
INTRODUCTION
Members of the Academy, colleagues and friends, I am honored to be here this afternoon to deliver the 44th Maude Abbott Lecture not only because this lecture marks a highlight of our meeting but also because it pays tribute to a woman who was so much a pillar of our Academy. Maude Abbott was, by all accounts, an impressive woman (Figs. 1,2). Orphaned as a young child, she excelled in school and graduated as valedictorian from McGill University. One might think this would have entitled her to entry to McGill Medical School, but enormous prejudice existed against accepting women into medicine, and she was forced to attend the smaller and less prestigious Bishop Medical School. Despite setbacks of this type, Maude Abbott remained throughout life an optimistic woman beloved by both colleagues and friends. Her most important work was a meticulous classification of congenital malformations of the heart, displayed in a collection of pathologic specimens and elegantly illustrated atlas. After the death of Sir William Osler, her colleague and friend, she organized his papers for McGill University. In 1902, she became the first Secretary-Treasurer of the International Association of Medical Museums, an organization that over time evolved into our present Academy. With a deep sense of appreciation for what this remarkable woman did for this Academy and for women physicians in general, I am honored to present this lecture. I am dedicating this lecture to the memory of Dr. Rodger Haggitt (Fig. 3), whose Maude Abbott lecture last year epitomized what a Maude Abbott lecture should be—an exquisite blend of science, medicine, and personal wisdom. One would expect no less from Rodger, who moved between the often-disparate worlds of basic science and clinical practice with an easy grace and intellectual elegance. He is deeply missed by us, but his spirit and his love for our discipline persist.

Maude Abbott as a young woman.

Maude Abbott in her older years.

Dr. Rodger C. Haggitt.
In deciding what to present this afternoon, I asked friends and colleagues for suggestions. Several said that I should begin with some autobiographical remarks. In so doing, one treads a fine line between self-consciousness and self-indulgence. Nonetheless, I have decided to begin in this way not only to frame my career and interest in soft tissue pathology, but also to assure the younger members of our Academy that career paths are not neatly planned, but follow circuitous routes, and that self-discovery often occurs serendipitously.
I was born in the city of Lynn, just north of Boston, the eldest of six children in a peripatetic military family for whom the only constancy was change itself (Fig. 4). My father was a charismatic Army surgeon who placed great value on education. He insisted that each of his children, whether son or daughter, receive a good education and, therefore, firmly supported my desire to attend Wellesley College, even though it meant considerable financial hardship for our family. I entered college intending to be an archaeologist, but by my junior year my interests began to change. I enrolled in a cellular biology course that featured no textbooks but rather a series of original scientific articles. My first assignment was to read and analyze the now-classic article by Marilyn Farquhar (the 1988 Maude Abbott Lecturer) and Nobelist George Palade on junctions in epithelial cells (1; Fig. 5). Struck by the beauty of the electron micrographs and the harmonious marriage of structure with function, I began to understand that my true love was to be in those sciences that enjoyed strong morphologic underpinnings.

The Whelan family in 1986. Back row: Thomas and Normal Whelan (parents). Front row: Rosemary, Sally, Michael, Sharon, Thomas, and Christal (children).

Title page and electron micrograph from classic article by Farquhar and Palade (1) describing structure of junctional complexes in epithelial cells.
In the fall of 1967, I entered Johns Hopkins School of Medicine, determined to become a pathologist. Not until my third year of medical school, which included a Saturday morning surgical pathology course taught by the director of surgical pathology, William Shelley (Fig. 6), was I convinced I would become a surgical pathologist. Dr. Shelley was a charming southern gentleman whose diagnostic brilliance was admired by clinicians and fellow pathologists alike. Those of us who stayed at Johns Hopkins Hospital to train in pathology were disappointed by his departure that same year to return to his home state of North Carolina. Nonetheless, my residency was marked by an association with intellectually stimulating colleagues such as Jean Olsen, a co-author of Heptinstall’s renal textbook, David Keren, a preeminent clinical pathologist, and Stanley Hamilton, chair of pathology at M.D. Anderson Cancer Hospital, to name only a few. (Fig. 7) My four years also had its amusing moments, like Stan Hamilton’s first day in surgical pathology, when he broke the cryostat, convincing us he was destined for a chairmanship. Our chairman, Dr. Robert Heptinstall, or “Heppy” as he was called, was very fond and proud of his residents, although he worked hard to conceal this at times. He had perfected a brilliant means of dealing with our requests for new “toys” by claiming that brains and equipment were inversely related. Needless to say, we asked for very little in those days. My arrival at the Armed Forces Institute of Pathology (AFIP) shortly after residency marked the beginning of a decade-long association with my mentor, Dr. Franz Enzinger, one of the generation of great diagnostic pathologists, the likes of which we may never see again. This tenure also spawned a lifelong interest in sarcomas. This afternoon, I would like to share with you a piece of my life’s interest and tell you the story of pleomorphic sarcomas, a topic around which several of my early publications centered. As a group, pleomorphic sarcomas present a formidable challenge to surgeon and oncologist and at the same time tell us much about ourselves as pathologists—how we embrace new ideas, cling to old ones, and deal with the interesting interface of science and medicine.

Dr. William Shelley, director of surgical pathology, Johns Hopkins Hospital in the mid to late 1960s.

House staff in pathology at Johns Hopkins Hospital, 1975, with Chair Robert Heptinstall (center).
A pleomorphic sarcoma can be defined simplistically as one in which the cells display marked atypia (Fig. 8). It is not altogether synonymous with undifferentiated because many pleomorphic sarcomas display evidence of differentiation. As a group, pleomorphic sarcomas occur in adults and, therefore, contrast with poorly differentiated sarcomas occurring in children, which are usually round cell lesions. Some pleomorphic sarcomas arise de novo, whereas others evolve through the process of dedifferentiation (e.g., dedifferentiated liposarcoma; 2, 3) through a series of mutagenic events not well understood. Although many types of sarcoma occasionally assume a pleomorphic appearance, there are some that consistently or even definitionally have a pleomorphic appearance. These include malignant fibrous histiocytoma (4, 5), dedifferentiated (3) and pleomorphic liposarcoma (6, 7, 8) (Fig. 9), pleomorphic rhabdomyosarcoma (9, 10, 11) (Fig. 9), and pleomorphic leiomyosarcoma (11, 12). For purposes of this lecture, we will consider all pleomorphic sarcomas as high grade, although there are some exceptions.

Malignant fibrous histiocytoma as the prototype of the pleomorphic sarcoma.

Pleomorphic liposarcoma and pleomorphic rhabdomyosarcoma, two forms of pleomorphic sarcoma showing evidence of differentiation. The former (A) is characterized by pleomorphic lipoblasts and the latter (B), by rhabdomyoblasts containing muscle filaments and cross-striations.
All sarcomas, pleomorphic and nonpleomorphic, share certain common properties, and James Ewing should be credited with many of these early important observations put forward in his classic textbook on tumors published in 1918 (13; Fig. 10). His observations were amazingly sophisticated for his time, especially when compared with competing textbooks, such as the early general textbook of pathology written by MacCallum. Like many, Ewing recognized that sarcomas were derived from mesenchymal tissues, but unlike many, he understood that most spindle cell tumors that arose from epithelial organs were not sarcomas but rather carcinomas assuming a spindled appearance. He described beautifully the concept of “pseudoencapsulation,” or the notion that progressive growth of sarcomas produces a compressed rim of normal tissue that gives the deceptive appearance of a capsule—a concept that even today eludes many surgeons. He clearly appreciated the role that angiogenesis played in the continued growth of tumors and even suggested that tumors themselves induced the vascular proliferation, a view that we currently embrace. Although he never proposed a formal classification of sarcomas in this book, he believed that the cells of most sarcomas were closely related to the fibroblast and that the level of differentiation varied. In fact, it appears that he viewed this as a continuum, with some lesions becoming very pleomorphic—in effect, his view of pleomorphic sarcomas was that most were fibroblastic, a view that we will come to see is not very different from our current view.

Dr. James Ewing.
After Ewing, the focus on sarcomas shifted to a neighboring institution, Columbia-Presbyterian Hospital, and its director of surgical pathology, Arthur Purdy Stout, arguably the father of American soft tissue pathology (Fig. 11). Although he published on nearly every soft tissue tumor, his contribution to the understanding of pleomorphic sarcomas began with two sequential publications in the early 1960s (14, 15; Fig. 12). Explanting cells from pleomorphic sarcomas (and other lesions) into tissue culture, he noted that they grew cell processes, phagocytosed carbon particles, and moved in an amoeboid fashion, suggesting to him that they were “histiocytic” tumors. Further observing the ability of the cells to produce collagen, he proposed that histiocytes function as “(facultative) fibroblasts” and hence, coined the term malignant fibrous xanthoma for these tumors. Admittedly, there was great subjectivity to this work, and ultimately his concept proved incorrect, but the zeal with which he embraced new technologies would prove to be precedent setting for our discipline.

Dr. Arthur Purdy Stout (left) and his protégé, Dr. Raffaele Lattes. (Courtesy of Dr. Raffaele Lattes).

Title page and illustrations from the classic Stout articles on malignant fibrous histiocytoma.
The concept of malignant fibrous histiocytoma was greeted with enthusiasm by Franz Enzinger, the newly appointed chair of soft tissue pathology at the AFIP, who placed it in his 1969 World Health Organization classification of soft tissue tumors (16; Fig. 13). This classification represented a quantum leap in the classification of soft tissue tumors and was accompanied by a glass slide set of nearly 200 cases, no doubt contributing greatly to the rapid assimilation of the classification.

Dr. Franz Enzinger, chair of soft tissue pathology, Armed Forces Institute of Pathology, and author of the World Health Organization’s classification of soft tissue tumors.
In 1975, while still a resident, I ventured to the AFIP to study with Dr. Enzinger, an association that lasted over a decade. I began my tenure there with a massive study of several hundred pleomorphic sarcomas retrieved from the vast archives of this institution, which at that time represented a nearly unique resource in the area of diagnostic consultations. I soon discovered that the majority of pleomorphic sarcomas displayed no differentiation by light microscopy, nor did they occur widely in the general population but developed almost exclusively in adults usually after the age of 50 years and segregated in the proximal extremities and retroperitoneum (4). These tumors were often characterized by an inflammatory infiltrate, a phenomenon now known to be associated with cytokine production and occasionally paraneoplastic symptoms (17, 18). It was for this group of pleomorphic, undifferentiated sarcomas of adults that we employed the term, malignant fibrous histiocytoma. Many questioned Stout’s histiocytic concept, and this term over the years waxed and waned in popularity. In fact, Dr. Lauren Ackerman at one point claimed that the malignant fibrous histiocytoma was so rampant that it had become a public health problem in Washington, D.C., requiring his singular efforts to stamp it out (Fig. 14). But the value of this term, in my mind, was not so much in suggesting a relationship to a histiocyte but rather in segregating a group of high-grade relatively “undifferentiated” adult sarcomas that could systematically be studied over the ensuing years.

Dr. Lauren V. Ackerman, an early critic of the entity of malignant fibrous histiocytoma, taken at a meeting of the International Skeletal Society in Salzburg in the early 1990s. Photograph illustrates the phenomenon that things apart from tissue specimens get mislabeled.
This belief was reflected in two of my earliest papers in soft tissue pathology, in which I was less concerned with the line of differentiation than in defining prognostic parameters. Although size of a sarcoma had always been viewed as important, the depth of a sarcoma and its relationship to outcome was relatively unexplored. Impressed by the work of Dr. Wallace Clark relating depth of invasion in melanomas to outcome, I reasoned that the same might apply to sarcomas. In fact, this proved to be true. Malignant fibrous histiocytomas located superficial to the fascia had an excellent prognosis compared with those deep to the fascia (4, 5; Fig. 15).

Diagram showing relationship between depth and metastasis in malignant fibrous histiocytoma.
Over the ensuing decade, numerous excellent studies appeared from worldwide groups; these studies served to define the biologic profile of malignant fibrous histiocytoma, the prototype of the pleomorphic sarcoma (19, 20, 21, 22, 23, 24, 25, 26). Size was a continuous variable directly related to metastatic risk, with small lesions (<5 cm) having a significantly lower risk of metastasis than large (>5 cm) ones. Depth, too, emerged as a significant prognostic factor. Collectively, these observations have directly contributed to the recent American Joint Committee on Cancer staging system of sarcomas, which employs 3 parameters (grade, size, and depth), in contrast to earlier systems, which employed only grade supplemented by size (27). In this new staging system, large, deep, high-grade sarcomas are Stage III, or high-risk, sarcomas (Fig. 16). They grow by extending tendrils into surrounding tissue, such that excising only gross tumor leaves microscopic deposits. Local recurrences develop in 20–35% of patients as a result of regrowth of residual tumor (Fig. 17). In the experience of some, microscopically positive margins also correlate with an increased risk of distant metastasis and diminished survival (28), but this is not borne out by all studies (29). Although one might surmise that local recurrence in a high-risk sarcoma would be associated with distant metastasis because it would serve as a source of metastasis, this point has been controversial. Some studies have been unable to show significant differences in metastasis between patients who have had or not had a local recurrence, whereas others contend that there is an association (30, 31) but that it is likely noncausal (29, 32) and simply serves as a surrogate marker of a more aggressive tumor (29). There is no dispute, however, that distant metastasis is the primary reason that patients succumb to their tumor. Metastasis is greatly time dependent and occurs in 30–40% of patients within 2–3 years of diagnosis, after which the risk appears to decrease. In fact, after 5 years, the metastatic risk between low- and high-risk sarcomas seems to equilibrate.

Definition of high-risk (Stage III) sarcomas by American Joint Committe on Cancer staging system.

Clinical behavior of high-risk (Stage III) sarcomas.
Concurrent with the plethora of clinical studies, pathologists continued to investigate the lineage of this tumor. On the basis of a large body of literature, it gradually became clear that the cells of the malignant fibrous histiocytoma possessed neither the ultrastructural nor immunophenotypic profile of the histiocyte but rather were more closely related to primitive mesenchymal cells or fibroblasts (33, 34, 35, 36). By immunohistochemistry, they lacked the usual enzymes (e.g., lysozyme; 34, 35) and surface markers (CD45, CD14,CD15; 35) associated with histiocytes and possessed, according to electron microscopy, prominent branching endoplasmic reticulum similar to fibroblasts (36; Fig. 18). Moreover, features often attributable to histiocytes, such as prominent primary and secondary lysosomes, which were observed in some cells in malignant fibrous histiocytoma, were easily explained by the phagocytic properties often acquired by neoplastic cells. However, study of the intermediate filaments in these lesions disclosed the unexpected presence of keratin, desmin, and even neural filaments (37, 38), raising the question of whether these unusual profiles reflected aberrant expression or a subtle form of differentiation not previously appreciated.

Electron micrograph illustrating fibroblastic features of a neoplastic cell in a malignant fibrous histiocytoma.
These observations provided the stage for the seminal paper by Dr. Christopher Fletcher, that suggested that with meticulous ultrastructural and immunohistochemical analysis, most pleomorphic soft tissue tumors could be subclassified (39). Although this paper has been interpreted by many as deconstructing the entity of malignant fibrous histiocytoma, others have believed that it reaffirmed the entity by demonstrating that, indeed, some sarcomas (the exact percentage was debated) resisted classification even after careful morphologic, immunohistochemical, and ultrastructural analysis. However, in my mind, its true importance lay in demonstrating that as diagnostic technologies become more sophisticated, we will invariably uncover more differences in things that we previously considered the same, leading inevitably to a more fundamental question about the significance of differences. If we approach the problem pragmatically, as expressed in the words of Gertrude Stein, we might say that “a difference to be a difference must make a difference.” Let us then reflect on the possible reasons for classifying pleomorphic sarcomas.
-
1
Personal satisfaction. It makes us feel good to bring a case to closure with a name or label. Clinicians like that too. Dr. David Dahlin once quipped that clinicians request labels on tumors because, in the unlikely event they cure one, they like to know the name of what it was they cured.
-
2
Behavior. Most of us want to believe that the various labels that we place on pleomorphic sarcomas translate into differences in behavior. Unfortunately, it is difficult to make a strong case that the various pleomorphic sarcomas are behaviorally significantly different from one another. Much of the problem stems from the fact that aside from malignant fibrous histiocytoma, other pleomorphic sarcomas are rare and the data relating to outcome limited. Even if we were able to pool or combine studies, there are significant barriers to overcome. Pathologists would have to agree to certain common diagnostic guidelines. For example, when immunohistochemical data seemingly conflicts with light-microscopic observations, which do we believe? Even experts may differ on the weight that they place on various features. In a exercise of classifying pleomorphic sarcomas, presented at the International Academy of Pathology meeting in Nagoya, Japan, in 2000, six international experts (Drs. Brooks, Fisher, Iwasaki, Kindblom, Meis-Kindblom, and Weiss) agreed on the classification of pleomorphic soft tissue lesions in most (80%), but not all cases. I would expect this figure to be lower among those with less experience. We would have to decide which of the several grading systems in place would be used and whether the system could be reproducibly employed in general practice. In comparing pleomorphic sarcomas, what end points will we compare—metastatic rate, 5-year survival, or disease-free survival? Factors such as size, depth, and location would have to be kept nearly constant across groups so as to isolate differentiation as the only variable. It is, therefore, not surprising that our information is fragmentary at best. Although it might appear from one large study that dedifferentiated liposarcoma, with a metastatic rate of 17% (3), has a better prognosis than other pleomorphic sarcomas (4), one needs to bear in mind that metastases in that study included only clear-cut metastases in organs such as liver and lung and did not include intra-abdominal lesions, which may well represent local metastases. Furthermore, the apparent difference in behavior between dedifferentiated liposarcoma and other pleomorphic sarcomas might well reflect the volume differences in the undifferentiated component rather than intrinsic differences related to phenotype. Finally, even if we overcame all of these barriers and were able to show differences that were “statistically significant,” at what point would they become “clinically significant”?
-
3
Treatment. Is there evidence that the various types of pleomorphic sarcoma are treated differently? For better or worse, all high-grade sarcomas are treated with wide local excision, combined with either pre- or postoperative radiation. Chemotherapy may be added, although its benefits have been debated. A recent large meta-analysis suggests that chemotherapy improves outcome, but not at a level that achieves statistical significance (40). There is some agreement that certain sarcomas (e.g., leiomyosarcoma) are more chemoresistant than others, however (40). Therefore, although we may labor hard to subclassify a high-grade pleomorphic sarcoma, it is doubtful that this piece of information influences therapy in the majority of cases.
-
4
Cytogenetic differences. One might also hope that each of the pleomorphic sarcomas would harbor a unique cytogenetic abnormalities that would allow each to be studied and understood more mechanistically, as has occurred with many of the nonpleomorphic sarcomas, such as myxoid liposarcoma, Ewing’s sarcoma, alveolar rhabdomyosarcoma, and clear cell sarcoma. To date, no recurring specific differences have been noted among pleomorphic sarcomas (41). All are characterized by numerous and complex genetic alterations (Fig. 19), although there appears to be a correlation between the degree of complexity and the grade (41). One exception is the dedifferentiated liposarcoma, which retains giant marker and/or ring chromosomes as a vestige of its origin from a well-differentiated liposarcoma (42) (Fig. 20). This is not to say that in time, these complex cytogenetic aberrations will not be deciphered into meaningful or specific patterns.
FIGURE 19 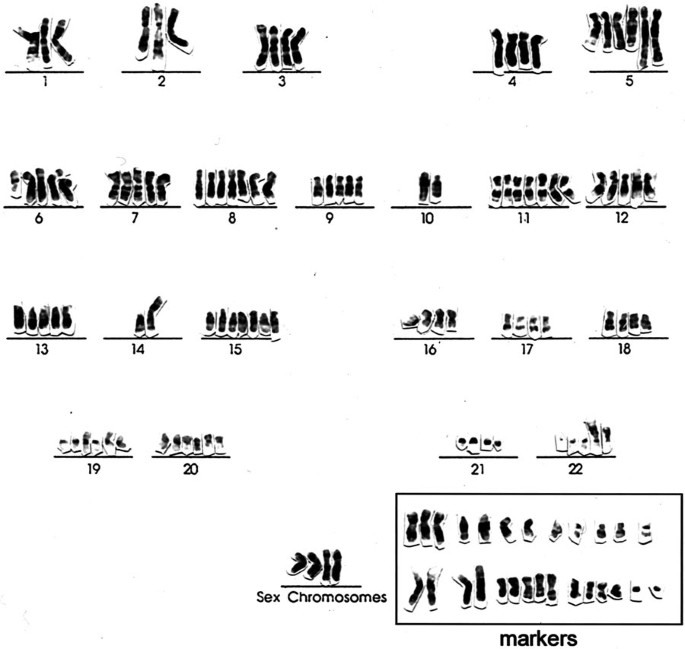
Karyotype of a malignant fibrous histiocytoma illustrating numerous complex chromosomal aberrations (courtesy of Dr. Jonathan Fletcher).
FIGURE 20 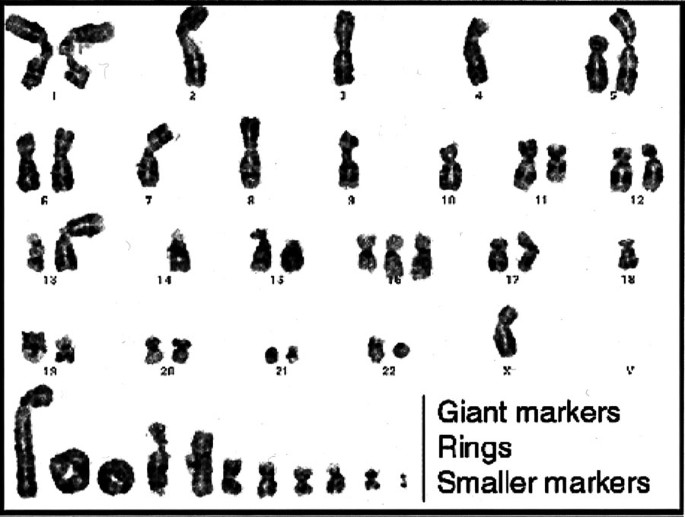
Karyotype of a dedifferentiated liposarcoma showing giant marker and ring chromosomes, reflective of its origin from a well-differentiated liposarcoma (atypical lipomatous tumor), in addition to numerous complex alterations. (Courtesy of Dr. Jonathan Fletcher.)


To conclude on the note that we ought not to classify pleomorphic sarcomas because we currently have no use for this information would be unduly pessimistic. It will become increasingly important to preserve histologic differences among tumors for future work and therapies, which may well be tailored along histogenetic lines. There is no better example of this phenomenon than the gastrointestinal stromal tumor (GIST), a tumor that was reproducibly recognized by surgical pathologists for decades before its similarity to the interstitial cell of Cajal was noted and long before mutations in the tyrosine kinase receptor c-kit and overexpression of its protein were described. Although the molecular and immunohistochemical features of this tumor have since become virtually definitional for this neoplasm, the groundwork that surgical pathologists provided by segregating these lesions as a special and distinctive lesion facilitated their systematic study by the scientific community. Even more gratifying has been the promising work showing that the tyrosine kinase inhibitor STI 571 can inhibit the constitutively activated kit kinase (43) by binding to the kinase region of the protein (44) and affecting downstream regulators of proliferation and apoptosis, with dramatic clinical results in patients (45). Although the long-term effects of this drug remain uncertain because of mutational changes in tumors (46), our hopes are, of course, that other such paradigms exist and that we as pathologists will play a role in their elucidation.
I am at the end of my story for now. What lessons have we learned from this story? First, as a discipline, we have a rich and deep tradition of research and investigation that has gone hand in hand with clinical practice. We have been fast to seize on new technologies and apply them to our clinical problems, as evidenced by use of tissue culture techniques, followed by electron microscopy, immunohistochemistry, cytogenetics, genomics, and, now, proteomics. Surgical pathology is a discipline that prefers order and certainty over disorder and uncertainty. As a result, we fastidiously label tumors. But we must remember that the point of labeling tumors is to accurately communicate, not obfuscate and confuse. When we change names or invent new ones, we must do so responsibly and in a way that communicates effectively with those with whom we work. We have much to learn just in terms of the basic biology of the tumors we have described. To do that, we must partner with each other and with our clinical colleagues to achieve consensus on basic issues of diagnosis, grading, and staging.
The challenges ahead are great, but most important among these is the challenge of leaving a legacy of talented young pathologists to succeed us and to participate in the exciting work that we have begun. Each of us has experienced moments of doubt or uncertainty because of the difficulties that we have faced in attracting a cadre of bright innovative individuals to our discipline. We blame the length of our training programs, loss of control over medical student curriculum, and insufficient funding for research, to name a few. To be sure, all enter the equation. But, we, too, bear some responsibility. I submit to you that we have allowed our discipline to be labeled as unattractive and undesirable. Consider this book on how to choose a medical specialty (available at most bookstores), with the following quotation describing the specialty of emergency medicine (47): “the new kid on the block. The fastest growing medical specialty. The opportunity to be a real doctor:…one of the most popular residency choices among top-ranked graduates of medical school.” Now compare it with the description of pathology: “the pathologist is the unknown man of medicine. Called the ‘doctor’s doctor,’ the pathologist acts as a consultant to clinicians. Therefore, the only channels by which his or her services reach the patient are controlled by other doctors. Some characterize pathologists as reclusive, insecure, uncomfortable, ill at ease with others, and inept at interpersonal communication” (Fig. 21). If you were a medical student, which would you choose? You might as well ask, “would you like to write the great American novel or ride a bus?” If you will permit me, I would like to close with one last autobiographical note. When I was very young, I used to ask my father what should I become when I grew up. His answer was always the same: “do what you enjoy, everything else will follow.” With some minor refinements, this has always served me well. I would add one other comment to that advice. If you enjoy your life’s work, share that joy, share that love, share that passion with those around you. This is one of the surest and most personal of ways to show that our discipline is both exciting and viable. Thank you.

A recent book providing capsular descriptions of various medical specialties.
References
Farquhar MG, Palade GE . Junctional complexes in epithelia. J Cell Biol 1963; 17: 375–412.
Weiss SW, Rao VK . Well differentiated liposarcoma (atypical lipomatous tumors) of deep soft tissues of the extremities, retroperitoneum, and miscellaneous sites: a follow up study of 92 cases with analysis of the incidence of dedifferentiation. Am J Surg Pathol 1992; 16: 1051–1058.
Henricks WH, Chu YC, Goldblum JR, Weiss SW . Dedifferentiated liposarcoma: a clinicopathologic analysis of 155 cases with a proposal for an expanded definition of dedifferentiation. Am J Surg Pathol 1991; 21: 271–281.
Weiss SW, Enzinger FM . Malignant fibrous histiocytoma: an analysis of 200 cases. Cancer 1978; 41: 2250–2266.
Weiss SW, Enzinger FM . Myxoid variant of malignant fibrous histiocytoma. Cancer 1977; 39: 1672–1685.
Mentzel T, Bosenberg M, Fletcher CDM . Pleomorphic liposarcoma: clinicopathologic and prognostic analysis of 31 cases [abstract]. Mod Pathol 1999; 12: 55A.
Downes KA, Goldblum JA, Montgomery EA, Fisher C . Pleomorphic liposarcoma: a clinicopathologic analysis of 19 cases. Mod Pathol 2001; 14: 179–184.
Miettinen M, Enzinger FM . Epithelioid variant of pleomorphic liposarcoma: a study of 12 cases of a distinctive variant of high grade liposarcoma. Mod Pathol 1999; 12: 722–728.
Gaffney EF, Dervan PA, Fletcher CD . Pleomorphic rhabdomyosarcoma in adulthood: analysis of 11 cases with definition of diagnostic criteria. Am J Surg Pathol 1993; 17: 601–609.
Furlong MA, Mentzel T, Fanburg-Smith JC . Pleomorphic rhabdomyosarcoma in adults: a clinicopathologic study of 38 cases with emphasis on morphologic variants and recent muscle-specific markers. Mod Pathol 2001; 14: 295–603.
Schuerch W, Begin LR, Seemayer TA, et al. Pleomorphic soft tissue myogenic sarcomas of adulthood: a reappraisal in the mid 1990’s. Am J Surg Pathol 1996; 20: 131–147.
Oda Y, Miyajima K, Kawaguchi K, et al. Pleomorphic leiomyosarcoma: clinicopathologic and immunohistochemical study with special emphasis on its distinction from ordinary leiomyosarcoma and malignant fibrous histiocytoma. Am J Surg Pathol 2001; 25: 1030–1038.
Ewing J . Neoplastic diseases: a textbook on tumors. Philadelphia: W.B. Saunders; 1919.
O’Brien JE, Stout AP . Malignant fibrous xanthomas. Cancer 1964; 17: 1445–1451.
Ozzello L, Stout AP, Murray MR . Cultural characteristics of malignant histiocytomas and fibrous xanthomas. Cancer 1963; 16: 331–334.
Enzinger FM, Lattes R, Torloni H . Histologic typing of soft tissue tumors. Geneva: World Health Organization; 1969.
Hamada T, Komiya S, Hiraoka K, et al. IL-6 in a pleomorhic type of malignant fibrous histiocytoma presenting high fever. Hum Pathol 1998; 29: 758–761.
Melhem MF, Meisler AJ, Saito R, et al. Cytokines in inflammatory malignant fibrous histiocytoma presenting with leukemoid reaction. Blood 1993; 82: 2038–2044.
Enjoji M, Hashimoto H, Tsuneyoshi M, Iwasaki H . Malignant fibrous histiocytoma: a clinicopathologic study of 130 cases. Acta Pathol Jpn 1980; 30: 727–741.
Le Doussai V, Coindre JM, Leroux A, et al. Prognostic factors for patients with localized primary malignant fibrous histiocytoma: a multicenter study of 216 patients with multivariate analysis. Cancer 1996; 77: 1823–1830.
Kearney MM, Soule EH, Ivins JC . Malignant fibrous histiocytoma: a retrospective study of 167 cases. Cancer 1980; 45: 167–178.
Pezzi CM, Rawlings MS, Esgro JJ, Pollock RE, et al. Prognostic factors in 227 patients with malignant fibrous histiocytoma. Cancer 1992; 69: 2098–2103.
Pritchard DJ, Reiman HM, Turcotte RE, Ilstrup DM . Malignant fibrous histiocytoma of the soft tissues of the trunk and extremities. Clin Orthop 1993; 289: 58–65.
Rooser B, Willen H, Gustafson P, et al. Malignant fibrous histiocytoma of soft tissue: a population based epidemiological and prognostic study of 137 patients. Cancer 1991; 67: 499–505.
Salo JC, Lewis JJ, Woodruff JM, et al. Malignant fibrous histiocytoma of the extremity. Cancer 1999; 85: 1765–1772.
Zagars GK, Mullen JR, Pollack A . Malignant fibrous histiocytoma: outcome and prognostic factors following conservation surgery and radiotherapy. Int J Radiat Oncol Biol Phys 1996; 34: 983–994.
Soft tissue sarcoma.In: Fleming ID, Cooper JS, Henson DE, et al., editors. AJCC cancer staging handbook. Philadelphia: Lippincott Williams and Wilkins; 1997.p. 139–146.
Heslin MJ, Woodruff J, Brennan MF . Prognostic significance of a positive microscopic margin in high-risk extremity sarcoma: implications for management. J Clin Oncol 1996; 14: 473–478.
Trovik CS, Bauer HCF, Alvegard TA, et al. Surgical margins, local recurrence and metastasis in soft tissue sarcomas: 559 surgically treated patients from the Scandinavian Sarcoma Group Register. Eur J Cancer 2000; 36: 710–716.
Pisters PWT, Leung DHY, Woodruff JM, et al. Analysis of prognostic factors in 1041 patients with localized soft tissue sarcomas of the extremities. J Clin Oncol 1996; 14: 1679–1689.
Lewis JJ, Leung D, Heslin M, Woodruff JM, Brennan MF . Association of local recurrence with subsequent survival in extremity soft tissue sarcoma. J Clin Oncol 1997; 15: 646–652.
Trovik CS . Local recurrence of soft tissue sarcoma: a Scandinavian Sarcoma Group project. Acta Orthop Scand 2001; 72(300 Suppl): 22–26.
Fu Y-S, Gabbiani G, Kaye GI, Lattes R . Malignant soft tissue tumors of probable histiocytic origin (MFH): general considerations and electron microscopic and tissue culture studies. Cancer 1975; 35: 176–198.
Wood GS, Beckstead JH, Turner RR, et al. Malignant fibrous histiocytoma tumor cells resemble fibroblasts. Am J Surg Pathol 1986; 10: 323–335.
Iwasaki H, Isayama T, Johzaki H, Kikuchi M . Malignant fibrous histiocytoma: evidence of perivascular mesenchymal cell origin immunocytochemical studies with monoclonal anti-MFH antibodies. Am J Pathol 1987; 1218: 528–537.
Erlandson RA, Woodruff JM . Role of electron microscope in the evaluation of soft tissue neoplasms, with emphasis on spindle cell and pleomorphic tumors. Hum Pathol 1998; 29: 1372–1381.
Hirose T, Kudo E, Hasegawa T, et al. Expression of intermediate filaments in malignant fibrous histiocytomas. Hum Pathol 1989; 20: 871–877.
Lawson CW, Fisher C, Gatter KC . An immunohistochemical study of differentiation in malignant fibrous histiocytoma. Histopathology 1987; 11: 375–383.
Fletcher CDM . Pleomorphic malignant fibrous histiocytoma fact or fiction: A critical appraisal based on 159 tumors diagnosed as pleomorphic sarcoma. Am J Surg Pathol 1992; 16: 213–228.
Tierney JF, Steward LA, Parmar MKB . Adjuvant chemotherapy for localized resectable soft-tissue sarcoma of adults: meta-analysis of individual data. Lancet 1997; 35: 1647–1654.
Mertens F, Fletcher CDM, Dal Cin P, et al. Cytogenetic analysis of 46 pleomorphic soft tissue sarcomas and correlation with morphologic and clinical features: a report of the CHAMP study group. Genes Chromosom Cancer 1998; 22: 16–25.
Fletcher JA . Cytogenetic analysis of soft tissue tumors.In: Weiss SW, Goldblum JR. Enzinger and Weiss’s Soft tissue tumors. 4th ed. Philadelphia: Mosby-Harcourt Brace; 2001.
Heinrich MG, Griffith DJ, Druker BJ, et al. Inhibition of c-kit receptor tyrosine kinase activity by STI 571, a selective tyrosine kinase inhibitor. Blood 2000; 96: 925–932.
Schindler T, Bornmann W, Pellicena P, Miller TW, Clarkson B, Kuriyan J . Structural mechanism for STI-571 inhibition of Abelson tyrosine kinase. Science 2000; 289: 1938–1942.
Joensuu H, Roberts PJ, Sarlomo-Rikala M, et al. Effect of the tyrosine kinase inhibitor STI571 in a patient with a metastatic gastrointestinal stromal tumor. N Engl J Med 2001; 344: 1052–1056.
Gorre ME, Mohammed M, Ellwood K, et al. Clinical resistance to STI-571 cancer therapy caused by BCR-ABL gene mutation or amplification. Science 2001; 293: 876–880.
Taylor AD . How to choose a medical specialty. 2nd ed. Philadelphia: W.B. Saunders; 1993.p. 57, 149.
Acknowledgements
I would like to gratefully acknowledge the assistance of Bob Santoianni in the preparation of the photographs for this article. This work is dedicated to the memory of Dr. Rodger Haggitt.
Author information
Authors and Affiliations
Corresponding author
Additional information
Presented at the 90th Annual Meeting of the United States and Canadian Academy of Pathology, Atlanta, March 6, 2001.
Rights and permissions
About this article
Cite this article
Weiss, S. 44th Maude Abbott Lecture. Mod Pathol 15, 77–86 (2002). https://doi.org/10.1038/modpathol.3880494
Accepted:
Published:
Issue date:
DOI: https://doi.org/10.1038/modpathol.3880494
