
Abstract
Background
While advanced gastrointestinal stromal tumors (GISTs) are primarily treated with tyrosine kinase inhibitors (TKIs), acquired resistance from specific mutations in KIT or PDGFRA frequently occurs. We aimed to assess the utility of circulating tumor DNA (ctDNA) as a modality of therapeutic decision-making in advanced GIST.
Methods
We conducted a pooled analysis of SCRUM-Japan studies for advanced GIST patients. We compared patient characteristics analyzed with tissue and blood samples, assessed gene alteration profiles, and evaluated prognostic implications from ctDNA status.
Results
In 133 patients, tissue and blood samples were analyzed for 89 and 44 patients, respectively. ctDNA was detected in 72.7% of cases; no prior treatment or progressive disease was significantly associated with ctDNA-positivity. ctDNA-positive patients had significantly shorter progression-free survival compared with ctDNA-negative patients (hazard ratio = 3.92; P = 0.007). ctDNA genotyping revealed a complex landscape of gene alterations, characterized by multi-exonic mutations in KIT, compared with tissue-based analysis. Patients who received TKIs matched to the identified KIT mutation in ctDNA demonstrated significantly longer PFS than those with unmatched treatment (median, 8.23 vs. 2.43 months; P < 0.001).
Conclusions
ctDNA-based analysis facilitates assessment of disease status and genomic profiles, thus potentially assisting in identifying optimal therapeutic strategies for advanced GIST patients.
Similar content being viewed by others

Introduction
Gastrointestinal stromal tumors (GISTs), arising from the interstitial cells of Cajal, represent a rare but predominant soft-tissue sarcoma within the gastrointestinal tract [1]. The main causative events for GIST involve gain-of-function mutations in the KIT or PDGFRA genes, encoding receptor tyrosine kinases, that are present in approximately 70% and 15% of cases, respectively [2, 3]. In the remaining 10% to 15% of patients, other driver genes such as BRAF, NF1, and SDH are implicated; some patients also lack specific genetic mutations [4].
The development of imatinib, a small molecule that inhibits the activity of the KIT tyrosine kinase, has brought about a remarkable paradigm shift in the clinical management of cancer patients. Other tyrosine kinase inhibitors (TKIs) such as sunitinib and regorafenib are current standard-of-care treatments [5, 6]. However, the majority of patients that undergo imatinib therapy commonly experience drug resistance, known as secondary resistance [7, 8]. The primary cause of secondary resistance (approximately 70% of cases) is secondary mutations in KIT or PDGFRA, specifically in the ATP-binding pocket encoded by exons 13 and 14 or in the activation loop encoded by exons 17 and 18 [9,10,11,12]. The distinct therapeutic effects by TKIs can be attributed to the differences in the exons harboring these mutations. While clinical guidelines provide recommendations concerning the sequence of TKI treatment, a therapeutic strategy targeting secondary gene mutations has not yet been established [13].
There has been a recent surge of interest in the investigation of circulating tumor DNA (ctDNA) as a less-invasive liquid biopsy technique compared with tissue sampling [14,15,16]. Longitudinal ctDNA testing may allow the detection of acquired resistance variants generated from selective pressure from kinase inhibitors. While this method may aid in the best selection of the next line of therapy determined by the acquired resistance genotype in GIST patients, its utility has not been elucidated [17, 18].
The present study aimed to identify gene alteration profiles in GIST using data derived from tissue and blood samples separately, evaluate the impact of ctDNA on prognostication, and identify resistance-associated alterations in patients with advanced GIST. In pursuit of these objectives, we integrated the clinical trials on the SCRUM-Japan platform, encompassing GI-SCREEN, GOZILA, MOSTAR-SCREEN, and MONSTAR-SCREE-2, which are nationwide molecular profiling programs in Japan [19, 20].
Patients & methods
Patients
This study included patients with advanced GIST who participated in SURUM-Japan trials, including GI-SCREEN (UMIN000016344), GOZILA (UMIN000029315), MONSTAR-SCREEN (UMIN000036749), and MONSTAR-SCREEN-2 (UMIN000043899). The study protocol was approved by the Institutional Review Board of National Cancer Center Hospital East (UMIN000049334).
GI-SCREEN included patients with advanced gastrointestinal cancers between February 2015 and April 2019, and tumor tissue samples were sequenced. GOZILA is a plasma ctDNA profiling study initiated in January 2018. Patients with advanced gastrointestinal cancers who had disease progression during systemic chemotherapy or targeted therapy were enrolled, and plasma ctDNA was sequenced. MONSTAR-SCREEN enrolled patients with advanced solid tumors between May 2019 and February 2021 and performed longitudinal plasma ctDNA genotyping before and after systemic therapy. MONSTAR-SCREEN-2 was launched in May 2021, and whole-exome and transcriptome analyses of tumor tissue and plasma have been conducted in patients with advanced solid tumors.
Analysis of tissue samples
In GI-SCREEN, archival formalin-fixed paraffin-embedded (FFPE) tumor tissues were collected and analyzed between February 2015 and March 2017 using the Oncomine Comprehensive Assay v1 (Thermo Fisher Scientific, Inc., Waltham, MA), a multi-biomarker next-generation sequencing (NGS) assay that covers 143 of the most relevant cancer genes. The Oncomine Comprehensive Assay v3 covering 161 of the most relevant cancer genes was used between April 2017 and April 2019. These assays identify relevant single nucleotide variants (SNVs), copy number variations, gene fusions, and indels in a streamlined workflow. MONSTAR-SCREEN-2 uses the CARIS MI Profile assay (CARIS Life Sciences, Phoenix, AZ), a whole-exome and whole-transcriptome sequencing assay of tumor tissue samples. This technology includes whole-exome sequencing for DNA mutations, copy number alterations, insertions/deletions, and genomic signatures for loss of heterogeneity, microsatellite instability (MSI), and tumor mutational burden (TMB). The targeted gene panels in these assays include KIT and PDGFRA.
Analysis of blood samples
In GOZILA, plasma ctDNA was analyzed using Guardant360 (Guardant Health, Inc., Redwood City, CA.), a comprehensive ctDNA sequencing encompassing 74 genes. MONSTAR-SCREEN profiled ctDNA using FoundationOne Liquid CDx (Foundation Medicine, Inc., Cambridge, MA), which targets alterations of 324 genes and complex biomarkers, such as MSI, TMB, and tumor fraction. The targeted genes in these assays include KIT and PDGFRA. Somatic cell-free DNA alterations were identified using a proprietary bioinformatics pipeline. ctDNA fraction was measured by the maximum variant allelic fraction (VAF). According to the assay’s technical information, the detection sensitivity for genomic alterations is delineated as follows: In FoundationOne Liquid CDx, for short genomic variants, the assay exhibits median Limits of Detection (LOD) of 0.82% and 0.40% VAF for standard and enhanced sensitivity, respectively. In contrast, the Guardant360 assay demonstrates established LODs ranging from 1.0% to 1.5% mutant allele frequency with a 5 ng cell-free DNA input and 0.2% mutant allele frequency with a 30 ng input. To adjust and estimate the clonality for somatic SNVs and indels, relative clonality was initially defined as alteration VAF divided by maximum somatic VAF in the sample. We then conducted a comparison of the ctDNA clonality of KIT or PDGFRA mutations by relative clonality.
Statistical analysis
Clinical characteristics were compared using the Chi-squared test for categorical variables and the Mann–Whitney U test for continuous variables. Patients were classified into three groups on the basis of disease status at the time of enrollment: “no prior targeted therapy”, “progressive disease” (which was characterized by a change in treatment within one month following enrollment), and “on treatment” (in which treatment was continued for more than one month post-enrollment). Regarding the therapeutic spectrum of TKIs for KIT or PDGFRA mutational status in this study, we defined imatinib as effective for KIT exon 11 and PDGFRA exon 18 (other than D842V) mutations, sunitinib as effective for KIT exon 9/11/13/14 mutations, and regorafenib as effective for KIT exon 9/13 (other than V654A)/14/17/18 mutations, as suggested by previous reports [21, 22]. We summarized the associations between the mutational status and the efficacy of different TKIs in the present study (Supplementary Fig. 1). Overall survival (OS) was estimated as the time from the date of enrollment to the date of death from any cause. Progression-free survival (PFS) was estimated for each treatment after enrollment and defined as the time from the date of treatment initiation to either the date of disease progression or death from any cause. OS and PFS were calculated using the Kaplan–Meier method and assessed using the log-rank test. Cox proportional hazards models were employed for both univariate and multivariate analyses. P-values of <0.05 were considered statistically significant. Statistical analyses were performed using the R Statistics software program, version 4.2.3. The data cut-off date for the analyses was June 30, 2023.
Results
Patient characteristics associated with tissue genotyping failure and ctDNA detection
A total of 133 patients with advanced GIST were included in SCRUM-Japan studies, including 79 of 5743 enrolled in GI-SCREEN, 36 of 5499 enrolled in GOZILA, 8 of 2224 in MONSTAR-SCREEN, and 10 of 1897 enrolled in MONSTAR-SCREEN-2 (Supplementary Fig. 2). Tissue genotyping was not performed in seven patients because of insufficient sample quality. Of the remaining 126 patients, 82 and 44 patients underwent tissue and ctDNA genotyping, respectively.
The baseline characteristics of patients examined by tissue genotyping are shown in Table 1. Among the 82 patients who underwent tissue genotyping, 65 (79.3%) had successfully sequenced results, whereas sequencing failed in 17 (20.7%). Specimens collected from a metastatic site and prior treatment were significantly associated with tissue genotyping failure (P = 0.047 and P = 0.033, respectively).
The baseline characteristics of the 44 patients who underwent ctDNA analysis are shown in Table 2. ctDNA was not detected in 12 (27.3%) patients. Patients on treatment were not likely to have detectable ctDNA compared with patients with those on progressive disease or those with no prior targeted therapy at the time of blood sampling (P = 0.027). The median duration from ctDNA analysis to the subsequent manifestation of clinical disease status was 8.0 days (95% confidence interval, 2.0–14.5) for patients classified as progressive disease and 127.5 days (95% confidence interval, 42.5–287.5) for those classified as on treat, respectively. Notably, factors such as tumor location, presence of primary tumor, metastatic organs, and the number of metastatic organs were not associated with ctDNA positivity.
Survival analysis in accordance with ctDNA status
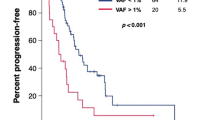
We evaluated PFS and OS in accordance with ctDNA status. PFS was significantly worse in the ctDNA-positive group compared with the ctDNA-negative group (HR, 3.92; 95% CI, 1.34–11.5; log-rank P = 0.007) (Fig. 1a). OS in the ctDNA-positive group tended to be less favorable compared with the ctDNA-negative group, but without statistical significance (HR 2.17; 95% CI 0.60–7.88; log-rank P = 0.231) (Fig. 1b). Cox multivariate analysis, integrating potential confounding variables, demonstrated that ctDNA negativity was an independent prognostic factor for PFS (HR 3.46; 95% CI 1.07–11.2; P = 0.038) (Supplementary Table 1). Furthermore, we analyzed PFS, focusing on 39 patients clinically determined to be PD (PD and on treatment statuses) by physicians after treatment. Consequently, PFS was significantly longer in ctDNA-negative patients (HR 3.15; 95% CI 1.06–9.31; log-rank P = 0.038), consistent with the overall sample analysis (Supplementary Fig. 3).

a Progression-free survival in accordance with ctDNA status: PFS was significantly shorter in patients with ctDNA-positive compared to those with ctDNA-negative. b Overall survival in accordance with ctDNA status: patients with ctDNA-positive exhibited a trend towards less favourable OS compared to those with ctDNA-negative.
Landscape of genomic alterations identified by tissue and ctDNA genotyping
We examined the landscape of genomic alterations identified through tissue genotyping (Fig. 2a). The presence of KIT exon 11 mutation (50.8%) was observed, followed by mutations in KIT exons 9 (9.2%) and 17 (6.2%). Notably, each of these mutations occurred as sole exon mutations. Among other pathogenic mutations, mutations in genes related to the cell cycle or cell differentiation pathways (12.3%), particularly RB1 mutations (6.2%), were frequently observed.

a Landscape of genomic alterations identified with tissue samples in patients with advanced GIST: mutation profiles in tissue samples are depicted separately for pre- and post-chemotherapy timepoints. Specific mutations in KIT exons are highlighted, with other alterations categorised by gene function, including cell cycle and differentiation, chromatin modulation, DNA repair-related genes, and others. b Landscape of genomic alterations identified with ctDNA analysis in patients with advanced GIST: genomic alterations identified in ctDNA are grouped into three stages: progressive disease, prior to chemotherapy, and on treatment. The maximum somatic variant allele frequency for each patient is depicted at the bottom. Mutations in KIT and PDGFRA are illustrated with a gradient representing relative clonality. Alterations in genes other than KIT and PDGFRA are categorised by function into driver genes, receptor tyrosine kinases, cell cycle and differentiation, chromatin modulators, and growth signalling pathways. Gene amplifications are marked in orange. c Comparison between tissue and ctDNA genotyping regarding KIT and PDGFRA mutations: the proportion of various mutation patterns in KIT and PDGFRA are shown cumulatively for both tissue genotyping and ctDNA genotyping. d Comparison between maximum somatic VAF of ctDNA with and without KIT and PDGFRA mutations: ctDNA harbouring KIT or PDGFRA mutations exhibits a significantly higher maximum somatic VAF compared to samples without these mutations. e Relative ctDNA clonality of KIT and PDGFRA mutations: KIT mutations other than exons 9/11 demonstrate significantly lower ctDNA clonality compared to mutations in KIT exons 9/11. f Dynamic change of ctDNA clonality of KIT and PDGFRA mutations in Patient #1: upper CT images illustrate the dynamic response of liver metastases to treatment, while the line graph below shows the corresponding changes in relative ctDNA clonality.
ctDNA genotyping, performed in the 32 patients with ctDNA positivity, revealed a different KIT mutational pattern from that obtained with tissue-based genotyping (Fig. 2b). Sole KIT exon 9 (9.4%) or 11 (15.6%) mutations were less prevalent; instead, multi-exonic mutations in KIT encompassing exons 9, 10, 11, 13, 17, 18, and 20 were more prevalent (25.0%). Among other pathogenic mutations, TP53 mutations were most frequently observed, accounting for 18.8% of patients. No patients exhibited high microsatellite instability. In cases where mutations in genes other than KIT display high VAF, resulting in a relatively lower VAF for the KIT gene mutation, this would consequently lead to a higher maximum VAF and a proportionately lower clonality for the KIT mutation. This phenomenon is illustrated in row 9, which presents a KIT exon 11 mutation with a VAF of 0.36% and an STK11 mutation with a maximum somatic VAF of 50.1%, yielding a calculated relative clonality of 0.007 for the KIT mutation. We next compared KIT and PDGFRA alterations between tissue and blood genotyping results (Fig. 2c). The proportion of KIT and PDGFRA mutations in tissue and blood samples was similar, comprising 69.2% in tissue (45/65) and 68.8% in blood (22/32). However, blood samples exhibited a more diverse spectrum of KIT mutations, particularly evident in exon 11/13 (0% vs. 14.3%, P < 0.01) and exon 13/17 (0% vs. 9.5%, P < 0.01).
ctDNA clonality and the dynamic change of KIT and PDGFRA mutations
To elucidate the ctDNA clonality patterns related to KIT and PDGFRA mutations, as well as other alterations, we compared the maximum somatic VAF of ctDNA carrying KIT or PDGFRA mutations against those harboring other mutations (Fig. 2d). The maximum somatic VAF of ctDNA with KIT or PDGFRA mutations was significantly higher than that without these mutations (median: 8.36 vs. 0.33, P = 0.009). We next compared the ctDNA clonality of KIT exon 9/11 mutations and KIT mutations other than exon 9/11 (Fig. 2e). The median clonality of KIT exon 9/11 mutations was 100% (range, 2.0–100), while the KIT mutations other than exon 9/11 had a significantly lower median ctDNA clonality of 36.0% (range, 0.6–100) (P = 0.044).
To investigate the dynamic change in ctDNA alterations throughout TKI therapy, we focused on two patients who underwent repeated ctDNA assessment and had detectable KIT or PDGFRA. Details of the pathogenic mutation status from multiple blood samplings for three patients are provided in Supplementary Table 2. Patient #1 had advanced GIST with multiple liver metastases and received partial gastrectomy for peritonitis because of perforation of the primary lesion. ctDNA testing was performed prior to imatinib and 17.9 months after initiation of imatinib. Compared with the pretreatment result, the clonality of the KIT exon 11 mutation decreased after imatinib treatment from 43.7% to 14.5%, whereas KIT exon 13 mutation newly emerged with a clonality of 9.1%. The disease remained stable at 12 months from the second ctDNA evaluation but progressed markedly after 18 months; the patient was switched to sunitinib treatment, which is currently ongoing (Fig. 2f). Patient #2 underwent an initial ctDNA test before the initiation of imatinib and a second ctDNA test after disease progression on imatinib with an interval of 2.4 months. In the case of patient #2, avapritinib, which is the preferred therapeutic option for treating GIST harboring the PDGFRA D842V mutation in Western countries, has not yet received regulatory approval in our healthcare system. Consequently, imatinib is commonly employed as the standard treatment, irrespective of the underlying mutational profile. Furthermore, the specific alteration identified in this patient was D842_S847 > EF, a unique molecular event that is distinct from the well-characterized non-D842V mutations. Given these considerations, the decision was made to initiate treatment with imatinib, in line with the prevailing clinical practice in our country. The clonality of PDGFRA exon 18 mutation increased along with disease progression, and a novel mutation within KIT exon 20 emerged (Supplementary Fig. 4).
The relationships between KIT or PDGFRA mutation status and prior and subsequent TKIs in patients with ctDNA positivity
Out of the 35 samples with ctDNA positivity, which included 3 patients with multiple samples, 18 samples had KIT or PDGFRA mutations at progressive disease status. We evaluated the association between the efficacy of preceding TKIs and KIT or PDGFRA mutational status (Table 3). Within this sample subset, 14 (77.8%) harbored KIT exon mutations resistant to the preceding TKIs. Furthermore, we assessed the efficacy of subsequent TKIs for 17 patients harboring KIT or PDGFRA mutations in accordance with their mutational status (Fig. 3a). Patients who were subsequently treated with TKIs that matched their mutational status had a significantly longer PFS compared with those who received TKIs that were not matched [median PFS, 8.23 vs. 2.43 months; hazard ratio (HR), 0.09; 95% confidence interval, 0.02–0.43; log-rank P < 0.001] (Fig. 3b).

a Swimmer plot of treatment duration and clinical outcome in accordance with mutational status matched or unmatched to subsequent therapy: swimmer plot depicted treatment duration and clinical outcomes for patients receiving TKIs matched to their mutational status (top section) and those treated with non-matched TKIs (bottom section). Disease status at treatment initiation, therapeutic agents used, and continuity of treatment are indicated for each patient. b Progression-free survival in accordance with the suitability of TKIs for KIT or PDGFRA mutations: patients treated with TKIs matched to their specific KIT or PDGFRA mutation profiles exhibited significantly longer PFS compared to those who received mismatched TKIs.
Discussion
In the present study, we performed comprehensive genomic profiling of advanced GIST using the SCRUM-Japan platform. Our findings suggest that genomic characterization via liquid biopsies offers an enhanced understanding of the genetic diversity, heterogeneity, and dynamic genomic changes in GIST throughout therapeutic intervention compared with tissue-based analyses. Importantly, patients administered TKIs specifically tailored to mutations identified through ctDNA genotyping demonstrated markedly improved therapeutic outcomes relative to those receiving non-tailored TKIs. Due to the small sample size and potential background biases, caution should be exercised when interpreting these results. Further validation in larger cohorts is needed to confirm the clinical utility of this approach. Additionally, we discerned a robust association between ctDNA-positivity and progressive disease status, indicating ctDNA-positivity as a pivotal prognostic factor. These observations imply that ctDNA-based analysis can delineate intricate genetic profiles, which reflect not only the therapeutic lineage but also the contemporaneous disease status and help guide the establishment of optimal treatment strategies for patients with advanced GIST.
The success rate for tissue sample analysis in this study was approximately 80%. The collection of specimens from metastatic lesions and prior treatment were significant risk factors for analysis failure. These samples chiefly comprised archival tissues, and a success rate lower than expected may be from an insufficient quantity of samples because of minuscule metastatic lesions or compromised DNA quality. This is consistent with a previous report suggesting the importance of DNA quality and integrity for achieving successful sequencing [23]. Conversely, the ctDNA positivity of 72.7% in this study (including 36 and 8 patients evaluated with Guardant 360 and FoundationOne Liquid CDx, respectively) was superior to the 45–56.3% reported in retrospective studies [24, 25] and comparable to the 77.3% observed in the phase III INTRIGUE trial using Guardant 360 [18], but inferior to the 86.3% reported in the phase III VOYAGER trial [17], where Guardant 360 was used for ctDNA assessment in patients with heavily treated advanced GIST. These observations suggest that ctDNA positivity is contingent upon the tumor burden at the time of blood sampling. Furthermore, our investigation indicates that disease status at the time of sampling is significantly associated with ctDNA positivity, mirroring previous reports that showed that localized disease or prior treatment was linked to ctDNA negativity [26, 27]. Thus, it is feasible to conduct ctDNA testing at disease progression in clinical practice to select the optimal therapeutic approach.
Our results showed a disparity in the genetic landscapes between tissue and ctDNA genotyping. Tissue-based analysis revealed a simpler KIT mutation status, primarily characterized by sole exon 11 mutations. In contrast, ctDNA genotyping displayed a more heterogeneous spectrum of KIT mutations, including the combination of KIT ATP-binding pocket and activation-loop mutations, reported as the predominant region of secondary mutations [9,10,11]. This phenomenon is likely attributable to the following factors: approximately 80% of the tissue specimens were archival samples derived from surgical procedures at the point of initial treatment, in contrast, the majority of blood samples were obtained during the phase of progressive disease or on treat after therapeutic interventions. We posit that this discrepancy underlies the observed intricate mutational status in blood samples within our investigation. Moreover, ctDNA genotyping revealed that ctDNA harboring KIT exon 9/11 mutations exhibited higher VAF and clonality compared with those harboring other mutations. Furthermore, ctDNA devoid of KIT or PDGFR mutations exhibited significantly diminished VAF. These observations suggest that therapeutic interventions may attenuate the prevalence of KIT-mutated cancer cells responsive to treatment, thereby facilitating the emergence of cells harboring mutations associated with acquired resistance. Additionally, ctDNA shedding from cells sensitive to therapeutic interventions may be less detectable when the tumor fraction is diminished by the effective treatment. Indeed, our dynamic examination of ctDNA indicates that VAF and clonality are subject to treatment-induced fluctuations and that novel mutational clones may emerge over time, as previously reported by Wada et al [28]. The phenomenon of clonal evolution occurring throughout the treatment among patients with GIST has also been posited in previous reports [29,30,31]. Consequently, ctDNA analysis holds potential as a tool for tracking the clonal evolution of cancer cells.
There are the distinct differences between imatinib, sunitinib, and regorafenib due to the mutational status in KIT exons. In alignment with previous reports, our data indicated that approximately 80% patients with ctDNA-positivity displayed resistant mutations relevant to the preceding TKIs [21, 22]. Moreover, our findings indicate the potential of ctDNA analysis to enhance therapeutic efficacy and prolong prognosis by the application of TKIs tailored to the KIT or PDGFRA mutations. While clinical guidelines, including those published by the National Comprehensive Cancer Network (NCCN) and the European Society of Medical Oncology (ESMO), provide recommendations concerning the sequence of TKIs, our results suggest the possibility of selecting therapeutic interventions on the basis of genetic mutations identified through ctDNA analysis, thereby personalizing the treatment strategy [32, 33]. While, regarding the patients with mutations for which both sunitinib and regorafenib has been considered ineffective, ripretinib, as endorsed by the NCCN and ESMO guidelines, or alternatively, the selection of pimitespib, which has garnered approval in Japan, emerge as therapeutic options according to its approval status [34, 35]. Additionally, avapritinib, identified as treatment strategy for GIST patients with PDGFRA D842V mutation, was recently reported to have antitumour activity in patients with a broad range of KIT mutations [36,37,38]. Furthermore, the utility of combination targeted therapy with TKIs has recently been reported [39, 40], and a phase III trial comparing combination vs. sunitinib alone is currently underway (NCT05208047). Thus, it may be possible to develop ctDNA-guided therapeutic strategies, in which single-agent or combination TKI is selected on the basis of specific mutation profiles identified through ctDNA analysis (Supplementary Fig. 5). We recognize that the categorization of ‘matched mutational status’ and ‘unmatched status’ is based on limited evidence and is not widely employed in clinical practice. Furthermore, the small sample size, encompassing various treatment lines, may introduce significant background biases, which could limit the robustness of the survival analysis. Nevertheless, our findings are supported by a recent biomarker analysis of the INTRIGUE phase III trial, published by Heinrich et al. [41]. The authors reported that ctDNA sequencing may improve the prediction of the efficacy of single-drug RTK therapies in patients with GIST. While our study provides novel insights into the potential utility of liquid biopsies in GIST management, we acknowledge the need for further validation in larger cohorts to confirm our findings and address the limitations associated with the small sample size and potential biases. Furthermore, this study also demonstrated the significant prognostic impact of ctDNA status on PFS in patients with advanced GIST. In line with research indicating ctDNA-positivity as a critical poor prognostic factor in various malignancies [42, 43], this study confirmed the profound impact of ctDNA positivity on disease trajectory and its utility as a biomarker for therapeutic decision-making in advanced GIST patients with acquired resistance to preceding TKIs.
This study has several limitations. One of the major limitations of our study is the lack of paired tissue and blood analyses for each patient. Due to the retrospective nature of our cohort and the limited availability of matched samples, we were unable to perform a comprehensive comparison of tissue and ctDNA findings at the individual patient level. This shortcoming hinders our ability to draw definitive conclusions regarding the concordance between the two approaches and may have obscured important associations between tissue and ctDNA profiles. Future prospective studies with larger cohorts and systematically collected paired samples are necessary to validate our findings and provide a more robust understanding of the relationship between tissue and ctDNA analyses in the context of GIST. Second, this study was retrospective and constrained by a limited sample size, precluding definitive conclusions. However, it constitutes an integrated analysis of four prospective trials in the SCRUM-Japan platform, and therefore, potential selection bias was mitigated. Conversely, the fact that analyses were conducted using different assay systems from four distinct trials cannot preclude the possibility that this variability might have impacted the outcomes. In sum, the sample size of this study is not sufficiently large to eliminate bias, rendering the formulation of assertive conclusions challenging. Third, because tumor size, metabolic tumor volume, or pathological characteristics, all of which are potential indicators that could influence ctDNA positivity in GIST, have not been evaluated, their impact on ctDNA positivity or KIT mutational status could not be assessed in this study. While we provided information on the number of metastatic organs, metastatic sites, and the presence or absence of primary tumor resection in Table 2, we did not observe any significant differences in these parameters. Future studies should aim to collect detailed information on tumor burden and investigate its association with ctDNA VAF, as this could provide valuable insights into the clinical utility of ctDNA analysis in GIST. A better understanding of the relationship between ctDNA levels and tumor volume or metabolic activity could help refine the interpretation of ctDNA results and guide treatment decision-making in patients with GIST. Finally, there was only a limited number of patients underwent longitudinal ctDNA analysis. This limitation hinders our capacity to draw robust, generalized conclusions, thus affecting the applicability of our findings. While the proportion of patients with KIT exon 9/11 mutations in ctDNA analysis was lower compared with previous reports, we cannot evaluate the paired pre-treatment tissue samples. KIT mutation at initial diagnosis was not eradicated by treatment interventions [44]. However, our findings confirmed the low maximum VAF of ctDNA without KIT or PDGFRA mutations, leading us to hypothesize that clonal evolution related to acquired resistance mechanisms may shed light on this phenomenon, potentially indicating treatment response and the low tumor fraction harboring original alterations. Additional studies are required to elucidate the influence of KIT mutation change or diminishing of pre-existing mutations on the sensitivity of ctDNA analyses.
In conclusion, the present study illuminated the significant correlation between ctDNA status and disease status at sampling time in advanced GIST patients and indicated ctDNA status as an independent prognostic factor. Furthermore, ctDNA-based analysis facilitated real-time evaluation of genomic profiles, including acquired mutations, emergent mutations resistant to ongoing treatments, and convergent alterations associated with malignant progression. These findings further support the utility of ctDNA in GIST, highlighting its cardinal role in therapeutic decision-making for advanced GIST patients and its potential to instigate a paradigm shift in precision medicine for this rare population, informed by molecular profiling through ctDNA testing.
Data availability
All other data are available from the corresponding author upon reasonable request. Due to privacy laws, access to raw data is restricted. Those requests will be reviewed by a study steering committee to verify whether the request is subject to any intellectual property or confidentiality obligations.
References
Hirota S, Isozaki K, Moriyama Y, Hashimoto K, Nishida T, Ishiguro S, et al. Gain-of-function mutations of c-kit in human gastrointestinal stromal tumors. Science. 1998;279:577–80. https://doi.org/10.1126/science.279.5350.577.
Heinrich MC, Corless CL, Duensing A, McGreevey L, Chen CJ, Joseph N, et al. PDGFRA activating mutations in gastrointestinal stromal tumors. Science. 2003;299:708–10. https://doi.org/10.1126/science.1079666.
Blay JY, Kang YK, Nishida T, von Mehren M. Gastrointestinal stromal tumours. Nat Rev Dis Primers. 2021;7:22 https://doi.org/10.1038/s41572-021-00254-5.
Corless CL, Barnett CM, Heinrich MC. Gastrointestinal stromal tumours: origin and molecular oncology. Nat Rev Cancer. 2011;11:865–78. https://doi.org/10.1038/nrc3143.
von Mehren M, Joensuu H. Gastrointestinal Stromal Tumors. J Clin Oncol. 2018;36:136–43. https://doi.org/10.1200/jco.2017.74.9705.
Demetri GD, van Oosterom AT, Garrett CR, Blackstein ME, Shah MH, Verweij J, et al. Efficacy and safety of sunitinib in patients with advanced gastrointestinal stromal tumour after failure of imatinib: a randomised controlled trial. Lancet. 2006;368:1329–38. https://doi.org/10.1016/s0140-6736(06)69446-4.
Nishida T, Hirota S, Taniguchi M, Hashimoto K, Isozaki K, Nakamura H, et al. Familial gastrointestinal stromal tumours with germline mutation of the KIT gene. Nat Genet. 1998;19:323–4. https://doi.org/10.1038/1209.
Shankar S, vanSonnenberg E, Desai J, Dipiro PJ, Van Den Abbeele A, Demetri GD. Gastrointestinal stromal tumor: new nodule-within-a-mass pattern of recurrence after partial response to imatinib mesylate. Radiology. 2005;235:892–8. https://doi.org/10.1148/radiol.2353040332.
Wardelmann E, Merkelbach-Bruse S, Pauls K, Thomas N, Schildhaus HU, Heinicke T, et al. Polyclonal evolution of multiple secondary KIT mutations in gastrointestinal stromal tumors under treatment with imatinib mesylate. Clin Cancer Res. 2006;12:1743–9. https://doi.org/10.1158/1078-0432.Ccr-05-1211.
Liegl B, Kepten I, Le C, Zhu M, Demetri GD, Heinrich MC, et al. Heterogeneity of kinase inhibitor resistance mechanisms in GIST. J Pathol. 2008;216:64–74. https://doi.org/10.1002/path.2382.
Heinrich MC, Corless CL, Blanke CD, Demetri GD, Joensuu H, Roberts PJ, et al. Molecular correlates of imatinib resistance in gastrointestinal stromal tumors. J Clin Oncol. 2006;24:4764–74. https://doi.org/10.1200/jco.2006.06.2265.
Joensuu H, Vehtari A, Riihimäki J, Nishida T, Steigen SE, Brabec P, et al. Risk of recurrence of gastrointestinal stromal tumour after surgery: an analysis of pooled population-based cohorts. Lancet Oncol. 2012;13:265–74. https://doi.org/10.1016/s1470-2045(11)70299-6.
Koo DH, Ryu MH, Kim KM, Yang HK, Sawaki A, Hirota S, et al. Asian Consensus Guidelines for the Diagnosis and Management of Gastrointestinal Stromal Tumor. Cancer Res Treat. 2016;48:1155–66. https://doi.org/10.4143/crt.2016.187.
Heitzer E, Haque IS, Roberts CES, Speicher MR. Current and future perspectives of liquid biopsies in genomics-driven oncology. Nat Rev Genet. 2019;20:71–88. https://doi.org/10.1038/s41576-018-0071-5.
Serrano C, George S. Gastrointestinal Stromal Tumor: Challenges and Opportunities for a New Decade. Clin Cancer Res. 2020;26:5078–85. https://doi.org/10.1158/1078-0432.Ccr-20-1706.
Gómez-Peregrina D, García-Valverde A, Pilco-Janeta D, Serrano C. Liquid Biopsy in Gastrointestinal Stromal Tumors: Ready for Prime Time? Curr Treat Options Oncol. 2021;22:32. https://doi.org/10.1007/s11864-021-00832-5.
Serrano C, Bauer S, Gómez-Peregrina D, Kang YK, Jones RL, Rutkowski P, et al. Circulating tumor DNA analysis of the phase III VOYAGER trial: KIT mutational landscape and outcomes in patients with advanced gastrointestinal stromal tumor treated with avapritinib or regorafenib. Ann Oncol. 2023. https://doi.org/10.1016/j.annonc.2023.04.006.
Trent JC, Jones RL, George S, Gelderblom H, Schöffski P, von Mehren M, et al. Outcomes in patients with advanced gastrointestinal stromal tumor who did not have baseline ctDNA detected in the INTRIGUE study. J Clin Oncol. 2023;41:11536. https://doi.org/10.1200/JCO.2023.41.16_suppl.11536.
Nakamura Y, Fujisawa T, Taniguchi H, Bando H, Okamoto W, Tsuchihara K, et al. SCRUM-Japan GI-SCREEN and MONSTAR-SCREEN: Path to the realization of biomarker-guided precision oncology in advanced solid tumors. Cancer Sci. 2021;112:4425–32. https://doi.org/10.1111/cas.15132.
Nakamura Y, Okamoto W, Denda T, Nishina T, Komatsu Y, Yuki S, et al. Clinical Validity of Plasma-Based Genotyping for Microsatellite Instability Assessment in Advanced GI Cancers: SCRUM-Japan GOZILA Substudy. JCO Precis Oncol. 2022:e2100383. https://doi.org/10.1200/PO.21.00383.
Napolitano A, Vincenzi B. Secondary KIT mutations: the GIST of drug resistance and sensitivity. Br J Cancer. 2019;120:577–8. https://doi.org/10.1038/s41416-019-0388-7.
Serrano C, Mariño-Enríquez A, Tao DL, Ketzer J, Eilers G, Zhu M, et al. Complementary activity of tyrosine kinase inhibitors against secondary kit mutations in imatinib-resistant gastrointestinal stromal tumours. Br J Cancer. 2019;120:612–20. https://doi.org/10.1038/s41416-019-0389-6.
Kuwata T, Wakabayashi M, Hatanaka Y, Morii E, Oda Y, Taguchi K, et al. Impact of DNA integrity on the success rate of tissue-based next-generation sequencing: Lessons from nationwide cancer genome screening project SCRUM-Japan GI-SCREEN. Pathol Int. 2020;70:932–42. https://doi.org/10.1111/pin.13029.
Xu H, Chen L, Shao Y, Zhu D, Zhi X, Zhang Q, et al. Clinical Application of Circulating Tumor DNA in the Genetic Analysis of Patients with Advanced GIST. Mol Cancer Ther. 2018;17:290–6. https://doi.org/10.1158/1535-7163.Mct-17-0436.
Arshad J, Roberts A, Ahmed J, Cotta J, Pico BA, Kwon D, et al. Utility of Circulating Tumor DNA in the Management of Patients With GI Stromal Tumor: Analysis of 243 Patients. JCO Precis Oncol. 2020;4:66–73. https://doi.org/10.1200/po.19.00253.
Bettegowda C, Sausen M, Leary RJ, Kinde I, Wang Y, Agrawal N, et al. Detection of circulating tumor DNA in early- and late-stage human malignancies. Sci Transl Med. 2014;6:224ra24. https://doi.org/10.1126/scitranslmed.3007094.
Maier J, Lange T, Kerle I, Specht K, Bruegel M, Wickenhauser C, et al. Detection of mutant free circulating tumor DNA in the plasma of patients with gastrointestinal stromal tumor harboring activating mutations of CKIT or PDGFRA. Clin Cancer Res. 2013;19:4854–67. https://doi.org/10.1158/1078-0432.Ccr-13-0765.
Wada N, Kurokawa Y, Takahashi T, Hamakawa T, Hirota S, Naka T, et al. Detecting Secondary C-KIT Mutations in the Peripheral Blood of Patients with Imatinib-Resistant Gastrointestinal Stromal Tumor. Oncology. 2016;90:112–7. https://doi.org/10.1159/000442948.
Klug LR, Khosroyani HM, Kent JD, Heinrich MC. New treatment strategies for advanced-stage gastrointestinal stromal tumours. Nat Rev Clin Oncol. 2022;19:328–41. https://doi.org/10.1038/s41571-022-00606-4.
Namløs HM, Boye K, Mishkin SJ, Barøy T, Lorenz S, Bjerkehagen B, et al. Noninvasive Detection of ctDNA Reveals Intratumor Heterogeneity and Is Associated with Tumor Burden in Gastrointestinal Stromal Tumor. Mol Cancer Ther. 2018;17:2473–80. https://doi.org/10.1158/1535-7163.Mct-18-0174.
Jilg S, Rassner M, Maier J, Waldeck S, Kehl V, Follo M, et al. Circulating cKIT and PDGFRA DNA indicates disease activity in Gastrointestinal Stromal Tumor (GIST). Int J Cancer. 2019;145:2292–303. https://doi.org/10.1002/ijc.32282.
Casali PG, Blay JY, Abecassis N, Bajpai J, Bauer S, Biagini R, et al. Gastrointestinal stromal tumours: ESMO-EURACAN-GENTURIS Clinical Practice Guidelines for diagnosis, treatment and follow-up. Ann Oncol. 2022;33:20–33. https://doi.org/10.1016/j.annonc.2021.09.005.
von Mehren M, Kane JM, Riedel RF, Sicklick JK, Pollack SM, Agulnik M, et al. NCCN Guidelines® Insights: Gastrointestinal Stromal Tumors, Version 2.2022. J Natl Compr Canc Netw. 2022;20:1204–14. https://doi.org/10.6004/jnccn.2022.0058.
Kurokawa Y, Honma Y, Sawaki A, Naito Y, Iwagami S, Komatsu Y, et al. Pimitespib in patients with advanced gastrointestinal stromal tumor (CHAPTER-GIST-301): a randomized, double-blind, placebo-controlled phase III trial. Ann Oncol. 2022;33:959–67. https://doi.org/10.1016/j.annonc.2022.05.518.
Bauer S, Jones RL, George S, Gelderblom H, Schöffski P, von Mehren M, et al. Mutational heterogeneity of imatinib resistance and efficacy of ripretinib vs sunitinib in patients with gastrointestinal stromal tumor: ctDNA analysis from INTRIGUE. J Clin Oncol. 2023;41:397784. https://doi.org/10.1200/JCO.2023.41.36_suppl.397784.
Heinrich MC, Jones RL, von Mehren M, Schöffski P, Serrano C, Kang YK, et al. Avapritinib in advanced PDGFRA D842V-mutant gastrointestinal stromal tumour (NAVIGATOR): a multicentre, open-label, phase 1 trial. Lancet Oncol. 2020;21:935–46. https://doi.org/10.1016/s1470-2045(20)30269-2.
Jones RL, Serrano C, von Mehren M, George S, Heinrich MC, Kang YK, et al. Avapritinib in unresectable or metastatic PDGFRA D842V-mutant gastrointestinal stromal tumours: Long-term efficacy and safety data from the NAVIGATOR phase I trial. Eur J Cancer. 2021;145:132–42. https://doi.org/10.1016/j.ejca.2020.12.008.
Heinrich MC, Li J, Zhang X, Jones RL, George S, Trent JC, et al. Clinical efficacy of avapritinib in gastrointestinal stromal tumors (GIST) with different KIT genotypes: Post hoc analysis of the phase 1 NAVIGATOR and phase 1/2 CS3007-101 trials. J Clin Oncol. 2023;41:11523. https://doi.org/10.1200/JCO.2023.41.16_suppl.11523.
Wagner AJ, Severson PL, Shields AF, Patnaik A, Chugh R, Tinoco G, et al. Association of Combination of Conformation-Specific KIT Inhibitors With Clinical Benefit in Patients With Refractory Gastrointestinal Stromal Tumors: A Phase 1b/2a Nonrandomized Clinical Trial. JAMA Oncol. 2021;7:1343–50. https://doi.org/10.1001/jamaoncol.2021.2086.
Xia Y, Zhang X, Guo X, Huang S, Wu C, Jiang A, et al. Combination targeted therapy with avapritinib and sunitinib in patients with refractory gastrointestinal stromal tumors after failure of standard treatments: A small prospective pilot study. J Clin Oncol. 2023;41:11538. https://doi.org/10.1200/JCO.2023.41.16_suppl.11538.
Heinrich MC, Jones RL, George S, Gelderblom H, Schöffski P, von Mehren M, et al. Ripretinib versus sunitinib in gastrointestinal stromal tumor: ctDNA biomarker analysis of the phase 3 INTRIGUE trial. Nat Med. 2024;30:498–506. https://doi.org/10.1038/s41591-023-02734-5.
Wang W, He Y, Yang F, Chen K. Current and emerging applications of liquid biopsy in pan-cancer. Transl Oncol. 2023;34:101720. https://doi.org/10.1016/j.tranon.2023.101720.
Kotani D, Oki E, Nakamura Y, Yukami H, Mishima S, Bando H, et al. Molecular residual disease and efficacy of adjuvant chemotherapy in patients with colorectal cancer. Nat Med. 2023;29:127–34. https://doi.org/10.1038/s41591-022-02115-4.
Nishida T, Kanda T, Nishitani A, Takahashi T, Nakajima K, Ishikawa T, et al. Secondary mutations in the kinase domain of the KIT gene are predominant in imatinib-resistant gastrointestinal stromal tumor. Cancer Sci. 2008;99:799–804. https://doi.org/10.1111/j.1349-7006.2008.00727.x.
Acknowledgements
The authors thank the patients, investigators, site staff who participated in this study, and Translational Research Support Office of the National Cancer Center Hospital East for study management and data Center support (all the National Cancer Center Hospital East Translational Research Support Office members). We thank Gabrielle White Wolf, PhD, from Edanz (https://jp.edanz.com/ac) for editing a draft of this manuscript.
Funding
This work was supported by SCRUM-Japan Funds (http://www.scrum-japan.ncc.go.jp/index.html).
Author information
Authors and Affiliations
Contributions
T. Hashimoto: Conceptualization, data curation, formal analysis, investigation, visualization, writing–original draft, project administration. Y. Nakamura: Conceptualization, resources, formal analysis, visualization, writing–original draft, project administration. Y. Komatsu: Resources, writing–review and editing. S. Yuki: Resources, writing–review and editing. N. Takahashi: Resources, writing–review and editing. N. Okano: Resources, writing–review and editing. H. Hirano: Resources, writing–review and editing. K. Ohtsubo: Resources, writing–review and editing. T. Ohta: Resources, writing–review and editing. E. Oki: Resources, writing–review and editing. T. Nishina: Resources, writing–review and editing. H. Yasui: Resources, writing–review and editing. H. Kawakami: Resources, writing–review and editing. T. Esaki: Resources, writing–review and editing. N. Machida: Resources, writing–review and editing. A. Doi: Resources, writing–review and editing. S. Boku: Resources, writing–review and editing. T. Kudo: Resources, writing–review and editing. Y. Yamamoto: Resources, writing–review and editing. A. Kanazawa: Resources, writing–review and editing. T. Denda: Resources, writing–review and editing. M. Gotoh: Resources, writing–review and editing. N. Iida: Writing–review and editing. H. Ozaki: Writing–review and editing. T. Shibuki: Writing–review and editing. M. Imai: Writing–review and editing. T. Fujisawa: Resources, supervision, writing–review and editing. H. Bando: Conceptualization, supervision, project administration, writing–review and editing. Y Naito: Resources, writing–review and editing. T. Yoshino: Conceptualization, supervision, funding acquisition, project administration, writing–review and editing.
Corresponding author
Ethics declarations
Competing interests
TH declare no potential conflicts of interest. YN reports grants from Taiho Pharmaceutical Co., Ltd., Chugai Pharmaceutical Co., Ltd., Daiichi Sankyo Co., Ltd., Guardant Health, Inc., Genomedia, Inc., Roche Diagnostics, and Seagen, Inc.; and honoraria from Chugai Pharmaceutical Co., Ltd., and Guardant Health AMEA outside the submitted work. YK reports grants from Novartis, Yakult Honsha Co., Ltd., Pfizer, Bayer, and Taiho Pharmaceutical Co., Ltd. outside the submitted work. SY reports honoraria from Eli Lilly Japan K.K., Chugai Pharmaceutical Co., Ltd., Taiho Pharmaceutical Co., Ltd., Bayer Yakuhin Ltd., Bristol-Myers Squibb K.K., Takeda Pharmaceutical Co., Ltd., Merck Biopharma Co., Ltd., MSD K.K., Ono Pharmaceutical Co., Ltd., Nippon Boehringer Ingelheim Co., Ltd., Daiichi Sankyo Co., Ltd. outside the submitted work; NT reports honoraria from Taiho Pharmaceutical Co., Ltd., Ono Pharmaceutical Co., Ltd., Bristol-Myers Squibb K.K. outside the submitted work; NO reports honoraria from Taiho Pharmaceutical Co., Ltd., Eli Lilly Japan, Eisai, Bayer Yakuhin, Chugai Pharmaceutical Co., Ltd., Ono Pharmaceutical, Takeda Pharmaceutical Co., Ltd., Daiichi Sankyo Co., Ltd., AstraZeneca, MSD, and Incyte outside the submitted work.; HH reports honoraria from Bristol-Myers Squibb Japan, Chugai Pharmaceutical Co., Ltd., Novartis, Taiho Pharmaceutical Co., Ltd., Fujifilm Pharmaceuticals, Teijin Pharma, and Ono Pharmaceutical outside the submitted work; TO reports grants from Takeda Pharmaceutical Co. Ltd. and honoraria from Eli Lilly, Taiho Pharmaceutical Co., Ltd., Chugai Pharmaceutical Co., Ltd., Takeda Pharmaceutical Co., Ltd., Daiichi Sankyo Co., Ltd., EA Pharma Co., Ltd., Bristol-Myers Squibb K.K., Ono Pharmaceutical, Eisai Co., Ltd., Yakult Honsha Co., Ltd., and Otsuka Pharmaceutical Co., Ltd. outside the submitted work; EO reports research funding from Guardant Health, Inc. and reports honoraria from Chugai Pharmaceutical Co., Ltd., Eli Lilly, Takeda Pharmaceutical Co., Ltd., Bristol and Ono Pharmaceutical outside the submitted work; T. Nishina reports honoraria from Daiichi Sankyo Co., Ltd., Ono Pharmaceutical Co., Ltd., Bristol Myers Squibb, Eli Lilly Japan K.K., Takeda Pharmaceutical Co., Ltd., Merck Serono Pharmaceutical Co., Ltd., Chugai Pharmaceutical Co., Ltd., Taiho Pharmaceutical Co., Ltd and Yakult Honsha Co., Ltd. outside the submitted work; H.K reports grants from Eisai Co. Ltd., Taiho Pharmaceutical Co. Ltd., Kobayashi Pharmaceutical. Co., Ltd., Bristol-Myers Squibb Co. Ltd., Daiichi Sankyo Co. Ltd. consulting fees from Daiichi Sankyo Co. Ltd., and honoraria from Bristol-Myers Squibb Co. Ltd., Bayer Yakuhin Ltd., Eli Lilly Japan K.K., MSD K.K., Ono Pharmaceutical Co. Ltd., Chugai Pharmaceutical Co. Ltd., Daiichi Sankyo Co. Ltd., Merck Biopharma Co., Ltd., Takeda Pharmaceutical Co. Ltd., Yakult Pharmaceutical Industry, Teijin Pharma Ltd., Taiho Pharmaceutical Co. Ltd., and Otsuka Pharmaceutical Co., Ltd. outside the submitted work; TE reports grants from MSD, Daiichi Sankyo Co., Ltd., Pfyzer, Astellas, Quintiles, SyneosHealth, Chugai Pharmaceutical Co., Ltd., Amgen, Ono, Novartis, Astellas Amgen Biopharma, Asahikasei Pharma, and IQVIA, and honoraria from Chugai Pharmaceutical Co., Ltd., Daiichi Sankyo Co., Ltd., Taiho Pharmaceutical Co., Ltd., Eli Lilly, Bristol, and MSD outside the submitted work; YY reports honoraria from Ono Pharmaceutical, Taiho Pharmaceutical Co., Ltd., Bristol Myers Squibb, Servier, Yakult, Takeda Pharmaceutical Co., Ltd., Chugai Pharmaceutical Co., Ltd., Daiichi Sankyo Co., Ltd., Lily, Astrazeneca, Bayer, and Incyte outside the submitted work; TD reports research funds from MSD, Amgen, Bristol Myers Squibb Foundation, Pfizer, and Ono Pharmaceutical and honoraria from Sysmex, Ono Pharmaceutical, Sawai Pharmaceutical Co and Daiichi Sankyo Co., Ltd., outside the submitted work; MG reports grants from Nippon Kayaku CO., Ltd., Chugai Pharmaceutical CO., Ltd., Taiho Pharmaceutical Co., Ltd., Ono Pharmaceutical CO., Ltd., Eli Lilly Japan K.K., and MSD K.K. outside the submitted work; MI reports honoraria from Caris Life Sciences and consulting fee from Sumitomo Corp. outside the submitted work; TF reports honoraria from Amelief outside the submitted work; HB reports research funding from Ono Pharmaceutical and honoraria from Taiho Pharmaceutical Co., Ltd. Pharmaceutical and Eli Lilly, Japan outside the submitted work; YN reports grants from ABBVIE, Ono, Daiichi Sankyo Co., Ltd., Taiho Pharmaceutical Co., Ltd., Pfizer, Boehringer Ingelheim, Eli Lilly, Eisai, AstraZeneca, Chugai Pharmaceutical Co., Ltd., and Bayer, and honoraria from AstraZeneca, Eisai, Ono, Guardant Health, Inc., Takeda Pharmaceutical Co., Ltd., Eli Lilly, Novartis, Pfizer, Chugai Pharmaceutical Co., Ltd., PDR pharma, Nihon Kayaku, Taiho Pharmaceutical Co., Ltd., Bristol, Bayer, Daiichi Sankyo Co., Ltd., and MSD outside the submitted work; TY reports honoraria from Chugai Pharmaceutical Co., Ltd., Takeda Pharmaceutical Co., Ltd., Merck, Bayer Yakuhin, Ono Pharmaceutical and MSD K.K; consulting fee from Sumitomo Corp.; and research grant from Amgen, Chugai Pharmaceutical Co., Ltd., Daiichi Sankyo Co., Ltd., Eisai, FALCO Biosystems, Genomedia Inc., Molecular Health, MSD, Nippon Boehringer Ingelheim, Ono, Pfizer, Roche Diagnostics, Sanofi, Sysmex and Taiho Pharmaceutical Co., Ltd., outside the submitted work. No disclosures were reported by the other authors.
Ethics approval and consent to participate
This study included patients with advanced GIST who participated in SURUM-Japan trials, including GI-SCREEN (UMIN000016344), GOZILA (UMIN000029315), MONSTAR-SCREEN (UMIN000036749), and MONSTAR-SCREEN-2 (UMIN000043899). The study protocol was approved by the Institutional Review Board of National Cancer Center Hospital East (UMIN000049334).
Consent for publication
All subjects have written informed consent.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Hashimoto, T., Nakamura, Y., Komatsu, Y. et al. Different efficacy of tyrosine kinase inhibitors by KIT and PGFRA mutations identified in circulating tumor DNA for the treatment of refractory gastrointestinal stromal tumors. BJC Rep 2, 54 (2024). https://doi.org/10.1038/s44276-024-00073-7
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1038/s44276-024-00073-7

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}