
Abstract
Inactivation of the transcription factor AP-2β in a genetically mixed C57BL/6/129S1 mouse strain resulted in perinatal lethality as a consequence of massively enhanced apoptotic death of renal epithelial cells (Genes Dev 1997;11:1938–1948). Recently, we observed that the phenotype is modulated by genetic background because AP-2β mutant mice, backcrossed onto 129P2 background, survive approximately 2 weeks after birth, allowing for a detailed analysis of kidney function. Here we show that kidneys reveal varying amounts of cysts derived from all tubular structures (proximal and distal tubuli, collecting ducts). However, all mice died irrespective of the degree of cyst formation. Serum analysis of AP-2β mutant animals revealed defective tubular secretory function and ion homeostasis including severe hypocalcemia, hyperphosphatemia, and hyperuremia. Because hormonal calcium regulation was not impaired, the mice developed secondary renal hyperparathyroidism as typically observed in patients with terminal renal failure. We further demonstrate that molecular defects in the collecting duct system lead to insufficient water retention and urinary concentration. In summary, our studies reveal essential, nonredundant roles of AP-2β in renal tubular functions.
Similar content being viewed by others


Introduction
AP-2β belongs to a family of four closely related transcription factors, AP-2α, AP-2β, AP-2γ, and AP-2δ, which execute essential functions during mammalian embryonic development (Moser et al, 1995; Oulad-Abdelghani et al, 1996; Williams et al, 1988; Zhao et al, 2001). AP-2 genes have also been cloned from nonmammalian organisms including chicken, Xenopus, and Drosophila (Bauer et al, 1998; Mitchell et al, 1991; Monge and Mitchell, 1998; Shen et al, 1997; Snape et al, 1991), revealing considerable evolutionary conservation. All AP-2 proteins are characterized by an N-terminal proline- and glutamine-rich transactivation domain followed by a short, positively charged DNA-binding region and two α-helical structures separated by a span region. The alpha helices and the span region mediate dimerization of AP-2 proteins. Both homo- and heterodimers have been described (Williams and Tjian, 1991a, 1991b), leading to a large variety of conceptual AP-2 complexes in tissues with overlapping expression patterns.
During murine development, the previously analyzed AP-2 genes, AP-2α, AP-2β, and AP-2γ, are expressed in temporally and spatially overlapping patterns in neuroectodermal, ectodermal, and urogenital tissues (Chazaud et al, 1996; Mitchell et al, 1991; Moser et al, 1995, 1997b). Coexpression of AP-2 genes is particularly observed in neural crest cells, which migrate from the dorsal neural tube and give rise to a number of different tissues and structures including facial bones, the peripheral nervous system, and melanocytes. In addition, these studies demonstrate specific AP-2 gene expression patterns in skin and urogenital tissues and also in the central nervous system and the limb bud mesenchyme. Expression-pattern specificity can best be seen in the developing brain, where AP-2γ is expressed in the forebrain, AP-2β in the mid- and hindbrain, and AP-2α almost exclusively in the cerebellum (Chazaud et al, 1995; Moser et al, 1997b; Zhao et al, 2001). In mammals as well as in evolutionarily lower animals like Drosophila, Xenopus, and some avian species, AP-2 is strongly expressed in facial structures, in the central nervous system, the epidermis, and neural crest cells, indicating a general functional importance of AP-2 genes for the development of these tissues (Bauer et al, 1998; Epperlein et al, 2000; Monge and Mitchell, 1998; Shen et al, 1997; Snape et al, 1991).
A recently performed mutagenesis screen to isolate DAP-2 mutant alleles in flies resulted in several lethal-point mutant alleles flies caused these flies to die as adults or late pupal stages and show reduced proboscis, shortened legs, absent tarsal joints, and disruption of the protocerebral central complex (Kerber et al, 2001; Monge et al, 2001). The functional importance of AP-2 genes for the development of craniofacial structures, limb formation, and kidney development was also shown by gene inactivation of AP-2α (Schorle et al, 1996; Zhang et al, 1996), AP-2β (Moser et al, 1997a), and AP-2γ (Aumann et al, 2002; Werling and Schorle, 2002) in mice. AP-2α knockout mice die perinatally and develop exencephaly, which becomes apparent at embryonic stage E9.5 when the cranial neural folds fail to oppose to form the anterior neural tube. In addition, increased apoptotic cell death in the midbrain, hindbrain, and migrating cranial neural crest cells contributes to the development of the cranial defects. AP-2γ knockout mice die very early during embryogenesis because of severe defects in extraembryonic tissues.
Although embryonic expression of AP-2α, AP-β, and AP-γ overlaps significantly during development, the phenotype of AP-2β knockout mice does not show any of the developmental defects of AP-2α or AP-2γ mutant mice. AP-2β knockout mice complete normal embryonic development but show enhanced apoptotic cell death in renal epithelial cells, enlarged cystic tubular structures, and die at birth (Moser et al, 1997a). Consistent with an undisturbed embryonic development, the overall renal morphology was well organized, and the expression of developmental markers for the induction and conversion of mesenchymal cells into tubular structures was unaltered.
Although AP-2β mutant kidneys resemble human autosomal recessive polycystic kidney disease, we have previously excluded AP-2β as a candidate gene (Mücher et al, 1998). Subsequent to these studies, a large multidomain protein, PKHD1, was recently identified as the autosomal recessive polycystic kidney disease gene in close proximity to the AP-2β locus on chromosome 6p21 (Onuchic et al, 2002; Ward et al, 2002).
In this study, we demonstrate that AP-2β deficient mice survive approximately 2 weeks after birth in a 129P2 background and can therefore be used for detailed physiologic analysis of the phenotype. Our data reveal that AP-2β mutant animals suffer from impaired tubular secretory function, ion homeostasis, and severe hypocalcemia. Because of kidney failure, AP-2β mice develop secondary hyperparathyroidism.
Results
Growth Retardation of AP-2β Mutant Mice
We have previously described that AP-2β deficient mice, generated on a mixed C57BL/6 × 129S1 genetic background, develop polycystic kidney disease and die perinatally (Moser et al, 1997a). In this study, we analyzed the same mutation in 129P2 mice and observed significant attenuation of the phenotype. For the experiments described in this study, heterozygous AP-2β mutant mice were mated with inbred 129P2 mice and then paired for eight further generations within the same line before further analysis. On this background, the Mendelian ratios are preserved prenatally, but only approximately 10% of homozygous mutant animals survive between 10 and 14 days after birth. AP-2β deficient mice were born and became significantly retarded in development about 4 days after birth (Fig. 1, A and B). Homozygous mutants were easily detected as small animals with dry and scaly skin and revealed a striking shivering and trembling behavior accompanied with tetanic cramps. However, these mice were able to feed because their stomach was filled with milk and digestion appeared normal upon internal inspection. Gross anatomy was entirely preserved, with the exception of an additional sixth digit representing postaxial hexadactyly of the anterior extremity (data not shown).

Retardation in growth and development of AP-2β mutant mice. AP-2β-/- and wild-type litter mates at Day 10 postnatal (A). A statistical analysis (B) reveals severe retardation in growth of the homozygote mutants. Values are derived from 8 (-/-), 32 (+/-), and 24 (+/+) animals, respectively.
Cysts Formation in Kidneys of AP-β Mutant Mice
In similarity to the previously characterized prenatal AP-2β mutant animals, postnatal organs also appeared histologically normal, with the exception of kidneys (Moser et al, 1997a). Renal sections from a number of different mutants showed varying numbers of dilated tubules and tubular or collecting duct cysts, ranging from only a few small isolated cysts to up to hundreds of cystic structures dispersed throughout the entire kidney (Fig. 2, A to D). To determine the cellular origin of the cystic structures, immunohistochemical and lectin stainings were performed using markers for proximal tubular epithelium and collecting ducts. Immunostaining of the aquaporin-2 water channel, which is strongly expressed in the collecting duct epithelium (Verkman and Mitra, 2000), revealed that a portion of the cysts, with flattened epithelium lining the fluid lumen, were derived from collecting ducts. Furthermore, the lectin Tetragonolobus purpureus was used to identify proximal tubular epithelium, which was also shown to give rise to cysts (Fig. 3, A and B). These cysts probably result from secondary defects of distal tubular and collecting ducts because AP-2β is not expressed in the proximal tubular epithelium, as shown by in situ hybridization with an AP-2β specific probe (Fig. 3C). Ultrastructurally, the cyst lining epithelia showed a very flat dedifferentiated phenotype, whereas epithelia in ectatic tubuli remained in a differentiated phenotype corresponding to the respective tubular segment (Fig. 4, A and B).

Variation of number and size of renal cysts in different AP-2β deficient mice. Hematoxylin-eosin staining of kidney sections from wild-type (A) and homozygous mutants (B to D) at Day 8 pn (magnification, ×16).

Immunostaining of proximal tubular epithelium and collecting-duct system. Tetragonolobus purpureus (A) and anti-aquaporin 2 (B) stainings (magnification, ×200). In situ hybridization of a 8 pn mouse kidney section with an AP-2β specific cDNA probe revealing specific expression pattern in distal tubular and collecting duct epithelium (C) (magnification, ×25).

Ultrastructure of ectatic tubuli and renal cysts in AP-2β deficient mice. Ultrastructurally, epithelia in ectatic tubuli remained in a differentiated columnar phenotype (A) whereas the cyst lining epithelia showed a very flat dedifferentiated phenotype (B).
In contrast with the phenotype of other cystic kidney diseases, in particular ADPKD, glomerular cysts were not observed. On the basis of examination by light microscopy and electron microscopy, the glomerular architecture was entirely normal (data not shown).
Defective Ion Homeostasis in AP-2β Deficient Mice
Because the degree of cyst formation differed significantly in AP-2β mutant mice and did not correlate with longer survival, we assessed kidney function by measuring a number of different serum parameters in 6- to 10-day mutants and controls. Whole blood was isolated by heart puncture and ion, urea, glucose, and protein concentrations were determined from serum. As summarized in Table 1, ion homeostasis was severely impaired. Serum calcium levels were significantly decreased and caused the nervous and trembling behavior of mutant animals. Indeed, tetanic cramps were easily provoked, even by relatively mild manual manipulations, and eventually the animals died between Days 10 and 14 from cardiac arrest. In addition, increased urea, creatinine, and inorganic phosphate concentrations indicated functional tubular defects. Normal serum protein levels in AP-2β deficient mice suggested undisturbed glomerular filtration of primary urine consistent with the normal histologic and ultrastructural morphology of glomeruli.
Because serum calcium concentration is strongly dependent on regulation by parathyroid hormone (PTH) and vitamin D, we analyzed the production of these hormones in AP-2β deficient animals. On the basis of the observation that AP-2β is expressed in branchial arches that give rise to the parathyroid glands and the fact that other mutant mouse strains with developmental defects leading to parathyroid gland agenesis have been identified (Gunther et al, 2000; Peters et al, 1998), we explored the possibility that hypocalcemia resulted from a failure to develop parathyroidal glands. Therefore, we prepared the throat region of AP-2β knockout mice and studied the morphology of parathyroidal glands on serial tissue sections. As shown in Figure 5, A, B, D, and E, AP-2β mutants developed parathyroidal glands that appeared slightly hyperplastic and expressed large amounts of PTH. Furthermore, the gene of a calcium sensing receptor protein involved in the regulation of PTH vesicle secretion was equally expressed in both mutant and wild-type parathyroidal glands, indicating that AP-2β mutant animals developed functional parathyroidal glands. This observation was further strengthened by quantitation of vitamin D and PTH serum levels. Using a quantitative radioimmunoassay for the active form of vitamin D (1,25-OH2 vitamin D) and PTH, we measured a significant increase in PTH serum concentration, indicating a physiologic response to the low serum calcium levels and also a slight increase in vitamin D concentration (Fig. 6).

AP-2β deficient animals develop functional parathyroid glands. In situ hybridization with specific probes for parathyroid hormone (PTH) (A, D) and calcium sensing receptor protein (CasR) (B, E) on wild-type (A, B) and AP-2β deficient mouse sections of the throat region. (C, F) Hematoxylin-eosin stainings of B and E, respectively (magnification, ×25).

AP-2β deficient animals develop secondary renal hyperparathyroidism. Radioimmunoassays were used to measure serum levels of 1,25(OH)2 vitamin D and PTH in wild-type (+/+), heterozygous (+/-), and AP-2β knockout mice (-/-).
In summary, the renal histology, the disturbance of ion homeostasis, and the subsequent increase in PTH serum concentrations indicated development of secondary renal hyperparathyroidism in AP-2β deficient mice.
Defects of Distal Tubular Epithelium and Collecting Duct System Results in Kidney Failure
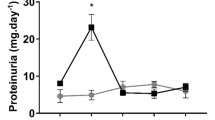
To test the capacity of the kidney to concentrate urine, bladder aspirates were collected from wild-type, heterozygous, and mutant animals to measure osmolarity. A decrease in urine osmolarity in AP-2β mutant mice as compared with controls indicated a failure in water retention and suggested that the renal water resorption in the distal tubular structures and in the collecting duct system was impaired (Fig. 7A). In addition, urinary calcium concentration was reduced in knockout mice as compared with wild-type controls, indicating that AP-2β deficient mice lose water, and thereby essential ions, by way of defective renal tubular epithelium (Fig. 7B). This observation corresponded with the dehydrated condition of the mutant animals and their dry and scaly skin.

Defects in water retention results in kidney failure. Urinary osmolarity (A) and Ca2+ concentration (B) in urine of wild-type (+/+), heterozygous (+/-), and AP-2β knockout mice (-/-) indicated impaired water resorption.
Discussion
Knockout studies unraveled essential functions of AP-2 transcription factors during embryonic development. Although AP-2 genes encode for highly homologous proteins, which are expressed at overlapping expression patterns in many embryonic tissues, the phenotypes of AP-2α, AP-2β, and AP-2γ mice are surprisingly different (Aumann et al, 2002; Moser et al, 1997a; Schorle et al, 1996; Werling and Schorle, 2002; Zhang et al, 1996). We showed previously that inactivation of the transcription factor AP-2β in a genetically mixed C57BL/6/129S1 mouse strain resulted in perinatal lethality as a consequence of massively enhanced apoptotic death of renal epithelial cells (Moser et al, 1997a). So far, other AP-2 genes have not been implicated in kidney function.
In this study, the AP-2β mutation was bred onto the 129P2 background, resulting in significant attenuation of the phenotype and allowing for the first time for a detailed analysis of the defects seen in the kidneys. Obviously, the phenotype is attenuated by modifier genes, and therefore a significant portion of the mice survive 2 weeks after birth. Interestingly, a modification of the phenotype has also been described for the AP-2α mutation, where a significant portion of heterozygous animals suffers from exencephaly when bred onto the 129P2 background (Kohlbecker et al, 2002). These findings suggest a more general role of AP-2 gene dosage because a slight deregulation might result in morphologic deficiencies.
Our data indicate that AP-2β gene activity is redundant in most AP-2β expressing tissues (peripheral and central nervous system, neural crest cell derivatives) except for kidneys. We have detected previously that AP-2β is the only member of the AP-2 genes that is expressed specifically during renal tubulogenesis, in particular in distal tubular and collecting duct epithelia (Chazaud et al, 1996; Moser et al, 1997b). Here, AP-2β deficiency cannot be compensated for through the action of other AP-2 family members.
Interestingly, data presented in this study clearly show that death of AP-2β deficient mice occurs independently of the number and size of cyst formation. Rather, AP-2β expression seems to be essential for maintaining functions of tubular and collecting duct epithelia in secretion of metabolites and water and ion homeostasis. Even animals without significant cyst formation develop renal failure and die within 2 weeks from massive hypocalcemia. Thus, our analyses point to a novel function of AP-2β in kidney metabolism and should trigger further studies of AP-2β expression in kidney diseases that involve tubular or collecting duct epithelia.
Materials and Methods
Histologic Analysis and Immunohistochemistry
Tissues were dissected from wild-type, heterozygous, and mutant animals at days 6 to 10 postnatal, fixed in 4% paraformaldehyde/PBS overnight and embedded in paraffin. Five micrometer tissue sections were stained with hematoxylin-eosin and used for immunohistochemistry or in situ hybridization.
Immunohistochemistry was performed as described in detail previously (Bosserhoff et al, 1999). Anti-aquaporin–2 antiserum (a generous gift from Dr. Aitner, Department of Internal Medicine II, RWTH Aachen, Germany) was incubated as a 1:100 dilution overnight and reacted with a secondary antirat antibody (DAKO, Hamburg, Germany) conjugated with horseradish peroxidase for 2 hours at room temperature, and then finally stained with diaminobenzidine (Sigma, Deisenhofen, Germany). HRP-conjugated lectin Tetragonolobus purpureus (Sigma) was diluted at 1:100, incubated for 2 hours at room temperature, and visualized with the substrates provided by the LSAB2-Kit (DAKO).
In Situ Hybridization
In situ hybridization was performed as described previously (Moser et al, 1995) using 33P-labeled sense and antisense cRNA riboprobes for parathyroid hormone, AP-2β, and calcium sensing receptor protein. Proteinase K pretreated slides (10 μg/ml) were acetylated in acetic anhydride diluted 1:400 in 0.1 M triethanolamine (pH 8) and hybridized at 50° C overnight in 50% formamide, 10% dextran sulfate, 10 mm Tris (pH 8), 10 mm NaPi (pH 7), 2xSSC, 5 mm EDTA (pH 8), 150 μg/ml tRNA, 10 mm DTT, and 10 mm β-mercaptoethanol supplemented with 5 × 104 cpm/μl of 33P-labeled sense or antisense riboprobes. The slides were washed twice in 50% formamide/2x SSC/20 mm β-mercaptoethanol, digested with RNase A (20 μg/ml) for 30 minutes at 37° C, and finally washed three times for 30 minutes at 50° C with the same washing buffer. After dehydrating, the slides were coated with Kodak NTB2 emulsion (Kodak, Rochester, New York) and exposed for 4 to 10 days. In situ hybridization was performed from tissues of four different animals of each genotype.
Determination of Serum and Urine Parameters
Whole blood was obtained by heart puncture from 6- to 10-day-old mice, and the serum was recovered after coagulation by centrifugation at 6000 rpm for 5 minutes. Urea, creatinine, ions, glucose, and protein concentrations were measured by an autoanalyzer (Johnson and Johnson, Neckarsgemünd, Germany). To determine PTH and 1,25(OH)2 vitamin D levels, both hormones were measured using radioimmunoassays against rat PTH or 1,25(OH)2 vitamin D (Meyer et al, 1996) (Immutopics, San Clemente, California). For both tests, sera from six knockout mice were pooled to obtain the required test volume of 100 μl. The assays were performed following the manufacturer’s instructions except that serum was diluted 1:1 with the supplied 0 standard.
For urine analyses, bladder aspiration was performed. Determination of urine osmolarity was measured in 50 μl of pooled urine from wild-type, heterozygous, and AP-2β knockout mice. Total urine calcium levels were measured by a commercially available assay (Sigma) using the o-cresolphthalein-complexone method.
All serum and urine parameters shown in Table 1, and Figures 5 to 7 represent mean values derived from multiple individuals of each genotype. Depending on availability, between 16 and 54 samples were analyzed for each mean value.
References
Auman HJ, Nottoli T, Lakiza O, Winger Q, Donaldson S, and William T (2002). Transcription factor AP-2gamma is essential in the extra-embryonic lineages for early postnatal development. Development 129: 2733–2747.
Bauer R, McGuffin ME, Mattox W, and Tainsky MA (1998). Cloning and characterization of the Drosophila homologue of the AP-2 transcription factor. Oncogene 17: 1911–1922.
Bosserhoff AK, Moser M, Hein R, Landthaler M, and Buettner R (1999). In situ expression patterns of melanoma-inhibiting activity (MIA) in malignant melanomas and breast cancers. J Pathol 187: 446–454.
Chazaud C, Oulad-Abdelghani M, Bouillet P, Decimo D, Chambon P, and Dolle P (1996). AP-2.2, a novel gene related to AP-2, is expressed in the forebrain, limbs and face during mouse embryogenesis. Mech Dev 54: 83–94.
Epperlein H, Meulemans D, Bronner-Fraser M, Steinbeisser H, and Selleck MA (2000). Analysis of cranial neural crest migratory pathways in axolotl using cell markers and transplantation. Development 127: 2751–2761.
Gunther T, Chen ZF, Kim J, Priemel M, Rueger JM, Amling M, Moseley JM, Martin TJ, Anderson DJ, and Karsenty G (2000). Genetic ablation of parathyroid glands reveals another source of parathyroid hormone. Nature 406: 199–203.
Kerber B, Monge I, Mueller M, Mitchell PJ, and Cohen S (2001). The AP-2 transcription factor is required for joint formation and cell survival in Drosophila leg development. Development 128: 1231–1238.
Kohlbecker A, Lee AE, and Schorle H (2002). Exencephaly in a subset of animals heterozygous for AP-2alpha mutation. Teratology 65: 213–218.
Meyer RA, Meyer MH, and Morgan PL (1996). Effects of altered diet on serum levels of 1,25-dihydroxyvitamin D and parathyroid hormone in X-linked hypophosphatemic (Hyp and Gy) mice. Bone 18: 23–28.
Mitchell PJ, Timmons PM, Hebert JM, Rigby PW, and Tjian R (1991). Transcription factor AP-2 is expressed in neural crest cell lineages during mouse embryogenesis. Genes Dev 5: 105–119.
Monge I, Krishnamurthy R, Sims D, Hirth F, Spengler M, Kammermeier L, Reichert H, and Mitchell PJ (2001). Drosophila transcription factor AP-2 in proboscis, leg and brain central complex development. Development 128: 1239–1252.
Monge I and Mitchell PJ (1998). DAP-2, the Drosophila homolog of transcription factor AP-2. Mech Dev 76: 191–195.
Moser M, Pscherer A, Imhof A, Bauer R, Amselgruber W, Sinowatz F, Hofstaedter F, Schule R, and Buettner R (1995). Molecular cloning and characterization of a second AP-2 transcription activator gene, AP-2β. Development 121: 2779–2788.
Moser M, Pscherer A, Roth C, Becker J, Mücher G, Zerres K, Dixkens C, Weis J, Guay-Woodford L, Buettner R, and Fässler R (1997a). Enhanced apoptotic cell death of renal epithelial cell in mice lacking transcription factor AP-2β. Genes Dev 11: 1938–1948.
Moser M, Rüschoff J, and Buettner R (1997b). Comparative analysis of AP-2α and AP-2β gene expression during murine embryogenesis. Dev Dyn 208: 1–12.
Mücher G, Becker J, Knapp M, Buettner R, Moser M, Dudnik-Schöneborn S, Somlo S, Germino G, Onuchic L, Avner E, Guay-Woodford L, and Zerre K (1998). Fine mapping of the autosomal recessive polycystic kidney disease locus (PKHD1) and the genes MUT, RDS, CSNK2β, and GSTA1 at 6p21-p12. Genomics 48: 40–45.
Onuchic LF, Furu L, Nagasawa Y, Hou X, Eggermann T, Ren Z, Bergmann C, Senderek J, Esquivel E, Zeltner R, Rudnik-Schöneborn S, Mrug M, Sweeney W, Avner ED, Zerres K, Guay-Woodford LM, Somlo S, and Germino GG (2002). PKHD1, the polycystic kidney and hepatic disease 1 gene, encodes a novel large protein containing multiple IPT domains and PbH1 repeats. Am J Hum Genetic 10: 1305–1317.
Oulad-Abdelghani M, Bouillet P, Chazaud C, Dolle P, and Chambon P (1996). AP-2.2: A novel AP-2-related transcription factor induced by retinoic acid during differentiation of P19 embryonal carcinoma cells. Exp Cell Res 225: 338–347.
Peters H, Neubuser A, Kratochwil K, and Balling R (1998). Pax9-deficient mice lack pharyngeal pouch derivatives and teeth and exhibit craniofacial and limb abnormalities. Genes Dev 12: 2735–2747.
Schorle H, Meier P, Buchert M, Jaenisch R, and Mitchell PJ (1996). Transcription factor AP-2 essential for cranial closure and craniofacial development. Nature 381: 235–238.
Shen H, Wilke T, Ashique AM, Narvey M, Zerucha T, Savino E, Williams T, and Richman JM (1997). Chicken transcription factor AP-2: Cloning, expression and its role in outgrowth of facial prominences and limb buds. Dev Biol 188: 248–266.
Snape AM, Winning RS, and Sargent TD (1991). Transcription factor AP-2 is tissue-specific in Xenopus and is closely related or identical to keratin transcription factor 1 (KTF-1). Development 113: 283–293.
Verkman AS and Mitra AK (2000). Structure and function of aquaporin water channels. Am J Physiol Renal Physiol 278: F13–F28.
Ward CJ, Hogan MC, Rossetti S, Walker D, Sneddon T, Wang X, Kubly V, Cunningham JM, Bacallao R, Ishibashi M, Milliner D, Torres VE, and Harris PC (2002). The gene mutated in autosomal recessive polycystic kidney disease encodes a large, receptor-like protein. Nat Genet 30: 1–11.
Werling U and Schorle H (2002). Transcription factor AP-2 gamma essential for early murine development. Mol Cell Biol 22: 3149–3156.
Williams T, Adamon A, Lüscher B, and Tjian R (1988). Cloning and expression of AP-2, a cell-type-specific transcription factor that activates inducible enhancer elements. Genes Dev 2: 1557–1569.
Williams T and Tjian R (1991a). Analysis of the DNA-binding and activation properties of the human transcription factor AP-2. Genes Dev 5: 670–682.
Williams T and Tjian R (1991b). Characterisation of a dimerisation motif in AP-2 and its function in heterologous DNA-binding proteins. Science 251: 1067–1071.
Zhang J, Hagopian-Donaldson S, Serbedzija G, Elsemore J, Plehn-Dujowich D, McMahon AP, Flavell RA, and Williams T (1996). Neural tube, skeletal and body wall defects in mice lacking transcription factor AP-2. Nature 381: 238–241.
Zhao F, Satoda M, Licht JD, Hayashizaki Y, and Gelb B (2001). Cloning and characterization of a novel mouse AP-2 transcription factor, AP-2δ, with unique DNA binding and transactivation properties. J Biol Chem 276: 40755–40760.
Acknowledgements
We are indebted to Dr. Aitner for providing the aquaporin 2 antiserum, to Dr. Quarles for providing the PTH cDNA probe, and to Dr. Lanske for providing the CasR cDNA probe.
This work was supported by grants from the Deutsche Forschungsgemeinschaft to RB.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Moser, M., Dahmen, S., Kluge, R. et al. Terminal Renal Failure in Mice Lacking Transcription Factor AP-2β. Lab Invest 83, 571–578 (2003). https://doi.org/10.1097/01.LAB.0000064703.92382.50
Received:
Published:
Issue date:
DOI: https://doi.org/10.1097/01.LAB.0000064703.92382.50
This article is cited by
-
Crucial role of TFAP2B in the nervous system for regulating NREM sleep
Molecular Brain (2024)
-
AP-2ε Expression in Developing Retina: Contributing to the Molecular Diversity of Amacrine Cells
Scientific Reports (2018)
-
Comparative transcriptomic analysis identifies evolutionarily conserved gene products in the vertebrate renal distal convoluted tubule
Pflügers Archiv - European Journal of Physiology (2017)
-
Standardized, systemic phenotypic analysis reveals kidney dysfunction as main alteration of Kctd1 I27N mutant mice
Journal of Biomedical Science (2017)
-
Familial Nonsyndromic Patent Ductus Arteriosus Caused by Mutations in TFAP2B
Pediatric Cardiology (2011)
