided dosing regimens and adaptive trial designs. Nevertheless, it is heartening to see that scientists are prepared to explore higher risk, more controversial alternatives to antibiotic therapy and, most importantly, can now garner support research funds from not-for-profit organisations dedicated to solving the superbug problem. Together, these initiatives may allow us to start to stem the tide of the spread of new resistance mechanisms, and eventually move beyond the ‘each new bug needs a new drug’ paradigm that has led us to the situation we face today.
Data availability
No datasets were generated or analysed during the current study.
References
Waring, M. J. et al. An analysis of the attrition of drug candidates from four major pharmaceutical companies. Nat. Rev. Drug Discov. 14, 475 (2015).
Shepherd, M. J. et al. Ecological and evolutionary mechanisms driving within-patient emergence of antimicrobial resistance. Nat. Rev. Microbiol. 22, 650–665 (2024).
Pooladvand, P., Kendal, J. R. & Tanaka, M. M. How cultural innovations trigger the emergence of new pathogens. Proc. Natl. Acad. Sci. 121, e2322882121 (2024).
Cocker, D. et al. Healthcare as a driver, reservoir and amplifier of antimicrobial resistance: opportunities for interventions. Nat. Rev. Microbiol. 22, 636–649 (2024).
Negatu, S. G., Arreguin, M. C., Jurado, K. A. & Vazquez, C. Being the Alice of academia: lessons from the Red Queen hypothesis. Pathog. Dis. 80, ftac034 (2022).
Murray, C. J. L. et al. Global burden of bacterial antimicrobial resistance in 2019: a systematic analysis. Lancet 399, 629–655 (2022).
Ikuta, K.S. et al. Global mortality associated with 33 bacterial pathogens in 2019: a systematic analysis for the Global Burden of Disease Study 2019. Lancet 400, 2221–2248 (2022).
Naghavi, M. et al. Global burden of bacterial antimicrobial resistance 1990-2021: a systematic analysis with forecasts to 2050. Lancet 404, 1199–1226 (2024).
Pérez Velasco, R. et al. Systematic Review of Economic Evaluations of Preparedness Strategies and Interventions against Influenza Pandemics. Plos One 7, e30333 (2012).
Spellberg, B. & Rex, J. H. The value of single-pathogen antibacterial agents. Nat. Rev. Drug Discov. 12, 963 (2013).
Michaelidis, C. I. et al. The hidden societal cost of antibiotic resistance per antibiotic prescribed in the United States: an exploratory analysis. BMC Infect. Dis. 16, 655 (2016).
Kwon, S., Schweizer, M. L. & Perencevich, E. N. National Institute of Allergy and Infectious Disease (NIAID) Funding for Studies of Hospital-Associated Bacterial Pathogens: Are Funds Proportionate to Burden of Disease?. Antimicrob. Resist Infect. Control 1, 5 (2012).
Wouters, O. J., McKee, M. & Luyten, J. Estimated Research and Development Investment Needed to Bring a New Medicine to Market, 2009-2018. JAMA 323, 844–853 (2020).
Krause, K. The Post-Approval Challenges of Antimicrobial Development.In National Academies of Sciences, Engineering and Medicine meeting for “Examining the Long-term Health and Economic Effects of Antimicrobial Resistance in the United States. https://www.nationalacademies.org/event/01-05-2021/examining-the-long-term-health-and-economic-effects-of-antimicrobial-resistance-in-the-united-states-meeting-3#sl-three-columns-efdc0833-2821-4f34-ac43-fd8cf7c2d044 (2021).
McKenna, M. The antibiotic paradox: why companies can’t afford to create life-saving drugs. Nature 584, 338–341 (2020).
Shlaes, D. M. The Economic Conundrum for Antibacterial Drugs. Antimicrob. Agents Chemother. 64, https://doi.org/10.1128/aac.02057-19 (2019).
Carr, A. & Springer, J. Antibiotic R&D, update 20. (Needham Biotechnology, New York, NY, 2019).
Rahman, S., Lindahl, O., Morel, C. M. & Hollis, A. Market concentration of new antibiotic sales. J. Antibiot. 74, 421–423 (2021).
Rex, J. H. et al. Progress in the Fight Against Multidrug-Resistant Bacteria 2005–2016: Modern Noninferiority Trial Designs Enable Antibiotic Development in Advance of Epidemic Bacterial Resistance. Clin. Infect. Dis. 65, 141–146 (2017).
Rex, J. In Praise of Non-Inferiority. AMR. Solutions Sept 19 2020. https://www.amr.solutions/2020/09/19/in-praise-of-non-inferiority/ (2025).
Dall, C. Plazomicin for tough infections: a tale of 2 clinical trials. CIDRAP News. https://www.cidrap.umn.edu/antimicrobial-stewardship/plazomicin-tough-infections-tale-2-clinical-trials (2025).
Eljaaly, K., Alharbi, A., Alshehri, S., Ortwine, J. K. & Pogue, J. M. Plazomicin: A Novel Aminoglycoside for the Treatment of Resistant Gram-Negative Bacterial Infections. Drugs 79, 243–269 (2019).
Rex, J. H. & Outterson, K. Clinical trial designs for non-traditional antibiotics. June 14 2018. CARB-X Resource website (https://carb-x.org/resources/presentations/), https://carb-x.org/wp-content/uploads/2018/06/RF-2018-06-14-Rex-Outterson-Duke-Margolis-Non-traditional-antibiotic-intro-.pdf (2018).
McDonnell, A., Dissanayake, R., Klemperer, K., Toxvaerd, F. & Sharland, M. The Economics of Antibiotic Resistance. CGD Working Paper 682. (Washington, DC: Center for Global Development, 2024).
McDonnell, A. et al. A New Grand Bargain to Improve the Antimicrobial Market for Human Health, Final Report of the CGD Working Group. (Washington, DC, and London, UK: Center for Global Development, 2023).
A, M. et al. Forecasting the Fallout from AMR: Economic Impacts of Antimicrobial Resistance in Humans – A report from the EcoAMR series. Paris (France) and Washington, DC (United States of America). World Organisation for Animal Health and World Bank, pp. 58. (2024).
Declaration by the Pharmaceutical, Biotechnology and Diagnostics Industries on Combating Antimicrobial Resistance. http://amr-review.org/sites/default/files/Declaration_of_Support_for_Combating_AMR_Jan_2016.pdf.
Pfizer antibacterial research moving to China, Fierce Biotech. Mar 16, 2011. https://www.fiercebiotech.com/biotech/pfizer-antibacterial-research-moving-to-china (2011).
Meiling, B. Housing AstraZeneca’s old R&D efforts in antibiotics, Entasis files for $86M IPO. https://endpts.com/housing-astrazenecas-old-rd-efforts-in-antibiotics-entasis-files-for-86m-ipo/.
Carroll, J. AstraZeneca-backed Entasis Therapeutics joins the IPO class of 2018 with a weak debut price. https://endpts.com/astrazeneca-backed-entasis-therapeutics-joins-the-ipo-class-of-2018-with-a-weak-debut-price/.
Entasis Therapeutics plummets 29% in worst biotech debut since 2002. https://www.nasdaq.com/article/entasis-therapeutics-plummets-29-in-worst-biotech-debut-since-2002-cm1028789.
Masson, G. Innoviva buys struggling antibiotic biotech Entasis, as the AstraZeneca spinout preps FDA filing. Fierce Biotech. May 23, 2022, https://www.fiercebiotech.com/biotech/innoviva-acquires-struggling-antibiotic-biotech-entasis-and-lead-drug-sul-dar(2024).
Carroll, J. UPDATED: Merck to shutter Cubist unit in May, axing most of the 128 R&D staffers. https://www.fiercebiotech.com/biotech/updated-merck-to-shutter-cubist-unit-may-axing-most-of-128-r-d-staffers (Fierce Biotech, accessed 16 December 2024).
Tong, A. Low sales, high cost: Melinta slashes HQ research staff as it struggles to grow antibiotics revenue; https://endpts.com/low-sales-high-cost-melinta-slashes-hq-research-staff-as-it-struggles-to-grow-antibiotics-revenue/) (2018).
Pflanzer, L. R. Allergan is selling off 2 of its non-core businesses in a bid to appease frustrated investors, Business Insider, https://www.businessinsider.com/allergan-selling-womens-health-and-infectious-disease-2018-5. Accessed Dec 16 2024.
Megget, K. Novartis exit from antibiotics a setback for race against resistance. https://www.chemistryworld.com/news/novartis-exit-from-antibiotics-a-setback-for-race-against-resistance/3009316.article. (Chemistry World, accessed 16 December 2024).
Despite Industry AMR Declaration commitments Sanofi quits R&D on anti-infectives. https://www.reactgroup.org/news-and-views/news-and-opinions/year-2018/despite-industry-amr-declaration-commitments-sanofi-quits-rd-on-anti-infectives/ (ReAct, accessed 16 December 2024).
Prescription for success: Emma Walmsley leads GSK transformation, Nov 29, 2018. Available from: https://www.europeanceo.com/profiles/prescription-for-success-emma-walmsley-leads-gsk-transformation.
Leaving the Lab: Tracking the Decline in AMR R&D Professionals. AMR Industry Alliance. https://www.amrindustryalliance.org/mediaroom/leaving-the-lab-tracking-the-decline-in-amr-rd-professionals/. (2024).
Dheman, N. et al. An Analysis of Antibacterial Drug Development Trends in the United States, 1980–2019. Clinical Infectious Diseases, online only, https://doi.org/10.1093/cid/ciaa859. (2020)
Wells, N., Nguyen, V.-K. & Harbarth, S. Novel insights from financial analysis of the failure to commercialise plazomicin: Implications for the antibiotic investment ecosystem. Humanit. Soc. Sci. Commun. 11, 941 (2024).
Tetraphase Pharmaceuticals Market Cap 2012-2019 | TTPH. Available from: https://www.macrotrends.net/stocks/charts/TTPH/tetraphase-pharmaceuticals/market-cap.
Decision of the Board of Directors to Appoint Administrators, Destiny Pharma Press Release, August 22, 2024. https://www.destinypharma.com/2024/08/22/decision-of-the-board-of-directors-to-appoint-administrators/. Accessed Dec 16 2024.
Ingelheim, B., Evotec and bioMérieux launch Aurobac, a joint venture to fight Antimicrobial Resistance. Boehringer Ingelheim press release, July 6 2022. https://www.boehringer-ingelheim.com/science-innovation/human-health-innovation/joint-venture-fight-antimicrobial-resistance. Accessed Dec 16 2024.
2020 Antimicrobial Resistance Benchmark, Access to Medicine Foundation, Jan 21 2020. https://accesstomedicinefoundation.org/resource/2020-antimicrobial-resistance-benchmarkAccessed Dec 16 2024.
Qpex Biopharma Announces Closing of $33 Million Series A Financing and Launch of Company to Address Worldwide Problem of Drug-Resistant Infections, PR Newswire, Oct 22, 2018. https://www.prnewswire.com/news-releases/qpex-biopharma-announces-closing-of-33-million-series-a-financing-and-launch-of-company-to-address-worldwide-problem-of-drug-resistant-infections-300734697.html. Accessed Dec 16 2024.
Member Spotlight: Qpex Biopharma, a Shionogi Group Company. Biocom California.July 11 2024. https://www.biocom.org/lifelines/member-spotlight-qpex-biopharma-a-shionogi-group-companymember-spotlight/. Accessed Dec 16 2024.
Theuretzbacher, U., Baraldi, E., Ciabuschi, F. & Callegari, S. Challenges and shortcomings of antibacterial discovery projects. Clin. Microbiol. Infect. 29, 610–615 (2023).
Engel, A. Snapshot of private financing into oncology-focused companies vs those focused on anti-biotics and anti-fungals. LinkedIn, 2024. https://www.linkedin.com/posts/aleks-engel_snapshot-of-private-financing-into-oncology-focused-activity-7100855831436374016-cfhe?utm_source=share&utm_medium=member_desktop. Accessed Dec 16 2024.
Sendek, J. Investing in Antibiotic Companies: Why the Sector is Compelling Now https://sperotherapeutics.com/investing-antibiotic-companies-sector-compelling-now/ (2019).
Baraldi, E., Årdal, C., Aho, E., Popescu, G.-A. & Melaku, T. The multifaceted nature of lack of access to antibiotics: types of shortage and specific causes, consequences, and solutions. Clin. Microbiol. Infect. 31(3), 333–338 (2024).
Policy and regulatory interventions to address antibiotic shortages in low and middle-income countries. (World Health Organization, accessed Dec 16 2024); https://www.who.int/publications/i/item/9789240100695.
Klein, E. Y. et al. Global trends in antibiotic consumption during 2016–2023 and future projections through 2030. Proc. Natl. Acad. Sci. 121, e2411919121 (2024).
How much does it cost? http://www.esa.int/Our_Activities/Human_and_Robotic_Exploration/International_Space_Station/How_much_does_it_cost (2019).
2019 Annual Contributions to CERN budget. https://fap-dep.web.cern.ch/rpc/2019-annual-contributions-cern-budget (2019).
Daniel, G. W., Schneider, M., Lopez, M. H. & McClellan, M. B. Implementation of a Market Entry Reward within the United States. J. Law Med. Ethics 46, 50–58 (2018).
Hager, N. Hatch, Casey Put Forward Bill to Encourage Antibiotic Innovation. Available from: https://www.finance.senate.gov/chairmans-news/hatch-casey-put-forward-bill-to-encourage-antibiotic-innovation (2018).
Sen. I., & Johnny R.-G. A., S.1712 - DISARM Act of 2019, 116th Congress (2019-2020), https://www.congress.gov/bill/116th-congress/senate-bill/1712. (2019).
Schnirring, L. Bipartisan bill proposes new ‘pull’ incentives for priority antibiotics, http://www.cidrap.umn.edu/news-perspective/2018/06/bipartisan-bill-proposes-new-pull-incentives-priority-antibiotics (2018).
Bennet, S.,& Michael F. [D-CO], S.4760 - The PASTEUR Act, 116th Congress (2019-2020), https://www.congress.gov/bill/116th-congress/senate-bill/4760/text?r=2&s=1. (2020).
Outterson, K. & Rex, J. PASTEUR Act (re)introduced: A delinked Pull award advances in the US!, September 30, 2020, https://amr.solutions/2020/09/30/pasteur-act-reintroduced-a-delinked-pull-award-advances-in-the-us/. (2020).
Wanted: a reward for antibiotic development. Nat. Biotechnol. 36, 555 (2018).
Outterson, K. A shot in the arm for new antibiotics. Nat. Biotechnol. 37, 1110–1112 (2019).
Antimicrobial resistance: UK launches 5-year action plan and 20-year vision. https://www.gov.uk/government/news/antimicrobial-resistance-uk-launches-5-year-action-plan-and-20-year-vision (2019).
Leonard, C. et al. Can the UK ‘Netflix’ Payment Model Boost the Antibacterial Pipeline?. Appl. Health Econ. Health Policy 21, 365–372 (2023).
Outterson, K. & Rex, J. H. Global Pull Incentives for Better Antibacterials: The UK Leads the Way. Appl. Health Econ. Health Policy 21, 361–364 (2023).
Dall, C. UK to test new payment model for antibiotics. http://www.cidrap.umn.edu/news-perspective/2019/07/uk-test-new-payment-model-antibiotics (2019).
Perkins, M. & Glover, D. How the ‘NHS model’ to tackle antimicrobial resistance (AMR) can set a global standard, https://www.england.nhs.uk/blog/how-the-nhs-model-to-tackle-antimicrobial-resistance-amr-can-set-a-global-standard/ (2020).
Duddy, C. “Netflix” for antimicrobials: The Antimicrobial Products Subscription Model in September 2024. https://commonslibrary.parliament.uk/netflix-for-antimicrobials-the-antimicrobial-products-subscription-model/ (House of Commons Library, accessed 16 December 2024).
Office of the Assistant Secretary for Planning and Evaluation (USA), National Action Plan for Combating Antibiotic-Resistant Bacteria 2020-2025. Federal Task Force on Combating Antibiotic-Resistant Bacteria, https://aspe.hhs.gov/pdf-report/carb-plan-2020-2025. (2020).
Adams, B. Small biotech the ‘true engine’ of antibiotic innovation, as pharma ‘spews nonsense’ on its R&D: Jim O’Neill, Mar 28 2019. Available from: https://www.fiercebiotech.com/biotech/small-biotech-true-engine-antibiotic-innovation-as-pharma-spews-nonsense-its-r-d?mkt_tok=eyJpIjoiTURFM01UVTNaalJpWkRoaSIsInQiOiJ2VURJNWNuYzhLWjZIKzRhMlJEdUp3V3UrNTl5OUV5XC9pVzBwWUdLUjQ1cFVCVm1PTTBBVStYSk81eFM3NjBhZGRNQlphRWg2bXpqcmRJTWthVmVKUkthbENRSGJ2YU1DM0lMbjByVG1YOXo1ZEJLOGJnY2wzSUNIbXZBTnRpeG4ifQ%3D%3D&mrkid=754719&utm_medium=nl&utm_source=internal.
Outterson, K. & Rex, J. H. Evaluating for-profit public benefit corporations as an additional structure for antibiotic development and commercialization. Transl. Res. 220, 182–190 (2020).
Rex, J. H. ND4BB: addressing the antimicrobial resistance crisis. Nat. Rev. Microbiol. 12, 231 (2014).
Outterson, K. et al. Accelerating global innovation to address antibacterial resistance: introducing CARB-X. Nat. Rev. Drug Discov. 15, 589 (2016).
Theuretzbacher, U., Savic, M., Årdal, C. & Outterson, K. Innovation in the preclinical antibiotic pipeline. Nat. Rev. Drug Discov. 16, 744 (2017).
Alm, R. A. & Gallant, K. Innovation in Antimicrobial Resistance: The CARB-X Perspective. ACS Infect. Dis. 6, 1317–1322 (2020).
BARDA Stories. BARDA Support Protects Against Drug-Resistant Threats. Antimicrobial programs address urgent and serious pathogens April 27, https://medicalcountermeasures.gov/stories/amr (2023).
Zeituni, E. M. & Raterman, E. L. NIAID’s Comprehensive Support Mechanisms for Antibiotic Development. ACS Infect. Dis. 6, 1299–1301 (2020).
National Institute of Allergy and Infectious Diseases, NIAID’S Antibiotic Resistance Research Framework: Current Status and Future Directions 2019, https://www.niaid.nih.gov/sites/default/files/AR2019.pdf. (2019).
Balasegaram, M. & Piddock, L. J. V. The Global Antibiotic Research and Development Partnership (GARDP) Not-for-Profit Model of Antibiotic Development. ACS Infect. Dis. 6, 1295–1298 (2020).
Kim, W., Prosen, K. R., Lepore, C. J. & Coukell, A. On the Road to Discovering Urgently Needed Antibiotics: So Close Yet So Far Away. ACS Infect. Dis. 6, 1292–1294 (2020).
Cooper, M. A. A community-based approach to new antibiotic discovery. Nat. Rev. Drug Discov. 14, 587 (2015).
Mullard, A. Preclinical antibiotic pipeline gets a pick-me-up. Nat. Rev. Drug Discov. 16, 741 (2017).
Blaskovich, M. A. T., Zuegg, J., Elliott, A. G. & Cooper, M. A. Helping Chemists Discover New Antibiotics. ACS Infect. Dis. 1, 285–287 (2015).
McGilvray, A. Compound screening: Fresh hunting ground. Nature 533, S65 (2016).
Zuegg, J., Hansford, K. A., Elliott, A. G., Cooper, M. A. & Blaskovich, M. A. T. How to Stimulate and Facilitate Early Stage Antibiotic Discovery. ACS Infect. Dis. 6, 1302–1304 (2020).
Wiecek, J. & Buckland-Merrett, G. A View from the Wellcome Trust Drug-Resistant Infections Priority Programme: Creating a Sustainable Research and Development Ecosystem to Meet the Global Need for Antibiotics. ACS Infect. Dis. 6, 1305–1307 (2020).
Beyer, P. & Paulin, S. The Antibacterial Research and Development Pipeline Needs Urgent Solutions. ACS Infect. Dis. 6, 1289–1291 (2020).
Engel, A. Fostering Antibiotic Development through Impact Funding. ACS Infect. Dis. 6, 1311–1312 (2020).
Enabling Breakthroughs in Antimicrobials, AMR Action Fund. https://www.amractionfund.com/.
Joint EFPIA-IFPMA Press Release Announcing the AMR Action Fund. July 9 2020 https://efpia.eu/news-events/the-efpia-view/statements-press-releases/joint-efpia-ifpma-press-release-announcing-the-amr-action-fund (Accessed 16 December, 2024).
Clancy, C. J. & Nguyen, M. H. Buying Time: The AMR Action Fund and the State of Antibiotic Development in the United States 2020. Open Forum Infect. Dis. 7, ofaa464 (2020).
Butler, M. S. & Cooper, M. A. Antibiotics in the clinical pipeline in 2011. J. Antibiot. 64, 413 (2011).
Butler, M. S., Blaskovich, M. A. & Cooper, M. A. Antibiotics in the clinical pipeline in 2013. J. Antibio. 66, 571 (2013).
Butler, M. S., Blaskovich, M. A. T. & Cooper, M. A. Antibiotics in the clinical pipeline at the end of 2015. J. Antibiot. 70, 3 (2016).
Butler, M. S. & Paterson, D. L. Antibiotics in the clinical pipeline in October 2019. J. Antibiot. 73, 329–364 (2020).
Butler, M. S., Henderson, I. R., Capon, R. J. & Blaskovich, M. A. T. Antibiotics in the clinical pipeline as of December 2022. J. Antibiot. 76, 431–473 (2023).
Theuretzbacher, U. et al. Analysis of the clinical antibacterial and antituberculosis pipeline. Lancet Infect. Dis. 19, e40–e50 (2019).
World Health Organization. 2023 Antibacterial agents in clinical and preclinical development: an overview and analysis. June 14 2024. ISBN: 978-92-4-009400-0 https://www.who.int/publications/i/item/9789240094000.
Jesudason, T. Antibacterial agents in preclinical and clinical development. Lancet Microbe 5, 100962 (2024).
Theuretzbacher, U. et al. Evaluating the innovative potential of the global antibacterial pipeline. Clin. Microbiol. Infect. 31, 903–909 (2023).
Antibiotics Currently in Global Clinical Development, March 2019. https://www.pewtrusts.org/-/media/assets/2019/03/antibiotics-currently-in-global-clinical-development.pdf?la=en&hash=078238EF15FACD9753ED2C4EBAB58F16B664B59E.
Lepore, C., Silver, L., Theuretzbacher, U., Thomas, J. & Visi, D. The small-molecule antibiotics pipeline: 2014-2018. Nat. Rev. Drug Discov. 18, 739 (2019).
Thomas, D. & Wessel, C. The State of Innovation in Antibacterial Therapeutics in February 2022. (Bio Industry Analysis, accessed 16 December, 2024); https://www.google.com/url?sa=t&source=web&rct=j&opi=89978449&url=https://www.bio.org/sites/default/files/2022-02/The-State-of-Innovation-in-Antibacterial-Therapeutics.pdf&ved=2ahUKEwjKu6SZsKuKAxUj4TgGHa7mBG4QFnoECBcQAQ&usg=AOvVaw1Hmr_r94sMK5cWPSCrB0nF.
Gigante, V. et al. Multi-year analysis of the global preclinical antibacterial pipeline: trends and gaps. Antimicrob. Agents Chemother. 68, e00535-24 (2024).
Theuretzbacher, U., Outterson, K., Engel, A. & Karlén, A. The global preclinical antibacterial pipeline. Nat. Rev. Microbiol. 18, 275–285 (2020).
Czaplewski, L. et al. Alternatives to antibiotics - a pipeline portfolio review. Lancet Infect. Dis. 16, 239–251 (2016).
Lakemeyer, M., Zhao, W., Mandl, F. A., Hammann, P. & Sieber, S. A. Thinking Outside the Box—Novel Antibacterials To Tackle the Resistance Crisis. Angew. Chem. Int. Ed. 57, 14440–14475 (2018).
CARB-X Complete Portfolio, as of 5/13/24. CARB-X. https://carb-x.org/wp-content/uploads/2024/06/CARB-X-Pipeline-2024.5.13.pdf (Accessed 2024).
Homer, J. A., Johnson, R. M., Koelln, R. A., Moorhouse, A. D. & Moses, J. E. Strategic re-engineering of antibiotics. Nat. Rev. Bioengineer. 3, 223–229 (2024).
Puttagunta, S. & Dunne, M. Antimicrobial Activity of Sulopenem Tested against Gram-positive and Gram-negative Organisms from the United States and Europe (2013–2015). Poster from ASM 2017. https://d1io3yog0oux5.cloudfront (New Orleans, 2017).
GARDP and Bugworks join forces to accelerate development of a novel antibiotic to treat serious bacterial infections. GARDP press release. July 28 2023. https://gardp.org/gardp-and-bugworks-join-forces-to-accelerate-development-of-a-novel-antibiotic-to-treat-serious-bacterial-infections/ (Accessed 2024).
Hameed, P. S. et al. BWC0977, a broad-spectrum antibacterial clinical candidate to treat multidrug resistant infections. Nat. Commun. 15, 8202 (2024).
Daruka, L. et al. ESKAPE pathogens rapidly develop resistance against antibiotics in development in vitro. Nat. Microbiol. 10, 313–331 (2025).
Feng, J. et al. A synthetic antibiotic class with a deeply-optimized design for overcoming bacterial resistance. Nat. Commun. 15, 6040 (2024).
Velkov, T. et al. Structure, Function, and Biosynthetic Origin of Octapeptin Antibiotics Active against Extensively Drug-Resistant Gram-Negative Bacteria. Cell Chem. Biol. 25, 380–391 (2018).
Payne, D. J., Gwynn, M. N., Holmes, D. J. & Pompliano, D. L. Drugs for bad bugs: confronting the challenges of antibacterial discovery. Nat. Rev. Drug Discov. 6, 29 (2006).
Tommasi, R., Brown, D. G., Walkup, G. K., Manchester, J. I. & Miller, A. A. ESKAPEing the labyrinth of antibacterial discovery. Nat. Rev. Drug Discov. 14, 529 (2015).
Hernandez, V. et al. Discovery of a novel class of boron-based antibacterials with activity against gram-negative bacteria. Antimicrob. Agents Chemother. 57, 1394–1403 (2013).
Anacor Pharmaceuticals, Inc. Says GlaxoSmithKline Stops Development of Antibiotic. Biospace, October 5, 2012 https://www.biospace.com/anacor-pharmaceuticals-inc-says-glaxosmithkline-stops-development-of-antibiotic. Accessed Dec 16 2024.
O’Dwyer, K. et al. Bacterial resistance to leucyl-tRNA synthetase inhibitor GSK2251052 develops during treatment of complicated urinary tract infections. Antimicrob. Agents Chemother. 59, 289–298 (2015).
Erwin, A. L. Antibacterial Drug Discovery Targeting the Lipopolysaccharide Biosynthetic Enzyme LpxC. Cold. Spring. Harbor. Perspect. Med. 6, a025304 (2016).
Tomaras, A. P. et al. LpxC Inhibitors as New Antibacterial Agents and Tools for Studying Regulation of Lipid A Biosynthesis in Gram-Negative Pathogens. mBio 5, e01551–14 (2014).
Raetz, C. R. Bacterial endotoxins: extraordinary lipids that activate eucaryotic signal transduction. J. Bacteriol. 175, 5745–5753 (1993).
Krause, K. M. et al. Potent LpxC Inhibitors with In Vitro Activity against Multidrug-Resistant Pseudomonas aeruginosa. Antimicrob. Agents Chemother. 63, e00977–19 (2019).
Cohen, F. et al. Optimization of LpxC Inhibitors for Antibacterial Activity and Cardiovascular Safety. ChemMedChem 14, 1560–1572 (2019).
Teng, M., Nammalwar, B., Taganov, K. & Puerta, D. T. US 10,611,747 B2 Pyrone Based Compounds for Treating Bacterial Infections. (Forge Therapeutics Inc. US 10, 611,747 B2, 2020).
Teng, M., Nammalwar, B., Taganov, K. & Puerta, D. T. US 10,414,735 B2 Substituted Hydroxypyrimidinones for Treating Bacterial Infections. (Forge Therapeutics Inc. US 10,414,735 B2, 2019).
Kalinin, D. V. & Holl, R. LpxC inhibitors: a patent review (2010-2016). Expert Opin. Ther. Pat. 27, 1227–1250 (2017).
Zhang, J. et al. Structure-based discovery of LpxC inhibitors. Bioorg. Med. Chem. Lett. 27, 1670–1680 (2017).
Kurasaki, H. et al. LpxC Inhibitors: Design, Synthesis, and Biological Evaluation of Oxazolidinones as Gram-negative Antibacterial Agents. ACS Med. Chem. Lett. 7, 623–628 (2016).
Yamada, Y. et al. Fragment-Based Discovery of Novel Non-Hydroxamate LpxC Inhibitors with Antibacterial Activity. J. Med. Chem. 63, 14805–14820 (2020).
Furuya, T. et al. N-Hydroxyformamide LpxC inhibitors, their in vivo efficacy in a mouse Escherichia coli infection model, and their safety in a rat hemodynamic assay. Bioorg. Med. Chem. 28, 115826 (2020).
Ennis, A. F. et al. Design and Evaluation of Pyridinyl Sulfonyl Piperazine LpxH Inhibitors with Potent Antibiotic Activity Against Enterobacterales. JACS Au 4, 4383–4393 (2024).
Lu, H. & Tonge, P. J. Inhibitors of FabI, an Enzyme Drug Target in the Bacterial Fatty Acid Biosynthesis Pathway. Acc. Chem. Res. 41, 11–20 (2008).
Pang, L., Weeks, S. D. & Van Aerschot, A. Aminoacyl-tRNA Synthetases as Valuable Targets for Antimicrobial Drug Discovery. Int J. Mol. Sci. 22, 1750 (2021).
Molasky, N. M. R. et al. A novel methionyl-tRNA synthetase inhibitor targeting gram-positive bacterial pathogens. Antimicrob. Agents Chemother. 68, e00745–24 (2024).
Butler, M. S., et al. A Review of Antibacterial Candidates with New Modes of Action. ACS Infect. Dis. 10, 3440–3474 (2024).
Vaara, M. Polymyxin Derivatives that Sensitize Gram-Negative Bacteria to Other Antibiotics. Molecules 24, 249 (2019).
Corbett, D. et al. Potentiation of Antibiotic Activity by a Novel Cationic Peptide: Potency and Spectrum of Activity of SPR741. Antimicrob. Agents Chemother. 61, e00200–e00217 (2017).
Rabanal, F. & Cajal, Y. Recent advances and perspectives in the design and development of polymyxins. Nat. Prod. Rep. 34, 886–908 (2017).
Mendes, R. E. et al. In Vitro Activity Analysis of a New Polymyxin, SPR741, Tested in Combination with Antimicrobial Agents against a Challenge Set of Enterobacteriaceae, Including Molecularly Characterized Strains. Antimicrob. Agents Chemother. 65, e00742–20 (2020).
Stainton, S. M. et al. Assessment of the In Vivo Activity of SPR741 in Combination with Azithromycin against Multidrug-Resistant Enterobacteriaceae Isolates in the Neutropenic Murine Thigh Infection Model. Antimicrob. Agents Chemother. 62, e00239–18 (2018).
Zurawski, D. V. et al. SPR741, an Antibiotic Adjuvant, Potentiates the In Vitro and In Vivo Activity of Rifampin against Clinically Relevant Extensively Drug-Resistant Acinetobacter baumannii. Antimicrob. Agents. Chemother. 61, e01239–17 (2017).
United States Securities and Exchange Commission. Form 10-K. Spero Therapeutics, Inc. For fiscal year ended Dec 31 2019. p73. https://www.annualreports.com/HostedData/AnnualReportArchive/s/NASDAQ_SPRO_2019.pdf.
X-factor to help antibiotics regain their spark. (University of Queensland, 2021).
Ogawara, H. Possible drugs for the treatment of bacterial infections in the future: anti-virulence drugs. J. Antibiot. 74, 24–41 (2021).
Mydock-McGrane, L. K., Hannan, T. J. & Janetka, J. W. Rational design strategies for FimH antagonists: new drugs on the horizon for urinary tract infection and Crohn’s disease. Expert Opin. Drug Discov. 12, 711–731 (2017).
Liu, F. et al. Identification of a Human Anti-Alpha-Toxin Monoclonal Antibody Against Staphylococcus aureus Infection. Front Microbiol 12, 692279 (2021).
Yu, X.-Q. et al. Safety, Tolerability, and Pharmacokinetics of MEDI4893, an Investigational, Extended-Half-Life, Anti-Staphylococcus aureus Alpha-Toxin Human Monoclonal Antibody, in Healthy Adults. Antimicrob. Agents Chemotherap. https://doi.org/10.1128/aac.01020-16 (2017).
Kollef, M. H. & Betthauser, K. D. Monoclonal antibodies as antibacterial therapies: thinking outside of the box. Lancet Infect. Dis. 21, 1201–1202 (2021).
François, B. et al. Efficacy and safety of suvratoxumab for prevention of Staphylococcus aureus ventilator-associated pneumonia (SAATELLITE): a multicentre, randomised, double-blind, placebo-controlled, parallel-group, phase 2 pilot trial. Lancet. Infect. Dis. 21, 1313–1323 (2021).
Lv, C. et al. Research Progress on Small Molecular Inhibitors of the Type 3 Secretion System. Molecules 27, 8348 (2022).
Salmond, G. P. C. & Fineran, P. C. A century of the phage: past, present and future. Nat. Rev. Microbiol. 13, 777 (2015).
Servick, K. Beleaguered phage therapy trial presses on. Science 352, 1506–1506 (2016).
Oechslin, F. Resistance Development to Bacteriophages Occurring during Bacteriophage Therapy. Viruses 10, 351 (2018).
Labrie, S. J., Samson, J. E. & Moineau, S. Bacteriophage resistance mechanisms. Nat. Rev. Microbiol. 8, 317 (2010).
Gómez, P. & Buckling, A. Bacteria-Phage Antagonistic Coevolution in Soil. Science 332, 106–109 (2011).
Gordillo Altamirano, F. et al. Bacteriophage-resistant Acinetobacter baumannii are resensitized to antimicrobials. Nat. Microbiol. 6, 157–161 (2021).
Lin, D. M., Koskella, B. & Lin, H. C. Phage therapy: An alternative to antibiotics in the age of multi-drug resistance. World J. Gastrointest. Pharm. Ther. 8, 162–173 (2017).
Dedrick, R. M. et al. Engineered bacteriophages for treatment of a patient with a disseminated drug-resistant Mycobacterium abscessus. Nat. Med. 25, 730–733 (2019).
Fox, A. Engineered phages stymie drug-resistant infection. Science 364, 518–519 (2019).
Servick, K. U.S. center will fight infections with viruses. Science 360, 1280–1281 (2018).
Kebriaei, R. et al. Bacteriophage AB-SA01 Cocktail in Combination with Antibiotics against MRSA-VISA Strain in an In Vitro Pharmacokinetic/Pharmacodynamic Model. Antimicrob. Agents Chemother. 65, e01863–20 (2020).
Kifelew, L. G. et al. Efficacy of Lytic Phage Cocktails on Staphylococcus aureus and Pseudomonas aeruginosa in Mixed-Species Planktonic Cultures and Biofilms. Viruses 12, 559 (2020).
Law, N. et al. Successful adjunctive use of bacteriophage therapy for treatment of multidrug-resistant Pseudomonas aeruginosa infection in a cystic fibrosis patient. Infection 47, 665–668 (2019).
Armata Pharmaceuticals Provides Update on Pseudomonas Respiratory Programs. January 2022. (Armata Pharmaceuticals Press Release, 16 accessed December, 2024); https://www.prnewswire.com/news-releases/armata-pharmaceuticals-provides-update-on-pseudomonas-respiratory-programs-301454261.html.
Hale, C. BiomX nets $32M to engineer bacteria-killing viruses for IBD and acne. February 2019. (Fierce Biotech, accessed 16 December, 2024); https://www.fiercebiotech.com/biotech/biomx-nets-32m-to-engineer-bacteria-killing-viruses-for-ibd-and-acne.
Vázquez, R. et al. Essential Topics for the Regulatory Consideration of Phages as Clinically Valuable Therapeutic Agents: A Perspective from Spain. Microorganisms 10, 717 (2022).
Barnard, A. M. L. & Fairhead, H. I. M. A commentary on the development of engineered phage as therapeutics. Drug Discov. Today 26, 2095–2098 (2021).
Barnard, A. M. L. & Cass, J. A. Targetable nano-delivery vehicles to deliver anti-bacterial small acid-soluble spore protein (SASP) genes. Emerg. Top. Life Sci. 5, 637–641 (2021).
Sharma, U., Vipra, A. & Channabasappa, S. Phage-derived lysins as potential agents for eradicating biofilms and persisters. Drug Discov. Today 23, 848–856 (2018).
Kåhrström, C. T. A killer hybrid. Nat. Rev. Microbiol. 10, 520 (2012).
Lukacik, P. et al. Structural engineering of a phage lysin that targets Gram-negative pathogens. Proc. Natl. Acad. Sci. (2012).
Larpin, Y. et al. In vitro characterization of PlyE146, a novel phage lysin that targets Gram-negative bacteria. PLOS ONE 13, e0192507 (2018).
Cisek, A. A., Dąbrowska, I., Gregorczyk, K. P. & Wyżewski, Z. Phage Therapy in Bacterial Infections Treatment: One Hundred Years After the Discovery of Bacteriophages. Curr. Microbiol. 74, 277–283 (2017).
Schuch, R., Khan, B. K., Raz, A., Rotolo, J. A. & Wittekind, M. Bacteriophage Lysin CF-301, a Potent Antistaphylococcal Biofilm Agent. Antimicrob. Agents Chemother. 61, e02666–16 (2017).
Schuch, R. et al. Combination Therapy With Lysin CF-301 and Antibiotic Is Superior to Antibiotic Alone for Treating Methicillin-Resistant Staphylococcus aureus–Induced Murine Bacteremia. J. Infect. Dis. 209, 1469–1478 (2013).
Waldron, J. Basilea opts not to take MRSA antibiotic into clinic due to stringent risk-return criteria. Feb 18. https://www.fiercebiotech.com/biotech/basilea-opts-not-take-mrsa-antibiotic-clinic-due-stringent-risk-return-criteria. (Fierce Biotech, accessed 25 February, 2025).
Ratner, M. Microbial cocktails raise bar for C. diff. treatments. Nat. Biotechnol. 38, 1366–1367 (2020).
Vendrik, K. E. W. et al. Periodic screening of donor faeces with a quarantine period to prevent transmission of multidrug-resistant organisms during faecal microbiota transplantation: a retrospective cohort study. Lancet Infect. Dis. 21, 711–721 (2021).
Hohmann, E. L. Faecal microbiota transplantation: more screening for old and new pathogens. Lancet Infect. Dis. 21, 587–589 (2021).
Garber, K. First microbiome-based drug clears phase III, in clinical trial turnaround. Nat. Rev. Drug Discov. 19, 655–656 (2020).
Suez, J., Zmora, N., Segal, E. & Elinav, E. The pros, cons, and many unknowns of probiotics. Nat. Med. 25, 716–729 (2019).
Nakatsuji, T. et al. Development of a human skin commensal microbe for bacteriotherapy of atopic dermatitis and use in a phase 1 randomized clinical trial. Nat. Med. 27, 700–709 (2021).
Sause, W. E., Buckley, P. T., Strohl, W. R., Lynch, A. S. & Torres, V. J. Antibody-Based Biologics and Their Promise to Combat Staphylococcus aureus Infections. Trends Pharmacol. Sci. 37, 231–241 (2016).
Zurawski, D. V. & McLendon, M. K. Monoclonal Antibodies as an Antibacterial Approach Against Bacterial Pathogens. Antibiotics 9, 155 (2020).
Verma, V. Leveraging monoclonal antibodies as therapeutics to address antimicrobial resistance in bacteria. J Appl Bio Biotech 11, 53–60 (2023).
Troisi, M. et al. A new dawn for monoclonal antibodies against antimicrobial resistant bacteria. Front. Microbiol. 13, 1080059 (2022).
Mariathasan, S. & Tan, M.-W. Antibody - Antibiotic Conjugates: A Novel Therapeutic Platform against Bacterial Infections. Trends Mol. Med. 23, 135–149 (2017).
Touti, F. et al. Antibody–Bactericidal Macrocyclic Peptide Conjugates To Target Gram-Negative Bacteria. ChemBioChem 19, 2039–2044 (2018).
Lehar, S. M. et al. Novel antibody–antibiotic conjugate eliminates intracellular S. aureus. Nature 527, 323 (2015).
Bray, N. Antibody–antibiotic conjugate tracks down hidden S. aureus. Nat. Rev. Drug Discov. 15, 18 (2015).
Hardt, W.-D. Homed to the hideout. Nature 527, 309 (2015).
Peck, M. et al. A Phase 1, Randomized, Single-Ascending-Dose Study To Investigate the Safety, Tolerability, and Pharmacokinetics of DSTA4637S, an Anti-Staphylococcus aureus Thiomab Antibody-Antibiotic Conjugate, in Healthy Volunteers. Antimicrob. Agents. Chemother. 63, e02588–18 (2019).
DiGiandomenico, A. et al. A multifunctional bispecific antibody protects against Pseudomonas aeruginosa. Sci. Transl. Med. 6, 262ra155–262ra155 (2014).
Rogers, J. V., Hall, V. L. & McOsker, C. C. Crumbling the Castle: Targeting DNABII Proteins for Collapsing Bacterial Biofilms as a Therapeutic Approach to Treat Disease and Combat Antimicrobial Resistance. Antibiotics 11, 104 (2022).
Conway, J. et al. Phase 1 study of the pharmacokinetics and clinical proof-of-concept activity of a biofilm-disrupting human monoclonal antibody in patients with chronic prosthetic joint infection of the knee or hip. Antimicrob. Agents Chemother. 68, e00655–24 (2024).
Xiong, Y. Q. et al. A Human Biofilm-Disrupting Monoclonal Antibody Potentiates Antibiotic Efficacy in Rodent Models of both Staphylococcus aureus and Acinetobacter baumannii Infections. Antimicrob. Agents Chemother. 61, e00904–e00917 (2017).
Estellés, A. et al. A High-Affinity Native Human Antibody Disrupts Biofilm from Staphylococcus aureus Bacteria and Potentiates Antibiotic Efficacy in a Mouse Implant Infection Model. Antimicrob. Agents Chemother. 60, 2292–2301 (2016).
Francois, B. et al. Interleukin-7 restores lymphocytes in septic shock: the IRIS-7 randomized clinical trial. JCI Insight 3, e98960 (2018).
Mullard, A. Sepsis researchers set sights on immunotherapeutic strategies. Nat. Rev. Drug Discov. 17, 381–383 (2018).
Peters van Ton, A. M., Kox, M., Abdo, W. F. & Pickkers, P. Precision Immunotherapy for Sepsis. Front Immunol. 9, 1926 (2018).
O’Neill, L. A. J., Bryant, C. E. & Doyle, S. L. Therapeutic targeting of Toll-like receptors for infectious and inflammatory diseases and cancer. Pharmacol. Rev. 61, 177–197 (2009).
Savva, A. & Roger, T. Targeting toll-like receptors: promising therapeutic strategies for the management of sepsis-associated pathology and infectious diseases. Front Immunol. 4, 387 (2013).
Mookherjee, N., Anderson, M. A., Haagsman, H. P. & Davidson, D. J. Antimicrobial host defence peptides: functions and clinical potential. Nat. Rev. Drug Discov. 19, 311–332 (2020).
Ganz, T. Defensins: antimicrobial peptides of innate immunity. Nat. Rev. Immunol. 3, 710–720 (2003).
Pazgier, M., Hoover, D. M., Yang, D., Lu, W. & Lubkowski, J. Human beta-defensins. Cell Mol. Life Sci. 63, 1294–1313 (2006).
Tjabringa, G. S., Ninaber, D. K., Drijfhout, J. W., Rabe, K. F. & Hiemstra, P. S. Human cathelicidin LL-37 is a chemoattractant for eosinophils and neutrophils that acts via formyl-peptide receptors. Int Arch. Allergy Immunol. 140, 103–112 (2006).
Kahlenberg, J. M. & Kaplan, M. J. Little peptide, big effects: the role of LL-37 in inflammation and autoimmune disease. J. Immunol. 191, 4895–4901 (2013).
Bitschar, K. et al. Lugdunin amplifies innate immune responses in the skin in synergy with host- and microbiota-derived factors. Nat. Commun. 10, 2730 (2019).
Narayana, J. L., Mishra, B., Lushnikova, T., Golla, R. M. & Wang, G. Modulation of antimicrobial potency of human cathelicidin peptides against the ESKAPE pathogens and in vivo efficacy in a murine catheter-associated biofilm model. Biochim Biophys. Acta Biomembr. 1861, 1592–1602 (2019).
Mehellou, Y. & Willcox, B. E. A two-pronged attack on antibiotic-resistant microbes. Nature 589, 517–518 (2021).
Singh, K. S. et al. IspH inhibitors kill Gram-negative bacteria and mobilize immune clearance. Nature 589, 597–602 (2021).
McEnaney, P. J., Parker, C. G., Zhang, A. X. & Spiegel, D. A. Antibody-Recruiting Molecules: An Emerging Paradigm for Engaging Immune Function in Treating Human Disease. ACS Chem. Biol. 7, 1139–1151 (2012).
McEnaney, P. J., Parker, C. G., & Zhang, A. X., Chapter Thirteen - Antibody-Recruiting Small Molecules: Synthetic Constructs as Immunotherapeutics. In Ann. Rep. Med. Chem. 481–518 (2017).
Kristian, S. A. et al. Retargeting pre-existing human antibodies to a bacterial pathogen with an alpha-Gal conjugated aptamer. J. Mol. Med. 93, 619–631 (2015).
Swanson, K. V., Deng, M. & Ting, J. P. The NLRP3 inflammasome: molecular activation and regulation to therapeutics. Nat. Rev. Immunol. 19, 477–489 (2019).
Ridker, P. M. et al. Effect of interleukin-1b; inhibition with canakinumab on incident lung cancer in patients with atherosclerosis: exploratory results from a randomised, double-blind, placebo-controlled trial. Lancet 390, 1833–1842 (2017).
Harrington, R. A. Targeting Inflammation in Coronary Artery Disease. N. Engl. J. Med 377, 1197–1198 (2017).
Karki, R. et al. Concerted activation of the AIM2 and NLRP3 inflammasomes orchestrates host protection against Aspergillus infection. Cell Host Microbe 17, 357–368 (2015).
Gorfu, G. et al. Dual role for inflammasome sensors NLRP1 and NLRP3 in murine resistance to Toxoplasma gondii. MBio 5, e01117–13 (2014).
Coll, R. C. et al. A small-molecule inhibitor of the NLRP3 inflammasome for the treatment of inflammatory diseases. Nat. Med 21, 248–255 (2015).
Mathur, A. et al. A multicomponent toxin from Bacillus cereus incites inflammation and shapes host outcome via the NLRP3 inflammasome. Nat. Microbiol 4, 362–374 (2018).
LaRock, C. N. et al. IL-1β is an innate immune sensor of microbial proteolysis. Sci. Immunol. 1, eaah3539–eaah3539 (2016).
Schaale, K. et al. Strain- and host species-specific inflammasome activation, IL-1β release, and cell death in macrophages infected with uropathogenic Escherichia coli. Mucosal. Immunol. 9, 124–136 (2016).
Kang, R. et al. A novel PINK1- and PARK2-dependent protective neuroimmune pathway in lethal sepsis. Autophagy 12, 2374–2385 (2016).
Cornelius, D. C. et al. NLRP3 inflammasome inhibition attenuates sepsis-induced platelet activation and prevents multi-organ injury in cecal-ligation puncture. PLoS ONE 15, e0234039 (2020).
Lin, L. et al. An NLRP3 inflammasome-triggered cytokine storm contributes to Streptococcal toxic shock-like syndrome (STSLS). PLoS Pathog. 15, e1007795 (2019).
Robinson, K. M. et al. The inflammasome potentiates influenza/Staphylococcus aureus superinfection in mice. JCI Insight 3, e97470 (2018).
Bikard, D. et al. Exploiting CRISPR-Cas nucleases to produce sequence-specific antimicrobials. Nat. Biotechnol. 32, 1146 (2014).
Citorik, R. J., Mimee, M. & Lu, T. K. Sequence-specific antimicrobials using efficiently delivered RNA-guided nucleases. Nat. Biotechnol. 32, 1141 (2014).
Ram, G., Ross, H. F., Novick, R. P., Rodriguez-Pagan, I. & Jiang, D. Conversion of staphylococcal pathogenicity islands to CRISPR-carrying antibacterial agents that cure infections in mice. Nat. Biotechnol. 36, 971 (2018).
Strich, J. R. & Chertow, D. S. CRISPR-Cas Biology and Its Application to Infectious Diseases. J. Clin. Microbiol. 57, e01307–e01318 (2019).
Greene, A. C. CRISPR-Based Antibacterials: Transforming Bacterial Defense into Offense. Trends Biotechnol. 36, 127–130 (2018).
Pursey, E., Sünderhauf, D., Gaze, W. H., Westra, E. R. & van Houte, S. CRISPR-Cas antimicrobials: Challenges and future prospects. PLOS Pathog. 14, e1006990 (2018).
Balcha, F. B. & Neja, S. A. CRISPR-Cas9 mediated phage therapy as an alternative to antibiotics. Anim. Dis. 3, 4 (2023).
Nath, A. et al. Phage delivered CRISPR-Cas system to combat multidrug-resistant pathogens in gut microbiome. Biomed. Pharmacother. 151, 113122 (2022).
Dall, C. CRISPR-based phage therapy shows promise in first human trial. June 1, 2023. https://www.cidrap.umn.edu/antimicrobial-stewardship/crispr-based-phage-therapy-shows-promise-first-human-trial. (CIDRAP, accessed 16 December 2024).
Gelman, M. GlaxoSmithKline backs a preclinical acne treatment from Eligo Bioscience that uses CRISPR to kill bacteria, Endpoint News. https://endpts.com/glaxosmithkline-backs-a-preclinical-acne-treatment-from-eligo-bioscience-that-uses-crispr-to-kill-bacteria/ (2021).
Gencay, Y. E. et al. Engineered phage with antibacterial CRISPR–Cas selectively reduce E. coli burden in mice. Nat. Biotechnol. 42, 265–274 (2024).
Mishra, R. P. N., Oviedo-Orta, E., Prachi, P., Rappuoli, R. & Bagnoli, F. Vaccines and antibiotic resistance. Curr. Opin. Microbiol. 15, 596–602 (2012).
Jansen, K. U., Knirsch, C. & Anderson, A. S. The role of vaccines in preventing bacterial antimicrobial resistance. Nat. Med. 24, 10 (2018).
Obolski, U. et al. Vaccination can drive an increase in frequencies of antibiotic resistance among nonvaccine serotypes of Streptococcus pneumoniae. Proc. Natl. Acad. Sci. 115, 3102–3107 (2018).
Wellcome Trust, Boston Consulting Group. Vaccines to tackle drug resistant infections. An evaluation of R&D opportunities. https://vaccinesforamr.org/read-the-report/ (2018).
Khanna, S. Defending against a difficult clostridioides with a vaccine. Lancet Infect. Dis. 21, 157–158 (2021).
de Bruyn, G. et al. Safety, immunogenicity, and efficacy of a Clostridioides difficile toxoid vaccine candidate: a phase 3 multicentre, observer-blind, randomised, controlled trial. Lancet Infect. Dis. 21, 252–262 (2021).
Berner, R. Group B streptococcus vaccines: one step further. Lancet Infect. Dis. 21, 158–160 (2021).
Prieto, A. et al. Targeting plasmid-encoded proteins that contain immunoglobulin-like domains to combat antimicrobial resistance. eLife 13, RP95328 (2024).
York, A. Targeting the spread of antimicrobial resistance plasmids. Nat. Rev. Microbiol. 22, 595–595 (2024).
Nontraditional Products for Bacterial Infections in Clinical Development. https://www.pewtrusts.org/-/media/assets/2019/03/nontraditional-products-for-bacterial-infections-in-clinical-development.pdf?la=en&hash=F98E4DBD536F730D3051F97D82F9B4E5C52B7A07 (2019).
Acknowledgements
There is no funding to acknowledge for this manuscript.
Ethics declarations
Competing interests
Both M.A.C. and M.A.T.B. have received extensive funding for antimicrobial research from various government grants and industry collaborative funding, are inventors on patents covering new antimicrobials, and have consulted for companies conducting antimirobial research.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Blaskovich, M.A.T., Cooper, M.A. Antibiotics re-booted—time to kick back against drug resistance. npj Antimicrob Resist 3, 47 (2025). https://doi.org/10.1038/s44259-025-00096-1
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s44259-025-00096-1
This article is cited by
-
Antimicrobial peptide resistance in Salmonella AMR: the role of surface binding and lipopolysaccharide remodelling: one health implications
World Journal of Microbiology and Biotechnology (2026)
