
Abstract
A method is described for multi-residue, high-throughput determination of trace levels of 22 organochlorine pesticides (OCPs) and 5 pyrethroid pesticides (PYPs) in Chinese medicinal (CM) health wines using a QuEChERS (quick, easy, cheap, effective, rugged, and safe) based extraction method and gas chromatography-electron capture detection (GC-ECD). Several parameters were optimized to improve preparation and separation time while still maintaining high sensitivity. Validation tests of spiked samples showed good linearities for 27 pesticides (R = 0.9909–0.9996) over wide concentration ranges. Limits of detection (LODs) and quantification (LOQs) were measured at ng/L levels, 0.06–2 ng/L and 0.2–6 ng/L for OCPs and 0.02–3 ng/L and 0.06–7 ng/L for PYPs, respectively. Inter- and intra-day precision tests showed variations of 0.65–9.89% for OCPs and 0.98–13.99% for PYPs, respectively. Average recoveries were in the range of 47.74–120.31%, with relative standard deviations below 20%. The developed method was then applied to analyze 80 CM wine samples. Beta-BHC (Benzene hexachloride) was the most frequently detected pesticide at concentration levels of 5.67–31.55 mg/L, followed by delta-BHC, trans-chlordane, gamma-BHC, and alpha-BHC. The validated method is simple and economical, with adequate sensitivity for trace levels of multi-class pesticides. It could be adopted by laboratories for this and other types of complex matrices analysis.
Similar content being viewed by others


Introduction
Chinese medicinal (CM) health wines have gained wide acceptance due to their high nutritive and medicinal values, unique flavor, and other health functions1,2, and are frequently consumed in Asia, Europe, and other countries. In particular, these wines are playing an ever increasing role in Asian life, culture, and diet, and consumers have strict requirements concerning their quality.
CM health wines are composed of various Chinese medicinal materials (CMMs) which are subject to insect pests or microbial contaminants during the growing process3. A wide range of pesticides are frequently applied during CMMs growth to control pests and increase yield and quality. However, if these harmful chemicals do not degrade naturally or are not completely removed, they are able to penetrate CMM plant tissues4,5. Even at low or ultra-low levels, upon transfer into CM health wines the pesticide residues are potential risk to human health, as they are particularly persistent and have a tendency for bio-accumulation6,7. At present, maximum residue levels (MRLs) for pesticides, including those used for CMMs growth have been established in some countries8,9. These limits, however, have not been applied to processed products, including CM health wines7,10. Consequently, it is currently mandatory to monitor pesticide residues in CM health wines for consumer protection, compliance with some limited standards, and fair trade certification. The large number and volume of pesticides, as well as low MRLs, necessitates highly sensitive and selective sensing methods.
Because CM health wines originate from medicinal plants and herbal materials, they represent very complicated matrices for analysis of pesticide residues. Furthermore, pesticides usually exist at trace (ppm) or lower levels, making the development of analytical methods and highly sensitive instrumentation a great challenge. In recent years, numerous analytical methods have been reported for screening low-levels of pesticide residues in CMMs and wines3,4,11. Among these, the most commonly used are chromatographic methods, such as liquid chromatography (LC) or gas chromatography (GC) coupled with a range of detectors3,4,7,10,11,12,13,14. Although gas chromatography coupled to (tandem) mass spectrometry (GC-MS or GC-MS/MS) has improved the detection procedure, unavoidable matrix effects during the extraction process often interfere with detection of target pesticides, making quantification and analysis of residues erroneous and ambiguous. Due to the wide differences in physical and chemical properties, GC coupled with an electron capture detector (GC-ECD) for organchlorine pesticides (OCPs) and pyrethroid pesticides (PYPs), and with a nitrogen phosphorous detector (GC-NPD) for phosphate pesticides, has been widely used and is officially listed in the Chinese Pharmacopoeia15.
As a rule, pesticide residue analysis in complex matrices is performed using multi-step methods of extraction and pre-concentration, meaning that sample preparation is a crucial step. A variety of classical sample preparation methods, such as solid phase extraction (SPE)16,17,18, liquid-liquid extraction (LLE)19, solid-phase micro-extraction (SPME)20,21, membrane assisted solvent extraction (MASE)22,23, along with several modern methods including stir bar sorptive extraction (SBSE)24,25, and single-drop micro-extraction (SDME)26,27, etc. have been used to analyze pesticides in complex matrices including wines. However, these methods are usually onerous, requiring large sample volumes, special sorbents, and multiple cleanup steps. In recent decades, extensive efforts have been focused on developing new sample preparation techniques that save time, labor, and solvent consumption with the goal of limiting small matrix interferences and thereby improving overall analytical performance. QuEChERS (quick, easy, cheap, effective, rugged and safe) has been accepted as the “gold standard” for analysis of pesticide residues in various sample matrices28,29,30. Several previous studies have reported on the application of QuEChERS-based methods for analysis of multi-pesticide residues in wines31,32,33. To the best of our knowledge, however, there is not been a previous report on the use of a QuEChERS-based sample preparation procedure for the extraction of pesticides from CM health wines. Furthermore, no analytical method has been outlined for trace determination of multi-pesticide residues with regards to OCPs and PYPs in CM health wines.
The primary objective of this work was to develop and validate a simple, fast, sensitive, and reliable analytical method based on a QuEChERS extraction procedure for the simultaneous identification and quantification of 27 pesticides, including 22 OCPs and 5 PYPs, using GC-ECD. Special attention was paid to optimizing the QuEChERS procedure and the GC-ECD conditions. In addition, an extensive validation study was used to evaluate analytical performance. The developed method was then assessed for its real-world applicability by analyzing multi-pesticide residues in 80 CM health wine samples collected from various Chinese markets.
Results and Discussion
Selection of sample preparation technique
Sample preparation is often the most critical issue of any method that deals with multiple residues because of the wide range of polarities, water solubilities, and volatilities of the pesticides that must be simultaneously extracted from the matrix. Co-extraction of interfering substances may lead to low recoveries of trace level pesticides, and should be avoided for efficient extraction. In the present study, two simple and common methods, namely QuEChERS and SPE, were compared for their efficacy in extracting pesticides from CM health wine samples. Results from repeated trials demonstrated that the peak numbers and intensities, together with the extraction efficacy, necessary cleanup and pesticide recovery rate using an SPE method were not better than the results of a QuEChERS technique (data were shown in Supplementary Figure 1). In addition, large amounts of organic solvents were used during the SPE extraction and cleanup, leading to higher costs and a higher possibility of pollution to the environment. Based on the results, extraction of pesticides from CM health wines was performed using a QuEChERS technique.
Optimization of a QuEChERS based sample preparation
Several parameters that influenced the performance of QuEChERS, such as the type of sorbent, the organic solvent, sample volume, and extraction time, were investigated in order to obtain the highest possible high recovery rates of the target pesticides.
Sorbent selection
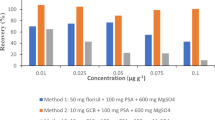
Because CM health wines are such complex matrices, one of the most important steps in the optimization of the QuEChERS procedure was to select an appropriate sorbent that could effectively remove interfering substances. Various sorbents were tested, including PSA, GCB, alumina oxide, and florisil, based on the knowledge that each sorbent is useful for specific purposes. After testing, florisil was chosen as a sorbent due to its low cost, easy cleanup, and high recoveries as compared to the other sorbents.
Next, the amount of sorbent needed to effectively remove target pesticides was determined. A large amount of sorbent can effectively purify pesticides in complex matrices, but with low overall recoveries. To determine the optimum sorbent level for purification efficiency, different amounts of florisil (100, 200, 300, 400 and 500 mg) were tested and compared. Results showed that using 400 mg florisil provided satisfactory purification efficiency as well as high recoveries for 27 pesticides in CM health wine samples.
Extraction solvent optimization
Selection of a suitable extraction solvent is essential for the development of a useful QuEChERS method. In order to achieve high extraction efficiency, organic solvents with different polarities and levels of water solubility were tested and ranked according to their extraction capacity and their overall behavior during GC-ECD analyses. Commonly used solvents with a wide range of polarities, including ethyl acetate, acetone, n-hexane, dichloromethane, or mixture of these solvents were tested. Specific mixtures that were tested included dichloromethane with n-hexane (1:1, v/v), and n-hexane with ethyl acetate (1:1, v/v). Results showed the following: (1) use of dichloromethane as an extraction solvent led to serious emulsification in the sample; (2) use of n-hexane led to a good GC-ECD chromatogram of target analytes (Fig. 1), however some PYP pesticides were adsorbed, leading to low extraction efficacy; (3) in the case of using ethyl acetate (Fig. 2A) or a dichloromethane:n-hexane (1:1, v/v) mixture (Fig. 2B) as an extraction solvent, precise quantification was difficult because the fortified samples were not satisfactorily purified and interfering peaks were observed simultaneously at the retention times of target pesticides; (4) a mixture of n-hexane and ethyl acetate (1:1, v/v) gave higher average peak areas along with better overall recovery rates (70–110%) (Fig. 2C), while also exhibiting the lowest relative standard deviation (RSD) values (data not shown) at the retention times of target analytes, and was therefore the preferred extraction solvent.

GC-ECD chromatograms of (a) standard stock solution, (b) fortified sample solution with standards extracted without cleanup, and (c) fortified sample solution with standards extracted after cleanup with n-hexane as extraction solvent.

GC-ECD chromatograms of fortified sample solutions with standards extracted with (A) ethyl acetate, (B) dichloromethane:n-hexane (1:1, v/v), and (C) ethyl acetate:n-hexane (1:1, v/v) as extraction solvent.
Solvent volume and extraction time optimization
Extraction volume and time both display a positive and significant effect on extraction efficiency of target analytes, and are therefore significant variables to investigate. Raising the volume of the n-hexane:ethyl acetate (1:1, v/v) solvent from 2.0 to 5.0 mL increased the recovery rates of 27 pesticides, while further increase the volume showed no significant improvement. Further experiments tested extraction times and showed that for the mixture of sample solution and n-hexane:ethyl acetate (1:1, v/v) solvent, a 2 min extraction procedure provided the highest recovery of all target pesticides in the sample. Based on the results, 5.0 mL of n-hexane:ethyl acetate (1:1, v/v) solvent and an extraction time of 2 min were chosen for further tests.
Optimization of GC-ECD conditions
Crucial chromatographic parameters including column type, temperature, flow rate, and injection volume were optimized in order to obtain peak specificity and sensitivity for trace amounts of pesticides in CM health wine samples. The overarching goals were satisfactory separation, sensitive detection, and accurate quantification of trace levels of multiple pesticides.
GC capillary column type
Various types of GC columns were tested and an Agilent J & W DB-1701 capillary column, a low/mid-polarity column made of (14%-Cyanopropyl- phenyl)-methylpolysilicone, was chosen as the preferred column because it reduced material loss and provided wonderful separation of the 27 target pesticides.
Column temperature
A GC method with a high initial oven temperature can save analysis and recycling time, as well as improve peak-to-peak separation efficiency and peak shapes. However, too high a temperature may lead to evaporation of volatile substances that can both interfere with targets and contaminate of the analytical column and detector. Therefore, different initial oven temperatures and ramping programs were tested. When the initial temperature increased from 90 to 150 °C there was indeed an obvious decrease in the total run time, however for any temperature above 120 °C, the peak widths of the more volatile pesticides increased significantly and expressed poor peak shapes. Based on the above observed facts and the results shown in Supplementary Table 1, 120 °C was chosen as the initial oven temperature with a ramping program rate of 20 °C/min.
Flow rate
Another essential parameter influencing the separation efficiency of multiple components in GC analysis is the flow rate of nitrogen. To determine the best flow rate, a series of different constant gas flow rates were tested by evaluating the chromatographic separation and signal-to-noise (S/N) ratio of each target pesticide. The best results were achieved using a flow rate of 1.0 mL/min.
Injection volume
Another parameter that needed to be optimized was the injection volume of the samples. Earlier studies have shown that a large injection volume of sample solution generally improves the detection rate in GC analysis34,35, but too large an injection volume exceeded the tolerable limit of the liner and also raised the pressure. After several tests, all further experiments were completed using an injection volume of 1 μL.
Method validation
The optimum QuEChERS and GC-ECD conditions described above were used to determine the presence of any quantity target analytes as a way to verify the real world applicability of the method. The validation procedure was performed following the SANCO/10684/2009 European Guidelines36. The analytical performance characteristics investigated included selectivity, calibration curve, ability to determine a linear calibration curve, the limit of detection (LOD) and quantification (LOQ), precision, stability, and trueness (expressed as recovery rate). Precision was determined by measuring the relative standard deviation (RSD), and was evaluated as inter- and intra-day precision. In this QuEChERS extraction technique, the volume of the extract phase was smaller than that of the sample, and led to an in-exhaustive extraction of the analytes. Therefore, in this case, pesticides with suitable recoveries (70–120%) were included in the validation process.
Selectivity
The selectivity of the proposed method was verified by comparing results from a control (pesticides-free) health wine sample and a sample fortified with 27 pesticides. As was shown in Fig. 3, no interfering peaks were observed at the retention times of individual compounds, proving that there was sufficient selectivity for the analysis of multiple pesticides at trace levels. Figure 3 also showed that the control samples did not give false-positive signals.

GC-ECD chromatograms of (A) fortified CM health wine sample with 27 OCP and PYP standards, and (B) control sample. Peaks 1. hexachlorobenzene, 2. alpha-BHC, 3. quintozene, 4. gamma-BHC, 5. heptachlor, 6. aldrin, 7. chlorothalonil, 8. beta-BHC, 9. delta-BHC, 10. heptachlor epoxide, 11. triadimefon, 12. alpha-endosulfan, 13. cis-chlordane, 14. trans-chlordane, 15. p.p′-DDE, 16. dieldrin, 17. endrin, 18. o.p′-DDT, 19. p.p′-DDD, 20. beta-endosulfan, 21. p.p′-DDT, 22. methoxychlor, 23. fenpropathrin, 24. permethrin, 25. cypermethrin, 26. flucythrinate, 27. decamethrin.
Calibration curve and linearity
Calibration curves were constructed using standard working solutions at ten concentration levels obtained by diluting the standard stock solution with n-hexane (0.001, 0.005, 0.01, 0.05, 0.1, 0.2, 0.4, 0.6, 0.8 and 1.0 ng/mL for 22 OCPs; 0.002, 0.01, 0.02, 0.1, 0.2, 0.4, 0.6, 0.8, 1.0, 2.0 ng/mL for 5 PYPs). These were then analyzed under the chromatographic conditions described previously. An external calibration method based on the peak area of each analyte was then used to quantify targets. A in the form of y = Ax + B was constructed by plotting the peak areas (y) against standard concentrations (x). The results shown in Table 1 demonstrated that calibration curves with excellent linearity were obtained for the 27 pesticides. The coefficients of determination (R) were higher than 0.9900 in a wide concentration range for all targets except permethrin (R = 0.9876 in the range of 0.001 ng/mL to 2.0 ng/mL).
LOD and LOQ
LODs were estimated using the minimum concentrations detected for all target analytes based on signal-to-noise (S/N) ratio of three and LOQs were set as ten times this ratio. As listed in Table 1, the LOD and LOQ values were found to be at the low ng/L level, with LODs ranging from 0.06 ng/L to 2 ng/L for OCPs and from 0.02 ng/L to 3 ng/L for PYPs, and from 0.2 ng/L to 6 ng/L for OCPs and from 0.06 ng/L to 7 ng/L for PYPs. These values were lower than the MRL of 10 μg/kg or 10000 ng/L established by European Union8, and therefore demonstrated that the developed method has sufficient sensitivity for simultaneous determination of multi-pesticide residues at low concentrations.
Precision and stability
To gage the precision of the method, intra- and inter-day variations were estimated and expressed as RSD of the signals or peak areas for each analyte following an analysis of 0.1 mg/L standard working solution injected six times consecutively on the same day and injected six times over six consecutive days. The results in Table 1 showed that inter-day variation of peak areas for 27 pesticides were in the range of 0.65–9.89%, and intra-day variations of 0.98–13.99%. These values were in compliance with the requirements of the SANCO document (≤20%). Stability was investigated by injecting a CM health wine sample spiked with 0.5 mg/L of 27 pesticides at 0, 2, 4, 6, 10, and 12 h. The RSD values were lower than 10.15%, as shown in Table 1. All of the above results indicated that the proposed method was precise and that the fortified samples were stable.
Trueness
The trueness of the developed method was determined through recovery studies using control samples of CM health wines. Ningxiahong, Chinese jing, and Yedaolugui wines were selected as control samples based on their variety of CM raw materials including raw material from single plant, raw materials from various plants, and raw materials from a mixture of animals and plants, respectively. These samples were fortified at three (high, medium and low) spiking levels, which were 0.5, 0.05, 0.005 mg/kg for OCPs, and 1, 0.1, 0.01 mg/kg for PYPs. Next, all samples were extracted and analyzed in triplicate following the previously described procedure. The recovery percentages were then calculated using the following equation:

As listed in Table 2, the recoveries for the 27 OCPs and PYPs in the three control samples ranged from 50.38–120.31% for Ningxiahong wine, 47.74–113.65% for Chinese jing wine, and 50.56–110.21% for Yedaolugui wine with RSD values in the range of 0.19–24.69%. These results demonstrated that for nearly all pesticides tested the optimized method achieved recoveries (70–120%) and RSD values (≤20%), in line with criteria set by EU guidelines36.
These results demonstrated that the developed method was precise, accurate, and sensitive enough for simultaneous determination of trace levels of 27 pesticide residues in CM health wines.
Real sample analysis
The validated QuEChERS based extraction procedure coupled with GC-ECD method was then applied to measure levels of 22 OCP and 5 PYP residues in 80 CM health wine samples purchased or collected from various Chinese markets in China. As listed in Table 3, residues of 5 pesticides with contents above the LOQ were detected in 9 samples (11.3%), while the other samples tested negative for pesticide residues. Examples of the positive samples included a home-made ginseng wine that was found to contain 4 pesticides (alpha-BHC, gamma-BHC, beta-BHC and delta-BHC), as well as one Yedaolugui wine and one Jiafang wine that both contained the same 2 pesticides (beta-BHC and trans-chlordane). Out of the detected pesticides, beta-BHC was the most predominant with concentration levels ranging from 5.67–31.55 mg/L, as well as the most common incidence in 8 samples (10%). The next highest concentration was delta-BHC (17.30 mg/L) in 1 sample (1.3%), followed by trans-chlordane (3.58–7.45 mg/L) in 3 samples (3.8%), gamma-BHC (3.83 mg/L) in 1 sample (1.3%), and alpha-BHC (2.48 mg/L) in 1 sample (1.3%). The pesticide concentrations measured using this method were all below the suggested permissible level8. It was worth noting that all of the identified residues belonged to OCPs, with no PYPs detected in the tested samples. This may be due to the fact that PYPs have lower stability and are more easily degraded compared to OCPs. In addition, although BHC pesticide has low toxicity, low cost, and is highly effect, it was also the most commonly found OCP in the tested samples due to its chemical stability. The findings of this study reiterate the importance of maintaining strict control of pesticide use and not ignoring the potential harm pesticides may cause to human health. Further work will focus on measuring the pesticide levels in Chinese medicinal raw materials of used to make the wine samples that tested positive in order to better control their contents.
Conclusion
Analysis of multi-class pesticide residues is a current topic of interest in the field of analytical chemistry. Preparation of samples is often the main constraint for successful analysis. In this study, a simple and rapid method combining a QuEChERS based extraction procedure with GC-ECD was developed and validated for simultaneous monitoring and identifying trace levels of multi-pesticide residues. The QuEChERS procedure had the advantage of combining a simple isolation step for target pesticides with a single step sample clean-up method as compared with other sample preparation procedures16,17,18,19. Crucial parameters for QuEChERS extraction and chromatographic analysis were optimized and the developed method was validated. The final method provided a wide concentration range, satisfactory linearity, low LOD and LOQ, good precision, and a high recovery rate, which was comparable with other detection methods for trace levels of pesticides3,11,16,37. Next the method was used to simultaneously analyze 22 OCPs and 5 PYPs residues in 80 CM health wine samples. This successful real world application demonstrated that though the validated method used economical, cheap, and simplified extraction and clean-up procedures, it still maintained adequate sensitivity for detection of trace levels of pesticides and could easily be adopted by other laboratories for analysis of complex matrices.
To the best of our knowledge, this is the first report on simultaneous determination of multi-class pesticide residues in CM health wines in China. Total analysis time (less than 70 min, including 10 min for sample preparation plus 60 min for analysis) for 27 pesticides was shorter as compared to traditional methods3,16,37. Therefore high sample throughput could be achieved, and the method could be useful for pesticide monitoring programs that work with a large numbers of samples. This method not only lowered exposure to hazardous and toxic chemicals usually used for wine sample preparation, but also lowered the overall cost of the analysis. This method could therefore be used as a powerful reference for trace-analysis of multi-class contaminants in other complex matrices, including Chinese medicinal raw materials and related products, etc.
Materials and Methods
Chemicals and reagents
Twenty-seven pesticide standards (uncertainty μg/mL) including hexachlorobenzene (±0.05), alpha-BHC (±0.11), quintozene (±0.07), gamma-BHC (±0.25), heptachlor (±0.10), Aldrin (±0.17), chlorothalonil (±0.11), beta-BHC (±0.11), delta-BHC (±0.07), heptachlor epoxide (±0.08), triadimefon (±0.07), alpha-endosulfan (±0.10), cis-chlordane (±0.17), trans-chlordane (±0.11), p.p′-DDE (±0.11), dieldrin (±0.07), endrin (±0.08), o.p′-DDT (±0.10), p.p′-DDD (±0.17), beta-endosulfan (±0.11), p.p′-DDT (±0.11), methoxychlor (±0.07), fenpropathrin (±0.08), permethrin (±0.07), cypermethrin (±0.19), flucythrinate (±0.115), decamethrin (±0.07) were purchased from the Agro-Environment Protection Institute (Tianjin, China). They were stable over a period of at least three months and their chemical structures were shown in Fig. 4.

Chemical structures of 27 OCP and PYP pesticides.
Ethyl acetate, dichloromethane, n-hexane (HPLC grade) were obtained from Sinopharm Chemical Regent Co., Ltd (Beijing, China). HPLC-grade acetone was from MREDA (IL, USA). Florisil (60–100 mesh) was purchased from Sigma-Aldrich (Bellefonte, PA, USA).
Instrumentation
All analyses were performed using an Agilent 6890N gas chromatograph (Agilent Technologies, CA, USA) equipped with an ECD detector, an Agilent 7683 autosampler and an injector, connected to an HP ChemStation (Hewlett-Packard, Palo Alto, CA, USA) for instrument control and data analysis. A DB-1701 (30 m × 0.25 mm I.D., 0.25 μm) capillary column was used for chromatographic separation. Injector and detector temperatures were held at 220 °C and 300 °C, respectively. The oven temperature program was set as follows: initial 120 °C held for 1 min, ramped to 170 °C at 20 °C/min for 1 min, followed by ramped to 200 °C at 4 °C/min for 5 min, then ramped to 250 °C at 4 °C/min for 10 min and finally ramped to 270 °C at 10 °C/min for 20 min. The injection volume was 1.0 μL. Total run time was 60 min. Ultra-high purity nitrogen (over 99.99%) was selected as the carrier gas at a constant flow rate of 1.0 mL/min. Injection was performed at splitless mode with a purge time of 0.75 min. Quantification of the pesticides was performed using a external standard method based on the detected and integrated peak area.
Preparation of standard solution
A stock solution of mixed pesticide standards were prepared in n-hexane at the concentration of 100 mg/L and 2 mg/L and stored at −20 °C in a refrigerator. The standard working solutions were daily obtained by appropriate dilution of the stock solution.
Sampling
A total of 80 CM health wine samples, which could be divided to 25 types, such as Ningxiahong (n = 20), Chinese Jing wine (n = 14), Lotus white wine (n = 5), Yedaolugui wine (n = 5), Fenglin wine (n = 4), Diyi wine (n = 3), Yishebian wine (n = 1), Yisheshengbao wine (n = 1), Shiguogong wine (n = 1), Cordyceps sinensis wine (n = 1), Sanbian wine (n = 2), Rhodiola rasea wine (n = 1), Ginseng wine (n = 1), Lucid ganoderma wine (n = 1), Herba saussureae involucratae wine (n = 1), Tall gastrodia tuber wine (n = 1), Chinese Manoliavine wine (n = 1), Desertliving cistanche herb wine (n = 2), Ningxiner wine (n = 3), Zhuyeqing wine (n = 3), Jiafang wine (n = 2), Gucixiaotongye (n = 3), Guogong wine (n = 2), Huangjin wine (n = 1), Chinese Wolfberry wine (n = 1), were purchased or collected from various markets in China and stored at ambient temperature. Different Chinese medicinal raw materials are the main composition of them.
Sample preparation
QuEChERS
The basic QuEChERS method with moderate modifications was used for extraction of target OCPs and PYPs from CM health wine samples. Initially, a carefully measured 5.0 mL sample of the CM health wine together with 5.0 mL of n-hexane: ethyl acetate (1:1, v/v) were added into the extractor. The mixture was then shaken vigorously for 2 min in order to induce phase separation and pesticide extraction. It was allowed to rest for a moment until obvious stratification had occurred. The bottom layer (organic phase) was removed and evaporated under a steam of N2 at 40 °C to a final volume of 1 mL. The concentrated solution was precisely measured by transferring it into a 1.0 mL volumetric flask. Next, the solution was placed in an eppendorf tube containing 400 mg florisil and shaken for 30 s using a vortex machine. Finally,, the sample was centrifuged at 12000 rpm for 5 min and the supernatant was filtered through a 0.22 μm filter for injection into the GC-ECD system.
SPE
Solid-phase extraction was performed using florisil SPE cartridges that had been previously conditioned with elutriant. First, 5.0 mL samples of diluted CM health wines were percolated through the cartridges at a constant flow rate. Next, the cartridges were rinsed with 20.0 mL of elutriant and vacuum-dried for 10 min. The pesticides that had been retained in the cartridges were then eluted with 2 × 5.0 mL of n-hexane:ethyl acetate mixture (1:1, v/v) and the eluate was collected in a test tube and concentrated to a 1.0 mL volume. Finally, the concentrated solution was filtered through a 0.22 μm filter prior to injection into the GC-ECD system for analysis.
Additional Information
How to cite this article: Kong, W.-J. et al. Trace analysis of multi-class pesticide residues in Chinese medicinal health wines using gas chromatography with electron capture detection. Sci. Rep. 6, 21558; doi: 10.1038/srep21558 (2016).
References
Driver, J. H., Ginevan, M. E. & Whitmyre, G. K. Estimation of dietary exposure to chemicals: A case study illustrating of distributional analyses for food consumption data. Risk Anal. 16, 763–771 (1996).
Zhan, B. Y., Che, X. F. & Wei, B. C. Progress of Chinese medicine wine products. Inform Tradit Chin Med. 27, 120–122 (2010).
Łozowicka, B. et al. The evaluation of a fast and simple pesticide multiresidue method in various herbs by gas chromatography. J Nat Med. 68, 95–111 (2014).
Pelit, F. O., Ertaş, H., Seyrani, I. & Ertaş, F. N. Assessment of DFG-S19 method for the determination of common endocrine disruptor pesticides in wine samples with an estimation of the uncertainty of the analytical results. Food Chem. 138, 54–61 (2013).
Páleníková, A. et al. Multifamily determination of pesticide residues in soya-based nutraceutical products by GC/MS-MS. Food Chem. 173, 796–807 (2015).
Du, G. et al. Rapid determination of pesticide residues in herbs using selective pressurized liquid extraction and fast gas chromatography coupled with mass spectrometry. J Sep Sci. 35, 1222–1232 (2012).
Garcia, M. A. et al. Effect of fungicide residues on the aromatic composition of white wine inoculated with three Saccharomyces cerevisiae strains. J Agric Food Chem. 52, 1241–1247 (2004).
National Health and Family Planning Commission (NHFPC) of China. GB 2673–2014 National food safety standard-Maximum residue limits for pesticides in food. (2014). Available at: http://www.fas.usda.gov/data/china-maximum-residue-limits-pesticides-food. (Accessed: 20th March, 2014).
European Commission. EU Pesticides database. URL. Available at: http://ec.europa.eu/sanco_pesticides/public/?evnt=homepage&CFTOKEN=43965337&jsessionid=08a0960f96eefa47b013d254c362e4a4d491TR. (Accessed: 05.22.14).
Global MRL Database. URL. Available at: https://www.globalmrl.com/home (Accessed: August 2008).
Guo, Q., Lv, X., Tan, L. & Yu, B. Y. Simultaneous determination of 26 pesticide residues in 5 Chinese medicinal materials using solid-phase extraction and GC-ECD method. Chin J Nat Med. 7, 210–216 (2009).
Sen, K., Cabaroglu, T. & Yilmaz, H. The influence of fining agents on the removal of some pesticides from white wine of Vitis vinifera L. Cv. Emir. Food Chem Toxicol. 50, 3990–3995 (2012).
Yang, Y. H. et al. A multiresidue method for simultaneous determination of 44 organophosphorous pesticides in Pogostemon cablin and related products using modified QuEChERS sample preparation procedure and GC-FPD. J Chromatogr B 974, 118–125 (2015).
Martínez-Domínguez, G., Nieto-García, A. J., Romero-González, R. & Frenich, A. G. Application of QuEChERS based method for the determination of pesticides in nutraceutical products (Camellis sinensis) by liquid chromatography coupled to triple quadrupole tandem mass spectrometry. Food Chem. 177, 182–190 (2015).
Chinese Pharmacopoeia Commission. Chinese Pharmacopoeia, Pharmacopoeia of People’s Republic of China, Vol. I, Chemical Industry Press, Beijing, 2010.
Economou, A., Botitsi, H., Antoniou, S. & Tsipi, D. Determination of multi-class pesticides in wines by solid-phase extraction and liquid chromatography-tandem mass spectrometry. J Chromatogr A 1216, 5856–5867 (2009).
Wang, Y. F. & Du, R. Simultaneous extraction of trace organophosphorous pesticides from plasma sample by automated solid phase extraction and determination by gas chromatography coupled with pulsed flame photometric detector. Forensic Sci Int. 198, 70–73 (2010).
Al-Degs, Y. S., Al-Ghouti, M. A. & El-Sheikh, A. H. Simultaneous determination of pesticides at trace levels in water using multiwalled carbon nanotubes as solid-phase extractant and multivariate calibration. J Hazard Mater. 169, 128–135 (2009).
Rezaee, M. et al. Determination of organic compounds in water using dispersive liquid-liquid microextraction. J Chromatogr A 1116, 1–9 (2006).
Navarro, S., Barba, A., Navarro, G., Vela, N. & Oliva, J. Multiresidue method for the rapid determination-in grape, must and wine-of fungicides frequently used on vineyards. J Chromatogr A 882, 221–229 (2000).
Miao, Q., Kong, W. J., Yang, S. H. & Yang, M. H. Comparison of sample preparation method combined with gas chromatography with electron-capture detection for the analysis of multipesticide residues in lotus seeds. J Sep Sci. 36, 2010–2019 (2013).
Moeder, M., Bauer, C., Popp, P., van Pinxteren, M. & Reemtsma, T. Determination of pesticide residues in wine by membrane-assisted solvent extraction and high- performance liquid chromatography-tandem mass spectrometry. Anal Bioanal Chem. 403, 1731–1741 (2012).
Shi, X. Z. et al. Simultaneous analysis of polychlorinated biphenyls and organochlorine pesticides in seawater samples by membrane-assisted solvent extraction combined with gas chromatography-electron capture detector and gas chromatography-tandem mass spectrometry. J Chromatogr B 972, 58–64 (2014).
Giordano, A., Fernández-Franzón, M., Ruiz, M. J., Font, G. & Pico, Y. Pesticide residue determination in surface waters by stir bar sorptive extraction and liquid chromatography/ tandem mass spectrometry. Anal Bioanal Chem. 393, 1733–1743 (2009).
Farajzadeh, M. A., Djozan, D., Nouri, N., Bamorowat, M. & Shalamzari, M. S. Coupling stir bar sorptive extraction-dispersive liquid-liquid microextraction for preconcentration of triazole pesticides from aqueous samples followed by GC-FID and GC-MS determinations. J Sep Sci. 33, 1816–1828 (2010).
Garbi, A., Sakkas, V., Fiamegos, Y. C., Stalikas, C. D. & Albanis, T. Sensitive determination of pesticides residues in wine samples with the aid of single-drop microextraction and response surface methodology. Talanta. 82, 1286–1291 (2010).
dos Anjos, J. P. & de Andrade, J. B. Simultaneous determination of pesticide multiresidues in white wine and rosé wine by SDME/GC-MS. Microchem J. 120, 69–76 (2015).
CENT/TC 275, N236, Draft, Food of plant origin, Determination of pesticide residues using GC-MS and LC-MS/(MS) following acetonitrile extraction/partitioning and clean-up by dispersive SPE, QuEChERS method, European Committee for Standardisation, Stuttgart, Germany (2006).
Wilkowska, A. & Biziuk, M. Determination of pesticide residues in food matrices using the QuEChERS methodology. Food Chem. 125, 803–812 (2011).
Mol, H. G. J. et al. Identification in residue analysis based on liquid chromatography with tandem mass spectrometry: Experimental evidence to update performance criteria. Anal Chim Acta 873, 1–13 (2015).
Cunha, S. C., Fernandes, J. O., Alves, A. & Oliveira, M. B. P. P. Fast low-pressure gas chromatography-mass spectrometry method for the determination of multiple pesticides in grapes, musts and wines. J Chromatogr A 1216, 119–126 (2009).
Jiang, Y. P. et al. Multiresidue method for the determination of 77 pesticides in wine using QuEChERS sample preparation and gas chromatography with mass spectrometry. Food Addit Contam Part A. 26, 859–866 (2009).
Dasgupta, S. et al. Optimization of two-dimensional gas chromatography time-of-flight mass spectrometry for separation and estimation of the residues of 160 pesticides and 25 persistent organic pollutants in grape and wine. J Chromatogr A 1217, 3881–3889 (2010).
Kirchner, M., Matisová, E., Otrekal, R., Hercegová, A. & de Zeeuw, J. Search on ruggedness of fast gas chromatography-mass spectrometry in pesticide residues analysis. J Chromatogr A 1084, 63–70 (2005).
Łozowicka, B. et al. The evaluation of a fast and simple pesticide multiresidue method in various herbs by gas chromatography. J Nat Med. 68, 95–111 (2014).
European Commission, DG-SANCO. Method validation and quality control procedures for pesticide residues analysis in food and feed, Document No. SANCO/12495/2011 (2012).
Ravelo-Pérez, L. M., Hernández-Borges, J., Borges-Miquel, T. M. & Rodríguez-Delgado, M. Á. Solid-phase microextraction and sample stacking micellar electrokinetic chromatography for the analysis of pesticide residues in red wines. Food Chem. 111, 764–770 (2008).
Acknowledgements
The authors are very grateful for the support from the National Science Foundation of China (81473346, 81573595, 81274072) and Xiehe New Star Project.
Author information
Authors and Affiliations
Contributions
Q.Z.L. and M.H.Y. conceived and designed the experiments. Q.Z.L., Q.T.L. and D.D.K. performed the experiments. Q.Z.L. and W.J.K. analyzed the data. W.J.K. and Q.T.L. co-wrote the paper. All authors reviewed the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Supplementary information
Rights and permissions
This work is licensed under a Creative Commons Attribution 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/
About this article

Cite this article
Kong, WJ., Liu, QT., Kong, DD. et al. Trace analysis of multi-class pesticide residues in Chinese medicinal health wines using gas chromatography with electron capture detection. Sci Rep 6, 21558 (2016). https://doi.org/10.1038/srep21558
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/srep21558
This article is cited by
-
Toxicokinetic of cyphenothrin in rabbits
Environmental Science and Pollution Research (2022)
