
Abstract
Background
Liquid biopsy can evaluate minimally residual disease. Hotspot mutations are also common in non-coding regions among the MIBC patients. We evaluated the status of MIBC with hotspot mutations with cfDNA.
Methods
Tumor and blood from MIBC patients were collected prospectively. We evaluate the VAF of mutations (TERT, PLEKHS1, ADGRG6 and WDR74) with digital PCR in tumor and cfDNA as somatic mutation. We originally designed and validated primers and probes. VAF of cfDNA and clinical imaging were matched. This study was approved by the Institutional Review Board (#2022-157).
Result
37 MIBC patients were enrolled and 28 (76%) patients had any hotspot. Among the 21 patients of follow-up cohort, cfDNA predicted recurrence 58 days earlier than the diagnosis by CT scan. Furthermore, the detection of ctDNA at the first visit after radical cystectomy was associated with recurrence free survival (P = 0.0043) and overall survival (P = 0.017). The patient who received neoadjuvant chemotherapy (NAC) and diagnosed as ypT0 belonged to the nonrecurrence group with negative ctDNA.
Conclusion
Hotspot mutation is promising biomarker to predict earlier recurrence than CT-scan. Multiple detection of mutations in cfDNA contributes to reliable recurrence prediction.
Similar content being viewed by others


Background
Bladder cancer (BC) is one of the most frequently diagnosed cancers, with more than 570,000 new cases and 210,000 BC-related deaths worldwide in 2020 [1]. At initial presentation, 70% to 75% of patients have non-muscle invasive bladder cancer (NMIBC), 20% to 25% have muscle invasive bladder cancer (MIBC), and 5% have metastatic disease [2]. Neoadjuvant chemotherapy followed by radical cystectomy is associated with improved overall survival (OS) rates with pathological complete response achieved in 30–40% of cases. However, MIBC patients often have poor outcomes (5-year survival rate: 50–60%) [3]. Predicting the disease status of MIBC patients is important for proper therapeutic intervention, but there are currently no reliable tumor biomarkers to assess BC disease status. Therefore, earlier detection of disease recurrence before obtaining radiographic images and monitoring disease status are unmet needs for managing patients with advanced BC.
Liquid biopsy has recently been used for cancer molecular profiling to enable a precision oncology approach [4,5,6]. Circulating tumor DNA (ctDNA) analysis has been widely applied in preclinical research and clinical practice, although the sensitivity of detecting ctDNA varies depending on the cancer type and disease progression [7]. Recent accumulating evidence has demonstrated that ctDNA is a promising minimally invasive blood-based biomarker for early detection of metastasis recurrence and monitoring BC treatment efficacy [8,9,10,11,12,13].
For the analysis of cell-free DNA (cfDNA), ultra-deep sequencing and digital polymerase chain reaction (dPCR) methods have been used for their high sensitivity [14]. Ultra-deep sequencing could detect 0.01% of frequent mutations in certain genes or regions, even if the mutation was unknown. However, ultra-deep sequencing is an expensive method, with frequent testing therefore increasing the financial burden. The dPCR method shows similar excellent sensitivity as ultra-deep sequencing for detecting 0.01% of frequent mutations, but is relatively less expensive for analyzing cfDNA because it does not require a next-generation sequencing (NGS) platform [5]. However, specific primers and probes are required prior to conducting dPCR, which can take several weeks to design and validate. Therefore, it would be useful to establish a method to detect recurrent BC hotspot mutations in ctDNA samples using the dPCR method that is both time and cost efficient in clinical settings.
In BC, hotspot mutations in the telomerase reverse transcriptase (TERT) gene promoter region have been reported in 60% to 80% of patients [15,16,17,18]. In addition, hotspot mutations in the phosphatidylinositole-4,5-bisphosphate 3-kinase catalytic subunit alpha (PIK3CA) and fibroblast growth factor receptor 3 (FGFR3) genes were reportedly detected in 7% to 27% and 3% to 20% of BC patients, respectively [18,19,20,21]. However, the frequencies of hotspot mutations other than those in the TERT promoter region were not satisfactory, as the PIK3CA and FGFR3 mutations were detected in only one patient and no patients, respectively. Using more common hotspot mutations would increase the number of patients who could be monitored with the dPCR method. In this study, we aimed to identify hotspot mutations recurrently detected in BC, develop an assay to detect ctDNA using dPCR, and examine the utility of this approach for monitoring the disease status of advanced BC cases.
Materials and methods
Patients and clinical evaluation
This study included 37 patients diagnosed with BC at Kyushu University Hospital (Fukuoka, Japan) between 2019 and 2022. The follow-up cohort included 21 patients who were monitored using cfDNA analysis and computed tomography (CT) scans every 3–6 months after radical treatment. The cfDNA collection was terminated when local recurrence or distant metastasis was diagnosed by CT scan, or when cfDNA remained negative without recurrence for over 1 year after radical cystectomy in ypT0pN0 cases. Written informed consent was obtained from all patients. The study was approved by Kyusyu University Hospital Institutional Review Board (approval number 2022-157). The study was conducted in accordance with the principles described in the Declaration of Helsinki and the Ethical Guidelines for Epidemiological Research enacted by the Japanese Government.
Sample collection and DNA extraction
Briefly, tumor tissues were collected before formalin fixation in the operating room. For the five cases where this was not possible, DNA was extracted from formalin-fixed paraffin-embedded (FFPE) samples. Tumor tissues that were obtained by transurethral resection or radical cystectomy were stored at −80°C until DNA extraction. Tumor DNA was extracted from these tissues with a DNeasy blood and tissue kit (Qiagen, Hilden, Germany) following the manufacturer’s instructions. DNA from FFPE samples was extracted using the QIAamp DNA FFPE tissue kit (Qiagen). DNA samples were quantified using an ND-1000 spectrophotometer (NanoDrop Technologies, Wilmington, DE, USA). DNA quality was confirmed with a Tapestation (Agilent Technologies, Santa Clara, CA, USA).
Blood samples were collected in three tubes (7 mL EDTA-2Na or 7 mL Cell-Free DNA BCT (Streck, La Vista, NE, USA)). After centrifugation at 3000×g for 10 min at 4°C, buffy coat and plasma were transferred into new tubes. An additional centrifugation step was performed for the plasma samples to reduce pellets (16,000×g, 10 min, 4°C). Buffy coat and plasma samples were stored at −80°C until DNA extraction. The DNeasy blood and tissue kit (Qiagen) was used to extract germline DNA from buffy coat samples that were collected prior to any treatment. Plasma samples were thawed on ice, then cfDNA was extracted using the QIAamp circulating nucleic acid kit (Qiagen) according to the manufacturer’s suggested protocol.
DNA sequencing
The sequencing libraries were prepared with the Twist Library Preparation Kit, Mechanical Fragmentation with Covaris Focused-ultrasonicator S220 (Covaris, Woburn, MA, USA) for 2 × 80 seconds and Custom Target Enrichment Panel (Twist Bioscience, South San Francisco, CA, USA) according to the manufacturer’s instructions. The original panel of 245 genes, as listed in Supplementary Table 1, was designed to include the introns, 5’ untranslated region (UTR), and 3’ UTR. DNA (50–100 ng) samples from 37 tumors and matched germline DNA samples were used for library preparation. Cluster amplification and 151 bp paired-end sequencing was performed according to the manufacturer’s protocol (Illumina, San Diego, CA, USA) on the NovaSeq 6000 system (Illumina).
Sequencing data analysis
Sequencing adapters were removed bioinformatically using Picard v2.27.4 software (https://gatk.broadinstitute.org/hc/en-us/articles/360037052812-Mar). Trimmed reads were mapped to the human reference genome (hg19) using bwa v0.7.17-r1188 (https://bio-bwa.sourceforge.net/), with PCR duplicates flagged by Picard MarkDuplicates. The alignments were deduplicated (Call Duplex Consensus Reads -M110) by Unique Molecular Identifiers (UMI) using fgbio v2.0.2.5 (https://github.com/fulcrumgenomics/fgbi).
The single nucleotide variants (SNVs) and insertions/deletions (InDels) of each sample were identified using samtools v1.15 [22] and varscan2 v2.4.4 (http://varscan.sourceforge.net/using-varscan.html) with somatic mode, then annotated using VEP v.108 [23, 24] (GRCh37 + ToMMo38KJPN).
Hotspot mutation PCR primers and probes
The primer and probes for the TERT promoter 228 C > T and 250 C > T were purchased from BioRad (Watford, UK). We designed the primer and probe sequences to detect point mutations in the PLEKHS1 promoter, ADGRG6 promoter, and WDR74 promoter regions. To generate validation templates (wild-type, WT; mutant type, Mut), the PCR amplicons of tumor-derived template DNA were inserted into the pGEM-T Easy Vector using the pGEM T Easy Vector system (Promega, Madison, WI, USA). PCR primer information is listed in Table 1 and the PCR conditions are described in the Supplementary methods. HD5α competent cells (Takara, Kusatsu, Japan) were transformed with the plasmids and cultured monoclonality for pure plasmid amplification. Plasmid sequence accuracy was confirmed by Sanger sequencing using the Applied Biosystems 3500 Genetic Analyzer (Thermo Fisher Scientific, Waltham, MA, USA). A plasmid purity value of over 99.9% was confirmed using droplet dPCR assays. Mut plasmid was diluted in a multistep manner with WT plasmid to create plasmid solutions with a variant allele frequency (VAF) of 0.063–10%, which were used for the spike-in experiments. At least three independent experiments were conducted for each assay.
Droplet dPCR assay
Typical dPCR experiments for cfDNA were performed with negative and positive controls using WT and Mut plasmids, respectively. The DNA input was 20,000 copies of plasmid, 10 ng of tumor or germline DNA, or 2 µL (at least 0.5 ng) of cfDNA per reaction. Details of the PCR components and conditions are described in the Supplementary methods. The primer/probe sequences are listed in Table 1. A droplet generator (BioRad) was used to create droplets and a Veriti 96-well thermal cycler (Thermo Fisher Scientific) was used for PCR experiments. Analysis of the DNA and VAF calculations for each sample were performed using the QX200 droplet dPCR system and Quantasoft (BioRad). Each experiment contained three positive control wells (pure Mut plasmid) and three negative control wells (pure WT plasmid) for validation. The dPCR results were considered valid only if there were more than 10,000 droplets, with over 100 droplets containing DNA. If the number of DNA-containing droplets was below this threshold, then the DNA was deemed to be of low quality and the result was rejected. When a cfDNA VAF value of over 0.5% was detected, it was defined as ctDNA positive. The threshold of 0.5% was set based on the value obtained when more than 200 droplets containing DNA were detected, with at least one positive droplet present. For patients with multiple hotspot mutations, the highest VAF of all hotspot mutations was employed.
Statistical analysis
All statistical analyses were performed using EZR software (Saitama Medical Center, Jichi Medical University, Saitama, Japan), a graphical user interface for R 2.13.0. Statistical analyses related to VAF were performed using Student’s t-tests. Recurrence was defined as lymph node enlargement or metastasis to other organs detected by CT scan. For analysis of recurrence-free survival (RFS), recurrence and death from any cause were defined as the end events. For analysis of OS, death from any cause was defined as the end event. Patients with none of these events were censored at the last follow-up visit. The number of days from the date of radical therapy to the date of the earliest end event or censoring was calculated for analysis. Survival analyses were conducted using the Kaplan–Meier method and log-rank test. The Cox proportional hazards model was used to estimate hazard ratios (HRs) with 95% confidence interval (CI). All P-values are two-sided. P-values < 0.05 were considered statistically significant.
Results
Patient characteristics and hotspot mutation identification
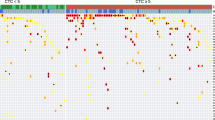
The clinicopathological characteristics of the 37 patients are shown in Table 2. The clinical course of the patients is shown in Fig. 1. Of the 37 patients, 23 underwent radical cystectomy or chemoradiation therapy as radical therapy. Nearly 50% of the patients had de novo or recurrent metastases. As a follow-up cohort, cfDNA was monitored (median follow-up duration: 2.7 years) in 21 patients (Fig. 1), among whom 15 patients underwent radical cystectomy.

NAC neoadjuvant chemotherapy, AC adjuvant chemotherapy.
First, we performed NGS analysis on tumor DNA samples from 37 patients. As shown in Supplementary Table 2, recurrent mutations in several genes were identified. Among them, we chose hotspot mutations in the TERT promoter (chr5:1,295,228 C > T [49%], chr5:1,295,250 C > T [8%]), PLEKHS1 promoter (chr10:115,511,590 G > A [16%], chr10:115,511,593 C > T [27%]), ADGRG6 promoter (chr6:142,706,206 G > A [27%], chr6:142,706,209 C > T [24%]), and WDR74 promoter (chr11:62,609,254 G > A [14%]). Among the 37 patients, at least one of the hotspot mutations shown above was detected in 28 (75.6%) individuals (Fig. 2).

TERT 228 C > T: TERT promoter (chr5:1,295,228 C > T), TERT 250 C > T: TERT promoter (chr5:1,295,250 C > T), PLEKHS1 590 G > A: PLEKHS1 promoter (chr10:115,511,590 G > A), PLEKHS1 593 C > T: PLEKHS1 promoter (chr10:115,511,593 C > T), ADGRG6 (206 G > A) ADGRG6 promoter (chr6:142,706,206 G > A), ADGRG6 209 C > T: ADGRG6 promoter (chr6:142,706,209 C > T), and WDR74 254 G > A: WDR74 promoter (chr11:62,609,254 G > A).
Original hotspot mutation assay development and validation
Next, we developed original assays to detect these hotspot mutations using the dPCR method, except for TERT 228 C > T and TERT 250 C > T, for which commercial dPCR primers and probes are not available. Subsequently, spike-in experiments using the WT and Mut plasmids were performed (Fig. 3). For all mutations, the VAF of pure plasmid was greater than 99.95%, and a mixture with 10% VAF was quantified accurately. For PLEKHS1 590 G > A, PLEKHS1 593 C > T, and ADGRG6 206 G > A, 0.125% VAF of mutants was statistically different from WT without mutants (P = 0.00296, 0.00916, and 0.00005, respectively). Furthermore, for GPR126 209 C > T and WDR74 254 G > A, 0.063% VAF of mutants was statistically different from WT without mutants (P = 0.00976 and 0.0027, respectively). Additionally, we confirmed that these primers and probes designed for hotspot mutations could detect these mutations in tumor DNA samples using dPCR, which was consistent with the NGS results. In contrast, cfDNA samples derived from healthy individuals were negative for these hotspot mutations (data not shown).

*P < 0.01, **P < 0.001.
Monitoring cfDNA by droplet dPCR for hotspot mutations in MIBC patients
Among the 21 patients in the follow-up cohort, hotspot mutations in tumor tissue-derived DNA samples were detected in 12 MIBC patients. Among them, eight patients (#03, #09, #14, #25, #05, #11, #13, and #24) underwent radical cystectomy for nonmetastatic disease. However, four patients (#03, #09, #14, and #25) experienced recurrence (non-regional lymph node in two cases and multiple metastases in two cases) during the follow-up period, while the other four patients (#05, #11, #13, and #24) did not. The four recurrent patients had positive cfDNA samples before recurrence diagnosis by CT scan (median: 58 days; range: 0–106 days) (Fig. 4a).

a Time course of the ctDNA fraction in the indicated cases who experienced recurrence (recurrent cases) or did not (non-recurrent cases). The x-axes in the recurrent cases and non-recurrent cases represent the days before the date of recurrence detection by computed tomography (CT) scans and the latest collection of cfDNA, respectively. Kaplan–Meier curves for b recurrence-free survival and c overall survival according to positive and negative ctDNA samples after radical cystectomy.
Furthermore, we compared the prognosis between patients whose ctDNA was positive at the first visit after radical cystectomy and those whose ctDNA was negative. The results showed that both RFS (P = 0.0043, Fig. 4b) and OS (P = 0.017, Fig. 4c) were significantly better in patients with negative ctDNA after radical cystectomy. Patients in whom ctDNA was detected after radical cystectomy all had pathological findings of pT3 (ypT3) or higher in their resected specimens, whereas those diagnosed as ypT0 after NAC belonged to the group of negative ctDNA.
Discussion
This study revealed that BC patients displayed hotspot mutations in the promoter regions of a known gene (TERT) and novel genes (PLEKHS1, ADGRG6, and WDR74). Accordingly, we developed ctDNA detection assays using dPCR, which showed an excellent detection ability with the threshold of 0.5% VAF. This method has been suggested to lead to early detection of tumor recurrence, evaluation of minimal residual disease (MRD), and monitoring tumor burden in MIBC more efficiently and at a lower cost compared with the NGS method.
Previously, hotspot mutations in the TERT promoter region (60− 80%), PIK3CA (7− 27%), and FGFR3 (3− 20%) have been observed in BC patients [15,16,17,18,19,20,21]. Consistently, hotspot mutations in the TERT promoter region (228 C > T and 250 C > T) were detected in 57% of patients in our study. In addition, novel hotspot mutations in the PLEKHS1 (590 G > A and 593 C > T), ADGRG6 (206 G > A and 209 C > T), and WDR74 (254 G > A) promoter regions were detected in 38%, 41%, and 14% of patients, respectively. Accordingly, at least one of these hotspot mutations was detected in 76% of patients, suggesting that these hotspot mutations could serve as individual tumor biomarkers.
In addition, this study demonstrated the clinical utility of using dPCR to monitor hotspot mutations in BC patient ctDNA samples. Several studies have reported that cfDNA analysis could predict the recurrence of urothelial carcinoma after radical therapy earlier than radiological imaging [9,10,11]. Our developed assay for detecting hotspot mutations by dPCR showed a similar ability to detect disease recurrence earlier than with radiography. Christensen et al. previously showed that MRD-represented positive ctDNA by NGS after radical cystectomy was predictive of metastatic recurrence [9]. Our study also showed different patient prognoses between those with postoperative negative ctDNA and positive ctDNA samples after radical cystectomy.
Following surgery or treatment with curative intent, detection of ctDNA may signal the presence of MRD even in the absence of any other clinical evidence of disease [25]. In the IMvigor010 study, postoperatively collected blood samples from patients with invasive urothelial carcinoma were analyzed. The results demonstrated that patients with positive ctDNA had significantly worse RFS and OS (RFS: 4.4 months versus not available; OS: 15.8 months versus not available) [12]. In our report, we analyzed the first postoperative blood sample, observing significant differences in RFS and OS with ctDNA detection.
The development of NGS and droplet dPCR and other recent technological innovations have supported the molecular analysis of cfDNA with high sensitivity. Although droplet dPCR provides a highly sensitive assay with a relatively low cost and short time, specific primer and probe sequences are required to detect a mutation. Additionally, this method can detect a limited number of mutations. Therefore, it is necessary to establish an assay system for somatic mutations that can be used in most patients. Previously, Tamura et al. reported the benefits of the individualized ctDNA monitoring system using tumor-specific gene mutations for the early detection of postoperative recurrence in upper tract urothelial carcinoma patients [26]. Although this method can provide ctDNA monitoring by dPCR in most patients, new primers and probes need to be designed and validated if none are available.
A major improvement of the method in this study is the assay system designed for pre-defined hotspot mutations. This system allows for immediate application in screening mutations in primary tumors and subsequent blood testing. Notably, it eliminates the need for NGS analysis, avoids the necessity of designing patient-specific primers and probes, and relies solely on digital PCR, making it feasible for implementation in various medical facilities, including community hospitals. These advantages are expected to reduce development costs, labor costs, and transportation expenses, potentially enabling cost-effective routine monitoring of ctDNA. In the current management of MIBC, where follow-up primarily relies on periodic imaging due to the absence of reliable tumor markers, this approach has the potential to serve as an innovative diagnostic support tool. By enabling early detection of recurrence, it may facilitate more timely and appropriate therapeutic interventions
This study has some limitations. First, the number of enrolled patients was small. Despite MIBC not being a rare disease, the number of patients was limited by the single-institution nature of the study. The number of patients who were eligible for hotspot analysis was even smaller. Although a larger cohort is needed to obtain more robust results, this study still demonstrated promising findings. A further prospective validation study would be required before clinical application. An additional limitation is the restricted scope of NGS. Because sequencing covers a large region beyond the coding region of a gene, the number of genes covered was limited. In the future, genes may be identified that should not have been excluded.
In conclusion, our findings demonstrated the feasibility of hotspot mutation dPCR assays in non-coding regions in cfDNA and the clinical usefulness for BC patients. This study will contribute to dPCR assay development for ctDNA monitoring.
Data availability
All data generated or analyzed during this study are included in this published article and its supplementary information files.
References
Sung H, Ferlay J, Siegel RL, Lavarsanne M, Soerjomataram I, Jemal A, et al. Global cancer statistics 2020: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancer in 185 countries. CA A Cancer J Clin 2021;71:209–49.
Lopez-Beltran A, Cookson MS, Guercio BJ, Cheng L. Advances in diagnosis and treatment of bladder cancer. BMJ. 2024;384:e076743.
Beckabir W, Wobker SE, Damrauer JS, Midkiff B, De la Cruz G, Makarov V, et al. Spatial relationships in the tumor microenvironment demonstrate association with pathologic response to neoadjuvant chemoimmunotherapy in muscle-invasive bladder cancer. Eur Urol. 2024;85:242–53.
Nikanjam M, Kato S, Kurzrock R. Liquid biopsy: current technology and clinical applications. J Hematol Oncol. 2022;15:131.
Kurma K, Eslami-S Z, Alix-Panabieres C, Cayrefourcq L. Liquid biopsy: paving a new avenue for cancer research. Cell Adhension Migr. 2024;18:1–26.
Bhalla S, Passarelli R, Biswas A, De S, Ghodoussipour S. Plasma-derived cell-free DNA as a biomarker for early detection, prognostication, and personalized treatment of urothelial carcinoma. J Clin Med. 2024;13:2057.
Bettegowda C, Sausen M, Leary RJ, Kinde I, Wang Y, Agrawal N, et al. Detection of circulating tumor DNA in early- and late-stage human malignancies. Sci Transl Med. 2014;6:224ra24.
Patel KM, van der Vos KE, Smith CG, Mouliere F, Tsu D, Morris J, et al. Association of plasma and urinary mutant DNA with clinical outcomes in muscle invasive bladder cancer. Sci Rep. 2017;7:1–12.
Christensen E, Birkenkamp-Demtröder K, Sethi H, Shchegrova S, Salari R, Nordentoft I, et al. Early detection of metastatic relapse and monitoring of therapeutic efficacy by ultra-deep sequencing of plasma cell-free DNA in patients with urothelial bladder carcinoma. J Clin Oncol. 2019;37:1547–57.
Birkenkamp-Demtroder K, Christensen E, Nordentoft I, Knudsen M, Taber A, Høyer S, et al. Monitoring treatment response and metastatic relapse in advanced bladder cancer by liquid biopsy analysis. Eur Urol. 2018;73:535–40.
Carrasco R, Infelmo-Torres M, Gómez A, Trullas R, L.Roldán F, Ajami T, et al. Cell-free DNA as a prognostic biomarker for monitoring muscle-invasive bladder cancer. Intern J Mol Sci. 2022;23:11732.
Powles T, Assaf ZJ, Davarpanah N, Banchereau R, Szabados BE, Yuen KC, et al. ctDNA guiding adjuvant immunotherapy in urothelial carcinoma. Nature. 2021;595:432–7.
Lindskrog SV, Birkenkamp-Demtröder K, Nordentoft I, Laliotis G, Lamy P, Christensen E, et al. Circulating tumor DNA analysis in advanced urothelial carcinoma: insights from biological analysis and extended clinical follow-up. Clin Cancer Res. 2023;29:4797–807.
Todenhöfer T, Struss WJ, Seiler R, Wyatt AW, Black PC. Liquid biopsy-analysis of circulating tumor DNA (ctDNA) in bladder cancer. Bladder Cancer. 2018;4:19–29.
Cheng L, Shang S, Wang M, Lopez-Beltran A. Biological and clinical perspectives of TERT promoter mutation detection on bladder cancer diagnosis and management. Hum Pathol. 2023;133:56–75.
Allory Y, Beukers W, Sagrera A, Flandez M, Marques M, Marquez M, et al. Telomerase reverse transcriptase promoter mutation in bladder cancer: High frequency across stages, detectionin urine, and lack of association with outcome. Eur Urol. 2014;65:360–6.
Hosen MI, Sheikh M, Zvereva M, Scelo G, Forey N, Durand G, et al. Urinary TERT promoter mutations are detectable up to 10 years prior to clinical diagnosis of bladder cancer: Evidence from the Golestan Cohort study. EBioMedicine. 2020;53:102643.
Hayashi T, Fujita K, Hayashi Y, Hatano K, Kawashima A, McConkey DJ, et al. Mutational landscape and environmental effects in bladder cancer. Int J Mol Sci. 2020;21:6072.
Ahanidi HE, Azzouzi ME, Arrouchi H, Alaoui CH, Tetou M, Bensaid M, et al. AKT1 and PIK3CA activating mutations in Moroccan bladder cancer patients’ biopsies and matched urine. Pan Afr Med J. 2022;41:59.
Knowles MA, Platt FM, Ross RL, Hurst CD. Phosphatidylinositol 3-kinase (PI3K) pathway activation in bladder cancer. Cancer Metastasis Rev. 2009;28:305–16.
Christensen E, Birkenkamp-Demtröder K, Nordentoft I, Høyer S, van der Keur K, van Kessel K, et al. Liquid biopsy analysis of FGFR3 and PIK3CA hotspot mutations for disease surveillance in bladder cancer. Eur Urol. 2017;71:961–9.
Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, et al. The sequence alignment/map format and SAMtools. Bioinformatics. 2009;25:2078–9.
McLaren W, Gil L, Hunt SE, Riat HS, Ritchie GRS, Thormann A, et al. The ensembl variant effect predictor. Genome Biol. 2016;17:122.
Fuse N, Sakurai-Yageta M, Katsuoka F, Danjoh I, Shimizu R, Tamiya G, et al. Establishment of integrated biobank for precision medicine and personalized healthcare: The Tohoku Medical Megabank Project. Jpn Med Assoc J. 2019;2:0014.
Riethdorf S, Soave A, Rink M. The current status and clinical value of circulating tumor cells and circulating cell-free tumor DNA in bladder cancer. Transl Androl Urol. 2017;6:1090–110.
Tamura D, Abe M, Hiraki H, Sasaki N, Yashima-Abo A, Ikarashi D, et al. Postoperative recurrence detection using individualized circulating tumor DNA in upper tract urothelial carcinoma. Cancer Sci. 2024;115:529–39.
Acknowledgements
We thank Ms. Noriko Hakoda, Ms. Eriko Gunshima, Ms. Harumi Inoue, Ms. Hishimi Izumi, and Ms. Kyoko Fukushi for their excellent technical assistance. We also thank Dr. Youichi Ide, Dr. Keisuke Kodama, and Dr. Ikuei Hiraka for their invaluable contributions to NGS assay design. We thank Dr. Kazuki Mori and Dr. Hiroko Hagiwara for analysis of NGS data. We thank J. Iacona, Ph.D., from Edanz (https://jp.edanz.com/ac for editing a draft of this manuscript.
Funding
This research was supported by a research grant from Denka Co., Ltd., and represents the results of a collaborative effort with the company.
Author information
Authors and Affiliations
Contributions
ST was responsible for conceptualization, data curation, formal analysis, investigation, methodology, resources, validation, visualization and writing manuscript. MS was responsible for conceptualization, data curation, formal analysis, investigation, methodology, project administration, resources, supervision, visualization and writing manuscript. TM was responsible for conceptualization, data curation, formal analysis, investigation, methodology, project administration, resources, supervision. DT responsible for investigation and resources. SN was responsible for data curation, investigation and resources. ND and SM were responsible for resources. MY was responsible for resources and supervision. TU was responsible for conceptualization, Formal analysis, methodology, project administration, resources, supervision, visualization. YK, DK and ME were responsible for resources and supervision.
Corresponding author
Ethics declarations
Competing interests
Masaki Shiota received honoraria from Janssen Pharmaceuticals, AstraZeneca, Astellas Pharma, Sanofi, and Bayer Yakuhin, as well as research funding support from Astellas Pharma. Masatoshi Eto received honoraria from Ono Pharmaceutical, Takeda Pharmaceuticals, Novartis Pharma, Pfizer, Bristol-Myers Squibb, Janssen Pharmaceuticals, MSD, Merck Biopharma, AstraZeneca, and Eisai, as well as research funding support from Bayer Yakuhin, Astellas Pharma, Ono Pharmaceutical, and Takeda Pharmaceuticals. Masaki Shiota is an Associate Editor at BJC Reports. He was not involved in any aspect of handling of this manuscript or any editorial decisions.
Ethics approval, consent to participate and publication
Written informed consent was obtained from all patients. The study was approved by Kyusyu University Hospital Institutional Review Board (approval number 2022-157). The study was conducted in accordance with the principles described in the Declaration of Helsinki and the Ethical Guidelines for Epidemiological Research enacted by the Japanese Government.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Tsukahara, S., Shiota, M., Matsumoto, T. et al. Monitoring circulating tumor DNA by recurrent hotspot mutations in bladder cancer. BJC Rep 3, 26 (2025). https://doi.org/10.1038/s44276-025-00143-4
Received:
Revised:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s44276-025-00143-4
