
Abstract
Intracellular pathogens such as Mycobacterium tuberculosis (Mtb) evade host defence mechanisms to infect and survive within host cells. Host-directed therapy (HDT) offers a promising alternative to antibiotics and may overcome antimicrobial resistance. Using high-content screening, we identified benztropine (BZT), an approved Parkinson’s disease drug, as a potent inhibitor of intracellular Mtb. BZT is active in both human and murine macrophages but is inactive in broth. In an aerosol Mtb mouse infection model, oral administration of BZT reduced the burden of Mtb in the lungs by up to 70%. BZT was also active against Salmonella enterica serovar Typhimurium (STm) in an abscess model of infection, significantly reducing size and bacterial load. Chemical competition assays, CRISPR knockouts, and siRNA silencing assays revealed that BZT’s activity against Mtb is mediated via macrophage histamine receptor 1 (HRH1). Our findings establish BZT as a promising repurposed candidate and a lead compound for developing HRH1-targeting antibacterial HDTs.
Similar content being viewed by others

Introduction
With an estimated 1.3 million annual deaths, tuberculosis (TB) remains the major cause of bacterial mortality worldwide1. Despite significant success in reducing mortality over the past few decades, complete TB control with existing antibiotics remains a considerable challenge. Current treatment for TB involves a combination of at least three different antibiotics that directly target Mycobacterium tuberculosis (Mtb), the causative agent of TB. Major shortcomings of existing TB drugs include failure to shorten treatment duration and limited efficacy against emerging multidrug-resistant (MDR) Mtb. To overcome these challenges, new therapies with novel modes of action are sought, amongst which host-directed therapy (HDT) is considered an alternative, novel and attractive approach to antibiotics2,3,4.
HDT leverages drugs that enhance the host’s ability to control invading pathogens without directly targeting the pathogen itself. Specifically, HDTs modulate metabolic, immunological, and signalling pathways that are altered during infection to support intracellular bacterial survival and replication. Mtb is an obligate intracellular pathogen that infects and persists within immune cells, primarily alveolar macrophages5. Consequently, HDTs hold promise as adjunctive therapies to existing antibiotic regimens, with the potential to improve TB treatment outcomes by enhancing the innate antimicrobial functions of phagocytes2, targeting drug-resistant Mtb6 or mitigating TB-associated inflammation7,8. A key advantage of HDTs is their reduced likelihood of driving Mtb drug resistance, as they target host machinery rather than the pathogen9. The concept of HDT is supported by substantial evidence demonstrating that host immune signalling plays a crucial role during TB pathophysiology and that 90% of infected individuals naturally contain the infection10.
Several drugs and investigational compounds have been proposed as HDTs for TB11,12,13,14,15, with most efforts directed towards drug repurposing—a strategy that identifies new therapeutic applications for existing, clinically-approved drugs originally developed for other indications16. Compared to de novo drug development, repurposing offers several advantages, including lower attrition rates, accelerated development timelines, and reduced development costs. However, despite several ongoing clinical trials with repurposed drugs, no HDTs have been approved for TB treatment to date. In some cases, clinical efficacy has not mirrored promising preclinical findings. This underscores the need for continued efforts to identify newer HDTs and further expand the clinical pipeline to improve the likelihood of success.
There is an immunopathogenic overlap between TB and COVID-1917. For instance, both TB and severe COVID-19 infection are characterised by elevated expression of Ficolin-1 (FCN1)- and secreted phosphoprotein 1 (SPP1)-expressing macrophages18, which were shown to drive inflammation and tissue damage through increased oxidative stress and impaired pathogen clearance. Similarly, Interferon-γ (IFN-γ) and tumour necrosis factor (TNF) signalling pathways are hyperactivated in both diseases, contributing to cytokine storms and acute respiratory distress in COVID-19 and granuloma instability and mycobacterial dissemination in TB patients18,19. Immunopathogenic overlaps suggest that the two diseases may share common therapeutic targets. Indeed, some drugs proposed for HDT of TB have been reported to demonstrate activity against COVID-1920. Similarly, drugs that demonstrated activity against COVID-19 could be repurposed for HDT of TB. Aiming to identify new HDT candidates for TB, we conducted a focused phenotypic high-content screening of a compound library against Mtb-infected human THP-1-derived macrophage models. The library, known as the COVID-Box, consists of FDA-approved drugs and investigational compounds previously shown to be active against SARS-CoV-2 and other coronaviruses21.
This study identified benztropine (BZT), a drug prescribed for treating tremors in Parkinson’s disease (PD), as a potential HDT active against Mtb and Salmonella enterica serovar Typhimurium (STm). Its efficacy was demonstrated in both human and murine macrophage cell lines, in human monocyte-derived macrophages (hMDMs) from healthy donors, as well as in vivo using animal infection models. The mode of action revealed that its activity in TB is mediated through blocking macrophage histamine receptor 1 (HRH1).
Results
Intracellular screening identified seven hit compounds active against Mtb
The COVID Box library21, was initially screened at a single-point concentration of 50 µM against THP-1-derived macrophages infected with Mtb H37Rv expressing a red fluorescent protein (RFP). Growth inhibition was quantified by normalising the RFP signal, as previously described in refs. 22,23. Seven hit compounds, exhibiting more than 70% inhibition of Mtb growth and less than 25% cytotoxicity in macrophages, were identified (Fig. 1a, b and Table S1). Notably, five of these compounds are approved drugs currently in clinical use, and to the best of our knowledge, none of them have been reported to possess antibacterial activity (Fig. 1c). These include the PD drug, BZT24, vascular and smooth muscle relaxants ethaverine25 and drotaverine26, and immunosuppressant dihydroorotate dehydrogenase inhibitors vidofludimus27 and brequinar28. The remaining two hits, doxycycline and tetracycline, are antibiotics with previously reported activity against Mtb29. A secondary assay was then conducted to assess dose dependency and potency of the identified hits, revealing that all five drugs exhibited concentration-dependent inhibition of intracellular Mtb growth within a low to moderate µM range (Figs. 1d and S1). Moreover, enumeration of colony-forming units (CFU) in BZT-treated infected macrophages showed a 1-log reduction of bacterial growth at 50 µM (Fig. 1e).

a Scatter plot showing the distribution of inhibitory effects of compounds (50 µM each) from the COVID Box library on intracellular Mtb in THP-1 macrophages 72 h post-infection. Green circles represent hits that inhibit Mtb growth by more than 70% with less than 25% cytotoxicity. b Seven hits with >70% intracellular Mtb growth inhibition (blue bars) and minimal cytotoxicity (red bars) were identified. Two are established antibiotics (doxycycline and tetracycline), while the other five were newly identified. c Chemical structures of the five newly identified hits. d Dose-dependent activity of BZT (IC50 = 15 µM) against intracellular Mtb. e CFU counts of Mtb after 72 h of infection in THP-1 macrophages. NT non-treated control. Data represent the mean ± SEM of at least two independent experiments. Statistical significance was determined using an unpaired T-test; **p < 0.005, *p < 0.05.
BZT exhibits host-directed activity against intracellular Mtb
The intracellular activity of the newly identified hits against Mtb could be attributed to either a direct antibacterial effect or be mediated through activation of pathogen control mechanisms in host macrophages. To assess this, Mthe tb growth inhibition characteristics of each inhibitor were tested in vitro in 7H9 broth media at the same concentration used in the intracellular assay. While ethaverine, drotaverine, brequinar and vidofludimus demonstrated direct activity against Mtb in broth culture, BZT did not inhibit Mtb growth in this medium (Figs. 2a and S2). To rule out that BZT may exhibit direct activity against Mtb at higher concentrations, we tested activity against Mtb in broth at higher concentrations. No growth inhibition was observed up to 100 µM (Fig. 2b), but at significantly higher concentrations (200 µM and 400 µM), BZT did show direct inhibition of Mtb growth in axenic media. To determine whether BZT’s direct anti-Mtb activity could be achieved by its uptake and concentration within the macrophage, we pretreated macrophages with BZT for 24 h, infected them with Mtb and monitored intracellular Mtb load following 96 h from infection. No significant difference in Mtb growth was observed between BZT-pretreated cells and the vehicle control (Fig. 2c), suggesting that macrophages do not concentrate sufficient BZT to achieve direct antimicrobial activity against Mtb. Furthermore, it is unlikely that BZT concentrations in macrophages exceed 90 µM, the IC50 against THP-1 cells, as higher levels would be toxic. Taken together, these results suggest that BZT’s observed intracellular activity against Mtb is confined to the intracellular environment within infected macrophages.

a Comparison of Mtb growth inhibition between intracellular infection (ex vivo) and direct antimicrobial activity in broth (in vitro) of the five identified hits. BZT showed no activity in broth against Mtb. b BZT did not inhibit Mtb growth in broth at concentrations up to 100 µM, whereas ethaverine and drotaverine exhibited concentration-dependent growth inhibition. c Pre-treatment of THP-1-derived macrophages with BZT (30 µM) for 24 h before Mtb infection did not result in significant inhibition of Mtb growth. Comparative analysis of the effect of BZT treatment on changes; d in intracellular Mtb growth, e percentage of infection THP-1 macrophages, and f the average number of Mtb per infected cells at the start of the infection (time 0) and 96 h post-infection. BZT exhibited bacteriostatic effects, leading to a gradual reduction of all three parameters, but still maintained higher levels than the basal level at time 0. In contrast, the TB drug bedaquiline (BDQ, 4 µM) showed bactericidal activity. NT non-treated controls. Data represent mean ± SEM of at least two independent experiments. Statistical significance was determined using a paired ratio T-test; *p < 0.05.
BZT is bacteriostatic against Mtb
To elucidate whether the intracellular activity of BZT against Mtb results from enhanced bacterial killing (bactericidal) or suppression of intracellular Mtb replication (bacteriostatic), we conducted a comparative analysis of Mtb infection at 0 h post-infection (p.i.) and at 96 h p.i. Specifically, we measured total Mtb load, the percentage of infected cells, and the average number of Mtb per infected cell. In the non-treated macrophages, all three parameters increased over time, reflecting some degree of intracellular Mtb growth (Fig. 2d, f). Conversely, treatment with the positive control drug bedaquiline resulted in a marked reduction in Mtb infection, percentage of infected cells, and average Mtb per infected cell at 96 h post-infection to levels below baseline, consistent with its well-established bactericidal activity (Fig. 2d, f). In BZT-treated macrophages, we observed a significant decrease in intracellular Mtb load (Fig. 2d), percentage of infected cells (Fig. 2e), and average number of Mtb per infected cell compared to vehicle-treated controls (Fig. 2f). However, these values remained higher at 96 h post-infection relative to baseline (0 h), indicating that BZT limits intracellular Mtb replication and survival without promoting bacterial killing. These findings suggest that BZT exerts a bacteriostatic host-directed activity.
BZT is active intracellularly against Mtb in murine- and human-derived macrophages
We further characterised the spectrum of host cells capable of harbouring BZT’s intracellular activity against Mtb. BZT activity was evaluated in Mtb-infected murine-derived macrophages (RAW 264.7) and hMDMs collected from three healthy donors. In murine-derived macrophages, BZT showed higher potency with an IC50 of 4 µM compared to 15 µM in THP-1 macrophages (Fig. 3a). BZT also exhibited dose- and time-dependent Mtb growth inhibition in infected hMDMs, with maximal effect occurring between days 5 and 10 (Fig. 3b, c). The comparable potency of BZT against Mtb in hMDMs (IC50 = 8.2 µM) and THP-1-derived macrophages further validates the suitability of the THP-1 macrophage infection model for screening HDT for TB. Interestingly, hMDMs from the three donors exhibited variability in their responses to Mtb infection, with cells from one donor notably controlling the infection effectively even without treatment (Fig. 3d), highlighting individual differences in innate Mtb control.

a BZT showed a dose-dependent intracellular activity against Mtb in RAW 264.7 cells. b hMDMs infected with GFP-expressing Mtb were monitored for two weeks using live-cell imaging and automated GFP fluorescence area analysis. Mtb-infected hMDMs were treated with two-fold serial dilutions of BZT. Data were normalised to cells treated with the vehicle control (1% DMSO), defined as 0% inhibition, and the positive control (1 µg/mL RIF + INH, R + I), defined as 100% inhibition. c BZT demonstrated a dose-dependent inhibition of Mtb growth in hMDMs. d Comparison of Mtb growth in hMDMs isolated from three healthy donors. The statistical analysis was conducted by pooling data across all donors. BZT showed dose-dependent activity against STm in e THP-1 macrophages and f RAW 264.7 cells. Data represent mean ± SEM of at least two independent experiments. NT non-treated control.
BZT demonstrates host-directed activity against Salmonella enterica STm
HDTs are often reported to possess cross-species activity against evolutionary distinct intracellular pathogens, as similar host pathways are exploited during infection2. To assess whether BZT’s intracellular activity extends beyond Mtb, we evaluated its effect against another pathogenic intracellular bacterium, STm. STm is a major cause of non-typhoidal salmonellosis (NTS), a leading bacterial cause of diarrhoeal disease, responsible for approximately 93 million cases and an estimated 77,500 deaths worldwide each year30. As with TB, the increasing prevalence of multidrug resistance in NTS, particularly resistance to key antibiotics such as fluoroquinolones and third-generation cephalosporins, poses a growing public health challenge31.
BZT exhibited even greater potency against STm in THP-1 macrophages and RAW 264.7 cells (Fig. 3e, f) with an IC50 value of 1.9 µM in THP-1 cells. Notably, BZT exhibited no direct antibacterial activity against STm at concentrations up to 100 µM (Fig. S2), indicating that its activity is entirely host-directed. BZT’s potent activity against STm through a host-targeted mechanism underscores the potential for repurposing to treat STm infections and offers a promising alternative approach to combat emerging drug resistance.
BZT’s activity is mediated through the host macrophage’s HRH1
Mechanistically, BZT interacts with three distinct molecular targets, acting as either an antagonist of the histamine and muscarinic receptors (HRH1 and M1, respectively) or as an inhibitor of the dopamine transporter (DAT)32,33. To determine whether any of these targets contribute to BZT’s activity against Mtb, pharmacological and genetic approaches were utilised.
As shown in Fig. 4a, BZT activity against Mtb was significantly reduced in the presence of histamine, the natural HRH1 ligand, whereas carbachol, an M1 receptor agonist, and dopamine, the DAT substrate, had no effect (Fig. S3). Moreover, consistent with previous observations34, histamine alone promoted Mtb growth within macrophages, while carbachol and dopamine had no effect compared to the untreated control.

In Mtb-infected THP-1 macrophages; a addition of histamine (HIS, 10 µM), the natural ligand of the H1 receptor, blocked BZT (30 µM)-mediated inhibition of intracellular Mtb growth and b pyrilamine (PYR, 50 µM), an HRH1 antagonist, exhibited intracellular activity against Mtb. Graphs showing % intracellular Mtb growth inhibition of; c eight FDA-approved H1 antagonists (50 µM each) and; d BZT (25 µM) and seventeen other structural analogues of BZT (25 µM each) in Mtb infected THP-1 macrophages all the tested compounds were added to the media 3 h post infection and Mtb growth inhibition was determined 72 h post infection. e Chemical structure of two of the BZT analogues with higher anti-Mtb potency than BZT. NT non-treated control. Data represent the mean ± SEM of at least two independent experiments. Statistical significance was determined using an unpaired T-test; ***p < 0.001, **p < 0.005, *p < 0.05; ns non-significant.
The further investigate receptor involvement, we tested the activities of target-specific antagonists with in vitro receptor binding affinities similar to BZT33,35,36. Of these, only the HRH1 blocker, pyrilamine, exhibited dose-dependent activity against intracellular Mtb, albeit with lower potency compared to BZT (Fig. 4b). In contrast, the M1 antagonist, atropine, and the DAT inhibitor, GBR-12935, had no effect at any tested concentration (Fig. S3).
As BZT is not classified as a conventional antihistaminic drug or HRH1 blocker, we tested a panel of known HRH1 blockers for their ability to inhibit Mtb intracellular growth. Four of the eight drugs tested—tripelennamine, fexofenadine, cetirizine and promethazine—were found to be active against intracellular Mtb; however, BZT remained the most potent (Fig. 4c). Interestingly, although cetirizine has been reported to attenuate the growth of intracellular Mtb at 10 µM34, it did not show significant activity under our experimental conditions.
The superior potency of BZT compared to classical antihistamines suggests that unique structural features may enable it to engage HRH1 differently or interact with an additional molecular target. To explore this, we screened 17 structurally related BZT analogues obtained from the National Cancer Institute/Discovery Therapeutics (NCI/DTP) Open Chemicals Repository (https://dtp.cancer.gov/repositories/) for intracellular activity against Mtb. Of these, seven analogues inhibited intracellular Mtb growth, with two compounds, NSC169431 and NSC22953, displaying higher potency than BZT (Fig. 4d and Table S2). Notably, NSC169431 corresponds to the FDA-approved antihistamine deptropine, while NSC22953 is a close structural analogue of BZT, differing only by a single methyl group at the 4-position of one of the phenyl rings (Fig. 4e). These findings suggest that while BZT’s antihistaminic activity contributes to its activity against Mtb, its specific structural features are also critical for achieving maximal potency.
Given that BZT’s antimicrobial activity in macrophages is mediated through HRH1, altering HRH1 gene expression should impact Mtb intracellular infection levels. Indeed, siRNA-mediated knockdown of HRH1 in macrophages (siHRH1) significantly reduced Mtb growth compared to siRNA scrambled controls (Fig. 5a). Moreover, treatment with BZT did not further decrease Mtb growth in the infected siHRH1, confirming that its activity is dependent on HRH1 expression.

a siRNA-mediated silencing of hrh1 transcription in THP-1 macrophages inhibited intracellular Mtb growth. The addition of BZT (30 µM) did not significantly affect Mtb growth, while the addition of exogenous HIS (10 µM) partially reversed Mtb inhibition caused by hrh1 siRNA. b Western blot of HRH1 protein expression levels in parental and HRH1 K/O cells. Actin is used as a loading control. c HRH1 CRISPR knockout THP-1 macrophage clones (HRH1 K/O) showed reduced intracellular Mtb levels. d Inhibition of Mtb growth in parental THP-macrophages was not significantly altered by BZT (10 µM) in HRH1 K/O. Colocalization of acidic phagosomes and pHrodo-labelled Mtb MC26206 auxotroph expressing GFP; e Heat-killed (HK) or live Mtb parental THP-1 macrophages that were either non-treated (NT) or treated with BZT (30 µM); f Heat-killed (HK) or live Mtb-infected parental THP-1 macrophages and live Mtb-infected HRH1 K/O; and g HK or live Mtb-infected HRH1 K/O that were either not treated (NT) or treated with BZT (30 µM). Data normalised to HK Mtb-infected cells, indicating 100% colocalization. Data represent the mean ± SEM of at least two independent experiments. Statistical significance was determined using unpaired T-test; ****p < 0.0001, ***p < 0.001, **p < 0.005, *p < 0.05; ns non-significant.
To further validate HRH1 involvement, we generated an hrh1 knockout in THP-1 cells using CRISPR. Knockout efficiency was validated by genomic sequencing (Fig. S4a), as well as by assessing HRH1 expression at both protein levels via western blot (Fig. 5b) and the mRNA levels using qPCR (Fig. S4b). Upon Mtb infection, the knockout cells (HRH1 K/O) resulted in significantly lower bacterial growth compared to the parental control (Fig. 5c). Consistently, BZT treatment did not significantly alter Mtb growth in the HRH1 K/O reinforcing that its activity is mediated via the macrophage HRH1 (Fig. 5c). Taken together, these findings demonstrate that BZT’s intracellular activity against Mtb is mediated through histamine signalling and its cognate HRH1 receptor.
Phagosomal acidification is associated with BZT treatment
Recent findings suggested that histamine, via HRH1, promotes Mtb growth by inhibiting phagosome acidification through the GRK2-p38MAPK signalling pathway34. Given that BZT targets HRH1, we hypothesised that BZT treatment may enhance phagosomal acidification, contributing to its intracellular activity. To evaluate this, we examined the effect of BZT on phagosome acidification using a pHrodo-based colocalization assay37. For this experiment, we infected THP-1 parental or HRH1 K/O with pHrodo-labelled Mtb MC26206 auxotroph expressing green fluorescent protein (GFP). The Mtb MC²6206 strain is auxotrophic for leucine and pantothenate and retains all known virulence genes and remains capable of establishing infection similar to Mtb38. Colocalization between acidified phagosomes and pHrodo-labelled Mtb was then analysed. As shown in Fig. 5d, BZT treatment is associated with increased colocalization between live Mtb and pHrodo in infected THP-1 macrophages, indicating enhanced acidification of vesicles containing the active bacteria. A similar increase of colocalization between Mtb and pHrodo in HRH1 K/O compared to parental controls (Fig. 5e) was observed. BZT treatment of Mtb-infected HRH1 K/O did not alter colocalization (Fig. 5f), suggesting that the increase in phagosomal acidification following BZT treatment is linked to inhibition of HRH1.
BZT reduces Mtb load in the lungs of TB-infected mice
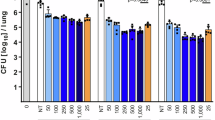
The in vivo efficacy of BZT against Mtb was evaluated using a mouse model of TB39. Mice were infected with a low-dose aerosol-based Mtb to mimic the natural course of infection in humans. Infected mice then received an oral dose of BZT (10 mg/kg or 20 mg/kg) alone as mono-therapy or BZT (10 mg/kg) in combination with the first-line anti-Mtb drug rifampin (RIF) (10 mg/kg or 25 mg/kg), for two weeks starting from day 14 post-infection. At the end of the treatment period, bacterial loads in the lungs, liver and spleen were quantified by CFU enumeration. Mice treated with BZT as a mono-therapy at either dose exhibited a significant reduction in pulmonary bacillary load (Fig. 6a). Notably, BZT at 20 mg/kg, resulted in a 70% reduction of Mtb load from the lungs of infected mice, an efficacy comparable to RIF at 10 mg/kg (80% reduction). BZT treatment had no impact on bacterial loads in the liver or spleen (Fig. 6b, c). Additionally, co-administration of BZT with RIF did not significantly enhance RIF’s activity (data not shown), indicating a lack of synergy in this mouse TB model.

a Oral treatment with 10 mg/kg and 20 mg/kg BZT reduced Mtb load in the lungs of Mtb-infected mice. Rifampin (RIF) at 10 mg/kg and 25 mg/kg, served as a positive control. BZT did not reduce Mtb levels in mouse; b livers; c spleens. BZT (5 mg/kg, subcutaneous) reduced; d abscess size; and e bacterial recovery from abscesses in a STm high-density abscess model. f BZT reduced clinical signs of morbidity resulting from high-density subcutaneous infection with STm. Data represent the mean ± SEM of two or three independent experiments containing 2–3 biological replicates each (N = 5–6). Statistical significance was determined using the Mann–Whitney U-test; ****p < 0.0001, ***p < 0.001, **p < 0.005, ns non-significant, NT non-treated controls.
BZT inhibits STm in a high-density abscess model of infection
The in vivo efficacy of BZT against STm was evaluated using a high-density subcutaneous (abscess) model of infection, as previously described40. Mice were subcutaneously infected with STm on the right dorsum. One hour post-infection, they received a subcutaneous injection of BZT (5 mg/kg) at the infection site. Abscess formation and disease progression were monitored over 72 h. At the experimental endpoint, abscess size (or visible dermonecrosis) was measured, and contents were harvested and homogenised for bacterial CFU enumeration. As shown in Fig. 6d, BZT demonstrated in vivo efficacy, reducing abscess size by 71.1% compared to the vehicle-treated mice. Similarly, BZT treatment resulted in a 1-log reduction in the median bacterial load within the abscesses (Fig. 4e), indicating its in vivo antimicrobial activity. Additionally, BZT treatment significantly reduced clinical signs of morbidity resulting from the infection (Fig. 6f).
Discussion
BZT, clinically approved in 1956 for the treatment of PD symptoms, is an orally active drug with well-characterised pharmacokinetics and safety profiles. Beyond its established use in PD, BZT has demonstrated potential for repurposing in other diseases, including multiple sclerosis35 and cancer36. Our discovery that BZT effectively inhibits intracellular Mtb and STm infections in various macrophage models, including hMDMs, coupled with its in vivo efficacy in mouse Mtb and STm models, highlights its promise as a candidate for HDT against TB and other intracellular bacterial infections. To our knowledge, this study provides the first evidence supporting BZT’s potential role as an HDT for TB and other intracellular pathogens.
In PD, BZT acts through antimuscarinic and dopamine-mimetic mechanisms32. Notably, genetic loci linked to PD are also associated with increased susceptibility to infections and inflammatory diseases, including leprosy and irritable bowel disease41,42. Furthermore, mutations in PD-associated genes, such as LRKK2 and PARK2, are linked with increased susceptibility to TB43,44. Some drugs developed to treat PD are also reported to be active for treating TB45,46. As such, initially, we assumed that BZT activity might be linked to Parkinson's disease. However, through pharmacological and genetic analyses, we demonstrated that BZT’s activity against Mtb is independent of its PD targets and instead operates via inhibition of the host macrophage HRH1 receptor.
Histamine production is induced upon Mtb infection, modulating inflammation and suppressing protective immunity. In HDC −/− mice, which lack histidine decarboxylase (HDC) and thus the ability to synthesise histamine from histidine, lower Mtb CFU in the lungs suggest a role for histamine in limiting bacterial clearance47. Additionally, histamine has been reported to promote Mtb growth in THP-1 macrophages through HRH134. Expanding on this, we demonstrated that inhibiting the HRH1 receptor with pyrilamine or BZT, or silencing hrh1 expression using siRNA and CRISPR knockout, significantly reduces Mtb intracellular growth. Furthermore, blocking HRH1 with BZT or knocking out hrh1 was found to be associated with enhanced acidification of Mtb-containing phagosomes, aligning with previously reported signalling pathways34.
Although histamine, through activation of HRH1, clearly promoted intracellular Mtb growth, not all the assessed HRH1 antagonists were effective in reducing Mtb replication within macrophages. The activity and potency of the tested HRH1 antagonists against Mtb appeared to be confined to a specific chemical subclass. Among them, BZT and its structural analogues, such as deptropine and NSC22953, exhibited the most pronounced effects in macrophages. This suggests that these compounds may engage HRH1 through a distinct binding mode or signalling cascade functionally linked to HRH1, or they may act on additional molecular targets.
In this study, we have also demonstrated that the HRH1-mediated effect of BZT against Mtb is distinct from its role in treating PD. This presents an opportunity to develop novel HRH1-selective BZT analogues with enhanced potency against Mtb while minimising M1- or DAT-mediated side effects. The design and synthesis of selective BZT analogues that differentiate between HRH1 and DAT receptors have been previously described33. Notably, the introduction of a bulky alkyl functional group at the nitrogen atom of the tropane ring favours DAT selectivity48, whereas small substituents (e.g., OH, F, CH₃, or CF₃) on one of the aromatic rings of the diphenylmethoxy moiety enhance selectivity for HRH1. Our screening of structurally related BZT analogues supports these findings, as HRH1-selective analogues with small substituents on the diphenylmethoxy group exhibited greater potency against Mtb than BZT (Fig. S4). This suggests that increased HRH1 selectivity enhances BZT’s antimycobacterial activity. Moreover, following similar structural optimisation strategies used in the development of non-sedating second-generation antihistamines, BZT analogues with reduced CNS side effects could be designed by introducing a carboxylic acid functional group49.
BZT demonstrated dose-dependent efficacy both in an ex vivo assay and as a monotherapy in vivo, achieving up to 70% reduction in Mtb CFU levels in the lungs of infected mice. As previously noted, most HDTs are intended for use as adjuvant treatments, and their antimicrobial properties are often under-characterised in the literature. Nevertheless, BZTs’ in vivo activity is comparable to or exceeds CFU reduction levels reported for other HDTs currently in clinical trials, including metformin50, statins51, imatinib52, and vitamin D53.
BZT dosages used in the mouse TB efficacy study followed previously established preclinical in vivo dosing for multiple sclerosis (10 mg/kg)35, taking into consideration BZT toxicity data from murine (intraperitoneal LD₅₀ = 65 mg/kg) and rat (oral LD₅₀ = 940 mg/kg) models54. In vivo TB efficacy of BZT, when administered via the oral route, suggests no significant pharmacokinetic barriers in achieving the required therapeutic concentrations at the target site. Oral bioavailability is a critical factor in TB drug development, where prolonged treatment is necessary. Effective oral delivery not only improves therapeutic outcomes but also supports patient adherence, which is essential for treatment success. Furthermore, no observable toxicity was detected with daily BZT administration at the highest tested dose (20 mg/kg), which remains well below the reported LD50 in mice (65 mg/kg), indicating the potential for dose escalation to enhance efficacy. However, rough human equivalent dose estimations using previously reported animal-to-human equivalent dose conversion models55 suggest that the dose used in the in vivo mouse study may exceed the maximum prescribed daily dose for PD treatment.
Beyond its efficacy against Mtb, BZT demonstrated effectiveness in treating STm infection in a mouse model, achieving over a 1-log reduction in bacterial CFU. This further underscores its potential for treating other intracellular bacterial infections. Notably, local STm infection in mice required an even lower BZT dose (5 mg/kg), which was administered as a single dose directly at the infection site.
Collectively, the combined in vitro and in vivo results demonstrate that BZT exhibits host-directed activity against two distinct intracellular pathogens—Mtb and STm—highlighting its potential for repurposing as an HDT for TB in combination with the existing antibiotics and STm infections. However, further clinical studies are necessary to confirm its efficacy and establish safe and effective dosing in human populations.
Methods
Screening library and drug preparation
The screening library, consisting of 160 compounds in a 96-well format, was generously provided by the Medicines for Malaria Venture (MMV) at no cost. These compounds, prepared as 10 mM DMSO stocks, were stored at −20 °C. BZT analogues were sourced from the National Cancer Institute’s Developmental Therapeutics Program (NCI/DTP) collection, also free of charge. All drugs and inhibitors used in this experiment, except for Vidofludimus and Ethaverine (Molnova Biotech), were purchased from Sigma-Aldrich. Compounds were routinely dissolved in DMSO at a concentration of 10 mM and stored at −20 °C until required.
Bacterial strains and culture conditions
The following strains were utilised: Mtb H37Rv (ATCC 27294), Mtb H37Rv expressing RFP (pTEC27)56, Mtb H37Rv expressing GFP (pFPV2)57, Mtb MC26206 auxotroph expressing GFP38, and STm (pCS26-Pac)58. Mycobacterial strains were cultured in Middlebrook 7H9 broth (Difco), supplemented with 10% (v/v) OADC (5% bovine albumin fraction, 2% dextrose, 0.004% catalase, 0.05% oleic acid, and 0.8% sodium chloride solution), 0.05% (v/v) Tween-80 (Sigma-Aldrich), the appropriate antibiotic, and +L-leucine +D-Pantothenate (for Mtb auxotroph) at 37 °C in standing cultures. STm (pCS26-Pac) was grown on Luria–Bertani (LB) agar plates or in LB broth with 50 µg/mL kanamycin at 37 °C.
Cell lines and culture conditions
THP-1 monocytes (ATTC TIB-202™) were grown in RPMI-1640 medium (Sigma-Aldrich) supplemented with 5% (v/v) foetal bovine serum (FBS), 2% (v/v) glutamine, and 1% (v/v) nonessential amino acids (NEAA) in T75 cell culture flasks. Cells were maintained at 37 °C in 5% CO2, with a density range of 0.25–1.0 × 106 cells/mL. Cultures were used for up to three months. For differentiation into macrophages, THP-1 monocytes were stimulated one day prior to experiments with RPMI-1640 medium supplemented with 40 ng/mL phorbol 12-myristate 13-acetate (PMA) (Sigma-Aldrich) at 37 °C and in 5% CO2. RAW 264.7 macrophage cells (Abelson murine leukaemia virus transformed) obtained from the ATCC® were maintained in Dulbecco’s Modified Eagle’s Medium (DMEM) supplemented with 10% (v/v) heat-inactivated FBS, 1% NEAA, and 1% sodium pyruvate. Cells were passaged at 80% confluence.
In vitro activity against Mtb
Test compounds were added at the required concentration to sterile 96-well (VWR®) flat-bottom plates containing 100 µL/well of 7H9 medium supplemented with 10% OADC, 0.05% Tween-80, and 50 µg/mL hygromycin. Subsequently, 100 µL of 5 × 105 cells/mL mycobacterial inoculum (OD600, 0.1 = 0.33 × 108 CFU/mL) was added to each well, bringing the final volume to 200 µL. The inoculum was prepared from a preculture grown in mid-log phase (OD600 = 0.3–0.7). Each plate also contained a negative control (1% DMSO) and a positive control (3 µM BDQ). The plates were sealed with parafilm, placed in a container with moist tissue, and incubated for six days at 37 °C. After incubation, fluorescence intensity in each well was measured using the BioTek Synergy H4 plate reader (excitation at 530 nm, emission at 590 nm) to determine the level of Mtb growth inhibition.
Intracellular assay of test compounds in Mtb-infected THP-1 macrophages
The intracellular activity of test compounds was assessed following a previously described method59. Briefly, THP-1 cells were differentiated into macrophages and infected with Mtb-RFP for 3 h at a multiplicity of infection (MOI) of 2:1. The infected cells were then incubated with the test compounds for 72 h at 37 °C, 5% CO2. After 72 h, the cells were washed once with RPMI-1640 media, fixed with 4% paraformaldehyde (PFA) in phosphate-buffered saline (PBS), and stained with 4’,6-diamidino-2-phenylindole (DAPI). Intracellular Mtb load was quantified using the CellInsight CX5 High Content platform (ThermoFisher Scientific) according to a previously described method22. Data were normalised to cells treated with the vehicle control (1% DMSO), defined as 0% inhibition, and the positive control bedaquiline (4 µM), defined as 100% inhibition. The activity assay in Mtb-infected RAW 264.7 cells was performed similarly, with the exception that the cells were not pre-differentiated and DMEM was used instead of RPMI.
Infection assay of Mtb-infected primary macrophages
hMDMs were isolated from peripheral blood mononuclear cells (PBMCs) of three healthy donors, as described previously22,60,61. Briefly, PBMCs were allowed to adhere for 2 h, after which non-adherent cells were washed away. The adherent monocytes were differentiated for seven days in complete DMEM containing 25 mM N-2-hydroxyethylpiperazine-N’-2-ethanesulfonic acid (HEPES), 100 U/mL penicillin, 100 μg/mL streptomycin, 2 mM L-glutamine (Gibco) and 10% active human serum, with media changes every 2–3 days. Macrophages were harvested by trypsinisation and seeded at 5000 cells/well in 30 μL of the above antibiotic-free (ABF) media in a 384-well plate and incubated overnight. Mtb H37Rv expressing GFP (pFPV2) was used to infect the hMDMs in equal volume at an MOI of 2 for 3 h. Compounds were diluted in ABF media containing 0.3 mM Draq7 (BD Biosciences) at five times the final concentration and added to the infected hMDMs to a final volume of 75 μL per well. Rifampicin and Isoniazid at 1 μg/mL each were used as a positive control for Mtb growth inhibition, while 0.1% DMSO served as the negative vehicle control. All incubation steps were carried out in a humidified environment at 37 °C with 5% CO2. Infected hMDMs were monitored using the Incucyte S3 (IncuCyte Live-Cell Analysis System, Sartorius) for 14 days post-infection, with images captured every 8 h (10×, 2 images/well). Bacterial growth was assessed by measuring the area of relative fluorescence (GFP signal).
Intracellular assay of test compounds in STm-infected THP-1 macrophages
STm expressing a luminescent reporter protein (pCS26-Pac)58 was cultured overnight on an LB agar plate. A broth culture was started several hours prior to infection. Once the culture reached an OD600 of 1, bacteria were pelleted by centrifugation and washed three times with RPMI media. The bacteria were then opsonised with 10% human serum for 30 min at 37 °C, then diluted in RPMI to a concentration of 1 × 106 CFU/mL. The opsonised bacteria were incubated with THP-1 macrophages seeded at 1 × 105 cells per well in a 96-well plate, which had been differentiated with PMA (40 ng/mL) for 24 h prior to infection. After a 30-min incubation, the infected cells were washed three times with fresh RPMI and incubated for an additional hour with 100 μg/mL gentamicin to eliminate remaining extracellular bacteria. The infected cells were then incubated with the tested compounds in the presence of 10 μg/mL of gentamicin. Intracellular bacterial growth was assessed after 72 h of infection by luminescence using a BMG FLUOstar Omega microplate reader (Mandel Scientific Inc.).
SiRNA-mediated gene silencing of hrh1
One day prior to transfection, THP-1 monocytes were seeded at 80,000 cells per well in 96-well clear flat-bottom plates (VWR®) and differentiated into macrophages. After differentiation, cells were transfected with 10 pmol siRNA (Integrated DNA Technologies) and 2 μL HiPerFect (Qiagen) transfection reagent per well, following the manufacturer’s instructions. Cells were then infected with Mtb pTEC27, and intracellular growth was quantified using the CellInsight CX5 (ThermoFisher Scientific) according to the protocol described above.
Quantitative real-time PCR analysis
For siRNA knockdown confirmation, THP-1 cells (80,000 cells/well) were transfected as described above with siRNA for 24 h, then washed and incubated for an additional 72 h to mimic infection time. RNA was extracted and purified using the Illustra RNAspin Mini RNA Isolation Kit (GE Healthcare), followed by a second purification step for reaction mixture clean-up per the manufacturer’s protocol. RNA yield and purity were assessed using a NanoDrop™ One Microvolume UV-Vis Spectrophotometer (Thermo Scientific™). For reverse transcription, 274 ng of RNA per condition and timepoint was used with the One Script Plus cDNA Synthesis Kit (ABM), using Oligo-(dT) primers as per the manufacturer’s instructions. Real-time PCR was performed on the CFX96 Real-Time System (BIO-RAD), with reactions carried out in 20-μL volumes containing 2× FastStart SYBR Green Master Mix (Roche), 5 μL of 5-fold diluted cDNA and 0.3 μM of each primer. Control reactions without reverse transcriptase were included to confirm the absence of genomic DNA contamination. The thermal cycling protocol included an initial denaturation at 95 °C for 10 min, followed by 35–41 cycles of denaturation at 95 °C for 15 s, annealing at 56 °C for 30 s, extension at 72 °C for 30 s, and a final extension at 72 °C for 1 min. For CRISPR knockout confirmation, two million parental and HRH1KO THP-1 monocytes were differentiated into macrophages over a two-day period. Total RNA was extracted as above and converted to cDNA using the iScript™ Select cDNA Synthesis Kit (Bio-Rad) with oligo(dT) primers, following the manufacturer’s protocol. Quantitative real-time PCR (RT-qPCR) was performed using the C1000 Touch Thermal Cycler (Bio-Rad) and FastStart Universal SYBR Green Master Mix (Roche Applied Science) as above, but in 40 μL reactions. All reactions were performed in technical triplicate. Relative-fold HRH1 expression levels were calculated using the delta-delta Ct method62, normalised to the GAPDH gene expression. All primers (forward: 5’-CCAGCGTCTAAGGAACAAGGT-3’, reverse: 5’-TGCCCTCTGTATACCAGACAC-3’, GAPDH forward: 5’-GCCTCAAGATCATCAGCAATGC-3’, GAPDH reverse: 5’-GTGGTCATGAGTCCTTCCACGA-3’) were ordered from Integrated DNA Technologies.
CRISPR interference of hrh1 in THP-1 macrophages
CRISPR knockouts in THP-1 cells were designed and synthesised at Synthego Corporation (Menlo Park, CA, USA) using single guide RNA (sgRNA) targeting hrh1 genes. sgRNA and Cas9 were transfected into THP-1 cells, generating two pools containing parental cells and various mutations in the individual hrh1genes. HRH1 knockout clones (HRH1 K/O) were isolated from each pool through the limiting dilution method63 and then clonally expanded. Clones were analysed by PCR and sequencing, as well as western blot.
HRH1 K/O clone sequencing
PCR was performed on the HRH1 K/O clones to isolate the hrh1 gene, which was sent for sequencing. The sequence of the clone was compared to the guide sequence with the Synthego ICE (Interface of CRISPR Edits) program (https://ice.editco.bio/#/) to determine the mutations in the hrh1 gene of the clone cell line.
Western blot analysis of HRH1 knockout in THP-1 macrophages
THP-1 cells were differentiated into macrophages and lysed in RIPA buffer with protease inhibitors (Roche). 30 μg of each lysate was separated on a 12.5% SDS-PAGE gel and transferred to nitrocellulose. Membranes were blocked (3% milk in TBS, 1 h, RT), incubated with HRH1 rabbit polyclonal antibody (Thermo Fisher, PA5-120691, 1:3000, 1 h, RT), followed by goat anti-rabbit HRP-conjugated secondary antibody (Sigma, 1706515, 1:3000, 1 h, RT). Detection was performed using SuperSignal West Pico PLUS (Thermo Fisher). For loading control, membranes were stripped, re-blocked, and probed with mouse anti-actin (Invitrogen, MA5-15739, 1:3000, overnight, 4 °C), followed by goat anti-mouse HRP-conjugated secondary antibody (Sigma, 1706516, 1:3000, 1 h, RT), and imaged using Azure 300 Imager (Azure Biosystems).
pHrodo colocalization assay in Mtb
Mtb MC26206 auxotroph expressing GFP was incubated with pHrodo dye (50 µM) at 37 °C for 30 min. The labelled bacteria were divided into two portions: one heat-killed (HK) at 65 °C for 45 min and the other kept at 37 °C. Both HK and live bacteria were opsonised in 10% human serum and added at an MOI of 20:1 to plates containing pre-differentiated THP-1 parental cells or HRH1 K/O clones, treated with either BZT (30 µM) or the vehicle (H2O). The infection proceeded for 1.5 h at 37 °C, after which cells were washed, fixed with 3.7% PFA, stained with Hoechst, and analysed for colocalization using the CellInsight CX5 High-Content platform (ThermoFisher Scientific).
In vivo Mtb infection mouse model
Forty female BALB/c mice (Charles River Laboratories, Wilmington, MA) were challenged intranasally with 1 × 102 CFU of Mtb H37Rv per mouse. One day post-challenge, a random subset (n = 5) of mice were euthanised, their lungs removed, homogenised and plated to enumerate CFU. Counts confirmed 9 × 10¹–1.5 × 10² CFU/lung. The remaining infected mice were monitored until day 14 post-challenge, at which point another random subset of mice (n = 5) were euthanised, and lungs, spleens and livers were harvested, homogenised using a gentleMACS dissociator (Miltenyi Biotech) and plated to again enumerate CFU. On average, bacterial burdens were 1 × 105 CFU/lung and 5 × 101–1 × 102 CFU/liver, with no Mtb detected in the spleens. The remaining infected mice were divided in to six treatment groups consisting of 5 mice/group receiving daily gavage for two weeks with either BZT (10 mg/kg or 20 mg/kg), rifampin (10 mg/kg or 25 mg/kg), or a combination of BZT (10 mg/kg) and rifampin (10 mg/kg) as well as the non-treated control. At 28 days post-challenge, all mice were euthanised, and lung, spleen, and liver homogenates were plated to confirm CFU enumeration. The experiment was done in two biological replicates.
High-density in vivo subcutaneous (abscess) STm infection mouse model
The in vivo activity of BZT against STm was tested using a subcutaneous abscess model, as previously described40. Female CD-1 mice were obtained from Charles River Laboratories, Wilmington, MA, aged 4–6 weeks and weighing 25 ± 5 g, were group-housed in cohorts of three to four. Each mouse was shaved and treated with a chemical depilatory to remove dorsal fur, followed by ethanol disinfection. STm (pCS26-Pac) (~1 × 108 CFU in 50 µL) was injected subcutaneously into the right dorsum. BZT (5 mg/kg) or the vehicle (DMSO) was administered subcutaneously to the formed abscess &&site, and disease progression was monitored daily for 72 h. At the experimental endpoint, abscess size (or visible dermonecrosis) was measured using callipers. Abscesses were then harvested in PBS, homogenised with a Mini-Beadbeater (BioSpec Products, Bartlesville, OK), and plated on LB agar for bacterial enumeration. Prior to efficacy testing, BZT and DMSO toxicities were assessed in uninfected mice and found to be non-toxic (data not shown). Three independent experiments, each with two or three biological replicates, were performed.
Statistical analyses
All statistical analyses were performed using GraphPad Prism 10.2.3. Data are presented as mean ± SEM. Statistical significance was determined using an unpaired T-test, unless otherwise specified. The threshold for statistical significance was set at p < 0.05. Statistical significance is indicated as follows: p < 0.05 (*), p < 0.005 (**), p < 0.001 (***), and p < 0.0001 (****). Specific details regarding the statistical tests and sample sizes for each experiment are provided in the relevant figure legends or text.
Data availability
All data are presented in the manuscript and supplementary material. Raw data that support the findings of this study are available from the corresponding author upon request.
References
WHO. Global Tuberculosis Report https://www.who.int/teams/global-programme-on-tuberculosis-and-lung-health/tb-reports/global-tuberculosis-report-2024 (2024).
Shapira, T., Christofferson, M. & Av-Gay, Y. The antimicrobial activity of innate host-directed therapies: a systematic review. Int J. Antimicrob. Agents 63, 107138. https://doi.org/10.1016/j.ijantimicag.2024.107138 (2024).
Kilinç, G., Saris, A., Ottenhoff, T. H. M. & Haks, M. C. Host-directed therapy to combat mycobacterial infections. Immunol. Rev. 301, 62–83, https://doi.org/10.1111/imr.12951 (2021).
Young, C., Walzl, G. & Du Plessis, N. Therapeutic host-directed strategies to improve outcome in tuberculosis. Mucosal Immunol. 13, 190–204, https://doi.org/10.1038/s41385-019-0226-5 (2020).
Cohen, S. B. et al. Alveolar macrophages provide an early Mycobacterium tuberculosis niche and initiate dissemination. Cell Host Microbe 24, 439–446.e4, https://doi.org/10.1016/j.chom.2018.08.001 (2018).
Zumla, A. et al. Host-directed therapies for tackling multi-drug resistant tuberculosis: learning from the pasteur-bechamp debates. Clin. Infect. Dis. 61, 1432–1438, https://doi.org/10.1093/cid/civ631 (2015).
Krug, S., Parveen, S. & Bishai, W. R. Host-directed therapies: modulating inflammation to treat tuberculosis. Front. Immunol. 12, 660916. https://doi.org/10.3389/fimmu.2021.660916 (2021).
Subbian, S. et al. Pharmacologic inhibition of host phosphodiesterase-4 improves isoniazid-mediated clearance of Mycobacterium tuberculosis. Front. Immunol. 7, 238, https://doi.org/10.3389/fimmu.2016.00238 (2016).
Torfs, E., Piller, T., Cos, P. & Cappoen, D. Opportunities for overcoming Mycobacterium tuberculosis drug resistance: emerging mycobacterial targets and host-directed therapy. Int. J. Mol. Sci. 20, 2868, https://doi.org/10.3390/ijms20122868 (2019).
Colangeli, R. et al. Mycobacterium tuberculosis progresses through two phases of latent infection in humans. Nat. Commun. 11, 4870. https://doi.org/10.1038/s41467-020-18699-9 (2020).
Lee, A., Xie, Y. L., Barry, C. E. & Chen, R. Y. Current and future treatments for tuberculosis. BMJ 368, m216, https://doi.org/10.1136/bmj.m216 (2020).
Dutta, N. K. et al. Adjunctive host-directed therapy with statins improves tuberculosis-related outcomes in mice. J. Infect. Dis. 221, 1079–1087, https://doi.org/10.1093/infdis/jiz517 (2020).
Rao, M. et al. Improving treatment outcomes for MDR-TB—novel host-directed therapies and personalised medicine of the future. Int. J. Infect. Dis. 80S, S62–S67, https://doi.org/10.1016/j.ijid.2019.01.039 (2019).
Naicker, N., Sigal, A. & Naidoo, K. Metformin as host-directed therapy for TB treatment: scoping review. Front. Microbiol. 11, 435, https://doi.org/10.3389/fmicb.2020.00435 (2020).
Peña-Díaz, S. et al. Glycogen synthase kinase 3 inhibition controls Mycobacterium tuberculosis infection. iScience 27, 110555. https://doi.org/10.1016/j.isci.2024.110555 (2024).
Mourenza, Á, Gil, J. A., Mateos, L. M. & Letek, M. Novel treatments against Mycobacterium tuberculosis based on drug repurposing. Antibiotics 9, 550, https://doi.org/10.3390/antibiotics9090550 (2020).
Sheerin, D., Abhimanyu, A., Wang, X., Johnson, W. E. & Coussens, A. Immunopathogenic overlap and shared therapeutic targets for Covid-19 and tuberculosis predicted from transcriptomic meta-analysis. SSRN Electron. J. https://doi.org/10.2139/ssrn.3815987 (2021).
Sheerin et al. Immunopathogenic overlap between COVID-19 and tuberculosis identified from transcriptomic meta-analysis and human macrophage infection. iScience 25, 104464. https://doi.org/10.1016/j.isci.2022.104464 (2022).
Booysen, P. et al. Immune interaction between SARS-CoV-2 and Mycobacterium tuberculosis. Front. Immunol. 14, 1254206. https://doi.org/10.3389/fimmu.2023.1254206 (2023).
Mishra, B. B., Essafi, M., Singh, R., Gupta, S. & Parihar, S. P. Editorial: repurposed drugs as immune-modulators to combat infectious diseases. Front. Immunol. 13, 848373. https://doi.org/10.3389/fimmu.2022.848373 (2022).
Almeida-Paes, R. et al. Medicines for malaria venture COVID box: a source for repurposing drugs with antifungal activity against human pathogenic fungi. Mem. Inst. Oswaldo Cruz 116, e210207. https://doi.org/10.1590/0074-02760210207 (2021).
Shapira, T. et al. High-content screening of eukaryotic kinase inhibitors identify CHK2 inhibitor activity against Mycobacterium tuberculosis. Front. Microbiol. 11, 553962. https://doi.org/10.3389/fmicb.2020.553962 (2020).
Sahile, H. A., Rens, C., Shapira, T., Andersen, R. J. & Av-Gay, Y. DMN-Tre labeling for detection and high-content screening of compounds against intracellular mycobacteria. ACS Omega 5, 3661–3669, https://doi.org/10.1021/acsomega.9b04173 (2020).
Connolly, B. S. & Lang, A. E. Pharmacological treatment of Parkinson disease: a review. JAMA 311, 1670–1683, https://doi.org/10.1001/jama.2014.3654 (2014).
Wang, Y. & Rosenberg, R. L. Ethaverine, a derivative of papaverine, inhibits cardiac L-type calcium channels. Mol. Pharm. 40, 750–755 (1991).
Tömösközi, Z., Finance, O. & Arányi, P. Drotaverine interacts with the L-type Ca2+ channel in pregnant rat uterine membranes. Eur. J. Pharm. 449, 55–60, https://doi.org/10.1016/S0014-2999(02)01993-3 (2002).
Muehler, A., Peelen, E., Kohlhof, H., Gröppel, M. & Vitt, D. Vidofludimus calcium, a next generation DHODH inhibitor for the Treatment of relapsing-remitting multiple sclerosis. Mult. Scler. Relat. Disord. 43, 102129. https://doi.org/10.1016/j.msard.2020.102129 (2020).
Peters, G. J. Re-evaluation of Brequinar sodium, a dihydroorotate dehydrogenase inhibitor. Nucleosides Nucleotides Nucleic Acids 37, 666–678, https://doi.org/10.1080/15257770.2018.1508692 (2018).
Gonzalo, X., Casali, N., Broda, A., Pardieu, C. & Drobniewski, F. Combination of amikacin and doxycycline against multidrug-resistant and extensively drug-resistant tuberculosis. Int J. Antimicrob. Agents 45, 406–412, https://doi.org/10.1016/j.ijantimicag.2014.11.017 (2015).
Stanaway, J. D. et al. The global burden of non-typhoidal salmonella invasive disease: a systematic analysis for the Global Burden of Disease Study 2017. Lancet Infect. Dis. 19, 1312–1324, https://doi.org/10.1016/S1473-3099(19)30418-9 (2019).
Amir, Y. et al. The prevalence of antimicrobial drug resistance of non-typhoidal Salmonella in human infections in sub-Saharan Africa: a systematic review and meta-analysis. Expert Rev. Anti Infect. Ther.22, 761–774, https://doi.org/10.1080/14787210.2024.2368989 (2024).
Brocks, D. R. Anticholinergic drugs used in Parkinson’s disease: an overlooked class of drugs from a pharmacokinetic perspective. J. Pharm. Pharm. Sci. 2, 39–46 (1999).
Kulkarni, S. S., Kopajtic, T. A., Katz, J. L. & Newman, A. H. Comparative structure-activity relationships of benztropine analogues at the dopamine transporter and histamine H1 receptors. Bioorg. Med. Chem. 14, 3625–3634, https://doi.org/10.1016/j.bmc.2006.01.017 (2006).
Mo, S. et al. Mycobacterium tuberculosis utilizes host histamine receptor H1 to modulate reactive oxygen species production and phagosome maturation via the p38MAPK-NOX2 axis. mBio 13, e0200422. https://doi.org/10.1128/mbio.02004-22 (2022).
Deshmukh, V. A. et al. A regenerative approach to the treatment of multiple sclerosis. Nature 502, 327–332, https://doi.org/10.1038/nature12647 (2013).
Sogawa, C. et al. Antiparkinson drug benztropine suppresses tumor growth, circulating tumor cells, and metastasis by acting on SLC6A3/dat and reducing STAT3. Cancers12, 523, https://doi.org/10.3390/cancers12020523 (2020).
Kapellos, T. S. et al. A novel real time imaging platform to quantify macrophage phagocytosis. Biochem. Pharmacol. 116, 107–119 (2016).
Vilchèze, C. et al. Rational design of biosafety level 2-approved, multidrug-resistant strains of Mycobacterium tuberculosis through nutrient auxotrophy. mBio 9, e00938-18 (2018).
Kramnik, I. & Beamer, G. Mouse models of human TB pathology: roles in the analysis of necrosis and the development of host-directed therapies. Semin Immunopathol. 38, 221–237, https://doi.org/10.1007/s00281-015-0538-9 (2016).
Pletzer, D., Mansour, S. C., Wuerth, K., Rahanjam, N. & Hancock, R. E. W. New mouse model for chronic infections by gram-negative bacteria enabling the study of anti-infective efficacy and host-microbe interactions. mBio 8, e00140-17. https://doi.org/10.1128/mBio.00140-17 (2017).
Mira, M. T. et al. Susceptibility to leprosy is associated with PARK2 and PACRG. Nature 427, 636–640, https://doi.org/10.1038/nature02326 (2004).
Fava, V. M., Dallmann-Sauer, M. & Schurr, E. Genetics of leprosy: today and beyond. Hum. Genet. 139, 835–846, https://doi.org/10.1007/s00439-019-02087-5 (2020).
Härtlova, A. et al. LRRK2 is a negative regulator of Mycobacterium tuberculosis phagosome maturation in macrophages. EMBO J. 37, e98694. https://doi.org/10.15252/embj.201798694 (2018).
Patrick, K. L., Bell, S. L., Weindel, C. G. & Watson, R. O. Exploring the “multiple-hit hypothesis” of neurodegenerative disease: bacterial infection comes up to bat. Front. Cell Infect. Microbiol. 9, 138, https://doi.org/10.3389/fcimb.2019.00138 (2019).
Jiang, Y. M. et al. Effective treatment of manganese-induced occupational parkinsonism with p-aminosalicylic acid: a case of 17-year follow-up study. J. Occup. Environ. Med. 48, 644–649, https://doi.org/10.1097/01.jom.0000204114.01893.3e (2006).
Maitra, A. et al. Repurposing-a ray of hope in tackling extensively drug resistance in tuberculosis. Int. J. Infect. Dis. 32, 50–55, https://doi.org/10.1016/j.ijid.2014.12.031 (2015).
Carlos, D. et al. Histamine plays an essential regulatory role in lung inflammation and protective immunity in the acute phase of Mycobacterium tuberculosis infection. Infect. Immun. 77, 5359–5368, https://doi.org/10.1128/IAI.01497-08 (2009).
Agoston, G. E. Novel N-substituted 3α-[bis(4’-fluorophenyl)methoxy]tropane analogues: selective ligands for thedopamine transporter. J. Med. Chem 40, 4329–4339, https://doi.org/10.1021/jm970525a (1997).
Simon, F. E. R. & Simons, K. J. H1 antihistamines: current status and future directions. World Allergy Organ J. 1, 145–155, https://doi.org/10.1186/1939-4551-1-9-145 (2008).
Singhal, A. et al. Metformin as adjunct antituberculosis therapy. Sci. Transl. Med. 6, 263ra159, https://doi.org/10.1126/scitranslmed.3009885 (2014).
Parihar, S. P. et al. Statin therapy reduces the Mycobacterium tuberculosis burden in human macrophages and in mice by enhancing autophagy and phagosome maturation. J. Infect. Dis. 209, 754–763, https://doi.org/10.1093/infdis/jit550 (2014).
Napier, R. J. et al. Imatinib-Sensitive tyrosine kinases regulate mycobacterial pathogenesis and represent therapeutic targets against tuberculosis. Cell Host Microbe 10, 475–485, https://doi.org/10.1016/j.chom.2011.09.010 (2011).
Reeme, A. E. & Robinson, R. T. Dietary vitamin D3 suppresses pulmonary immunopathology associated with late-stage tuberculosis in C3HeB/FeJ mice. J. Immunol. 196, 1293304, https://doi.org/10.4049/jimmunol.1500931 (2016).
Cayman Chemical. Safety Data Sheet acc. to OSHA HCShttps://cdn.caymanchem.com/cdn/msds/33474m.pdf (2023).
Reigner, B. G. & Blesch, K. Estimating the starting dose for entry into humans: principles and practice. Eur. J. Clin. Pharm. 57, 835–845, https://doi.org/10.1007/s00228-001-0405-6 (2002).
Bardarov, S. et al. Specialized transduction: an efficient method for generating marked and unmarked targeted gene disruptions in Mycobacterium tuberculosis, M. bovis BCG and M. smegmatis. Microbiology148, 3007–3017, https://doi.org/10.1099/00221287-148-10-3007 (2002).
Kalsum, S. et al. A high content screening assay for discovery of antimycobacterial compounds based on primary human macrophages infected with virulent Mycobacterium tuberculosis. Tuberculosis135, 102222. https://doi.org/10.1016/j.tube.2022.102222 (2022).
Bjarnason, J., Southward, C. M. & Surette, M. G. Genomic profiling of iron-responsive genes in Salmonella enterica serovar Typhimurium by high-throughput screening of a random promoter library. J. Bacteriol. 185, 4973–4982, https://doi.org/10.1128/JB.185.16.4973-4982.2003 (2003).
Rankine-Wilson, L., Rens, C., Sahile, H. A. & Av-Gay, Y. Mycobacterium tuberculosis Infection of THP-1 cells: a model for high content analysis of intracellular growth and drug susceptibility. Methods Mol. Biol. 2427, 73–82, https://doi.org/10.1007/978-1-0716-1971-1_7 (2022).
Andersson, B., Nordvall, M. J., Welin, A., Lerm, M. & Schön, T. A novel mycobacterial growth inhibition assay employing live-cell imaging of virulent M. tuberculosis and monitoring of host cell viability. Tuberculosis124, 101977. https://doi.org/10.1016/j.tube.2020.101977 (2020).
Santucci, P. et al. Intracellular localisation of Mycobacterium tuberculosis affects efficacy of the antibiotic pyrazinamide. Nat. Commun. 12, 3816. https://doi.org/10.1038/s41467-021-24127-3 (2021).
Rao, X., Huang, X., Zhou, Z. & Lin, X. An improvement of the 2ˆ(-delta delta CT) method for quantitative real-time polymerase chain reaction data analysis. Biostat. Bioinforma. Biomath. 3, 71–85 (2013).
Giuliano, C. J., Lin, A., Girish, V. & Sheltzer, J. M. Generating single cell–derived knockout clones in mammalian cells with CRISPR/Cas9. Curr. Protoc. Mol. Biol. 128, e100. https://doi.org/10.1002/cpmb.100 (2019).
Acknowledgements
The work was funded by the Canadian Institute of Health Research (CIHR) under grants PUU-177960 and PJT-152931. Henok A. Sahile acknowledges support from CIHR for the Research Excellence, Diversity, and Inclusion (REDI) Early Career Transition Award (CIHR 202305DI1), which provided stipend and research funding for this work. We also acknowledge the CL-3 Facility, Facility for Infectious Disease and Epidemic Research (FINDER), within the Life Sciences Institute at the University of British Columbia. We thank Nina Maeshima and Virginia Pichler for critically reviewing our manuscript and Dr. Maria Lerm, Blanka Anderson, and Eva Danielson from Linköping University for performing the live imaging assays. Additionally, we acknowledge the support from the Chemical Biology Consortium Sweden (CBCS), Linköping University Node, a national research infrastructure funded by the Swedish Research Council (Dr.nr. 2021-00179) and SciLifeLab.
Author information
Authors and Affiliations
Contributions
H.A.S. conceived, designed and performed experiments, analysed data and wrote and edited the manuscript. M.C. conducted the CRISPR experiment, colocalization and ex vivo Mtb and STm experiments. M.A.A. designed and performed the in vivo STm experiments and assisted with manuscript writing. C.R. performed the ex vivo STm experiments. H.H. conducted the siRNA experiments and assisted in manuscript writing; J.D.C. assisted with siRNA experiments and data analysis, contributed to the in vivo Mtb assay and edited the manuscript. J.C. designed, supervised, and conducted the in vivo Mtb assay. G.L. conducted qPCR assays. R.E.W.H. supervised the in vivo STm work. Y.A.-G. conceived and designed the study, secured funding, supervised the work, and wrote and edited the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Ethics
The mouse Mtb and STm infection trials were conducted in accordance with Canadian Council on Animal Care (CCAC) guidelines and approved by the University of Saskatchewan Animal Research Ethics Board (Protocol #20230084) and the University of British Columbia Animal Care Committee (Protocol A23-0030-A002), respectively.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Sahile, H.A., Christofferson, M., Alford, M.A. et al. The Parkinson’s drug benztropine possesses histamine receptor 1-dependent host-directed antimicrobial activity against Mycobacterium tuberculosis. npj Antimicrob Resist 3, 70 (2025). https://doi.org/10.1038/s44259-025-00143-x
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s44259-025-00143-x
