
Abstract
It is unclear to what extent genetic risk offsets the protective effects of better premorbid cognitive health on the risk of Alzheimer’s disease (AD). We tested for associations between measures of premorbid cognitive health, apolipoprotein (APOE) e4 ‘risk’ genotype, and their interaction, with risk of incident AD and age of diagnosis, in UK Biobank participants aged ≥55 years at baseline, adjusted for potential confounders. During follow-up, 3,505/252,340 (1.39%) participants received an incident diagnosis of AD. There were significant associations between better performance on each cognitive test with lower risk of incident AD, and later age at diagnosis. However, the benefit of better baseline cognitive scores on AD risk was significantly attenuated in APOE e4 carriers. These data demonstrate that the association between premorbid cognitive health and subsequent risk of AD is influenced by APOE e4 genotype. This has implications for risk stratification and targeted intervention.
Similar content being viewed by others


Introduction
Alzheimer’s disease (AD) is a neurodegenerative disorder with a significant public health impact1. To date, strategies to identify effective therapies for AD have been largely inffective2,3. As such, there is an increasing focus on identifying measures to prevent and delay the onset of disease to reduce the overall burden. There is a necessity in that context for targeted intervention and stratification, and thus a need to identify populations at heightened risk. While multiple individual risk factors are recognised for AD4,5, their interactions are less well-understood.
The largest single risk factor for AD after increasing age is possession of an e4 allele in the apolipoprotein e (APOE) gene, where one copy (heterozygous) increases risk around three-fold and two copies (homozygous) increases risk around twelve-fold (versus neutral e3e3 genotype), and each associated with earlier age at onset6. APOE e4 may interact with known risk factors for cognitive impairment in ageing, although evidence for this is inconsistent7,8. Poorer non-demented cognitive abilities (generally) are a consistent predictor of worse later-life outcomes9, including mortality and AD risk1. This observation might be due to some of the following: worse cognitive scores reflecting prodromal decline (towards dementia)2; lower cognitive reserve10 (where people appear to show poor structural/functional brain health but are cognitively healthy); cognitive faculties that influence lifestyle11—which secondarily increase AD risk12—and/or that cognitive health reflects a broad ‘system integrity’ indicative of general physical health13.
Despite the known contribution of both premorbid cognitive health and APOE e4 genotype to AD risk, there is a significant gap in the literature regarding their interaction. The present study will test the hypothesis that, among people who possess an APOE e4 allele, there will be a weaker association between healthier baseline cognition (memory, reasoning and information processing speed) and risk of AD (in terms of incidence and age of onset) than among non-carriers. Specifically, we hypothesise that the association between better premorbid cognitive health with better AD outcomes—lower incidence and later age of diagnosis—will be significantly attenuated in e4 carriers (e3e4; e4e4) versus non-carriers (e3e3; e2e2; e2e3).
Results
Descriptive statistics
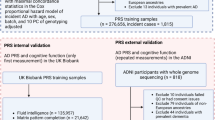
After described genetic quality-controlled (QC) exclusions, 252,340 participants aged ≥55 were included in this study, of whom 117,869 were male (46.7%), with an average baseline age of 62.13 (SD = 4.06). Following baseline assessment, 3505 participants were diagnosed with AD by end of follow-up, a median 11.45 years after baseline (range = 0.6–16.6; median 11.48 after excluding 20 participants with onset within 2 years of baseline) with an average age at diagnosis of 75.94 (SD = 4.44) In terms of APOE genotype, 61,325 participants (24.3%) had one e4 allele while 6178 (2.5%) were homozygous. Allele frequencies were similar when stratified by sex. Detailed demographics are shown in Supplementary Table 2.
Association of APOE e4 with incident Alzheimer’s disease
In fully adjusted models, APOE e4 allele presence was associated with increased risk of incident AD (HR = 4.70, 95% confidence interval (CI) = 4.38–5.04, P < 0.001). More specifically, this was dose-dependent: it was higher among those with homozygous e4e4 genotype (HR = 13.16, 95% CI = 11.85–14.61, P < 0.001; versus no e4 allele) than among those with a single copy of e4 (HR = 3.93,95% CI = 3.65–4.23, P < 0.001; versus no e4).
The relationship between the APOE e4 genotype and risk of AD diagnosis appeared sex-dependent. There was evidence of sex/APOE e4 genotype interaction (P < 0.001) where the magnitude of additional risk conferred by e4 presence was larger in females (HR = 5.64, 95% CI = 5.11–6.23; P < 0.001) compared with males (HR = 3.88, 95% CI = 3.51—4.29; P < 0.001). Female e4 carriers had the highest risk of all groups (see Fig. 1).

Adjusted for age at time of assessment, Townsend deprivation index, 10 genetic principal components for stratification, university/college degree, ever-smoking history, genotypic array and history of coronary heart disease, hypertension and diabetes. The Y-axis represents the relative probability of remaining disease-free.
An overall APOE e4 present (versus absent) effect on age of diagnosis was non-significant (unstandardised beta years = −0.20, 95% CI = −0.40 to 0.01, P = 0.062). There was a significant association between e4e4 genotype and earlier age at onset (unstandardised beta = −0.56 years, 95% CI = −0.87 to −0.26, P < 0.001; versus no e4). The average age of onset for a non-e4 carrier was 76.07 years (standard error [SE] = 0.08), and for e4e4 homozygotes, the average age of onset was 75.51 years (SE = 0.13).
Association of premorbid cognition with incident Alzheimer’s disease
There were consistent associations between premorbid cognitive scores and risk of incident AD (see Table 1). There were significant adjusted associations between cognitive test scores and age at diagnosis for log RT (unstandardised beta = −0.37 years, 95% CI = −0.47 to −0.28, P < 0.001) and verbal-numerical reasoning (0.58 years, 95% CI = 0.38–0.78, P < 0.001), but not memory scores (see Table 2). Higher RT scores reflect worse performance, while higher reasoning scores reflect better performance. This means that for each SD, better RT and reasoning scores, the age of diagnosis is on average ~0.4 and 0.6 years later, respectively. There was no e4 allele/sex interaction nor any e4 allele/cognitive score interactions (all P > 0.10), whether analysed as e4 present/absent or homozygous versus not.
APOE e4 attenuates the influence of premorbid cognition on the risk of incident AD diagnosis
There was significant interaction between e4 presence and verbal-numerical reasoning scores (P = 0.005) on risk of AD. This was such that the HRs for cognitive scores versus risk of AD were closer to 1 in participants who possessed an e4 allele: i.e. the association of better premorbid cognitive score, with lower likelihood of a subsequent AD diagnosis, was attenuated in e4 carriers. The beneficial association between better verbal-numerical reasoning with lower AD risk, was stronger in non-e4 carriers (HR = 0.64) compared with e4 carriers (HR = 0.79) (see Fig. 2). There was no interaction between memory scores and e4 genotype on AD incidence (P > 0.05), and only marginally with log RT (P = 0.048; above our P > 0.01 threshold).

Adjusted for age at time of assessment, sex, Townsend deprivation index, 10 genetic principal components for stratification, university/college degree versus not, ever-smoking history, genotypic array and history of coronary heart disease, hypertension and diabetes. ‘Good’ is >1 SD above the mean; ‘poor’ is ≤1 SD below the mean, and ‘average’ is intermediate. The Y-axis represents the relative probability of remaining disease-free.
Sensitivity analyses
Results were unchanged when we removed n = 20 participants diagnosed within 2 years of assessment; controlled for participants with a baseline neurological condition; and/or tested for cognitive/AD associations uncorrected for sociodemographic and lifestyle factors. The e4/AD associations were very similar univariately (i.e. controlling for only 10 PCs and array) as per fully adjusted. Fully adjusted models gave very similar results when self-reported conditions (e.g. diabetes) were supplemented with ICD9/10 data. Results were similar when we tested for interaction between APOE e4 allele dosage (0/1/2), baseline cognitive scores, and incident AD. This analysis was not possible for the reasoning task because no participant with the relatively rarer e4e4 genotype had baseline reasoning data, and subsequently developed AD. When we repeated analyses removing APOE e2e2 and e2e3 carriers (n = 33,892), i.e. specifically comparing e3e3 versus e4 carriers, all above findings were virtually identical. The only difference was that the previously marginal interaction between log RT and APOE e4 genotype on log RT scores (above), attenuated from P = 0.048 to P = 0.077.
Discussion
Premorbid cognitive health predicts the likelihood of subsequent AD, and APOE e4 is the largest AD risk factor after increasing age1,14. The utility of cognitive scores may reflect better baseline system integrity, healthier average lifestyles, cognitive reserve and/or lower current test scores due to some prodromal decline towards dementia10,15,16. Around 42% of AD can be statistically attributed to APOE e4 genotype17 (via population attributable fraction), potentially due to its highly pleiotropic effects across multiple biological pathways18 (e.g. amyloid beta clearance19).
Here, we show that, firstly, better baseline premorbid cognitive ability lowers the risk of subsequent AD incidence. Secondly, better cognitive health is associated with later diagnosis in those who were ultimately diagnosed with AD. Finally, we show that APOE e4 genotype presence attenuates the association between better baseline cognition and AD incidence: risk reduction goes from 36% (e4 absent) to 21% (e4 present) per one SD of better reasoning scores. We therefore provide evidence of APOE genotype impacting disease trajectories20. These findings have relevance for stratification of high-risk populations in whom targeted intervention (e.g. brain health clinics) may be more appropriate21. In particular, our findings show that the e4 genotype is a more important predictor than premorbid cognitive health. Participants with ‘good’ reasoning scores but who possess an e4 allele are at a higher average risk for AD than participants with ‘poor’ scores with no e4 allele.
The association between APOE e4 genotype and poorer later-life brain health appears primarily via accelerated longitudinal decline rather than cross-sectional abilities22,23. We have previously shown modest association between the APOE e4 genotype and cross-sectional cognitive abilities in the UK Biobank (UKB)8. This phenomenon—predominantly longitudinal rather than cross-sectional influence—aligns with the observation in this report that while better cognitive scores were associated with lower disease risk, this was partly mitigated in e4 carriers. We report additive individual significant associations between worse cognitive scores and separately APOE e4 with earlier onset (i.e. no interaction). Delayed onset of 6 months, for example, at the population level would have substantial public health benefits5. The tractable aspects of these observations could include aspects of APOE e4 genotypic modification19, including risk stratification, and/or modification of lifestyle risk factors, which may be part of the cognition/onset pathway12.
It is unclear if dementia can be fundamentally prevented: this assertion requires large-scale population data, including comprehensive data to the point of death, to demonstrate that an intervention or modifiable factor categorically prevents rather than delays dementia, and these data are relatively scarce5. Binary yes/no diagnosis is a broad, potentially coarse phenotype which may have misclassification biases and not capture detailed individual differences—there is a need to move towards more fine-grained, secondary dementia phenotyping24. There is some evidence of earlier onset of AD in APOE e4 carriers, although these studies tend to be relatively small25 where the largest studies to date are N = 126226 and N = 175027. Age of diagnosis in AD reflects a relatively more fine-grained phenotype than outright diagnosis25, and later average age of diagnosis is a potential manifestation of preserved cognitive function. Here we show that better cognitive ability appears to protect individuals from earlier diagnosis, on average.
Calvin et al.1 previously demonstrated the cognitive health/risk of AD association1 and here we extend that beyond outright AD risk, to later onset in people with better baseline cognitive health. This could reflect better cognitive health/reserve, or it may reflect a degree of decline in people in the AD prodrome2. Effects of APOE genotype on AD are well-observed14, and we reinforce prior observations that the e4 genotype has a deleterious effect on age of diagnosis25, although only in homozygotic participants here on average. There is potential distinction between the age of (symptom) ‘onset’ compared with the age at ‘diagnosis’ based on largely ICD-10 codes, where diagnosis would be later but more conservative. The relative lack of association from e4 present versus absent, compared with other samples which show stronger overall e4 effects, suggests differences between UKB versus other cohorts, whether in terms of ascertainment or other characteristics. It may reflect acknowledged ‘healthy bias’ in UKB, perhaps in a more age- or sex-specific manner than has been considered to date28. Ascertainment differs from some prior studies, which report age of earliest impairment, or reported onset5, whereas we primarily report formal diagnoses based on ICD9/10. There will be a degree of slight inaccuracy in our data because birthdates were set to the 15th of the month. This is unlikely to be of a magnitude to significantly affect estimates: participants can only be a maximum of 2 weeks from their ‘true’ age of diagnosis. There will be other variables influencing that phenotype, for example delays in seeking diagnosis and waiting times.
UKB has well-documented selection bias, which may influence exposure/outcome estimates28. Age of diagnosis was based on formal codes (largely from secondary hospital records), and this may not reflect the earliest sign of cognitive dysfunction. However, there is no clear reason that this should differ systematically by predictor status, and therefore average differences between predictors may at least be reflective of ‘real’ differences.
The UKB cognitive tests have certain limitations: they are bespoke, although they have shown reasonable validation with more traditional cognitive assessments, and have been shown to correlate with aspects of brain structure29. Some weaknesses include floor effects on the memory test, relatively brief duration (e.g. the reasoning test has a 2-min time limit), relatively few trials on the reaction time test, and that all tests have a ‘speed’ component where ideally a ‘pure’ isolated memory test would not30. While UKB has a range of cognitive assessments, including subsequent online assessments, we elected for the baseline cognitive data (memory errors; reaction time; fluid intelligence), because these are mostly in the full cohort, maximising sample size, duration of follow-up and consequent statistical power.
These findings reflect average level differences in cognitive health versus AD outcomes; there is likely to be significant inter-individual explanations for poorer cognitive health. We are therefore not yet at the point where an individual’s genetic and cognitive information combined can reliably predict clinical aspects of AD onset24.
There is likely some degree of reverse causality where participants have an amount of AD-caused reductions in cognitive scores at baseline, and subsequently became diagnosed. We have attempted to minimise this by looking at people aged 55 and over, i.e. a number of years before AD would be typically expected, and sensitivity analyses removing the small number of participants with AD less than 2 years after cognitive assessment. There is likely some degree of residual confounding in our findings. Future studies, particularly with additional follow-up and wider ascertainment of cases, e.g. from general practice data, may be appropriately powered to adjust for potential confounders such as historic stroke.
We have focussed on white British participants here because there is complex interplay between both allele frequency differences in APOE e genotypes31 as well as phenotypic exposure/outcome associations across ethnicities32. Belloy et al. demonstrated in a diverse cohort (N = 67,656) that AD risk conferred by APOE e genotypes varies by ethnicity. Our sample N and cohort composition is not appropriately powered for such subdivisions, and therefore we focussed on the more homogenous White British population33. Future studies should seek to replicate these findings in more diverse cohorts34, in terms of ethnicity, but also to a more representative cohort relative to the general population. Future studies may investigate if longitudinal cognitive change is a superior predictor of AD onset rather than snapshot cross-sectional abilities. Future studies may investigate the role of the protective e2 genotype (versus neutral e3e3), any modifying role of non-AD polygenic risk for dementia35, and/or non-AD dementias like vascular.
APOE e4 genotype significantly modified associations between reasoning scores and AD, but less so for processing speed (i.e. nominally significant at P < 0.05 but not our more conservative threshold), and not memory errors. This may reflect ‘true’ modification of the link between specific cognitive faculties and AD (i.e. reasoning particularly), or rather that the different tests vary in how well they measure differences in cognitive health. We have previously reported aspects of these psychometric properties30. The memory score has a recognised ‘floor effect’ (i.e. many participants scored zero errors), and the RT task is averaged over only four trials. In a subsample of ~20k UKB participants with repeated cognitive data, the reasoning test showed the best reliability (Pearson r = 0.7) compared with RT (r = 0.5–0.6) and memory errors (r = 0.2). Fawns-Ritchie and Deary36 demonstrated in a novel cohort (N = 160) that the reasoning test correlated consistently around r = 0.2–0.4 with a battery of established cognitive assessments, e.g. from the Wechsler Adult Intelligence Scale-IV. By contrast, the memory/RT scores were less consistently correlated with scores on established cognitive tests, and often were not significantly correlated. Alternatively, the reasoning test was completed in only around ~150k of the full 502k UKB participants because it was introduced mid-way through the baseline assessment; it may be that there is non-random bias in who completed this test, which influences the observation. Future study in the expanded battery of cognitive tests, which were conducted several years post-baseline, may be informative with regard to the specificity of APOE genotype modifying only the reasoning/AD association.
Non-demented cognitive health and APOE e genotype are established, significant factors in terms of AD risk. Here we demonstrate largely independent roles for the two on AD, as phenotyped in two ways: overall risk and age of diagnosis in cases. Adjusted for a range of potential confounders, we show that APOE e4 allele presence weakens some of the protective association between healthier cognition in early ageing, with a lower risk of AD. This may reflect a variety of pathways but has potential routes to mitigation strategies, e.g. via interventional brain health clinics in high-risk populations. Future studies may broaden this to wider genetic approaches, e.g., including non-APOE genetic variation.
Methods
Cohort
UKB is a prospective general population cohort, where ~502,000 participants attended one of 22 assessment centres in Scotland, England and Wales, between 2006 and 201037. Participants were aged 40–70 years at baseline, and non-demented participants were intentionally recruited; we excluded the minority of participants with a pre-baseline diagnosis (n = 11). This project was completed using UKB project 17689.
Demographics
Age, sex, ethnicity and educational attainment were self-reported. The Townsend deprivation index was derived from the postcode of residence38. This provides an area-based measure of socioeconomic deprivation derived from aggregated data on car ownership, household overcrowding, owner occupation, and unemployment. Higher Townsend scores equate to higher levels of area-based socioeconomic deprivation.
Cognitive test variables
We examined tests that were included as part of the bespoke UKB baseline cognitive assessment30. One of these was a task with thirteen logic/reasoning-type questions and a 2-min time limit labelled ‘fluid intelligence’ in the UKB protocol, but hereafter (commonly) referred to as verbal-numerical reasoning36. The maximum score was 13, where higher scores indicate better performance. ‘Pairs matching’ was a visuospatial memory test, where participants were asked to memorise the positions of six card pairs, and then match the pairs from memory while making as few errors as possible. We refer to pairs matching as the memory test from here on. Scores on the memory test are for the number of errors that each participant made, and higher scores are therefore worse. Participants completed a timed test of symbol matching similar to the common card game Snap, referred to hereafter as the reaction time task; scores are measured in milliseconds, with higher values indicating worse performance. The verbal-numerical reasoning task was added partway through the baseline assessments; this was completed in around one-third of baseline participants. These tests have been described and their reliability and validity reported previously30,36. We did not use the baseline cognitive tests of Prospective memory or Numeric memory, because the former had relatively little variation and the latter was in a relatively small subsample of participants.
Lifestyle
Participants self-reported their smoking history: current, past or never. We collated past and current smokers into ‘ever’ (vs. never). Using self-report, participants responded to the touch-screen question ‘Has a doctor ever told you that you have had any of the following conditions?’ (high blood pressure, hereafter ‘hypertension’; stroke; angina; heart attack), where each was coded as binary absent/present. We collated self-reported heart attack and angina into coronary heart disease (CHD). Participants were also asked ‘Has a doctor ever told you that you have diabetes?’ via a touch-screen questionnaire39.
Alzheimer’s disease ascertainment
AD was ascertained based on the ‘first occurrences’ UKB field, which collates diagnoses from self-report, hospital primary/secondary, death contributory/primary and death-only records. The majority of cases are ascertained from hospital primary/secondary data rather than death data (https://biobank.ndph.ox.ac.uk/ukb/field.cgi?id=42021). Cases were only analysed if they occurred after baseline (the vast majority of instances). The definitions are described in detail in an open-access document including classification codes (https://biobank.ndph.ox.ac.uk/ukb/refer.cgi?id=460).
Age of diagnosis was calculated from the date of diagnosis minus (the 15th of) month and the year of birth. The 15th was used because exact date of birth is a (UKB) protected phenotype, and the 15th is such that a participant’s birthday could only be a maximum of ~2 weeks away. Follow-up for all incidence analyses was censored at 31st October for England, 31st May for Wales, or 31st August 2022 for Scotland (https://biobank.ndph.ox.ac.uk/ukb/exinfo.cgi?src=Data_providers_and_dates), individual date of loss-to-follow-up (<1%) or all-cause death—whichever occurred earlier.
Genotyping
UKB genotyping was conducted by Affymetrix using a bespoke BiLEVE Axiom array for ~50,000 participants, and the remaining ~450,000 on the Affymetrix Axiom array40. All genetic data were QC and imputed centrally by UKB, e.g. principal components for stratification, genotypic batch/array, etc. The APOE e locus is directly genotyped based on two single-nucleotide polymorphisms: rs429358 and rs7412. Further information on the genotyping process is available on the UKB website (http://www.ukbiobank.ac.uk/scientists-3/genetic-data), which includes detailed technical documentation for QC and imputation.
Analyses
We used regressions models adjusting for age at time of assessment, sex, Townsend deprivation score, education (college or university degree; yes/no), 10 genetic principal components (PCs) for stratification, genotypic array, history of CHD, diabetes, hypertension, and history of ever-smoking because each of these are potential confounders or risk factors.
We report uncorrected P values throughout, and conservatively consider P < 0.01 nominally significant. We report Cox regressions for AD incidence (hazard ratios/HR, including 95% CIs), and linear regressions for predictors versus age at diagnosis (unstandardised and standardised betas, then translated into per-month differences where significant). Proportional hazard assumptions were checked with the post-estimate phtest Stata function. Stata version 18 was used for all analyses41. Variables with non-normal distributions were natural-log transformed: namely, reaction time and memory test errors (+1).
Exclusions
We excluded participants with a baseline age <55 years because this study aimed to investigate the role of premorbid, non-AD affected cognition in people who have a realistic possibility of late-onset AD by the end of study follow-up. We excluded participants with self-reported non-white British ancestry, self-report vs. genetic sex mismatch, putative sex chromosomal aneuploidy, excess heterozygosity, and allelic missingness rate >0.1. We focussed on white British participants (87% of the cohort) specifically because there is evidence that the APOE e genotypic effect on AD risk varies by ethnicity42, and the cohort was not appropriately powered to test for such granular interactions. We removed one random participant in cases where two individuals were 2nd cousins or closer, based on relatedness quotient. We removed participants with the relatively rare e2e4 genotype, which includes protective/deleterious variants, respectively. Neurological conditions in 5% of participants were controlled for as a sensitivity analysis, and these are listed in Supplementary Table 1.
Data availability
UKB is an open-access resource available to verified researchers upon application, at https://www.ukbiobank.ac.uk/.
Code availability
Analysis syntax is available upon request.
References
Calvin, C. M. et al. Predicting incident dementia 3-8 years after brief cognitive tests in the UK Biobank prospective study of 500,000 people. Alzheimers Dement.15, 1546–1557 (2019).
Sperling, R. A. et al. Toward defining the preclinical stages of Alzheimer’s disease: recommendations from the National Institute on Aging-Alzheimer’s Association workgroups on diagnostic guidelines for Alzheimer’s disease. Alzheimers Dement. 7, 280–292 (2011).
Dobson, R. et al. Eligibility for antiamyloid treatment: preparing for disease-modifying therapies for Alzheimer’s disease. J. Neurol. Neurosurg. Psychiatry 95, 796–803 (2024).
Livingston, G. et al. Dementia prevention, intervention, and care: 2024 report of the Lancet standing Commission. Lancet 404, 572–628 (2024).
Crenshaw, D. G. et al. Using genetics to enable studies on the prevention of Alzheimer’s disease. Clin. Pharmacol. Ther. 93, 177–185 (2013).
Tzioras, M., Davies, C., Newman, A., Jackson, R. & Spires-Jones, T. Invited Review: APOE at the interface of inflammation, neurodegeneration and pathological protein spread in Alzheimer’s disease. Neuropathol. Appl. Neurobiol. 45, 327 (2019).
Bos, M. M. et al. The ApoE ε4 Isoform: Can the risk of diseases be reduced by environmental factors?. J. Gerontol. Ser. A 74, 99–107 (2019).
Lyall, D. M. et al. Assessing for interaction between APOE ε4, sex, and lifestyle on cognitive abilities. Neurology 92. https://doi.org/10.1212/WNL.0000000000007551 (2019).
Deary, I. J., Hill, W. D. & Gale, C. R. Intelligence, health and death. Nat. Hum. Behav. 5, 416–430 (2021).
Stern, Y. How can cognitive reserve promote cognitive and neurobehavioral health?. Arch. Clin. Neuropsychol. 36, 1291–1295 (2021).
Corley, J., Cox, S. R. & Deary, I. J. Healthy cognitive ageing in the Lothian Birth Cohort studies: marginal gains not magic bullet. Psychol. Med. 48, 187–207 (2018).
Korologou-Linden, R. et al. The causes and consequences of Alzheimer’s disease: phenome-wide evidence from Mendelian randomization. Nat. Commun. 13, 1–14 (2022).
Deary, I. J. Looking for ‘system integrity’ in cognitive epidemiology. Gerontology 58, 545–553 (2012).
Riedel, B. C., Thompson, P. M. & Brinton, R. D. Age, APOE and sex: triad of risk of Alzheimer’s disease. J. Steroid Biochem. Mol. Biol. 160, 134–147 (2016).
Deary, I. J. et al. Apolipoprotein e gene variability and cognitive functions at age 79: a follow-up of the Scottish mental survey of 1932. Psychol. Aging 19, 367–371 (2004).
Deary, I. Why do intelligent people live longer?. Nature 456, 175–176 (2008).
Williams, D. M., Davies, N. M. & Anderson, E. L. The proportion of Alzheimer’s disease attributable to apolipoprotein E. medRxiv (2024).
Bu, G. Apolipoprotein E and its receptors in Alzheimer’s disease: pathways, pathogenesis and therapy. Nat. Rev. Neurosci. 10, 333–344 (2009).
Yu, J.-T., Tan, L. & Hardy, J. Apolipoprotein E in Alzheimer’s disease: an update. Annu. Rev. Neurosci. 37, 79–100 (2014).
Narasimhan, S. et al. Apolipoprotein E in Alzheimer’s disease trajectories and the next-generation clinical care pathway. Nat. Neurosci. https://doi.org/10.1038/S41593-024-01669-5 (2024).
Ritchie, C. W. et al. The Scottish Brain Health Service Model: rationale and scientific basis for a national care pathway of brain health services in Scotland. J. Prev. Alzheimer’s Dis. 9, 348–358 (2022).
Lyall, D. M. et al. Are APOE ɛ genotype and TOMM40 poly-T repeat length associations with cognitive ageing mediated by brain white matter tract integrity?. Transl. Psychiatry 4, e449 (2014).
Schiepers, O. J. G. et al. APOE E4 status predicts age-related cognitive decline in the ninth decade: longitudinal follow-up of the Lothian Birth Cohort 1921. Mol. Psychiatry 17, 315–324 (2011).
Lyall, D. M. et al. Artificial intelligence for dementia—applied models and digital health. Alzheimers Dement. 19, 5872–5884 (2023).
Lutz, M. W., Crenshaw, D. G., Saunders, A. M. & Roses, A. D. Genetic variation at a single locus and age of onset for Alzheimer’s disease. Alzheimers Dement. 6, 125–131 (2010).
van der Lee, S. J. et al. The effect of APOE and other common genetic variants on the onset of Alzheimer’s disease and dementia: a community-based cohort study. Lancet Neurol. 17, 434–444 (2018).
Smirnov, D. S., Galasko, D., Hiniker, A., Edland, S. D. & Salmon, D. P. Age-at-onset and APOE-related heterogeneity in pathologically confirmed sporadic Alzheimer disease. Neurology 96, E2272–E2283 (2021).
Lyall, D. M. et al. Quantifying bias in psychological and physical health in the UK Biobank imaging sub-sample. Brain Commun. https://doi.org/10.1093/BRAINCOMMS/FCAC119 (2022).
Cox, S. R., Ritchie, S. J., Fawns-Ritchie, C., Tucker-Drob, E. M. & Deary, I. J. Structural brain imaging correlates of general intelligence in UK Biobank. Intelligence https://doi.org/10.1016/j.intell.2019.101376 (2019).
Lyall, D. M. et al. Cognitive test scores in UK Biobank: data reduction in 480,416 participants and longitudinal stability in 20,346 participants. PLoS ONE 11, https://doi.org/10.1371/journal.pone.0154222 (2016).
Eisenberg, D. T. A., Kuzawa, C. W. & Hayes, M. G. Worldwide allele frequencies of the human apolipoprotein E gene: climate, local adaptations, and evolutionary history. Am. J. Phys. Anthropol. 143, 100–111 (2010).
Ntuk, U. E., Gill, J. M. R., Mackay, D. F., Sattar, N. & Pell, J. P. Ethnic specific obesity cut-offs for diabetes risk: cross-sectional study of 490,288 UK Biobank participants. Diab. Care 37, 2500–2507 (2014).
Belloy, M. E. et al. APOE genotype and Alzheimer disease risk across age, sex, and population ancestry. JAMA Neurol. 80, 1284–1294 (2023).
Perianayagam, A. et al. Cohort profile: the Longitudinal Ageing Study in India (LASI). Int. J. Epidemiol. 51, e167–e176 (2022).
Tank, R. et al. Association between polygenic risk for Alzheimer’s disease, brain structure and cognitive abilities in UK Biobank. Neuropsychopharmacology 47, 564–569 (2021).
Fawns-Ritchie, C. & Deary, I. J. Reliability and validity of the UK Biobank cognitive tests. PLoS ONE 15, e0231627 (2020).
Sudlow, C. et al. UK Biobank: an open access resource for identifying the causes of a wide range of complex diseases of middle and old age. PLoS Med. 12, e1001779 (2015).
Townsend, P. Townsend Deprivation Index. National Database for Primary Care Groupś and Trusts. (1998).
Lyall, D. M. et al. Associations between single andmultiple cardiometabolic diseases and cognitive abilities in 474 129 UK Biobank participants. Eur. Heart J. 38, 577–583 (2017).
Bycroft, C. et al. The UK Biobank resource with deep phenotyping and genomic data. Nature https://doi.org/10.1038/s41586-018-0579-z (2018).
StataCorp. Stata Statistical Software: Release 12. College Station, TX: StataCorp LP. http://www.stata.com/support/faqs/resources/citing-software-documentation-faqs/ (2011).
Belloy, E.M, Napolioni, V. & Greicius, M. D. Review: A quarter century of APOE and Alzheimer’s disease: progress to date and the path forward. https://doi.org/10.1016/j.neuron.2019.01.056 (2019).
Acknowledgements
This research has been conducted using the UK Biobank resource; we are grateful to UK Biobank participants and staff. UK Biobank was established by the Wellcome Trust medical charity, Medical Research Council (MRC), Department of Health, Scottish Government and the Northwest Regional Development Agency. It has also had funding from the Welsh Assembly Government and the British Heart Foundation. L.G. is funded by the King's College London DRIVE-Health Centre for Doctoral Training and the Perron Institute for Neurological and Translational Science. R.J.S. was supported by a University of Glasgow LKAS fellowship and a UKRI Innovation-HDR-UK Fellowship (MR/S003061/1). L.M.L. is funded by The John, Margaret, Alfred and Stewart Sim Fellowship, and a University of Glasgow Lord Kelvin Adam Smith Fellowship.
Author information
Authors and Affiliations
Contributions
Study concept and design: M.J. and D.M.L. Statistical analysis: D.M.L. Drafted the manuscript: M.J. and D.M.L. Critically revised content: M.J., J.F., A.K.K., R.T., L.G., E.R.R., L.M.L., J.W., J.J.A., R.J.S., T.Q., W.S., I.J.D., S.R.C. and D.M.L.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Ethical approval
This secondary-data analysis study was conducted under generic approval from the NHS National Research Ethics Service (approval letter dated 13th May 2016, ref 16/NW/0274). Written informed consent was obtained from all participants recruited to the UKB.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Jareebi, M.A., Fullerton, J., Kancheva, A.K. et al. Associations and interactions between premorbid cognitive health, apolipoprotein e4 genotype, and incident Alzheimer’s disease in UK Biobank (N = 252,340). npj Dement. 1, 8 (2025). https://doi.org/10.1038/s44400-025-00013-3
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s44400-025-00013-3
