
Abstract
The proliferation of publicly available single-cell RNA sequencing (scRNA-seq) data has created significant opportunities in biomedical research. However, the reuse of these resources is constrained by a series of preparatory steps, including metadata extraction from primary literature, retrieval of datasets from corresponding repositories, and the subsequent manual execution of standardized downstream analysis. These tasks often require manual scripting and rely on fragmented workflows, limiting accessibility, and increasing turnaround time. To address these challenges, we designed a two-component system consisting of an artificial intelligence (AI) agent coordinating an automated analysis pipeline. CellAtria (Agentic Triage of Regulated single-cell data Ingestion and Analysis) is an agentic AI framework that enables dialogue-driven, document-to-analysis automation through a chatbot interface. Built on a graph-based, multi-actor architecture, CellAtria integrates a large language model (LLM) with tool-execution capabilities to orchestrate the full lifecycle of data reuse. To support downstream analysis, CellAtria incorporates CellExpress, a co-developed pipeline that applies state-of-the-art scRNA-seq processing steps to transform raw count matrices into analysis-ready single-cell profiles. Thus, CellAtria provides computational skill-agnostic and time-efficient access to standardized single-cell data ingestion and analysis.
Similar content being viewed by others


Introduction
Single-cell RNA sequencing (scRNA-seq) has emerged as a central technique in biomedical research1,2,3,4, enabling high-resolution profiling of cellular heterogeneity across tissues, disease states, and therapeutic interventions5,6. Its adoption continues to grow across the pharmaceutical landscape7,8, where it supports applications in target discovery, biomarker stratification, and mechanism-of-action studies9,10,11,12,13. In parallel, public repositories have accumulated an unprecedented volume and diversity of scRNA-seq datasets, spanning multiple species, tissues, and experimental conditions14,15,16. This rapid expansion presents a unique opportunity to transform fragmented single-cell datasets into structured, decision-support workflows that enhance biological interpretation and accelerate translational research17,18.
Despite the availability of these datasets, integrating published studies into institutional and enterprise-level analysis workflows remains a slow and resource-intensive process (Fig. 1a). Data ingestion typically begins with manual interpretation of the source publication to establish biological context, followed by the extraction of key metadata—such as tissue type, species, disease condition, and accession identifiers—and culminates in the manual retrieval of datasets from corresponding public repositories. The process then continues with dataset processing and analysis, which often require adaptation to institutional conventions and custom scripting practices. These interpretation- and scripting-based tasks are often delegated to specialists with both domain knowledge and computational proficiency, resulting in high personnel costs and increased susceptibility to analyst-driven errors and inconsistent throughput due to user-dependent variability in single-cell data analysis. Given the repetitive and procedural nature of these tasks, shifting them left—from specialist bioinformaticians to bench scientists—would not only enable faster, more consistent execution of data workflows and reduce dependence on bespoke computational support, but also free up expert capacity for more exploratory or high-impact scientific efforts.

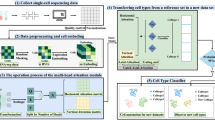
a Manual data onboarding cycle for public single-cell datasets. This panel illustrates the conventional workflow followed by investigators when integrating newly published datasets into internal research environments. The process spans from dataset announcement and assignment through metadata extraction, data validation, pipeline configuration, and analysis execution. b Architecture of agentic triage and execution. CellAtria employs a large language model (LLM) to mediate task dispatch across a graph-based, multi-actor execution framework, which is integrated with the co-developed CellExpress pipeline. High-level actions such as document parsing, metadata structuring, dataset retrieval, and file organization are coordinated through the LLM-mediated interface and mapped to appropriate backend tools for execution, ultimately producing analysis-ready outputs.
Agentic artificial intelligence (AI) systems19,20 offer a structured approach to automating complex biomedical workflows by coupling large language models (LLMs)21,22 with domain-specific computational toolchains23. These systems operate through modular, executable architectures in which predefined computational functions are dynamically composed in response to user prompts and contextual cues. The LLM serves as a semantic interface layer, interpreting natural language prompts and dispatching appropriate computational actions, thereby enabling dialogue-driven interaction with underlying infrastructure. Consequently, agentic AI systems, through their adaptive control flow, autonomous decision-making, and operational self-sufficiency, offer a scalable framework for minimizing manual intervention and democratizing access to bioinformatics infrastructures.
While some contemporary perspectives propose leveraging LLMs for dynamic, ad-hoc analytical code generation at runtime24,25,26,27, such strategies encounter obstacles in practice. The primary challenge with direct code generation for complex single-cell bioinformatics tasks lies in ensuring consistent output and scientific validity28. Such methods inherently struggle with reproducibility, as LLM outputs can vary based on model versions and prompting nuances29. More critically, the generated code carries a substantial risk of hallucinations30,31, logical errors32,33, incompatibility with state-of-the-art methods or software (due to the LLM’s training data temporal cutoff)34,35, and the misapplication of domain-specific parameters36. Any of these issues can lead to scientifically unsound analyses without extensive human oversight37 and debugging38. Most importantly, in clinical or highly regulated biomedical settings, the lack of protocol standardization inherent in ad-hoc code generation risks violating established quality control and analysis guidelines39. More recent developments, such as scExtract40 and SCassist41, have explored LLM-assisted pipelines. However, both rely on scripted or command-line execution and do not provide an agentic planner or a conversational interface to support interactive, multi-turn orchestration.
Moving beyond the risks in ad-hoc code-generation strategy, we adopted a critical architectural decision: an LLM-mediated tool-centric paradigm that balances flexibility with the imperative for scientific rigor and reproducibility. Here, the LLM’s role shifts from writing raw code to intelligently orchestrating a library of pre-vetted, robust analytical tools. We developed CellAtria, an agentic AI system that enables end-to-end, document-to-analysis automation in single-cell research (Fig. 1b). By combining natural language interaction with a graph-based, multi-actor architecture execution framework (Fig. 2a), CellAtria links tasks ranging from literature parsing, metadata extraction, and dataset retrieval to scRNA-seq processing via its co-developed companion pipeline, CellExpress, which applies state-of-the-art processing steps to transform raw count matrices into analysis-ready single-cell profiles (Fig. 2b). Utilizing CellAtria user interface (Supplementary Fig. S1), researchers interact with a language model that orchestrates pre-validated analytical tools (Supplementary Fig. S2), eliminating the need for manual scripting while ensuring standardized, reproducible analyses and accelerating the reuse of public single-cell resources.

a Language model-mediated orchestration of toolchains. Upon receiving a user prompt, the CellAtria interface transfers the request to the LLM agent, which interprets the user’s intent and autonomously invokes relevant tools. Outputs are then returned through the interface, completing a full cycle of context-aware execution. b Structure of the CellExpress pipeline. CellExpress implements a standardized single-cell RNA-seq workflow, encompassing project setup, quality control (QC), normalization, batch correction, dimensionality reduction, clustering, and cell type annotation. This pipeline is fully customizable and seamlessly integrated with the CellAtria orchestration layer, enabling automated execution from raw count matrices to interpretable results. It produces a comprehensive analytical package, including: (1) An interactive HTML report summarizing the workflow with key QC metrics and visualizations; (2) A finalized, fully annotated AnnData object for downstream analyses; (3) A machine-readable configuration export for full auditability; and (4) A QC-filtered AnnData object for alternative usages.
Results
A comprehensive description of the agentic system, CellAtria, including its architectural design and implementation details, is provided in the Methods section. To illustrate CellAtria’s capabilities, we implemented three prototypical use cases, each representing a common scenario in single-cell research.
CellAtria extracts metadata from article URLs and retrieves study-level datasets
In translational and early discovery research, investigators often encounter newly published studies whose associated datasets are highly relevant to institutional priorities.
To demonstrate CellAtria’s capability for literature-driven data acquisition and analysis execution, we selected a publicly available longitudinal transcriptomic study profiling immune responses in 2-month-old infants following routine vaccination42. Upon receiving the article URL, the agent conducts a multi-turn dialogue to parse the manuscript directly from the journal webpage, extract key structured metadata—including sample annotations and accession identifiers—and coordinate dataset retrieval from corresponding public repositories using GSE-level (GEO study-wide) accessioning (Supplementary Figs. S3–S10). Following user validation and direction, the agent proceeds to triage these data, executing essential organizational tasks and ensuring strict naming compatibility for seamless integration with downstream analytical functions (Supplementary Figs. S10, S11).
We note that this multi-turn interaction established a dynamic feedback loop, wherein the agent proactively surfaced context-aware downstream options, enabling real-time user validation and iterative adaptation (anticipate needs and suggest actions) aligned with the evolving agent strategic objective. This form of interactive and goal-conditioned reasoning exemplifies core agentic capabilities, which surpass the constraints of traditional rule-based approaches (e.g., static dashboard systems) by effectively handling uncertainty, enabling workflow reconfiguration on demand, and supporting user-driven exploration in a dialogic manner.
Thus, CellAtria effectively bridges literature discovery and structured dataset acquisition, establishing a foundation for automated, goal-directed workflows in single-cell research.
CellAtria parses scientific PDFs and retrieves sample-level datasets
Direct access to structured content on publisher websites can be restricted by technical barriers (e.g., dynamic page rendering, authentication walls) or constrained by licensing terms that limit programmatic scraping—even when institutional access rights are granted.
To address this limitation, the second scenario demonstrates metadata extraction from a locally stored PDF file in place of an article URL. CellAtria mitigates these access constraints by supporting direct parsing of PDF documents, enabling metadata retrieval in settings where web-based extraction is infeasible. We sought to demonstrate this capability by supplying CellAtria with an offline copy of a published study profiling T cell states across tumor, lymph node, and normal tissues in non-small cell lung cancer patients undergoing immune checkpoint blockade43. Upon uploading the PDF file, the agent engages in a multi-turn dialogue to extract structured metadata using a built-in document parser, enabling dataset retrieval even when the journal webpage scraping is infeasible (Supplementary Figs. S12, S13). In contrast to the first scenario, where datasets are retrieved at the study level, data acquisition here is carried out at the GEO sample level (GSM) using a task-specific tool, enabling fine-grained retrieval. The agent carries out each stage in response to user instructions conveyed through conversational natural language (Supplementary Figs. S14, S15).
We note that, to support verification and transparency, the real-time log viewer within the CellAtria interface continuously displays each issued prompt along with a status indicator confirming whether the associated tool invocation was successful. This persistent execution trace facilitates user comprehension and troubleshooting during live interactions (Supplementary Figs. S3–S15).
Thus, CellAtria enables flexible, fine-grained data acquisition even under restricted access conditions, while preserving full transparency and traceability through real-time execution logs.
CellAtria retrieves pre-identified datasets from public databases and manages file integration
In applied research settings, analysts often begin with pre-identified datasets—discovered through public single-cell portals or cross-study meta-analyses—where the source publication is already known or metadata extraction has been performed independently. In such cases, CellAtria supports direct ingestion of datasets from public repositories using user-supplied download URLs, bypassing upstream literature parsing and metadata extraction steps. This scenario was demonstrated using two curated collections (H5AD-formatted) from the CZ Cell by Gene Discover platform14: one integrating scRNA-seq data across 13 single-cell studies from 8 tumor types and normal tissues to delineate myeloid-derived cell states44, and another compiling scRNA-seq data from 223 patients across 9 cancer types to investigate cancer cell-specific responses to immune checkpoint blockade45. Upon receiving the dataset locations, the agent initiates a sequence of interactions with the user, subsequently retrieves the files, integrates them into the working directory, and prepares the necessary configuration files for downstream analysis (Supplementary Figs. S16–S18).
This operational mode demonstrates the agent’s ability to coordinate data acquisition and file integration through natural language prompts, enabling flexible invocation of tools at non-linear entry points within the agent’s engineered execution narrative.
CellAtria enables shell-level execution and file navigation within agent-guided workflows
Scientific agentic systems must balance automation with transparency and user oversight, particularly over file system operations, environment context, and task provenance. While language models can coordinate tool execution, these lower-level operations are more effectively managed through dedicated interface components that complement the conversational layer. To address this, CellAtria integrates a set of interactive panels (n = 4) that expose critical system-level functionality during live agent sessions (Supplementary Fig. S19).
To demonstrate the backend interpretability and user oversight of CellAtria during natural language interaction, we performed a series of targeted tests. Submitting the very simple prompt “Article title?” (implicitly requesting extraction of the publication title from a given URL42), we observed the agent’s internal reasoning, tool invocation, and model output displayed step-by-step in the agent backend panel (Supplementary Fig. S19a). Despite the minimal input, the agent accurately inferred the intended task, invoked the appropriate tool, and returned only the relevant information—faithfully aligning its output with the user’s request. This live trace offered direct insight into how natural language queries are processed and aligned with the agent’s internal tool logic. To verify the agent’s workspace context and access to relevant data, we then utilized the embedded terminal panel to navigate the project directory. This interaction confirmed the agent’s correct positioning within the expected working path and its access to necessary input files and subdirectories (Supplementary Fig. S19b). Complementing the terminal view, the file browser panel allowed us to visually inspect the same directory structure interactively, further reinforcing the consistency between user-issued commands and the agent-managed file system (Supplementary Fig. S19c). Finally, to ensure end-to-end provenance, the export utility provides two downloadable artifacts from each session: (i) a machine-readable conversation transcript capturing prompts, tool calls, and model response traces (Supplementary Figs. S19d, S20), and (ii) a structured LLM metadata file that records the backend language model configuration (Supplementary Figs. S19d, S21).
In practice, when a failure occurs, the live log viewer surfaces the specific error, while the agent backend panel links it to the exact agentic step that failed. Users can then inspect the cause, verify file presence or structure through the interactive file browser, and, where necessary, apply corrective actions using the embedded terminal, such as renaming, decompressing, or relocating files. All of these actions are possible without leaving the UI environment. Collectively, these interactive components establish CellAtria as a transparent, traceable, and auditable agentic system. By embedding low-level controls within a high-level dialogue framework, the system balances automation with user-in-the-loop oversight—a critical feature for fostering trust in AI-driven scientific discovery pipelines.
CellAtria enables end-to-end automation of single-cell RNA-seq processing through CellExpress
Having demonstrated the capabilities and task-specific functionalities of CellAtria, we postulated that by interacting with a fully automated downstream pipeline, CellAtria could further transform literature-based inputs into fully processed single-cell datasets—minimizing the need for hands-on user time and specialized analytic expertise by linking study discovery with standardized downstream analysis through a unified, dialogue-driven workflow.
To this end, we developed CellExpress, a companion computational pipeline that standardizes the processing of scRNA-seq data, transforming raw inputs into biologically interpretable, analysis-ready outputs (Fig. 2b). CellExpress builds on previously published and validated methods, ensuring that their application is carried out in a consistent, efficient, and unified workflow with minimal user intervention. A complete methodological description is provided in the Methods section. To facilitate integration with CellAtria, we also developed a suite of agent-triggered tools that support comprehensive pipeline configuration, execution control, and real-time monitoring—enabling the agent to coordinate the entire analytical workflow through natural language interaction (Supplementary Fig. S2).
To demonstrate CellExpress orchestration through CellAtria agent, we extended the previously initiated scenario using the peripheral blood scRNA-seq dataset from 2-month-old infants42. As with upstream tasks, the pipeline’s configuration, execution, and monitoring were conducted entirely through natural-language dialogue between the user and the agent, culminating in the successful operation of CellExpress pipeline (Supplementary Figs. S22–S28). Additionally, to evaluate the full analytical scope of CellExpress, we performed an autonomous pipeline execution with all modules enabled through complete argument specification. In this single-run execution (runtime: ~30 min), 18 samples were processed, yielding ~71,000 cells after quality control filtering, which included thresholds on gene count, UMI count, mitochondrial gene content, and doublet detection. Batch effects were subsequently corrected to account for sample-specific variation. Dimensionality reduction was conducted using both UMAP and t-SNE embeddings, followed by graph-based clustering. Automated cell type annotation showed high concordance between tissue-agnostic and tissue-specific models, both aligning with the original study. To quantify this alignment, we compared CellExpress-derived cell type compositions against the original expert annotations using label-harmonized compartments. The results showed strong agreement, with Pearson correlations of ~0.99, reflecting high concordance in major shared immune lineage frequencies (98%), including T cells, B cells, NK cells, and myeloid populations (Supplementary Fig. S29). Finally, cluster-level marker gene identification was performed to support downstream biological interpretation. All intermediate outputs are tabulated, visualized, and consolidated into an HTML summary report (the corresponding file is provided in the CellAtria GitHub repository; see Data Availability). In addition, all relevant artifacts, including the execution summary, sample metadata, and workflow configuration, are stored in a structured file to ensure auditability and reproducibility (Supplementary Fig. S30). A detailed description of the computational tools and models integrated into the CellExpress pipeline is provided in the Methods section.
Thus, CellExpress addresses persistent challenges in single-cell transcriptomic analysis by delivering a fully integrated, end-to-end workflow that ensures transparency and reproducibility through the use of rigorously benchmarked, field-standard components.
CellAtria enables full-lifecycle document-to-analysis execution through fully autonomous toolchain orchestration
The hallmark of robust agentic systems lies in their ability to autonomously synthesize complex operational sequences from a single, high-level command—abstracting procedural complexity while preserving fidelity to the intended outcome. Such generalizable and autonomous orchestration reflects a carefully designed, context-aware system prompt and an explicit, unambiguous definition of tool input/output (I/O) behaviors—elements that collectively underpin the system’s capacity for full-scope task execution.
To evaluate the extent of autonomous task coordination in CellAtria, we tested its ability to execute a complete document-to-analysis workflow using a single instruction, thereby eliminating the need for iterative user-agent interaction. In this scenario, the agent was provided with the primary article URL reporting longitudinal scRNA-seq data from 2-month-old infants42, with the objective of executing the CellExpress pipeline using the associated single-cell data. In response, and without further user input, CellAtria autonomously carried out the full underlying predesigned agentic workflow, leading to the successful execution of the CellExpress pipeline (Supplementary Fig. S31). In particular, CellAtria performed several key steps autonomously, including parsing the primary article, extracting structured metadata, retrieving the associated dataset, configuring the CellExpress pipeline with context-aware parameters, and dispatching the execution—all without manual intervention (Supplementary Fig. S32). Notably, this autonomous run, including the CellExpress pipeline’s runtime, completed all steps in under 10 min. This performance stands in stark contrast to the ~15 cumulative hours of manual effort typically required by a bioinformatics analyst for equivalent tasks, as per our internal benchmarks (including manual ingestion, metadata extraction, dataset retrieval, file reorganization, and fragmented script execution).
Hence, this assessment demonstrates CellAtria’s capability for strategic problem-solving by translating high-level objectives into a coherent sequence of tool invocations, marking its shift from prompt-driven response to an AI agent capable of autonomous workflow orchestration with minimal intervention and time-efficient performance.
CellAtria sustains scalable performance across multi-study compendia
To evaluate the robustness and scalability of CellAtria’s agentic automation, we benchmarked its end-to-end functionality across 25 publicly available human scRNA-seq datasets. These curated datasets span six major cancer types, including breast46,47,48,49,50 (n = 5), lung51,52,53 (n = 3), prostate54,55,56 (n = 3), colorectal57,58,59,60,61 (n = 5), ovary62,63,64,65,66 (n = 5), and pancreas67,68,69,70 (n = 4) (Supplementary Fig. S33). The cohort (290 samples) includes single-cell investigations of tumor-infiltrating immune remodeling (e.g., non-small cell lung cancer), resistance and phenotypic plasticity in metastatic settings (e.g., stage IV breast cancer), treatment-naive and relapsed disease states (e.g., prostate cancer), and immunologic shifts under checkpoint blockade (e.g., microsatellite instability high colorectal cancer). It also features high-resolution atlases of epithelial-immune crosstalk (e.g., breast and colorectal cancer) and spatial or multi-omics extensions (e.g., ovarian and pancreatic cancers). This biologically diverse panel was deliberately selected to challenge the agent across a range of disease etiologies, tissue microenvironments, and metadata contextual complexity, thereby offering a realistic and representative cross-section of biomedical use cases.
CellAtria autonomously executed the complete GEO-to-analysis workflow for each study using gpt-4o as the agentic controller. This agentic process involved dynamic metadata extraction from the GEO landing page (e.g., organism, disease label, tissue type, and single-cell GSM identifiers), automated dataset retrieval, and dynamic configuration and orchestration of the CellExpress pipeline. All benchmarking runs completed successfully without manual intervention (Supplementary Fig. S33).
Agent interaction metrics, focusing on the LLM’s internal reasoning and decision-making steps, revealed consistently low execution times, with an average of 1.45 ± 1.25 min. per task. Runtime variability was primarily driven by differences in dataset volume. To assess output consistency, two language model-specific indicators were used: the number of tokens generated per response (a proxy for content volume) and the response size in kilobytes (a proxy for serialized output size). These outputs averaged 6589 ± 1317 tokens and 20.5 ± 4.0 KB, respectively, with low Gini coefficients (≈0.10), indicating uniform verbosity and content balance across all runs (Supplementary Fig. S33).
We next sought to assess the agent performance when using a different LLM on the exact same dataset cohort. We therefore repeated the analysis using gpt-4o-mini as the agentic controller; all benchmarking runs again completed successfully without manual intervention. The average agentic task duration was 1.70 ± 1.70 min per dataset. The model generated 5439 ± 1369 tokens per run, corresponding to an average serialized output size of 16.9 ± 4.1 KB. The Gini coefficients for both token counts and output size were 0.14, indicating low dispersion and thus consistent verbosity across runs (Supplementary Fig. S33).
Across repeated runs, we observed minor lexical variation in the way biological descriptors were rendered (e.g., “PBMC” vs. “peripheral blood mononuclear cells,” or abbreviated disease labels vs. expanded forms). However, these differences were confined to the natural-language layer. CellExpress consumes the structured, schema-validated arguments produced by the supporting tool layer and is therefore agnostic to such surface-level term variability.
When integrated with the CellExpress pipeline, CellAtria supported full end-to-end processing of approximately one million post quality control (QC) cells, drawn from diverse input formats including 10× Genomics HDF5 (.h5), directory trios (matrix, barcodes, features), and plain text matrices (txt.gz). Average runtime was 3.16 min per dataset under default analysis settings. Each dataset contributed roughly 3.9 × 10⁴ post-QC cells (≈39k ± 32k), with memory usage scaling accordingly (10.1 ± 9.8GB per run). A moderately positive association was observed between post-QC cell count and memory consumption (Pearson r = 0.45, p < 0.05), indicating that RAM demand increases with dataset size, though not in a strictly linear fashion (Supplementary Fig. S33).
Hence, these results underscore CellAtria’s ability to handle complex and biologically rich single-cell datasets at scale. The agentic framework not only minimizes manual overhead but also maintains computational efficiency, output uniformity, and analytical robustness.
Discussion
Automation in single-cell analysis spans three trajectories: (i) targeted automation of labor-intensive, subjective steps (e.g., cell type annotation71,72), (ii) end-to-end pipelines assembled from automated modules73, and (iii) most recently, broad-scope, LLM-enabled code-generation workflows24. In this study, we unify these strands with an LLM-mediated, tool-centric paradigm that balances flexibility with scientific rigor and reproducibility. Specifically, we introduce CellAtria, an agentic system that enables dialogue-driven, document-to-analysis automation in single-cell research.
The system integrates a natural language interface with modular computational toolchains—including task-specific utilities that trigger the CellExpress pipeline (co-developed standalone standardized single-cell analysis pipeline)—to form a unified semantic layer that orchestrates data triage and analysis. Its composable architecture supports non-linear, context-aware interaction, allowing users to engage flexibly at different stages of data ingestion and preparation. This design enables researchers to process more studies with less effort, effectively improving their analytical capacity without sacrificing reproducibility or protocol adherence.
CellAtria’s orchestration-first, tool-centric strategy coordinates a pre-vetted modular toolchain (including the CellExpress pipeline), leveraging LLM strengths in intent interpretation and task delegation while eschewing free-form code synthesis. This approach guarantees that all executed analytical steps (including complex single-cell data analysis) adhere to established best practices, are fully transparent, and maintain the high level of reliability and auditability essential for robust scientific discovery, all while leveraging the full orchestration potential of LLMs.
Agentic systems operate according to an underlying execution narrative—a structured sequence of modular actions that defines how tasks are interpreted and fulfilled. While this narrative is inherently flexible and permits user-initiated entry at arbitrary points in the workflow, it remains anchored in a coherent logic that guides the agent’s behavior and goals. This design departs from traditional rule-based automation by allowing unconstrained user interaction while still aligning those inputs with predefined tools and execution pathways. In the case of CellAtria, this narrative integrates a four-stage operational sequence: (1) dynamic metadata extraction, (2) dataset acquisition, (3) file organization, and (4) downstream analysis execution. Each module can be invoked independently; however, this study focuses on the optimized execution path, which reflects the intended canonical workflow.
Agentic AI frameworks establish a principled division of responsibilities: domain experts engage with high-level analytical tasks through natural language interfaces, while system developers ensure the integrity and robustness of the underlying toolchains. The core reasoning engine of these agents—LLMs—while effective at semantically interpreting dynamic user prompts and aligning them with task objectives, remains inherently prone to hallucination30,31, often producing linguistically coherent yet semantically or factually incorrect outputs. To curb these vulnerabilities, CellAtria embeds safeguards at three levels: (1) tool-schema validation (rejecting ill-formed or non-existent actions), (2) restricted invocation patterns (permitting only vetted tool sequences and parameters), and (3) boundary-aware system prompts (explicitly steering the agent to decline or defer when capabilities are exceeded (Supplementary Fig. S34). Nevertheless, to safeguard analytical reliability and ensure interpretability, a human-in-the-loop paradigm, wherein a context-aware investigator actively evaluates, verifies, and contextualizes agent responses, remains indispensable.
Agentic AI systems ultimately inherit the strengths and weaknesses of the LLMs that power their reasoning layer; different LLMs may diverge in how they interpret edge cases, resolve ambiguous instructions, or render domain-specific concepts, because generation remains probabilistic rather than fully deterministic. This places a sustained burden on system designers to harden the tool layer, but it also means that users should treat agent outputs as decision support, not as authoritative replacements for expert review. At the same time, this dependency is a feature: as LLMs improve in grounding, factuality, and tool-use reliability, agentic workflows will gain robustness without requiring architectural rewrites.
CellAtria’s metadata extraction capabilities are optimized for scientific articles that conform to structured narrative conventions, such as standardized sectioning and consistent biomedical terminology. While the underlying language model has broad generalization capacity, the LLM may face ambiguity when applied to unstructured or idiosyncratic content, such as informal notes lacking hierarchical organization, inconsistent labeling, or documents with irregular phrasing (e.g., nonstandard abbreviations, ad hoc formatting, or domain-specific shorthand). For instance, when the agent encounters manuscripts that lack structured section headers or essential metadata fields (e.g., species, tissue type, disease), CellAtria defaults to marking those fields as unavailable. This safeguard is intentionally designed to prevent speculative inference and reduce hallucination risk.
A key design consideration for agentic systems is the LLM’s lack of direct internet access—a common constraint adopted to ensure security and maintain control over external interactions. Consequently, the system depends on deterministic, tool-mediated mechanisms for content retrieval and cannot independently browse or query online databases in real time. This reliance highlights the critical importance of standardized data management practices74: discrepancies between manuscript-reported metadata and repository-level annotations can hinder the reliability of agentic execution. We therefore advocate for closer harmonization between narrative metadata and structured repository schemas to better support scalable agentic applications.
The CellAtria framework’s objective was not to replicate outcomes from original publications, which would require access to exact computing infrastructure, parameter choices, and context-specific decisions, many of which are user-biased and often not publicly documented comprehensively. The variability introduced by manual, human-guided analysis reinforces the need for standardized frameworks like CellExpress. By enforcing schema-constrained execution and orchestrated automation, these tools mitigate analytic drift and interpretive subjectivity.
The modular design of CellAtria, combined with its graph-based execution architecture, enables extensibility: new decision-making and task-routing capabilities can be incorporated at the orchestration layer, while analytical and modality-specific logic is delegated to pluggable downstream pipelines. Therefore, the framework is pipeline-agnostic by construction, allowing alternative workflows to be integrated without redesigning the agentic core.
As automated frameworks increasingly interface with regulated data sources, ensuring ethical and compliant data handling becomes essential. When extracting metadata from scientific literature, CellAtria’s schema captures repository identifiers, publisher information, and author-reported conflicts of interest, providing legal and provenance context for each dataset. For proprietary or sensitive data, the Docker-based architecture of CellAtria enables reproducible execution within secure, auditable, and GxP-compliant environments, a critical requirement in clinical research.
In conclusion, CellAtria demonstrates how domain-informed agentic AI systems can operationalize scientific research processes in a computationally skill-agnostic manner, thereby accelerating discovery and supporting the transition toward next-generation, AI-integrated research ecosystems.
Methods
CellAtria interface and architectural framework
The CellAtria interface integrates seven principal components that provide fine-grained control over task execution and collectively facilitate rich agentic interaction (Supplementary Fig. S1): (1) Persistent Chatbot Window: Manages user–agent communication and maintains conversational continuity through an internal hidden state, supporting coherent, multi-turn exchanges. (2) User Input Panel: Accepts textual prompts and facilitates document uploads, with all inputs jointly processed through a unified execution handler. (3) Real-time Log Viewer: Displays user-agent transaction status. (4) Agent Backend Panel: Provides a live, step-by-step view of the agent’s internal reasoning, tool invocation sequence, and backend responses, directly supporting transparency and debugging. (5) Embedded Terminal Panel: Enables direct shell interaction within the agent’s runtime environment, facilitating robust system-level control without exiting the interface. (6) Interactive File Browser: Allows users to navigate project directories and inspect file contents within the active workspace, complementing terminal operations. (7) Export Utility: Captures session-level provenance by generating two structured artifacts: a machine-readable transcript of all user–agent interactions, and a metadata specification detailing the backend language model configuration.
These diverse interface elements are orchestrated atop a LangGraph-based75 backbone that encodes the agent’s execution flow logic as a directed graph of modular, state-aware functions, ensuring robust and interpretable coordination across heterogeneous toolchains and complex computational workflows.
CellAtria modular toolchain for task execution
To enable flexible and robust task execution, we developed a comprehensive suite of interoperable tools that encapsulate core agent functionalities across four principal operational domains (Supplementary Fig. S2): (1) Metadata Parsing and Semantic Structuring: Handles the extraction and organized representation of relevant information from diverse sources. (2) Programmatic Data Retrieval and Hierarchical Organization: Manages the automated acquisition of datasets and their structured arrangement within the workspace. (3) Standardized File Handling and Pre-processing: Ensures consistent management and preparation of data files for downstream analysis. (4) Automated Workflow Configuration and Execution Orchestration: Facilitates the dynamic setup and control of complex computational analysis pipelines. Each tool is precisely implemented as an atomic function with rigorously defined input/output (I/O) behavior, a design choice that fundamentally enables the agent to compose dynamic and reliable task sequences in response to user prompts. These tools are inherently embedded as graph nodes within CellAtria’s architectural backbone and accessed through natural-language interfaces, thereby facilitating intelligent, context-aware orchestration. This modular architecture not only accommodates user-initiated entry at arbitrary points within the workflow but also establishes a crucial foundation for tool reuse, adaptation, and the scalable extension of execution flows.
The LLM is responsible exclusively for unstructured content interpretation and structured metadata inference, as well as orchestrating execution of the pre‑vetted tools and analysis pipeline. All analytical decisions are governed by fixed logic or schema‑validated defaults, with no parameter choices delegated to the LLM. CellAtria does not currently employ Retrieval-Augmented Generation (RAG) or Model Context Protocol (MCP), as its modular toolchain and internal state tracking fulfill analogous roles in orchestrating dynamic task flows.
Web and PDF article ingestion for metadata extraction
As part of the metadata parsing and semantic structuring module, CellAtria implements lightweight content extraction routines for both web-based and local sources to facilitate automated ingestion of scientific literature. For journal URLs, the system programmatically retrieves HTML content using the “requests” library (v2.32.5) and isolates visible textual content via “BeautifulSoup” (v4.13.3), applying DOM-level (document object model) filtering to exclude non-informative elements such as scripts, metadata, and stylesheets. For local PDF documents, CellAtria leverages the “PyMuPDF” library “fitz” (v1.26.5) to extract paragraph-level text from the document’s text layer by iterating across pages. In both cases, the resulting structured text is passed to the language model for semantic parsing and downstream metadata field extraction. This approach is explicitly tool-mediated and does not rely on vector retrieval or RAG-style (Retrieval-Augmented Generation) pipelines.
Standardized single-cell data analysis via CellExpress
A cornerstone of CellAtria’s full-scale capabilities is the co-developed CellExpress, a standardized single-cell analysis pipeline - fully automated and engineered to deliver robust scRNA-seq analysis, from raw count matrices through comprehensive processing and report generation (Fig. 2b). Designed to lower bioinformatics barriers, CellExpress implements a comprehensive set of state-of-the-art, Scanpy-based76 processing stages, including: (1) quality control; performed globally or per sample, (2) data transformation; encompassing normalization, highly variable gene selection, and scaling, (3) dimensionality reduction; utilizing UMAP77 and t-SNE78, (4) graph-based clustering79, and (5) marker gene identification. Additional tools are seamlessly integrated to support advanced analysis tasks, such as doublet detection (Scrublet80), batch correction (Harmony81 and scVI82), and automated cell type annotation using both tissue-agnostic (SCimilarity83) and tissue-specific (CellTypist84) models.
All analytical steps are executed sequentially under centralized control, with parameters fully configurable via a comprehensive input schema. All arguments are made accessible through CellAtria’s agentic interface, allowing users to interact with and query their configurations using natural language (Supplementary Fig. S35). In addition, user-defined metadata (e.g., sex, disease status, tissue type, and other custom annotations) can be supplied alongside the dataset in a structured metadata table. These annotations are automatically incorporated into the analysis pipeline, enabling metadata-aware processing.
CellExpress implements a curated set of preprocessing steps and QC parameters informed by consensus practices in the field76,85,86,87,88,89,90. These defaults serve as a principled starting point rather than a fixed prescription and can be overridden when dataset-specific adjustment is warranted. Due diligence should be exercised by taking into account sample-specific factors, such as tissue type, dissociation protocol, platform-specific artifacts, and study objectives, when modifying pipeline parameters.
The pipeline natively supports a broad range of standard single‑cell inputs, including 10X Genomics Cell Ranger91 outputs (matrix, barcodes, and features), HDF5 (h5) matrices, and AnnData (H5AD) objects. In addition to 10X‑based chemistries, CellExpress also supports Parse Biosciences–formatted inputs92 (gene, metadata, and count matrix), enabling compatibility with non‑10X droplet‑based platforms. The pipeline also accepts generic count matrices supplied in plain‑text (txt.gz) or CSV‑style (csv.gz) format.
Upon execution, CellExpress processes a designated set of scRNA-seq samples and generates a comprehensive analytical package comprising four components: (1) a finalized, fully annotated AnnData object, directly suitable for downstream analysis; (2) a structured, publication-ready HTML report that captures a complete snapshot of the entire workflow, including key parameters, quality control metrics, dimensionality reduction, clustering results, and cell type annotations, presented through dynamics tables and graphical visualizations; (3) a complete export of all configuration settings in a machine-readable, standardized format to ensure auditability and full reproducibility; and (4) a quality control-filtered AnnData object generated for reuse in alternative or customized workflows.
CellExpress executes as a single-pass pipeline yet supports iterative workflows: intermediate outputs, such as QC visualizations, embeddings, clustering results, and annotations, are exposed in the generated HTML report, and parameters can be readily adjusted for seamless re-execution.
Designed for flexible deployment, CellExpress operates as a fully standalone pipeline for comprehensive scRNA-seq data analysis. It can be orchestrated either through an agentic system—as incorporated into the CellAtria framework—or via direct command-line execution. Furthermore, its Pythonic foundation directly addresses scalability constraints commonly associated with R-based pipelines (CellBrdige73), enabling more efficient handling of large single-cell datasets. We note that on-disk execution strategies, such as BPCell93, have improved the scalability of R-based workflows.
While CellExpress standardizes core processing, it intentionally delegates more specialized downstream analyses to the post-pipeline stage. This reflects the fact that such tasks often rely on study-specific priors, including subpopulation annotations, lineage assumptions, or temporal structure, which are not universally applicable. By restricting built-in methods to those broadly generalizable across datasets, CellExpress preserves flexibility for downstream analytical design to be tailored according to specific research objectives.
Preprocessing configuration for full CellExpress workflow demonstration
In the experiment benchmarking full pipeline execution of CellExpress using a peripheral blood scRNA‑seq dataset from 2‑month‑old infants (GSE213996), stringent quality control thresholds were applied: a minimum of 750 UMIs per cell, at least 250 detected genes per cell, and exclusion of genes expressed in fewer than 3 cells. Cells with mitochondrial gene content exceeding 15% were removed. Doublets were identified and filtered using Scrublet with a score cutoff of 0.25. Batch correction was performed using Harmony81, and cell type annotations were obtained using both SCimilarity83 and CellTypist84.
CellAtria containerized environment and CellExpress execution
The CellAtria runtime environment is fully containerized using Docker, enabling consistent and reproducible deployment across diverse computational infrastructures. This encapsulation strategy ensures environmental parity by isolating workflows from system-specific variability and mitigating software dependency conflicts, thereby facilitating seamless portability across local, cloud, and high-performance computing environments.
To preserve agent responsiveness during potentially long-running computations, CellAtria executes the CellExpress pipeline in a detached mode. Upon receiving a complete execution schema, the agent delegates the task to a background subprocess, decoupling it from the interactive session. Standard output and error streams are redirected to persistent log files for downstream inspection, and a unique process identifier is recorded to support real-time status tracking and diagnostics.
Large language model provider and computational environment
For all experiments, the LLM backend was provisioned via Azure OpenAI, specifically using managed deployments of gpt-4o (version as of 2024-11-20) and gpt-4o-mini (version as of 2024-07-18). No fine-tuning or domain-specific retraining was performed. The models were operated with a sampling temperature of 1.0 and a nucleus sampling parameter (top‑p) of 1.0. The CellAtria agent was built using the LangGraph (v0.5.4) orchestration framework. All operations were executed within a Docker container pinned to Python 3.12.9, running on an AWS EC2 r6i.32xlarge instance with 128 vCPUs and 1,024 GiB RAM. HTML reports produced by the CellExpress pipeline include an embedded version provenance section that programmatically records the exact versions of all R and Python packages used in the workflow.
Data availability
No new data were generated in this study. All datasets used in the demonstration use cases are publicly available, and the corresponding source publications are cited accordingly. The processed objects used in the third scenario are available via the CZ Cell by Gene Discover platform at the following links: https://cellxgene.cziscience.com/collections/3f7c572c-cd73-4b51-a313-207c7f20f188 and https://cellxgene.cziscience.com/collections/61e422dd-c9cd-460e-9b91-72d9517348ef. The CellExpress HTML summary report example, along with additional documentation, is available in the CellAtria GitHub repository at: https://github.com/AstraZeneca/cellatria/tree/main/docs.
Code availability
The source code, documentation, and all materials required to execute the agent are publicly available on the GitHub at https://github.com/AstraZeneca/cellatria.
References
Hao, Y. et al. Dictionary learning for integrative, multimodal and scalable single-cell analysis. Nat. Biotechnol. 42, 293–304 (2024).
Teichmann, S. & Efremova, M. Method of the year 2019: single-cell multimodal omics. Nat. Methods 17, 2020 (2020).
Molla Desta, G. & Birhanu, A. G. Advancements in single-cell RNA sequencing and spatial transcriptomics: transforming biomedical research. Acta Biochim. Polonica 72, 13922 (2025).
Haque, A., Engel, J., Teichmann, S. A. & Lönnberg, T. A practical guide to single-cell RNA-sequencing for biomedical research and clinical applications. Genome Med. 9, 1–12 (2017).
Cao, J. et al. The single-cell transcriptional landscape of mammalian organogenesis. Nature 566, 496–502 (2019).
Ghandi, M. et al. Next-generation characterization of the cancer cell line encyclopedia. Nature 569, 503–508 (2019).
Saw, P. E. & Song, E. Single-cell RNA sequencing (sc-RNAseq) in the development of biomedical therapeutics. In: RNA Therapeutics in Human Diseases (Springer, 2025).
Liu B., Hu S., Wang X. Applications of single-cell technologies in drug discovery for tumor treatment. Iscience 27, 110486 (2024).
Papalexi, E. & Satija, R. Single-cell RNA sequencing to explore immune cell heterogeneity. Nat. Rev. Immunol. 18, 35–45 (2018).
Lim, B., Lin, Y. & Navin, N. Advancing cancer research and medicine with single-cell genomics. Cancer Cell 37, 456–470 (2020).
Lim, J. et al. Transitioning single-cell genomics into the clinic. Nat. Rev. Genet. 24, 573–584 (2023).
Van de Sande, B. et al. Applications of single-cell RNA sequencing in drug discovery and development. Nat. Rev. Drug Discov. 22, 496–520 (2023).
Mathys, H. et al. Single-cell transcriptomic analysis of Alzheimer’s disease. Nature 570, 332–337 (2019).
Program, C. C. S. et al. CZ CELLxGENE discover: a single-cell data platform for scalable exploration, analysis and modeling of aggregated data. Nucleic Acids Res. 53, D886–D900 (2025).
Barrett, T. et al. NCBI GEO: archive for functional genomics data sets—update. Nucleic Acids Res. 41, D991–D995 (2012).
Regev, A. et al. The human cell atlas. elife 6, e27041 (2017).
Li, Q. et al. Progress and opportunities of foundation models in bioinformatics. Brief. Bioinforma. 25, bbae548 (2024).
Chen, Z., Wei, L. & Gao, G. Foundation models for bioinformatics. Quant. Biol. 12, 339–344 (2024).
Acharya, D. B., Kuppan, K. & Divya, B. Agentic AI: autonomous intelligence for complex goals–a comprehensive survey. IEEE Access 13, 18912–18936 (2025).
Shavit, Y. et al. Practices for Governing Agentic AI Systems. Research Paper (OpenAI, 2023).
Thirunavukarasu, A. J. et al. Large language models in medicine. Nat. Med. 29, 1930–1940 (2023).
Naveed, H. et al. A comprehensive overview of large language models. In ACM Transactions on Intelligent Systems and Technology (ACM, 2023).
Topsakal, O. & Akinci, T. C. Creating large language model applications utilizing langchain: a primer on developing LLM apps fast. In: International Conference on Applied Engineering and Natural Sciences (All Sciences Proceedings, 2023).
Huang, K. et al. Biomni: a general-purpose biomedical AI agent. Preprint at https://www.biorxiv.org/content/10.1101/2025.05.30.656746v1 (2025).
Xiao, Y. et al. Cellagent: an llm-driven multi-agent framework for automated single-cell data analysis. Preprint at https://arxiv.org/abs/2407.09811 (2024).
Xin, Q. et al. BioInformatics Agent (BIA): unleashing the power of large language models to reshape bioinformatics workflow. Preprint at https://www.biorxiv.org/content/10.1101/2024.05.22.595240v2 (2024).
Zhou, J. et al. An AI agent for fully automated multi-omic analyses. Adv. Sci. 11, 2407094 (2024).
Chen, Q. et al. A deep dive into large language model code generation mistakes: what and why? Preprint at https://arxiv.org/abs/2411.01414 (2024).
Guan, B., Roosta, T., Passban, P. & Rezagholizadeh, M. The order effect: investigating prompt sensitivity to input order in LLMs. Preprint at https://arxiv.org/abs/2502.04134 (2025).
Chen, Y. et al. Hallucination detection: robustly discerning reliable answers in large language models. In: Proceedings of the 32nd ACM International Conference on Information and Knowledge Management (ACM, 2023).
Sadat, M. et al. DelucionQA: detecting hallucinations in domain-specific question answering. Preprint at https://arxiv.org/abs/2312.05200 (2023).
Shen, X., Chen, Z., Backes, M. & Zhang, Y. In chatgpt we trust? measuring and characterizing the reliability of chatgpt. Preprint at https://arxiv.org/abs/2304.08979 (2023).
Tam, D. et al. Evaluating the factual consistency of large language models through news summarization. Findings of the Association for Computational Linguistics: ACL, 5220–5255 (2023).
Zhao, B., Brumbaugh, Z., Wang, Y., Hajishirzi, H. & Smith, N. A. Set the clock: temporal alignment of pretrained language models. Preprint at https://arxiv.org/abs/2402.16797 (2024).
Zhu, C. et al. Is your LLM outdated? A deep look at temporal generalization. Preprint at https://arxiv.org/abs/2405.08460 (2024).
Ye, W. et al. Assessing hidden risks of llms: an empirical study on robustness, consistency, and credibility. Preprint at https://arxiv.org/abs/2305.10235 (2023).
Tang, X. et al. Risks of AI scientists: prioritizing safeguarding over autonomy. Nat Commun 16, 8317 (2025).
Chong, C. J., Yao, Z. & Neamtiu, I. Artificial-intelligence generated code considered harmful: a road map for secure and high-quality code generation. Preprint at https://arxiv.org/abs/2409.19182 (2024).
Singh, R., Paxton, M. & Auclair, J. Regulating the AI-enabled ecosystem for human therapeutics. Commun. Med. 5, 181 (2025).
Wu, Y. & Tang, F. scExtract: leveraging large language models for fully automated single-cell RNA-seq data annotation and prior-informed multi-dataset integration. Genome Biol. 26, 174 (2025).
Nagarajan, V. et al. SCassist: an AI based workflow assistant for single-cell analysis. Bioinformatics 41, btaf402 (2025).
Nouri, N. et al. Young infants display heterogeneous serological responses and extensive but reversible transcriptional changes following initial immunizations. Nat. Commun. 14, 7976 (2023).
Pai, J. A. et al. Lineage tracing reveals clonal progenitors and long-term persistence of tumor-specific T cells during immune checkpoint blockade. Cancer Cell 41, 776–790.e777 (2023).
Guimarães, G. R. et al. Single-cell resolution characterization of myeloid-derived cell states with implication in cancer outcome. Nat. Commun. 15, 5694 (2024).
Gondal, M. N., Cieslik, M. & Chinnaiyan, A. M. Integrated cancer cell-specific single-cell RNA-seq datasets of immune checkpoint blockade-treated patients. Sci. Data 12, 139 (2025).
Joo, E. H. et al. Migratory tumor cells cooperate with cancer associated fibroblasts in hormone receptor-positive and HER2-negative breast cancer. Int. J. Mol. Sci. 25, 5876 (2024).
Otsuji, K. et al. Serial single-cell RNA sequencing unveils drug resistance and metastatic traits in stage IV breast cancer. NPJ Precis. Oncol. 8, 222 (2024).
Wu, S. Z. et al. A single-cell and spatially resolved atlas of human breast cancers. Nat. Genet. 53, 1334–1347 (2021).
Tokura, M. et al. Single-cell transcriptome profiling reveals intratumoral heterogeneity and molecular features of ductal carcinoma in situ. Cancer Res. 82, 3236–3248 (2022).
Timperi, E. et al. Lipid-associated macrophages are induced by cancer-associated fibroblasts and mediate immune suppression in breast cancer. Cancer Res. 82, 3291–3306 (2022).
Song, Q. et al. Dissecting intratumoral myeloid cell plasticity by single cell RNA-seq. Cancer Med. 8, 3072–3085 (2019).
Mitsuhashi, A. et al. Identification of fibrocyte cluster in tumors reveals the role in antitumor immunity by PD-L1 blockade. Cell Rep. 42, 112162 (2023).
Itahashi, K. et al. BATF epigenetically and transcriptionally controls the activation program of regulatory T cells in human tumors. Sci. Immunol. 7, eabk0957 (2022).
Zaidi, S. et al. Single-cell analysis of treatment-resistant prostate cancer: Implications of cell state changes for cell surface antigen–targeted therapies. Proc. Natl. Acad. Sci. 121, e2322203121 (2024).
Masetti, M. et al. Lipid-loaded tumor-associated macrophages sustain tumor growth and invasiveness in prostate cancer. J. Exp. Med. 219, e20210564 (2021).
Dong, B. et al. Single-cell analysis supports a luminal-neuroendocrine transdifferentiation in human prostate cancer. Commun. Biol. 3, 778 (2020).
Liu, X. et al. Th17 cells secrete TWEAK to trigger epithelial–mesenchymal transition and promote colorectal cancer liver metastasis. Cancer Res. 84, 1352–1371 (2024).
Guo, W. et al. Resolving the difference between left-sided and right-sided colorectal cancer by single-cell sequencing. JCI Insight 7, e152616 (2022).
Cortese, N. et al. High-resolution analysis of mononuclear phagocytes reveals GPNMB as a prognostic marker in human colorectal liver metastasis. Cancer Immunol. Res. 11, 405–420 (2023).
Li, J. et al. Remodeling of the immune and stromal cell compartment by PD-1 blockade in mismatch repair-deficient colorectal cancer. Cancer Cell 41, 1152–1169.e1157 (2023).
Zheng, X. et al. Single-cell transcriptomic profiling unravels the adenoma-initiation role of protein tyrosine kinases during colorectal tumorigenesis. Signal Transduct. Target. Ther. 7, 60 (2022).
Brand, J. et al. Fallopian tube single cell analysis reveals myeloid cell alterations in high-grade serous ovarian cancer. Iscience 27, 108990 (2024).
Geistlinger, L. et al. Multiomic analysis of subtype evolution and heterogeneity in high-grade serous ovarian carcinoma. Cancer Res. 80, 4335–4345 (2020).
Anadon, C. M. et al. Ovarian cancer immunogenicity is governed by a narrow subset of progenitor tissue-resident memory T cells. Cancer Cell 40, 545–557.e513 (2022).
Weber, L. M. et al. Genetic demultiplexing of pooled single-cell RNA-sequencing samples in cancer facilitates effective experimental design. Gigascience 10, giab062 (2021).
Anadon, C. M. et al. Protocol for the isolation of CD8+ tumor-infiltrating lymphocytes from human tumors and their characterization by single-cell immune profiling and multiome. STAR Protoc. 3, 101649 (2022).
Storrs, E. P. et al. High-dimensional deconstruction of pancreatic cancer identifies tumor microenvironmental and developmental stemness features that predict survival. npj Precis. Oncol. 7, 105 (2023).
Lin, W. et al. Single-cell transcriptome analysis of tumor and stromal compartments of pancreatic ductal adenocarcinoma primary tumors and metastatic lesions. Genome Med. 12, 80 (2020).
Chen, K. et al. Single-cell transcriptome profiling of primary tumors and paired organoids of pancreatobiliary cancer. Cancer Lett. 582, 216586 (2024).
Kim, S. et al. Integrative analysis of spatial and single-cell transcriptome data from human pancreatic cancer reveals an intermediate cancer cell population associated with poor prognosis. Genome Med. 16, 20 (2024).
Chamberlain, M. et al. Cell type classification and discovery across diseases, technologies and tissues reveals conserved gene signatures of immune phenotypes. J. Bioinform Syst. Biol. 6, 152 (2023).
Nouri, N., Gaglia, G., Kurlovs, A. H., de Rinaldis, E. & Savova, V. A marker gene-based method for identifying the cell-type of origin from single-cell RNA sequencing data. MethodsX 10, 102196 (2023).
Nouri, N., Kurlovs, A. H., Gaglia, G., de Rinaldis, E. & Savova, V. Scaling up single-cell RNA-seq data analysis with CellBridge workflow. Bioinformatics 39, btad760 (2023).
Puntambekar, S., Hesselberth, J. R., Riemondy, K. A. & Fu, R. Cell-level metadata are indispensable for documenting single-cell sequencing datasets. PLoS Biol. 19, e3001077 (2021).
LangGraph. Overview: https://langchain-ai.github.io/langgraph/concepts/why-langgraph/ (2025).
Wolf, F. A., Angerer, P. & Theis, F. J. SCANPY: large-scale single-cell gene expression data analysis. Genome Biol. 19, 1–5 (2018).
McInnes, L., Healy, J. & Melville, J. Umap: Uniform manifold approximation and projection for dimension reduction. Preprint at https://arxiv.org/abs/1802.03426 (2018).
Amir, E.-A. D. et al. viSNE enables visualization of high dimensional single-cell data and reveals phenotypic heterogeneity of leukemia. Nat. Biotechnol. 31, 545–552 (2013).
Setty, M. et al. Characterization of cell fate probabilities in single-cell data with Palantir. Nat. Biotechnol. 37, 451–460 (2019).
Wolock, S. L., Lopez, R. & Klein, A. M. Scrublet: computational identification of cell doublets in single-cell transcriptomic data. Cell Syst. 8, 281–291.e289 (2019).
Korsunsky, I. et al. Fast, sensitive and accurate integration of single-cell data with Harmony. Nat. Methods 16, 1289–1296 (2019).
Lopez, R., Regier, J., Cole, M. B., Jordan, M. I. & Yosef, N. Deep generative modeling for single-cell transcriptomics. Nat. Methods 15, 1053–1058 (2018).
Heimberg, G. et al. A cell atlas foundation model for scalable search of similar human cells. Nature 638, 1085–1094 (2025).
Domínguez Conde, C. et al. Cross-tissue immune cell analysis reveals tissue-specific features in humans. Science 376, eabl5197 (2022).
Good, J. D., Safina, K. R., Miller, T. E. & van Galen, P. Protocol for mitochondrial variant enrichment from single-cell RNA sequencing using MAESTER. STAR Protoc. 6, 103564 (2025).
Yates, J., Kraft, A. & Boeva, V. Filtering cells with high mitochondrial content depletes viable metabolically altered malignant cell populations in cancer single-cell studies. Genome Biol. 26, 91 (2025).
Luecken, M. D. & Theis, F. J. Current best practices in single-cell RNA-seq analysis: a tutorial. Mol. Syst. Biol. 15, e8746 (2019).
Kim, G. D., Lim, C. & Park, J. A practical handbook on single-cell RNA sequencing data quality control and downstream analysis. Molecules Cells 47, 100103 (2024).
Heumos, L. et al. Best practices for single-cell analysis across modalities. Nat. Rev. Genet. 24, 550–572 (2023).
Tian, L. et al. Benchmarking single cell RNA-sequencing analysis pipelines using mixture control experiments. Nat. Methods 16, 479–487 (2019).
10X-Genomics. Cell Ranger. https://www.10xgenomics.com/support/software/cell-ranger/latest (2025).
Filippov, I., Philip, C. S., Schauser, L. & Peterson, P. Comparative transcriptomic analyses of thymocytes using 10x Genomics and Parse scRNA-seq technologies. BMC Genom. 25, 1069 (2024).
Parks, B. & Greenleaf, W. Scalable high-performance single cell data analysis with BPCells. Preprint at https://www.biorxiv.org/content/10.1101/2025.03.27.645853v1 (2025).
Acknowledgements
We would like to extend our gratitude to the developers and maintainers of the various software packages and tools that were instrumental to this research. We thank Dr. Natasha Markuzon (Oncology Data Science & AI, AstraZeneca) for curating the list of datasets used in our benchmarking experiments. Funding provided by AstraZeneca US.
Author information
Authors and Affiliations
Contributions
N.N. and R.A.: Conceptualization. N.N.: Methodology, software development, and original manuscript drafting. N.N., R.A., and V.S.: Manuscript review, editing, and content refinement. V.S.: Supervision and strategic direction.
Corresponding authors
Ethics declarations
Competing interests
The authors are employees of AstraZeneca US.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Nouri, N., Artzi, R. & Savova, V. An agentic AI framework for ingestion and standardization of single-cell RNA-seq data analysis. npj Artif. Intell. 2, 8 (2026). https://doi.org/10.1038/s44387-025-00064-0
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s44387-025-00064-0
