
Abstract
The nuclear receptor (NR) superfamily consists of the orphan NR subgroup NR4A, of which Nur77, an immediate early response gene, plays a crucial role in liver physiology and pathophysiology. Nur77 acts as a transcription factor with genomic and non-genomic activities. This review summarises Nur77’s role in liver diseases and its potential as a therapeutic target, as numerous studies have explored the diverse therapeutic implications of targeting Nur77 across different liver diseases.
Similar content being viewed by others

The nuclear receptors superfamily
The human genome encompasses 48 distinct NR transcription factor members, which are structurally similar and depend on ligands to function, thus controlling different aspects of development by both positively and negatively regulating gene expression1,2. These receptors are intricately linked to the development and progression of different pathological conditions, such as metabolic diseases, cancer, and the regulation of central nervous system functions3,4. Although NRs were initially believed to localize into the nucleus, it is now common knowledge that in addition to the nucleus, NRs can also be found at the plasma membrane, in the mitochondria, and endoplasmic reticulum (ER)5.
For most of the NRs, the ligands are identified and are important in regulating NRs' biological activity derived from the endogenous metabolism or external sources such as diet, and include diverse classes like fatty acids, terpenoids, porphyrins, and amino acid derivatives. Upon ligand binding, the NRs undergo conformational changes, changing gene transcription, thus affecting cellular metabolism and function6.
Nuclear receptors, like other transcription factors, are organized into modular units with five distinct functional domains. The N-terminal domain varies in length and amino acid composition and is responsible for interacting with other transcription factors. This region also enables transactivation through its ligand-independent activation function-1 (AF-1). Following this is the DNA-Binding Domain (DBD), which is highly conserved and allows NRs to identify specific target sequences to initiate gene expression7. The DBD contains two zinc fingers and a C-terminal extension (CTE) that are necessary for interaction with DNA-specific sequences or hormone response elements (HREs). The flexible hinge works as a connector between the DBD and the Ligand-Binding Domain (LBD)/C-terminal AF-2 Domain domains. The fifth region is the LBD, which is predominantly hydrophobic and plays a critical role in identifying ligands. The LBD functions as a molecular switch, transitioning the receptor into an active state for transcription upon ligand binding, and the C-terminal AF-2 Domain is characterized by a highly variable sequence6,7,8.
Nuclear receptors in liver physiology
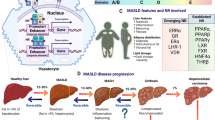
Nuclear receptors are vital in managing liver physiology and diseases as they regulate key metabolic functions including lipid metabolism, drug metabolism, bile acid regulation, liver regeneration, inflammation, fibrosis and cancer9. Among the key NRs involved in liver health are the Peroxisome Proliferator-Activated Receptors (PPARs), which regulate fat metabolism and energy homeostasis; the Farnesoid X Receptor (FXR), which is critical for bile acid regulation, and the Liver X Receptors (LXRs) that manage cholesterol levels and promote its excretion from the body. Other important NRs include the Pregnane X Receptor (PXR) and the Constitutive Androstane Receptor (CAR), which safeguard the liver from environmental toxins and drugs through their role in detoxification processes. Furthermore, dysregulation and genetic variations of these NRs can result in liver disorders, positioning NRs as crucial targets for innovative treatments in conditions such as fatty liver disease and drug-induced liver injury9. Figure 1 summarizes the key physiological roles of nuclear receptors in the liver.

This schematic summarizes selected roles of NRs across major hepatic processes. Lipid metabolism & steatosis: PPARα promotes hepatic fatty-acid β-oxidation during fasting101. LXRα promotes de novo lipogenesis (via SREBP-1c) and regulates cholesterol homeostasis102. HNF4α regulates lipoprotein metabolism/VLDL secretion103; FXR protects from steatosis by repressing hepatic lipogenesis and reducing intestinal lipid absorption104. Bile-acid homeostasis: FXR acts as a central regulator of bile acids105; PXR induces bile-acid detoxification programs106; VDR senses secondary bile acids and drives their detoxification107; CAR promotes detoxification of bile acids and xenobiotics108. Inflammation: PPARα exerts anti-inflammatory effects in liver109; LXR activation suppresses TLR → NF-κB–driven cytokine expression in hepatic mononuclear/Kupffer cells110; FXR represses NF-κB signaling111; VDR is immunomodulatory and anti-inflammatory in liver macrophages112. Fibrosis: PPARγ inhibits HSC activation113; VDR shows anti-fibrotic activity114; GR reduces TGF-β expression/signaling in HSCs115; Nurr1/NR4A2 inhibits HSC proliferation and extracellular-matrix genes via MAPK pathways116. Hepatocellular carcinoma (HCC): FXR is downregulated in HCC and low expression associates with aggressive clinicopathologic features117; PPARα has a dual role, protective against steatosis but chronic activation promotes HCC in rodents118; RORγ inhibits HCC growth and metastasis119.
In recent years, many NRs have been identified as dysregulated in liver diseases, and pharmacological ligand agonists have been proposed as treatment, such as the FXR agonist obeticholic acid in metabolic-associated steatohepatitis (MASH), although studies showed no improvement in MASH patients10. In contrast, Resmetirom® (Rezdiffra™), a Thyroid Hormone Receptor Beta (THRB) agonist, showed a significant benefit in MASH patients and was approved by the Food and Drug Administration (FDA). However, the improvement in MASH resolution and in liver fibrosis by at least one stage, as well as improved health-related quality of life occurred in less than 50% of patients10,11,12. Furthermore, Lanifibranor®, a Pan-PPAR agonist, demonstrated in the phase 2b study NATIVE, an improvement in MASH resolution and fibrosis improvement13, and recently showed to improve cardiometabolic health (CMH)14. It has now advanced to the Phase3 NATiV3 trial15.
Furthermore, Sex hormone NRs Androgen receptor (AR) and Estrogen Receptor alpha (ERa); and REV-ERBα (NR1D1), REV-ERBβ (NR1D2), and Retinoic Acid Related Orphan Receptor A (RORA) are NRs regulating central and peripheral circadian clocks and TGFβ1 signaling pathway, and all have emerged as possible targets in liver diseases11. The progression from Resmetirom’s FDA approval to Lanifibranor advancing into Phase 3 demonstrates the emerging success in targeting NRs. Building on the emerging success in targeting NRs, Nur77 also emerges as a promising therapeutic candidate in liver diseases due to its broad regulatory roles in metabolism, inflammation, and fibrosis16,17.
Orphan nuclear receptors: NR4A subfamily
Orphan NR are receptors for which no endogenous ligand has been identified so far. Unlike the ligand-activated steroid hormone receptors, NR4A receptors exhibit significant constitutive transcriptional activity and influence different physiological processes through alternative mechanisms18. The NR4A subfamily is composed of three closely related members, each with distinct biological roles: Nur77 (NR4A1) (Nuclear Receptor Subfamily 4 Group A Member 1), Nurr1 (Nuclear receptor related 1) (NR4A2), and NOR1 (Neuron-derived orphan receptor 1) (NR4A3). The NR4A family shares significant structural conservation, with their LBDs exhibiting 60–65% similarity, while the activation function-1 (AF-1) domains show greater divergence at 20–30% similarity, reflecting receptor-specific functional specialization19,20. The NR4A subfamily is known to respond dynamically to a variety of stressors and signaling molecules such as growth factors, cytokines, glucose, fatty acids21, and inflammatory molecules including prostaglandins, Tumor Necrosis Factor-alpha (TNF-α), Lipopolysaccharide (LPS), Interferon-gamma (IFN-γ)22, and granulocyte-macrophage colony-stimulating factor (GM-CSF)23.
Genomic location, structure, and functional domains of Nur77
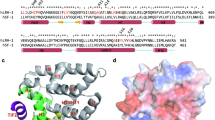
The NR4A1 gene, known for encoding the transcription factor Nur77, is located on chromosome 12 at the 12q13.13 region24. Historically, this gene has been referred to by several symbols, including HMR and GFRP1, and aliases such as TR3, N10, NAK-1, and NGFI-B. Nur77 was initially identified as Nerve Growth Factor Induced Clone B (NGFI-B) and later classified as an orphan NR25. Nur77 protein, consisting of 598 amino acids, features an N-terminal activation function region corresponding to the A/B region, a central DNA-binding domain (C region), a flexible hinge area (D region), and an E region, the C-terminal ligand-interacting domain. Further, Nur77 contains several functional domains, including an AF-1 region, DBD, and a C-terminal LBD (Fig. 2)26. Unique to Nur77 is the AF-1 domain, facilitating transcriptional activity by recruiting coactivators such as Steroid Receptor Coactivator-2 (SRC-2), p300, and P300/CBP-associated factor PCAF, a role typically attributed to the LBD in other NRs. The DBD in Nur77 specifically recognizes and binds to the NGFI-B Response Element (NBRE; sequence: AAAGGTCA) within target gene promoters, ensuring precise gene regulation27.

The schematic shows AF-1, DBD, hinge, and LBD contains AF-2. Brackets indicate sequence similarity among NR4A family (Nur77/NR4A1, Nurr1/NR4A2, NOR-1/NR4A3): AF-1 26–28%, DBD 94–95%, LBD (incl. AF-2) 58–65%120. The DBD has two C4 zinc-finger modules for DNA binding, and the LBD has the canonical 12-helix nuclear-receptor fold121. NR4A LBD: occluded pocket; atypical AF-228.
Crystallographic analysis has shown that the LBD of Nur77 lacks a conventional ligand-binding cavity, as bulky hydrophobic residues tightly pack the domain, occupying the space where a pocket would normally reside28. This distinctive architecture suggests that Nur77 is unlikely to rely on classical ligand binding for activation. Instead, its activity is regulated by expression level, subcellular distribution, including nuclear-to-cytoplasmic translocation29, and post-translational modifications such as phosphorylation30. Nevertheless, certain exogenous small molecules, such as Cytosporone B, TMPA, and THPN, can interact with alternative binding surfaces on the LBD to modulate its function30,31.
Nur77 as a Transcription Factor – genomic and non-genomic actions
Nur77, along with other members of the NR4A family, demonstrates diverse DNA-binding capabilities. It can bind individually as a monomer to the NGFI-B response element (NBRE; sequence AAAGGTCA), where a single receptor binds DNA. Additionally, Nur77 functions as part of a homodimer or heterodimer with other NR4A family members, binding to a synthetic consensus Nur-response element (NurRE; sequence 5′-TGACCTTTx6AAAGGTCA-3′)28. For example, Nur77 and Nurr1 can form heterodimers with the retinoid X receptor (RXR), activating transcription through direct repeat 5 (DR5) retinoid response elements2. Further, Nur77 can act as a cofactor, thus significantly enhancing the activity of specificity protein (Sp) transcription factors such as Sp1 and Sp4 at GC-rich promoter sequences, which enhances the transcription of several critical genes crucial for cell survival and proliferation32.
Numerous studies have demonstrated Nur77’s nuclear translocation to the cytosol upon stress or exposure to specific agents, thereby impairing mitochondrial integrity and facilitating apoptosis by enabling cytochrome c release in the cytosol33. The identification of this non-genomic action of Nur77 provided a new perspective on its involvement in apoptosis. For example, Nur77’s nuclear translocation is induced during ischemia/reperfusion, resulting in oxidative stress and calcium influx34. Further research revealed that Nur77 interacts with Bcl-2, triggering a conformational change exposing Bcl-2’s BH3 domain, thereby transforming Bcl-2 from an anti-apoptotic molecule into a pro-apoptotic factor35.
Furthermore, Nur77 translocates from the nucleus to the ER in response to ER stress, a process triggered by agents like CD437 D437/AHPN (6-[3-(1-adamantyl)-4-hydroxyphenyl]-2-naphthalene carboxylic acid), a synthetic retinoid and RARγ-selective agonist. This translocation enables Nur77 to specifically interact with ER-targeted Bcl-2, initiating an early release of calcium (Ca2+) from the ER resulting in apoptosis through the activation of ER-specific caspase-4. Nur77’s translocation to the ER depends on the nuclear export receptor Chromosome Region Maintenance 1 (CRM1) recognizing a nuclear export signal on Nur77, thus facilitating its movement out of the nucleus27,36. Compounds like 12-O-tetradecanoylphorbol-13-acetate (TPA) or CD437, induce translocation of Nur77 to the ER, where Nur77 binds Translocon-Associated Protein subunit gamma (TRAPγ). This interaction results in a depletion of ER calcium, inducing ER stress and triggering apoptosis in HepG2 hepatoma cell line37. Nur77’s non-genomic functions extend beyond apoptosis to other cellular processes. For instance, Nur77 can modulate p38α MAPK pathway38, interact with p53 to regulate its stability, and sequester liver kinase B1 (LKB1) in the nucleus, which suppresses phosphorylation of AMPKα39. It also plays a role in modulating the activity of the mTORC1 complex involved in cell growth and metabolism40.
Nur77: a key regulator connecting metabolism, inflammation, and fibrosis in liver disease
Nur77 plays a multifaceted role in maintaining liver homeostasis, acting as a critical nexus that integrates metabolic control, inflammatory responses, and fibrogenesis. Its diverse functions position Nur77 as a promising therapeutic target, and this broad spectrum of actions is illustrated in the integrated matrix of mechanisms shown in Fig. 3.

This schematic cartoon illustrates Nur77’s regulatory functions across liver pathologies, highlighting its therapeutic relevance. In liver inflammation, Nur77 inhibits NF-κB signaling, reduces pro-inflammatory cytokines, and supports anti-inflammatory M2 macrophage polarization. In fibrosis, Nur77 inhibits HSC activation and antagonizes the TGF-β–Smad2/3/4–ZEB signaling pathway, leading to reduced extracellular matrix proteins, e.g., fibronectin, α-SMA. In lipid metabolism and steatosis, Nur77 attenuates lipid uptake and lipogenesis while enhancing fatty acid oxidation through FGF21, thereby reducing hepatic triglyceride and cholesterol levels. In the context of HCC and tumor immunity, Nur77 exerts tumor-suppressive effects by modulating glycolysis (via PEPCK1, WFDC21P) and inhibiting β-catenin, reduces HK1 palmitoylation in HSCs ( ↓ glycolytic EVs), restores CAR-T/NK cell function, promotes immunosuppressive TME via high TI-Treg expression, and its inhibition boosts anti-tumor immunity via IFN-I^Mo/NK cell activation50,51,52,53,55,56,57,60,61,65,72,75,76.
Nur77’s key role in regulating hepatic metabolism
Nur77 is an essential regulator of hepatic metabolic processes, profoundly influencing both glucose and lipid metabolism by modulating specific genes critical for mitotic clonal expansion, adipogenic differentiation, and broader lipogenic and metabolic regulation21. Significantly, Nur77 also regulates metabolic homeostasis outside the liver, particularly in adipose tissue41. It exerts significant anti-adipogenic and anti-lipidemic effects, making it key in delaying obesity and its associated metabolic conditions like MASLD and hyperlipidemia42. In the liver, Nur77 actively participates in maintaining glucose homeostasis, especially under metabolic stress. An adenoviral overexpression of Nur77 in mice liver demonstrated a significant upregulation of genes essential for glucose production, such as glucose-6-phosphatase (G6pc), fructose bisphosphatase (Fbp1), and glucose transporter 2 (Glut2), resulting in increased glucose production and elevated plasma glucose levels43. In contrast, the absence of Nur77 led to increased insulin resistance and hepatic steatosis in mice on a high-fat diet, a condition accompanied by an upregulation of lipogenic genes in the liver, contributing to worsened glucose tolerance44. Src homology domain 3 binding kinase 1 (SBK1) phosphorylates Nur77 at serine 344 in hepatocytes, enhancing its transcriptional activity. This promotes FGF21 expression and inhibits hepatic lipogenic genes like Acetyl-CoA carboxylase 1 (ACACA) and Fatty Acid Synthase (FASN), observed in Hepa1-6 mouse hepatoma cells and high-fat diet-fed mice45. Additionally, Nur77 has demonstrated to protect against hyperhomocysteinemia (HHcy)-induced hepatic steatosis by inhibiting lipid accumulation through the suppression of fatty acid translocase (CD36) and Fatty Acid Transport Protein 2 (FATP2) expression, modulating histone acetylation, and broadly reducing hepatic lipid accumulation, both in vitro and in vivo46. Furthermore, Nur77 affects lipid homeostasis by regulating genes involved in lipid metabolism; for example, Nur77 hepatic overexpression in mice with adenovirus encoding human Nur77 (Ad.Nur77) suppresses transcription factor Sterol Regulatory Element-Binding Protein 1c (SREBP1c) activity, reduces hepatic triglyceride levels, and alters plasma lipid profiles47. Nur77 modulates hepatic cholesterol metabolism by impacting the expression levels of low-density lipoprotein receptor (LDLR) and 3-hydroxy-3-methyl-glutaryl-coenzyme A reductase (HMG-CoA), with decreased levels of Nur77 being associated with elevated expression of these enzymes and vice versa, as shown in HepG2 cells48. These metabolic roles underscore Nur77’s protective capacity against lipotoxicity and glucose dysregulation, which are foundational to liver disease progression.
Nur77’s role in mitigating liver inflammation
Beyond its direct metabolic functions, Nur77 critically modulates inflammatory processes, primarily by suppressing excessive pro-inflammatory responses through the inhibition of nuclear factor kappa B (NF-κB) signaling49. Its overexpression increases IκBα levels, which prevents NF-κB activation, thereby reducing the expression of adhesion molecules such as ICAM-1 and VCAM-1, limiting immune cell binding. Concurrently, Nur77 plays a pivotal role in promoting the anti-inflammatory M2 macrophage phenotype50. It fundamentally shapes macrophage plasticity by limiting pro-inflammatory activation and, when absent, shifts macrophages toward a more M1-like state; moreover, Nur77 suppresses pro-inflammatory gene expression and modulates macrophage mechanics including adhesion, migration and phagocytosis51. This regulation extends to suppressing pro-inflammatory gene expression and modulating cellular mechanisms within macrophages51. Evidence from liver injury models highlights Nur77’s indispensable role in inflammation control. In models of acute liver inflammation induced by lipopolysaccharide (LPS) and D-galactosamine (D-GalN), Nur77 was essential for the anti-inflammatory effects of celastrol; its absence in Nur77-null mice markedly diminished celastrol’s ability to reduce serum ALT/AST and pro-inflammatory cytokines such as IL-1β and IL-6 and to alleviate hepatic architecture disruption, p65 nuclear translocation, and IκBα degradation. Celastrol promotes Nur77’s mitochondrial translocation and triggers TRAF2-dependent ubiquitination of Nur77, leading to mitophagy and alleviating inflammation52. While celastrol shows promise in Nur77-mediated anti-inflammatory pathways, its effectiveness across different etiologies and stages of liver disease remains unclear, due to its unknown hepatoxicity. Similarly, in MAFLD models, the flavonoid hyperoside relies on Nur77 expression to induce M2 macrophage polarization and consequently mitigates inflammation, hepatic steatosis, and insulin resistance. However, these effects are absent in Nur77-deficient models53. Furthermore, Nur77 mediates the suppression of chronic liver inflammation in high-fat diet (HFD)–induced liver injury models. This is achieved as treatments with DPA-EA and DHA-EA bind to Nur77, leading to Nur77-induced anti-inflammatory effects through the NF-κB pathway, and reducing lipid synthesis while enhancing antioxidative responses54. In acute liver failure, early glucocorticoid treatment has been shown to reduce liver damage by upregulating Nur77 expression in Kupffer cells, leading to the suppression of pro-inflammatory cytokines and improved liver pathology through a glucocorticoid receptor (GR)-dependent mechanism55. Notably, Nur77 also controls monocyte differentiation, as demonstrated by the absence of anti-inflammatory Ly6C^Lo non-classical monocytes in Nur77 knockout models, leading to dysregulated immune responses and impaired liver repair, as demonstrated in biliary atresia56. These findings collectively establish Nur77 as a crucial orchestrator of hepatic immune responses, reducing the inflammation to protect against liver injury. However, the immunomodulatory role of Nur77 in T cells remains more complex. A recent study showed that a dual knockout of Nur77 and Nurr1 in T cells in a MASH in vivo model led to enhanced Treg expansion and immunosuppressive function, resulting in reduced liver inflammation and fibrosis57. This raises the possibility that in certain immune cell subsets, such as Tregs, NR4A receptors may limit protective functions57. However, using a dual knockout model makes it difficult to determine which receptor drives the observed effects. To better understand Nur77’s specific role in Treg regulation and fibrotic progression, future studies should use single-gene knockouts. This would clarify whether targeting Nur77 alone is sufficient or if dual inhibition is necessary for therapeutic benefit. Together, these findings suggest that Nur77’s effect on inflammation could vary based on disease stage and immune cell type. Further research is needed to assess Nur77’s expression and function in Tregs across MASLD/MASH progression and other liver diseases to clarify its potential as a therapeutic target in inflammation control.
Nur77’s influence on fibrogenesis and disease progression
Building upon its metabolic and anti-inflammatory roles, Nur77 shapes liver fibrogenesis through direct modulation of HSC activity. HSCs are central players in liver fibrogenesis; thus, identifying the regulation of dysregulated signaling pathways to halt HSC activation and fibrosis is a major research focus58. This is crucial given the established role of TGF-β signaling in advancing liver fibrosis and cirrhosis59, and previous studies have solidified Nur77 as a key regulator of this pro-fibrogenic pathway60,61. The lack of active Nur77 consequently results in continuous TGF-β activation and increased fibrogenesis61,62. Nur77 activity appears reduced in primary human HSCs carrying the PNPLA3 I148M variant, contributing to upregulated TGF-β1 signaling and increased MASLD/MASH susceptibility63,64. Additionally, in the context of viral hepatitis, exosomes isolated from HBV‑infected HepG2‑NTCP hepatocytes were transferred to HSC‑T6 cells in vitro, delivering miR‑506‑3p that downregulates Nur77 and activates fibrogenic gene expression. Further, lower Nur77 levels were observed in liver tissue from HBV-infected patients, confirming clinical relevance65. Complementing these direct inhibitory roles, external molecules also leverage Nur77’s fibrotic actions. Glucagon-like peptide 2 (GLP-2) has shown hepatoprotective and anti-fibrotic effects in sclerosing cholangitis models by enhancing Nur77 expression and activity, increasing its nuclear binding, and counteracting TGF-β-induced Nur77 suppression66. Similarly, oxytocin, a neuropeptide, reverses liver fibrosis in vivo by driving a Ly6C^high/Ly6C^low macrophage phenotype, with KEGG analysis revealing Nur77 activation. Oxytocin induces through calcium influx the MAPK pathway, promoting CREB transcription factor activity, which then binds to the Nur77 promoter to regulate its phenotypic switch67. Nur77 activation demonstrates suppression of pro-fibrogenic activation in rat HSC-T6 cells by inhibiting the TGF-β-Smad2/3/4-ZEB pathway, reducing the expression of fibronectin, vimentin and α-SMA, while enhancing Smad7 and E-cadherin expression60. However, this study did not include silencing or knockout of Nur77 gene, so the observed anti-EMT and anti-fibrotic effects cannot be definitively linked to Nur77. These findings feature the capacity of Nur77 to mitigate liver fibrogenesis and inhibit prof-fibrotic signaling pathways. Indeed, two prototype cytokines central to HSC activation during liver injury are TGF-β and platelet-derived growth factor (PDGF)68. While the research on the interaction between Nur77 and TGF-β is growing, its relationship with PDGF remains less clear and needs further investigation.
Nur77’s role in hepatocellular carcinoma and immune responses
In a combined DEN and CCl4-induced mouse hepatocarcinogenesis model, Nur77 knockout mice displayed increased hepatic fibrosis and promoted tumor development. Nur77 primarily modulates glucose metabolism by enhancing gluconeogenesis and inhibiting glycolysis through its crucial interaction with phosphoenolpyruvate carboxykinase 1 (PEPCK1). This interaction stabilizes PEPCK1 by reducing its SUMOylation and preventing its degradation. This leads to ATP depletion and cancer cell growth arrest. Clinically, lower Nur77 levels are linked with HCC development (stage I-III) and correlate with poor prognosis in HCC regardless of age and sex, and a Nur77 deficiency in mice promotes HCC development69. Furthermore, Nur77 transcriptionally upregulates lncRNA WFDC21P, which is typically reduced in HCC tissues. WFDC21P inhibits HCC cell proliferation, tumor growth, and tumor metastasis both in vitro and in vivo. WFDC21P inhibits glycolysis by simultaneously interacting with Phosphofructokinase (PFKP) and Pyruvate Kinase (PKM2), two key enzymes in glycolysis. These interactions abrogate the tetramer formation of PFKP to impede its catalytic activity and prevent the nuclear translocation of PKM2 to suppress its function as a transcriptional coactivator. Cytosporone B (Csn-B), an agonist for Nur77, can stimulate WFDC21P expression and suppress HCC in a WFDC21P-dependent manner. The Nur77-WFDC21P-PFKP/PKM2 axis has been identified as a new HCC suppressor mechanism connecting glycolytic remodeling70.
Furthermore, Nur77 inhibits β-catenin expression and activity through direct interaction, with decreased Nur77 and increased β-catenin levels observed in hepatoblastoma tissues. Treatment with Nur77 agonists not only inhibits cancer cell behavior but also enhances the effectiveness of cisplatin, suggesting potential therapeutic applications71. Beyond direct intracellular effects, Nur77 critically regulates intercellular communication in the tumor microenvironment. The intercellular communication is mediated by extracellular vesicles (EV) with TGF-β stimulating palmitoylation of hexokinase 1 (HK1) in HSCs, driving HK1 secretion via large, TSG101-dependent EV, a process demonstrated in both human LX-2 HSC cell line and mouse primary HSCs/moHSCs. HCC cells then hijack these HK1-containing EVs, accelerating glycolysis and promoting HCC progression, as confirmed in human HCC cell lines and various mouse orthotopic xenograft models. Nur77 in HSCs counters this by transcriptionally activating the depalmitoylase ABHD17B, which inhibits HK1 palmitoylation and thus reduces HK1 release, a mechanism investigated in LX-2 cells and validated through HSC-specific Nur77 knockout in mouse models. This protective Nur77 action is opposed by TGF-β-activated Akt, inducing Nur77 phosphorylation and degradation, as demonstrated in LX-2 cells. A small molecule, PDNPA, binds Nur77, creating steric hindrance to block Akt targeting and thereby preserving Nur77’s inhibition of HK1 release, ultimately inhibiting HCC progression, as shown in LX-2 cells, primary HSCs, and various mouse HCC models72.
In HCC, Nur77 expression is significantly elevated in tumor-infiltrating regulatory T cells (TI-Tregs) compared to Tregs from peripheral blood or normal tissues, and conventional CD4⁺ and CD8⁺ T cells. This enrichment in TI-Tregs contributes to an immunosuppressive tumor microenvironment73. Chronic inflammation, a hallmark of HCC, drives persistent antigenic stimulation74, thus leading to Nur77 upregulation in T cells73. The inhibition of Nur77 has been demonstrated to restore cytotoxic function and enhance tumor clearance, especially in models of chronic infection and CAR-T cell therapy73,75. Moreover, Nur77 acts as a negative regulator of IFN-γ–induced intermediate monocytes (IFN-IMo), which are known to produce IL-27 and CXCL9 and activate NK cells. In Nur77-deficient models, enhanced IFN-IMo activity leads to greater NK cell activation and reduced tumor immunosuppression, suggesting that Nur77 inhibition could promote anti-tumor immune responses76. This critical role in immune suppression positions Nur77 as a potential target for improving T cell-based immunotherapies, particularly CAR T-cell treatments, which often face challenges in solid tumors like HCC due to immune suppression. For instance, CAR T-cell therapy targeting Glypican-3 (GPC3) has demonstrated promise in HCC, with studies showing that GPC3-targeted CAR T-cells can promote tumor regression and enhance antitumor responses77. Further research is important to fully define Nur77’s complex and context-dependent roles in HCC, to balance its identified pro-tumorigenic immune functions with its beneficial tumor-suppressive actions in cancer cells. Specifically, future investigations are needed to ensure that targeting Nur77’s immunosuppressive effects in immune cells does not compromise Nur77's protective non-genomic pro-apoptotic functions or beneficial homeostatic roles.
Nur77 in liver regeneration, ischemia, and repair
Following the role of Nur77 in limiting liver inflammation, Nur77 exhibits dual context-dependent functions in liver regeneration and ischemia-reperfusion injury (IRI). During liver regeneration after two-thirds hepatectomy, Nur77 modulates regenerative responses and limits excessive hepatocyte proliferation. In Nur77 knockout mice, a dysregulated regeneration is obtained due to the acceleration in hepatocyte proliferation, and this occurs alongside significant liver injury, necrosis, elevated serum ALT, and high levels of pro-inflammatory cytokines such as IL-6, IL-12, IL-23, and CCL2, due to enhanced NF-κB and STAT3 signaling78. Nur77 expression is upregulated in in vitro hepatocyte models of hypoxia-reoxygenation, leading to suppression of the protective LKB1/AMPK signaling pathway, whereas silencing Nur77 restores AMPK activity, improving cell viability, reducing oxidative stress and inflammation, and decreasing apoptosis79. These findings are supported by in vivo studies where Nur77 knockdown reduced hepatic enzyme levels and inflammatory cytokines such as TNF-α and IL-1β after IRI. Mechanistically, Nur77 activates Cysteine-rich angiogenic inducer 61 (CYR61), which triggers NF-κB and TGF-β1 expression, worsening hepatic damage80. Taken together, these findings emphasize the context-specific role of Nur77. While it restrains uncontrolled proliferation during regeneration, it contributes to liver injury under acute ischemic stress. This complexity reflects Nur77’s cell-type-specific actions, for example, in hepatocytes, Nur77 activates injury pathways, whereas in immune cells such as macrophages, it promotes anti-inflammatory resolution. This duality is consistent with broader analyses of Nur77’s tissue- and context-dependent behavior81,82. It also aligns with its immunomodulatory effects, including Nur77’s ability to promote M2 macrophage polarization, regulate Ly6C^Lo monocyte differentiation, and suppress inflammatory signaling. Nur77’s role in Tregs further illustrates its cell-specific functions, particularly in the context of liver inflammation, as discussed earlier.
Nur77 as a therapeutic target in liver pathology
Over the last two decades, a variety of natural and chemical compounds and constructs have been identified that target Nur77 in liver disease, highlighting its significant therapeutic potential across multiple pathologies (Tables 1 and 2). The current therapeutic landscape for Nur77 largely comprises preclinical investigations, providing crucial proof-of-concept for diverse mechanisms of action.
Autophagy enhancement and lysosomal biogenesis
Recent studies demonstrated that targeting Nur77 can modulate autophagy to suppress HSC activation. A novel carbonyl-hydrazine-1-carboxamide derivative, designed to bind Nur77-LBD with reduced toxicity, was found to inhibit TGF-β1–induced HSC activation in LX-2 HSC. In vivo, in CCl₄-induced liver fibrosis mouse models, this compound markedly attenuated fibrosis by enhancing autophagic flux and promoting lysosomal biogenesis, which are essential mechanisms for HSC deactivation and dependent on Nur77 activity83. Although promising in early preclinical studies, these findings require further validation, particularly regarding the sustainability of autophagy modulation.
Metabolic reprogramming via Glutaminolysis inhibition
Enhancing Nur77 nuclear translocation may offer a therapeutic route to modulate the ERK/glutaminolysis axis in liver fibrosis. Emodin, a natural anthraquinone compound, increases Nur77 nuclear translocation in LX-2 cells (20 μM for 24 h). It achieves this by inhibiting Nur77 phosphorylation, as phosphorylation by kinases like ERK is required for Nur77’s nuclear export. In the nucleus, Nur77 interacts with DNA Methyltransferase 3 Beta (DNMT3b), which leads to increased methylation of the glutaminase 1 (GLS1) promoter and a subsequent blockade of glutaminolysis. In vivo, emodin–vitamin A liposome treatment (10 mg·kg−1, via tail vein) reduced fibrosis in a CCl₄-induced mouse model by interrupting this ERK/Nur77/glutaminolysis positive feedback loop. This mechanism ultimately leads to HSC senescence and alleviates liver fibrosis84. Although this mechanism offers promising anti-fibrotic effects, enhancing Nur77 nuclear localization and activity, raises questions about broader physiological implications. Given Nur77’s diverse role in cellular functions and its regulation of various gene programs, sustained modulation of its nuclear presence and epigenetic activity could influence parallel pathways. Further studies are needed to assess these wider effects and ensure long-term safety and specificity.
Inflammatory pathway suppression
Nur77’s anti-inflammatory actions also contribute to its anti-fibrotic potential. Mechanistically, Nur77 reduces inflammation by inhibiting NF-κB signaling and promoting M2 macrophage polarization, as shown in the section Nur77’s Role in Mitigating Liver Inflammation. Building on this mechanistic background, the therapeutic interventions that activate Nur77 suggest promising therapeutic potential. For example, Ginsenoside Rc activates Nur77 in both cultured rat HSCs and CCl₄-injured mice (10 mg/kg or 20 mg/kg). This activation suppressed the TLR4–MyD88 signaling pathway, reducing hepatic inflammation and fibrosis85. Many compounds target the TLR4–MyD88 signaling pathway in liver fibrosis86, although Ginsenoside Rc represents a notable compound uniquely reported to activate Nur77, marking a distinct Nur77-dependent anti-inflammatory strategy in this context85. Similarly, Nodakenin protects against alcoholic liver disease (ALD) through Nur77 activation, which suppresses the P2X7r signaling pathway, which reduces liver injury, inflammation, and lipid accumulation in ALD mouse models. Nodakenin treatment (10 mL/kg) reduced liver injury, inflammation, and lipid accumulation. Nur77 activation downregulated P2X7r, NLRP3, cleaved caspase-1, IL-1β, IL-6, IL-23, Lipin-1, and SREBP1. These effects were Nur77-dependent, as silencing Nur77 eliminated Nodakenin’s benefits87. Both Ginsenoside Rc and Nodakenin exhibit anti-fibrotic and anti-inflammatory effects via Nur77 activation, targeting the TLR4–MyD88 and P2X7r–NLRP3 pathways, respectively. In conditions like ALD and MASH, where both Pathogen-Associated Molecular Patterns (PAMP) and Damage-Associated Molecular Patterns (DAMP) signals are co-activated, targeting a single pathway may be insufficient. This underscores Nur77’s potential as a dual regulator, suggesting that combining such strategies may offer more effective control of inflammation. However, the broader immunological effects of sustained Nur77 activation, especially on T cells and NK cells, remain unclear and require further investigation. As previously discussed, sustained Nur77 activation may have detrimental effects on immune subsets such as Tregs and NK cells, which are critical in liver immune regulation.
Modulation of MAPK Signaling
Spinosin, a natural flavonoid-C-glycoside, interacts with Nur77 at residues R515 and R563. Spinosin has demonstrated to deactivate HSCs and, as a consequence, improves liver function in fibrotic mice by modulating the Nur77–ASK1–p38 MAPK axis, reducing collagen deposition and inflammatory cytokines such as IL-6 and IL-1β. Although the molecular targeting is well characterized, challenges remain in validating its specificity and evaluating potential off-target effects88.
Post-translation modification and macrophages reprogramming
Post-translational modifications (PTM) are crucial for regulating Nur77’s function. Phosphorylation is commonly linked to Nur77 inactivation by facilitating its export from the nucleus to the cytoplasm30. Studies in HSCs revealed that TGF-β1 stimulation activates the PI3K/Akt pathway, leading to Akt-dependent phosphorylation of Nur77 and its nuclear export, thereby impairing its antifibrotic function64. Akt primarily mediates Nur77 phosphorylation in PNPLA3-SNP wild-type HSCs, while Erk is involved in PNPLA3-1148M HSCs. Notably, this indicates that phosphorylation mechanisms can vary depending on the cellular context64. In addition to phosphorylation, acetylation by p300 stabilizes Nur77, while Histone Deacetylase 1 (HDAC1)-mediated deacetylation promotes its degradation, as demonstrated in hepatic IRI models89. Further, in MASH, modulating Nur77 through phosphorylation has emerged as a promising anti-fibrotic strategy. A study identified SH3 Domain Binding Kinase 1(SBK1) as a kinase that phosphorylates Nur77 at serine 344, resulting in increased FGF21 production and suppression of lipid anabolism in hepatocytes. This SBK1–Nur77–FGF21 axis was demonstrated in vitro using AML12 mouse liver cells and validated in vivo in high-fat diet–fed mice, where SBK1 overexpression enhanced FGF21 expression and improved hepatic lipid metabolism45. As phosphorylation represents a post-translational modification, these findings support the broader therapeutic strategy of modulating Nur77 through PTMs. Nonetheless, further studies are needed to define optimal dosing, validate biomarkers, and evaluate long-term safety in humans.
Moreover, recent preclinical research demonstrated that Nur77 acts as a molecular switch, orchestrating the transformation of macrophages from a pro-fibrotic to an anti-fibrotic state. This was shown in two in vivo MASH-fibrosis models: the Gubra Amylin NASH (GAN)-ob/ob model and a Western diet (WD)-CCl4 model. Treatment with PsTag-FGF21, a fusion protein containing a long-acting label with human FGF21, (2.42 mg·kg−1 PsTag-FGF21 twice a week via i.p. injection) reduced liver fibrosis in these models by promoting Nur77’s nuclear translocation in macrophages. Once in the nucleus, Nur77 binds to the insulin-like growth factor 1 (IGF-1) promoter and upregulates IGF-1 expression. PsTag-FGF21 administration significantly decreased the in vivo expression of HSCs activation markers. The anti-fibrotic effect of PsTag-FGF21 was primarily mediated by macrophages, which regulated the phenotypic switch from Ly6C^hi to Ly6C^lo cells, thereby inhibiting HSC activation. Nur77 was identified as a key factor underlying this PsTag-FGF21-induced Ly6C phenotypic switch and macrophage–HSC crosstalk. The study further identified Nur77’s N-Terminal intrinsically disordered region (IDR) as the key domain responsible for its function in this pathway, demonstrating that directly delivering Nur77’s IDR into the nucleus could mimic the anti-fibrotic effect of PsTag-FGF21, suggesting a promising therapeutic strategy for MASH90. The study provides strong preclinical support for PsTag-FGF21 and Nur77’s IDR in treating MASH-related fibrosis but lacks toxicity and long-term safety data. Using two in vivo models and multiple methods adds strength. The use of Nur77’s IDR is a novel approach, yet further research is needed to assess safety and potential immune responses. Together, these studies demonstrate the therapeutic potential of modulating Nur77 through distinct post-translational mechanisms, including phosphorylation and IDR-based strategies, which may offer novel ways to reprogram liver immune responses and reduce fibrosis progression in MASH.
Nur77 as a target for apoptosis and cancer therapy
As previously discussed, Nur77 exerts non-genomic functions, including its translocation from the nucleus to the mitochondria, where it interacts with Bcl-2 to induce apoptosis. This mechanism has drawn significant interest in therapeutic exploitation, particularly in cancer. Among the early pharmacological approaches, Csn-B, a natural Nur77 agonist isolated from the endophytic fungus Dothiorella sp. HTF3, demonstrated high binding affinity (IC₅₀ = 0.278 nM) to Nur77’s LBD and was shown to activate its transactivation function, thus triggering apoptosis through the Nur77–Bcl-2–Bax axis91. Further studies demonstrated Nur77’s mitochondrial translocation as a key step in apoptosis induction. For example, Cryptomeridiol (Bkh126), a compound derived from Magnolia officinalis, upregulates Nur77 expression and triggers ER stress-induced apoptosis in HCC cells. It promotes Nur77’s mitochondrial translocation, where it interacts with Bcl-2 and activates Bax, leading to cancer cell death, both in vitro and in vivo92 In a broader analysis of Nur77 modulators, as shown in Table 2, compounds like N-Butylidenephthalide and Bkh126 have been shown to induce Nur77-dependent apoptosis via mitochondrial targeting, triggering cytochrome-c release and caspase-3 activation, and are considered promising compounds to enhance treatment outcomes in HCC89,90. The compound 5-((4-(pyridin-3-yl)pyrimidin-2-yl) amino)-1H-indole-2-carboxamide derivatives (8b) also demonstrates good potency against various liver cancer cell lines with lower toxicity compared to the known Nur77 ligand, celastrol, suggesting a significant therapeutic advantage in developing safer agents. While these findings suggest that compound 8b functions as a selective Nur77 modulator with limited impact on RXRα and PPARγ94, a more comprehensive evaluation across a wider panel of NRs would be valuable to fully confirm its selectivity and reduce the risk of potential off-target effects. In contrast, Chromodomain-helicase-DNA-binding protein 1-like (CHD1L) directly binds Nur77 and retains Nur77 in the nucleus, thereby blocking its mitochondrial translocation and apoptotic function, specifically by blocking the release of cytochrome c into the cytoplasm and inhibiting the activity of caspase 3 and 9. This ultimately promoting tumor survival in HCC95. This critical interaction was further investigated by studies that demonstrated CHD1L silencing inhibits gastric cancer cell proliferation, invasion, and migration, and induces apoptosis. Targeting the CHD1L–Nur77 interaction may thus represent a therapeutic strategy to restore Nur77 translocation and enhance apoptosis in cancer cells96. However, studies exploring this therapeutic implication of the CHD1L–Nur77 interaction in the context of HCC remain limited. Additionally, the Nur77-derived peptide NuBCP-9 has shown promising anti-cancer effects by converting Bcl-2 from an anti-apoptotic into a pro-apoptotic protein. This novel mechanism selectively induces apoptosis in cancer cells, emphasizing its potential for therapeutic application. Similarly, compound 10 g 5-((8-methoxy-2- methylquinolin-4-yl)amino)-1H-indole-2-carbohydrazide derivatives binds to Nur77 and promotes its translocation to both the ER and mitochondria. This action induces Nur77-dependent ER stress and autophagy, which ultimately leads to apoptosis via caspase-3/9 activation. In vivo studies further confirmed that 10 g reduces tumor size and weight with good tolerability, marking it as a promising candidate for HCC therapy97. There is a growing interest in targeting Nur77’s non-genomic apoptotic functions as a potential therapeutic approach in HCC, as shown in Table 2; however, these strategies remain at the preclinical stage.
PROTAC-mediated Nur77 degradation
A significant advance in Nur77-targeted cancer therapy is the development of NR-V04, a proteolysis-targeting chimera (PROTAC) designed to degrade Nur77. NR-V04 promotes its selective degradation, disrupting the immunosuppressive tumor microenvironment. This results in enhanced infiltration of effector immune cells such as B cells and memory CD8⁺ T cells, while reducing myeloid-derived suppressor cells (MDSCs), thereby improving anti-tumor immunity. NR-V04 PROTAC presents a promising therapeutic avenue as demonstrated in in vivo models of colon cancer and melanoma cancer in in vivo wild-type (WT) or NR4A1−/− (KO) mice. Although NR-V04 has not yet been evaluated in liver disease models, Nur77 expression is significantly elevated in tumor-infiltrating Tregs in HCC compared with blood or normal tissues98, raising the possibility that targeting Nur77 could modulate the immunosuppressive microenvironment in liver cancer. While it effectively demonstrates Nur77’s pro-tumorigenic role in immune evasion, this contrasts with Nur77’s established tumor-suppressive functions in HCC by modulating glycolysis, and its role in alleviating inflammation50, and fibrogenesis67. Therefore, a key concern is whether Nur77 degradation could eliminate these beneficial roles when using NR-V04 PROTAC in liver fibrosis and cancer. Also, as a part of its non-genomic pro-apoptotic function, Nur77 translocates to mitochondria and ER to promote apoptosis92 or autophagy52. Hence, systemic degradation by PROTACs would block this pathway and impair a key tumor-suppressive mechanism. Furthermore, Nur77’s diverse, cell-type-specific functions, such as promoting anti-inflammatory M2 macrophage polarization51 for its role in Ly6C^Lo monocytes for liver repair56, raise questions about disrupting Nur77’s beneficial homeostatic roles. Despite an excellent safety profile in mice and its selectivity among the NR4A family, the systemic degradation of a pleiotropic protein like Nur77, given its celastrol known toxicities in the liver99, necessitates cautious evaluation for off-target effects in human translation100. The clinical application of NR-V04 PROTAC in liver diseases needs research.
Conclusions
NRs are a family of transcription factors that play important roles in the regulation of many aspects of development, and control important hepatic functions in liver physiology and pathophysiology. In this review, we specifically focused on Nur77, one of the NR orphan receptors with its genomic and non-genomic activities, and demonstrated diverse and context-dependent key roles in liver diseases. Novel concepts related to Nur77’s role in liver physiology and pathophysiology have been explored and are in progress to be successfully integrated into future effective therapies. As an example, a recently developed PROteolysis Targeting Chimaera-mediated degradation of Nur77 (NR-V04), which might prove of primary interest to suppress pro-oncogenic Nur77 activities. Or using a fusion protein containing a long-acting label, such as PsTag with human FGF21 targeting hepatocytes and macrophages, leading to reduced steatosis and fibrosis, possibly in combination with Resmetirom® an agonist that targets the dominant THR-β receptor isoform in hepatocytes. Furthermore, the recent developments of various multi-omics and spatial biology approaches will empower our knowledge of Nur77’s activity.
Data availability
No datasets were generated or analyzed during the current study.
References
Evans, R. M. & Mangelsdorf, D. J. Nuclear receptors, RXR, and the Big Bang. Cell 157, 255–266 (2014).
Mangelsdorf, D. J. & Evans, R. M. The RXR heterodimers and orphan receptors. Cell 83, 841–850 (1995).
Gustafsson, J.-A. Historical overview of nuclear receptors. J. Steroid Biochem. Mol. Biol. 157, 3–6 (2016).
Levin, E. R. & Hammes, S. R. Nuclear receptors outside the nucleus: extranuclear signalling by steroid receptors. Nat. Rev. Mol. cell Biol. 17, 783–797 (2016).
Burris, T. P. et al. International union of basic and clinical pharmacology CXIII: Nuclear receptor superfamily—Update 2023. Pharmacol. Rev. 75, 1233–1318 (2023).
Tao, L. J., Seo, D. E., Jackson, B., Ivanova, N. B. & Santori, F. R. Nuclear Hormone Receptors and Their Ligands: Metabolites in Control of Transcription. Cells 9, 2606 (2020).
Brélivet, Y., Rochel, N. & Moras, D. Structural analysis of nuclear receptors: from isolated domains to integral proteins. Mol. Cell. Endocrinol. 348, 466–473 (2012).
Dubois, V., Lefebvre, P., Staels, B. & Eeckhoute, J. Nuclear receptors: pathophysiological mechanisms and drug targets in liver disease. Gut 73, 1562–1569 (2024).
Wagner, M., Zollner, G. & Trauner, M. Nuclear receptors in liver disease. Hepatology 53, 1023–1034 (2011).
Younossi, Z. M. et al. Health-Related quality of life (HRQL) assessments in a 52-Week, Double-Blind, randomized, Placebo-Controlled phase 3 study of resmetirom (MGL-3196) in patients with metabolic dysfunction associated steatohepatitis (MASH) and fibrosis. Hepatology, 10.1097 (2024).
Crouchet, E. et al. Targeting the liver clock improves fibrosis by restoring TGF-β signaling. J. Hepatol. 82, 120–133 (2025).
Younossi, Z. M. et al. Obeticholic acid for the treatment of non-alcoholic steatohepatitis: interim analysis from a multicentre, randomised, placebo-controlled phase 3 trial. Lancet 394, 2184–2196 (2019).
Harrison, S. A. et al. A phase 3, randomized, controlled trial of resmetirom in NASH with liver fibrosis. N. Engl. J. Med. 390, 497–509 (2024).
Francque, S. M. et al. A randomized, controlled trial of the pan-PPAR agonist lanifibranor in NASH. N. Engl. J. Med. 385, 1547–1558 (2021).
Sabatini, S. & Gastaldelli, A. Metabolic effects and mechanism of action of the pan-PPAR agonist lanifibranor. J. Hepatol. 82, 950–952 (2025).
Kleberg, J. et al. Targeting Lineage-Specific Functions of NR4A1 for Cancer Immunotherapy. Int. J. Mol. Sci. 26, 5266 (2025).
Wang, Y., Li, N., Guan, W. & Wang, D. Controversy and multiple roles of the solitary nucleus receptor Nur77 in disease and physiology. FASEB J. 39, e70468 (2025).
de Vera, I. M. S. Vol. 1 134-137 (ACS Publications, 2018).
Zollner, G. & Trauner, M. Nuclear receptors as therapeutic targets in cholestatic liver diseases. Br. J. Pharmacol. 156, 7–27 (2009).
Maruyama, K. et al. The NGFI-B subfamily of the nuclear receptor superfamily. Int. J. Oncol. 12, 1237–1280 (1998).
Xu, Y. et al. Knockout of Nur77 leads to amino acid, lipid, and glucose metabolism disorders in zebrafish. Front. Endocrinol. 13, 864631 (2022).
Xie, P. et al. Emodin protects against lipopolysaccharide-induced acute lung injury via the JNK/Nur77/c-Jun signaling pathway. Front. Pharmacol. 13, 717271 (2022).
Birari, P. et al. Nur77 influences immunometabolism to regulate the release of proinflammatory cytokines and the formation of lipid bodies during Mycobacterium tuberculosis infection of macrophages. Pathog. Dis. 81, ftad033 (2023).
Chawnshang, C., Kokontis, J., Shutsung, L. & Yijan, C. Isolation and characterization of human TR3 receptor: a member of steroid receptor superfamily. J. Steroid Biochem. 34, 391–395 (1989).
Wu, L. & Chen, L. Characteristics of Nur77 and its ligands as potential anticancer compounds. Mol. Med. Rep. 18, 4793–4801 (2018).
Eells, J., Witta, J., Otridge, J., Zuffova, E. & Nikodem, V. Structure and function of the Nur77 receptor subfamily, a unique class of hormone nuclear receptors. Curr. Genom.1, 135–152 (2000).
Wansa, K. S. A., Harris, J. M. & Muscat, G. E. The activation function-1 domain of Nur77/NR4A1 mediates trans-activation, cell specificity, and coactivator recruitment. J. Biol. Chem. 277, 33001–33011 (2002).
Flaig, R., Greschik, H., Peluso-Iltis, C. & Moras, D. Structural basis for the cell-specific activities of the NGFI-B and the Nurr1 ligand-binding domain. J. Biol. Chem. 280, 19250–19258 (2005).
Li, H. et al. Cytochrome c release and apoptosis induced by mitochondrial targeting of nuclear orphan receptor TR3. Science 289, 1159–1164 (2000).
Pekarsky, Y. et al. Akt phosphorylates and regulates the orphan nuclear receptor Nur77. Proc. Natl. Acad. Sci. 98, 3690–3694 (2001).
Safe, S. et al. Nuclear receptor 4A (NR4A) family–orphans no more. J. Steroid Biochem. Mol. Biol. 157, 48–60 (2016).
Hedrick, E., Li, X. & Safe, S. Penfluridol represses integrin expression in breast cancer through induction of reactive oxygen species and downregulation of Sp transcription factors. Mol. Cancer Ther.16, 205–216 (2017).
Upadhyay, S. et al. Bis-Indole derivatives as dual nuclear receptor 4A1 (NR4A1) and NR4A2 ligands. Biomolecules 14, 284 (2024).
Cheng, Z. et al. Mitochondrial translocation of Nur77 mediates cardiomyocyte apoptosis. Eur. Heart J. 32, 2179–2188 (2011).
Lin, B. et al. Conversion of Bcl-2 from protector to killer by interaction with nuclear orphan receptor Nur77/TR3. Cell 116, 527–540 (2004).
Liang, B. et al. Involvement of TR3/Nur77 translocation to the endoplasmic reticulum in ER stress-induced apoptosis. Exp. Cell Res. 313, 2833–2844 (2007).
Chen, H. -z, Wen, Q., Wang, W. -j, He, J. -p & Wu, Q. The orphan nuclear receptor TR3/Nur77 regulates ER stress and induces apoptosis via interaction with TRAP. Int. J. Biochem. Cell Biol. 45, 1600–1609 (2013).
Liu, J. et al. Modulation of the Nur77-Bcl-2 apoptotic pathway by p38α MAPK. Oncotarget 8, 69731 (2017).
Zhan, Y. -y et al. The orphan nuclear receptor Nur77 regulates LKB1 localization and activates AMPK. Nat. Chem. Biol. 8, 897–904 (2012).
Wang, R. H. et al. The orphan receptor TR3 participates in angiotensin II-induced cardiac hypertrophy by controlling mTOR signalling. EMBO Mol. Med. 5, 137–148 (2013).
Zhang, Y. et al. Targeting nuclear receptor NR4A1–dependent adipocyte progenitor quiescence promotes metabolic adaptation to obesity. J. Clin. Investig. 128, 4898–4911 (2018).
Jung, Y.-S. et al. Dual targeting of Nur77 and AMPKα by isoalantolactone inhibits adipogenesis in vitro and decreases body fat mass in vivo. Int. J. Obes. 43, 952–962 (2019).
Pei, L. et al. NR4A orphan nuclear receptors are transcriptional regulators of hepatic glucose metabolism. Nat. Med. 12, 1048–1055 (2006).
Chao, L. C. et al. Insulin resistance and altered systemic glucose metabolism in mice lacking Nur77. Diabetes 58, 2788–2796 (2009).
Ahuja, P. et al. Src homology 3 domain binding kinase 1 protects against hepatic steatosis and insulin resistance through the Nur77–FGF21 pathway. Hepatology 77, 213–229 (2023).
Liang, H. et al. Orphan nuclear receptor NR4A1 suppresses hyperhomocysteinemia-induced hepatic steatosis in vitro and in vivo. FEBS Lett. 593, 1061–1071 (2019).
Pols, T. W. et al. Nur77 modulates hepatic lipid metabolism through suppression of SREBP1c activity. Biochem Biophys. Res Commun. 366, 910–916, https://doi.org/10.1016/j.bbrc.2007.12.039 (2008).
Zhang, P., Hu, Y., Yang, J., Zheng, L. & Wang, Q. The orphan nuclear receptor Nur77 regulates hepatic cholesterol metabolism through the suppression of LDLR and HMGCR expression. Mol. Med. Rep. 5, 1541–1547 (2012).
Rodríguez-Calvo, R., Tajes, M. & Vázquez-Carrera, M. The NR4A subfamily of nuclear receptors: potential new therapeutic targets for the treatment of inflammatory diseases. Expert Opin. Ther. targets 21, 291–304 (2017).
You, B., Jiang, Y.-Y., Chen, S., Yan, G. & Sun, J. The orphan nuclear receptor Nur77 suppresses endothelial cell activation through induction of IκBα expression. Circ. Res. 104, 742–749 (2009).
Lith, S. C. et al. Nuclear receptor Nur77 regulates immunomechanics of macrophages. Eur. J. Cell Biol. 103, 151419 (2024).
Hu, M. et al. Celastrol-induced Nur77 interaction with TRAF2 alleviates inflammation by promoting mitochondrial ubiquitination and autophagy. Mol. cell 66, 141–153. e146 (2017).
Sun, B., Zhang, R., Liang, Z., Fan, A. & Kang, D. Hyperoside attenuates non-alcoholic fatty liver disease through targeting Nr4A1 in macrophages. Int. Immunopharmacol. 94, 107438 (2021).
Fang, H. et al. Lipidome remodeling activities of DPA-EA in palmitic acid-stimulated HepG2 cells and the in vivo anti-obesity effect of the DPA-EA and DHA-EA mixture prepared from algae oil. Front. Pharmacol. 14, 1146276 (2023).
Deng, J. -w et al. Early use of dexamethasone increases Nr4a1 in Kupffer cells ameliorating acute liver failure in mice in a glucocorticoid receptor-dependent manner. J. Zhejiang Univ. Sci. B 21, 727 (2020).
Hanna, R. N. et al. The transcription factor NR4A1 (Nur77) controls bone marrow differentiation and the survival of Ly6C− monocytes. Nat. Immunol. 12, 778–785 (2011).
Aki, D. et al. The Nr4a family regulates intrahepatic Treg proliferation and liver fibrosis in MASLD models. J. Clin. Investig. 134, e175305 (2024).
Trivedi, P., Wang, S. & Friedman, S. L. The power of plasticity—metabolic regulation of hepatic stellate cells. Cell Metab. 33, 242–257 (2021).
Wang, X.-L., Yang, M. & Wang, Y. Roles of transforming growth factor-β signaling in liver disease. World J. Hepatol. 16, 973 (2024).
Huang, Q. et al. NR4A1 inhibits the epithelial–mesenchymal transition of hepatic stellate cells: Involvement of TGF-β–Smad2/3/4–ZEB signaling. Open Life Sci. 17, 447–454 (2022).
Palumbo-Zerr, K. et al. Orphan nuclear receptor NR4A1 regulates transforming growth factor-β signaling and fibrosis. Nat. Med. 21, 150–158 (2015).
Gao, L. et al. The roles of orphan nuclear receptor 4 group A1 and A2 in fibrosis. Int. Immunopharmacol. 139, 112705 (2024).
Martins, M. C. et al. Role of PNPLA3 in hepatic stellate cells and hepatic cellular crosstalk. Liver Int. 45, e16117 (2025).
Caon, E. et al. Exploring the impact of the PNPLA3 I148M variant on primary human hepatic stellate cells using 3D extracellular matrix models. J. Hepatol. 80, 941–956 (2024).
Yin, M. et al. Exosomes from hepatitis B virus-infected hepatocytes activate hepatic stellate cells and aggravate liver fibrosis through the miR-506-3p/Nur77 pathway. J. Biochem. Mol. Toxicol. 37, e23432 (2023).
Fuchs, C. D. et al. GLP-2 improves hepatic inflammation and fibrosis in Mdr2-/-mice via activation of NR4a1/Nur77 in hepatic stellate cells and intestinal FXR Signaling. Cell. Mol. Gastroenterol. Hepatol. 16, 847–856 (2023).
Zhai, X. et al. Oxytocin alleviates liver fibrosis via hepatic macrophages. JHEP Rep. 6, 101032 (2024).
Sánchez, A. & Fabregat, I. Growth factor-and cytokine-driven pathways governing liver stemness and differentiation. World J. Gastroenterol.16, 5148 (2010).
Bian, X. -l et al. Nur77 suppresses hepatocellular carcinoma via switching glucose metabolism toward gluconeogenesis through attenuating phosphoenolpyruvate carboxykinase sumoylation. Nat. Commun. 8, 14420 (2017).
Guan, Y. -f et al. Nur77-activated lncRNA WFDC21P attenuates hepatocarcinogenesis via modulating glycolysis. Oncogene 39, 2408–2423 (2020).
Zhou, J. et al. Nur77 inhibition of β-catenin expression mediates Hepatoblastoma progression and enhances cisplatin’s therapeutic effect. Gene 908, 148292 (2024).
Chen, Q. -t et al. HK1 from hepatic stellate cell–derived extracellular vesicles promotes progression of hepatocellular carcinoma. Nat. Metab. 4, 1306–1321 (2022).
Lith, S. C., van Os, B. W., Seijkens, T. T. & de Vries, C. J. ‘Nur’turing tumor T cell tolerance and exhaustion: novel function for Nuclear Receptor Nur77 in immunity. Eur. J. Immunol. 50, 1643–1652 (2020).
Refolo, M. G., Messa, C., Guerra, V., Carr, B. I. & D’Alessandro, R. Inflammatory mechanisms of HCC development. Cancers 12, 641 (2020).
Liu, X. et al. Genome-wide analysis identifies NR4A1 as a key mediator of T cell dysfunction. Nature 567, 525–529 (2019).
Wang, R. et al. Intermediate monocytes induced by IFN-γ inhibit cancer metastasis by promoting NK cell activation through FOXO1 and interleukin-27. J. ImmunoTher. Cancer 10, e003539 (2022).
Marofi, F. et al. CAR T cells in solid tumors: challenges and opportunities. Stem Cell Res. Ther. 12, 1–16 (2021).
Hu, Y. et al. Accelerated Partial Hepatectomy–induced liver cell proliferation is associated with liver injury in Nur77 knockout mice. Am. J. Pathol. 184, 3272–3283 (2014).
Zheng, Y., Tao, Y., Zhan, X. & Wu, Q. Nuclear receptor 4A1 (NR4A1) silencing protects hepatocyte against hypoxia-reperfusion injury in vitro by activating liver kinase B1 (LKB1)/AMP-activated protein kinase (AMPK) signaling. Bioengineered 13, 8349–8359 (2022).
Cao, J. et al. NR4A1 knockdown confers hepatoprotection against ischaemia-reperfusion injury by suppressing TGFβ1 via inhibition of CYR61/NF-κB in mouse hepatocytes. J. Cell Mol. Med. 25, 5099–5112 (2021).
Pei, L., Castrillo, A., Chen, M., Hoffmann, A. & Tontonoz, P. Induction of NR4A orphan nuclear receptor expression in macrophages in response to inflammatory stimuli. J. Biol. Chem. 280, 29256–29262 (2005).
Safe, S., Jin, U.-H., Hedrick, E., Reeder, A. & Lee, S.-O. Minireview: role of orphan nuclear receptors in cancer and potential as drug targets. Mol. Endocrinol. 28, 157–172 (2014).
Hu, H. et al. Design, synthesis, and biological evaluation of carbonyl-hydrazine-1-carboxamide derivatives as anti-hepatic fibrosis agents targeting Nur77. Bioorg. Chem. 140, 106795 (2023).
Chen, L. et al. Emodin promotes hepatic stellate cell senescence and alleviates liver fibrosis via a nuclear receptor (Nur77)-mediated epigenetic regulation of glutaminase 1. Br. J. Pharmacol. 180, 2577–2598 (2023).
Qin, B.-F. et al. Regulation of Nur77-TLR4/MyD88 signaling pathway is required for Ginsenoside Rc ameliorates hepatic fibrosis regression by deactivating hepatic stellate cells. Acta Histochem.125, 152079 (2023).
Ren, G., Bai, C., Yi, S., Cong, Q. & Zhu, Y. Mechanisms and therapeutic strategies for MAFLD targeting TLR4 signaling pathways. J. Innate Immun. 16, 45–55 (2024).
Song, J. et al. Regulation of the Nur77-P2X7r signaling pathway by Nodakenin: A potential protective function against alcoholic liver disease. Molecules 29, 1078 (2024).
Lin, G. et al. Spinosin inhibits activated hepatic stellate cell to attenuate liver fibrosis by targeting Nur77/ASK1/p38 MAPK signaling pathway. Eur. J. Pharmacol. 966, 176270 (2024).
Kang, S.-A. et al. Regulation of Nur77 protein turnover through acetylation and deacetylation induced by p300 and HDAC1. Biochemical Pharmacol. 80, 867–873 (2010).
Ji, Y. et al. A long-acting FGF21 attenuates metabolic dysfunction-associated steatohepatitis-related fibrosis by modulating NR4A1-mediated Ly6C phenotypic switch in macrophages. Br. J. Pharm. 181, 2923–2946 (2024).
Zhan, Y. et al. Cytosporone B is an agonist for nuclear orphan receptor Nur77. Nat. Chem. Biol. 4, 548–556 (2008).
Li, X. et al. Orphan nuclear receptor Nur77 mediates the lethal endoplasmic reticulum stress and therapeutic efficacy of cryptomeridiol in hepatocellular carcinoma. Cells 11, 3870 (2022).
Chen, Y.-L. et al. The induction of orphan nuclear receptor Nur77 expression by n-butylenephthalide as pharmaceuticals on hepatocellular carcinoma cell therapy. Mol. Pharmacol. 74, 1046–1058 (2008).
Qin, J. et al. Discovery of 5-((4-(pyridin-3-yl) pyrimidin-2-yl) amino)-1H-indole-2-carboxamide derivatives as novel anti-cancer agents targeting Nur77. Eur. J. Med. Chem. 244, 114849 (2022).
Chen, L. et al. Chromodomain helicase/adenosine triphosphatase DNA binding protein 1–like (CHD1l) gene suppresses the nucleus-to-mitochondria translocation of nur77 to sustain hepatocellular carcinoma cell survival. Hepatology 50, 122–129 (2009).
Li, D. et al. Chromodomain-helicase-DNA-binding protein 1-like (CHD1L) silencing inhibits gastric cancer cell proliferation, invasion, and migration. Transl. Cancer Res. 9, 6660 (2020).
Li, B. et al. Design, synthesis, and biological evaluation of 5-((8-methoxy-2-methylquinolin-4-yl) amino)-1H-indole-2-carbohydrazide derivatives as novel Nur77 modulators. Eur. J. Med. Chem. 204, 112608 (2020).
Wang, L. et al. PROTAC-mediated NR4A1 degradation as a novel strategy for cancer immunotherapy. J. Exp. Med. 221, https://doi.org/10.1084/jem.20231519 (2024).
Wu, M. et al. Celastrol aggravates LPS-induced inflammation and injuries of liver and kidney in mice. Am. J. Transl. Res. 10, 2078 (2018).
Jin, C., Wu, Z., Wang, L., Kanai, Y. & He, X. CYP450s-activity relations of celastrol to interact with triptolide reveal the reasons of hepatotoxicity of Tripterygium wilfordii. Molecules 24, 2162 (2019).
Kersten, S. et al. Peroxisome proliferator-activated receptor alpha mediates the adaptive response to fasting. J. Clin. Invest 103, 1489–1498 (1999).
Ducheix, S., Montagner, A., Theodorou, V., Ferrier, L. & Guillou, H. The liver X receptor: a master regulator of the gut–liver axis and a target for non alcoholic fatty liver disease. Biochem. Pharmacol. 86, 96–105 (2013).
Xu, Y. et al. A metabolic stress-inducible miR-34a-HNF4α pathway regulates lipid and lipoprotein metabolism. Nat. Commun. 6, 7466 (2015).
Clifford, B. L. et al. FXR activation protects against NAFLD via bile-acid-dependent reductions in lipid absorption. Cell Metab. 33, 1671–1684. e1674 (2021).
Stofan, M. & Guo, G. L. Bile acids and FXR: novel targets for liver diseases. Front. Med. 7, 544 (2020).
Kliewer, S. A. & Willson, T. M. Regulation of xenobiotic and bile acid metabolism by the nuclear pregnane X receptor. J. Lipid Res. 43, 359–364 (2002).
Makishima, M. et al. Vitamin D receptor as an intestinal bile acid sensor. Science 296, 1313–1316 (2002).
Cai, X., Young, G. M. & Xie, W. The xenobiotic receptors PXR and CAR in liver physiology, an update. Biochim. Biophys Acta (BBA)-Mol. Basis Dis. 1867, 166101 (2021).
Lalloyer, F. et al. Peroxisome proliferator–activated receptor-α gene level differently affects lipid metabolism and inflammation in apolipoprotein E2 knock-in mice. Arterioscler. Thromb. Vasc. Biol. 31, 1573–1579 (2011).
Endo-Umeda, K. et al. Liver X receptors regulate hepatic F4/80+ CD11b+ Kupffer cells/macrophages and innate immune responses in mice. Sci. Rep. 8, 1–14 (2018).
Wang, Y. D. et al. Farnesoid X receptor antagonizes nuclear factor κB in hepatic inflammatory response. Hepatology 48, 1632–1643 (2008).
Dong, B. et al. Vitamin D receptor activation in liver macrophages ameliorates hepatic inflammation, steatosis, and insulin resistance in mice. Hepatology 71, 1559–1574 (2020).
Hazra, S., Miyahara, T., Rippe, R. A. & Tsukamoto, H. PPAR gamma and hepatic stellate cells. Comp. Hepatol. 3, S7 (2004).
Abramovitch, S. et al. Vitamin D inhibits development of liver fibrosis in an animal model but cannot ameliorate established cirrhosis. 308, G112–G120 (2015).
Bolkenius, U. et al. Glucocorticoids decrease the bioavailability of TGF-β which leads to a reduced TGF-β signaling in hepatic stellate cells. Biochem. Biophys. Res. Commun. 325, 1264–1270 (2004).
Chen, P. et al. Orphan nuclear receptor NR4A2 inhibits hepatic stellate cell proliferation through MAPK pathway in liver fibrosis. PeerJ 3, e1518 (2015).
Su, H. et al. Downregulation of nuclear receptor FXR is associated with multiple malignant clinicopathological characteristics in human hepatocellular carcinoma. 303, G1245–G1253 (2012).
Zhao, Y., Tan, H., Zhang, X. & Zhu, J. Roles of peroxisome proliferator-activated receptors in hepatocellular carcinoma. J. Cell. Mol. Med. 28, e18042 (2024).
Liu, Q. et al. Targeting RORγ inhibits the growth and metastasis of hepatocellular carcinoma. Mol. Ther. 32, 749–765 (2024).
Kurakula, K., Koenis, D. S., van Tiel, C. M. & de Vries, C. J. NR4A nuclear receptors are orphans but not lonesome. Biochim. Biophys. Acta (BBA)-Mol. Cell Res. 1843, 2543–2555 (2014).
Pawlak, M., Lefebvre, P. & Staels, B. General molecular biology and architecture of nuclear receptors. Curr. Top. Med Chem. 12, 486–504 (2012).
Acknowledgements
No funding was granted.
Author information
Authors and Affiliations
Contributions
O.S.: drafting of the manuscript; critical revision of the manuscript for important intellectual content. K.R.: Conceptualization, drafting of the manuscript; critical revision of the manuscript for important intellectual content, supervision. All authors reviewed the manuscript.
Corresponding author
Ethics declarations
Competing interests
Ola Alsharif has no conflicts of interest relevant to this article to disclose. Krista Rombouts owns shares or receives stock options in Engitix Therapeutics Ltd. and receives consultancies from Engitix Therapeutics Ltd.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Alsharif, O., Rombouts, K. Mechanisms of orphan nuclear receptor Nur77 in liver health, disease, and therapeutic potential: narrative review. npj Gut Liver 2, 24 (2025). https://doi.org/10.1038/s44355-025-00037-9
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s44355-025-00037-9
