
Abstract
Small local populations of Silene alba, a short-lived herbaceous plant, were sampled in 1994 and again in 1999. Sampling included estimates of population size and genetic diversity, as measured at six polymorphic allozyme loci. When averaged across populations, there was very little change between samples (about three generations) in population size, measures of within-population genetic diversity such as number of alleles or expected heterozygosity, or in the apportionment of genetic diversity within and among populations as measured by Fst. However, individual populations changed considerably, both in terms of numbers of individuals and genetic composition. Some populations doubled in size between samples, while others shrank by more than 75%. Similarly, expected heterozygosity and allele number increased by more than two-fold in individual populations and decreased by more than three-fold in others. When population-specific change in number and change in measures of genetic diversity were considered together, significant positive correlations were found between the demographic and genetic variables. It is speculated that some populations were released from the demographic consequences of inbreeding depression by gene flow.
Similar content being viewed by others

Introduction
How genetics and demography interact to influence population viability has been a long-standing question in conservation biology. There is broad consensus that as populations become fragmented into increasingly smaller habitat patches, their continued viability is determined by both these factors, and possibly by their interaction (Ellstrand and Elam, 1993; Frankham, 1995; Young and Clarke, 2000; Amos and Balmford, 2001). Basic population genetic theory tells us that in small populations, genetic diversity erodes with time unless there are connections through gene flow to other populations. When decreases in genetic diversity are correlated with reductions in lifetime fitness components, there can be a positive feedback between population size and genetic diversity that can increase the probability of population extinction. If populations are also cut off from new input through immigration, they are deprived of a source of both new genetic diversity and the additional breeding individuals that could ameliorate the consequences of small size. Thus, theoretical studies have also emphasized the importance of migration in reducing the probability of extinction by dampening both genetic and demographic stochasticity (Goodman, 1987; Brown and Kodric-Brown, 1977).
Widely cited in conservation literature as the ‘extinction vortex’ or ‘mutational meltdown’ (Gilpin and Soulé, 1986; Lynch et al, 1995), the interaction of genetics and demography could also influence population persistence in common species, because it is generally accepted that even many abundant species are not uniformly distributed, but are structured into local breeding groups. Both the heterogeneity of habitat and the ecology of dispersal and gene flow influence the scale of this structure. Therefore, the viability of many naturally fragmented populations may also be influenced by the interaction of genetics and demography (Richards, 2000a). This could be true at the level of the local population or at the level of the metapopulation, depending on the number and connectivity of local populations (Thrall et al, 1998; Higgins and Lynch, 2001).
While the relation between the loss of genetic variation and population decline has been studied in experimental and natural populations (Menges, 1991; van Treuren et al, 1991; Oostermeijer et al, 1994; Heschel and Paige, 1995; Newman and Pilson, 1997; Fischer and Matthies, 1998; Hanski and Ovaskainen, 2000), the dynamical relation between genetic diversity and demography is less well-documented (but see Westemeier et al, 1998). Studies of genetic erosion and demographic decline in natural populations often compare classes of populations that differ in size, age, or degree of isolation (Menges, 1991; van Treuren et al, 1991; Oostermeijer et al, 1994; Ouborg and van Treuren, 1995). For example, we might predict that smaller populations would contain relatively low levels of genetic diversity, and that their component individuals would be less fit than would be the case in large populations. However, the prior history of the study populations is rarely known and populations with different prior histories are indistinguishable. For instance, a population could be small because it was recently founded, or because it had suffered a long decline. Perhaps, therefore, it would also be useful to track populations through time and ask whether there is any correlation between changes in genetic composition and changes in population size or individual fitness. If genetics and demography interact, one might predict that in populations in which genetic diversity is maintained or increased by gene flow, population size would remain stable or increase, whereas population size and genetic diversity might decrease over time in populations cut off from sources of gene flow.
In this study, we have focused on the genetic and demographic dynamics within populations of Silene latifolia, a short-lived perennial plant that has been the object of a long-term study of a regional metapopulation (Antonovics et al, 1994). Previous studies of this species, a comparison using nuclear allozyme loci, showed that younger populations of S. latifolia displayed higher genetic differentiation (using Wright's Fst) than older populations (McCauley et al, 1995). This suggests that the significant genetic structure accompanies founding events but is reduced by the homogenizing effects of gene flow in the years following colo-nization (but also see Ingvarsson and Giles, 1999). Additional studies have examined the factors affecting gene flow among small populations and the consequences of localized inbreeding (Richards et al, 1999; Richards, 2000b). This work has demonstrated that both the size and spatial isolation of the local population determines the rate of gene flow, and also the degree to which local populations suffer from inbreeding depression. Thus, individual populations could have very different demographic and genetic trajectories depending on their individual circumstances. It would therefore be informative to do comparative studies of these populations through time, in addition to analyzing static snapshots of genetic and demographic patterns, in order to better understand the relation between genetic diversity, demography, and population persistence.
We selected 11 populations of S. latifolia newly colonized in 1994 and have tracked their population size and genetic diversity over 6 years. Our interests in this study are two-fold. First, we ask how the populations vary in their individual genetic and demographic trajectories. Secondly, we evaluate the correlation across populations between changes in their growth rate and changes in their genetic diversity.
Materials and methods
Silene latifolia (=S. alba) is a dioecious short-lived perennial herb, originally introduced to the US from Europe in the mid-1800s, which has since spread throughout eastern North America and Canada (McNeill, 1977). The species is insect-pollinated by a variety of generalist insect pollinators including moths, hover flies, and bees (Altizer et al, 1998). Silene latifolia has been the object of a long-term study in Giles County, VA, tracking metapopulation dynamics and genetic structure (Antonovics et al, 1994; McCauley, 1994,1997; McCauley et al, 1995,2001; Richards, 2000b). This has involved a survey of a 25×25-km area adjacent to Mountain Lake Biological Station, in which there were hundred of S. latifolia populations. This survey, held annually in June since 1988, records the location, size, and sex of S. latifolia individuals within 40 m intervals along ∼150 km of roadside habitat.
The current study sampled 11 S. latifolia populations twice over a 6-year period, in 1994 and then again in 1999. In 1994, these same 11 populations (colonized in 1990–1994) were selected for the study based on small size (McCauley et al, 1995). The 11 populations are each at least 50 m from the nearest neighboring S. latifolia population and are separated by 1–20 km from one another. In addition, samples were taken in 1997 from eight of the original 1994 sites for DNA extraction.
The populations were sampled in 1994 by censusing the number of individuals of each sex and by taking leaf tissue from each flowering and vegetative individual. Seedlings were not studied. This method of sampling gave us information on the adult size of each population and provided us with tissue needed to obtain a six-locus allozyme genotype for each individual. Identical methods were used to sample each population in 1999. Other studies (Richards, 1997) have estimated average generation time in natural populations of S. latifolia to be approximately 1.5 years by direct census of marked plants, so there was most likely a three-generation interval between samples.
Allozyme electrophoresis was conducted at Mountain Lake Biological Station and at Vanderbilt University on both the 1994 and 1999 samples. Electrophoresis was carried out according to Werth (1985) and McCauley et al (1995). Individuals were scored for six loci (with their EC numbers); PGM (5.4.2.2), GPI (5.3.1.9), MDH (1.1.1.37), 6-PGD (1.1.1.44), SKDH (1.1.1.25), LAP (3.4.11.1). All loci were used in this study were known to be polymorphic, with two to six alleles segregating at each locus for a total of 27 alleles pooled across loci. Plants with known allozyme genotypes propagated clonally in the Vanderbilt greenhouse were used throughout the study to help maintain consistency in naming alleles.
The data from the 1994 sample are a subset of a larger data set reported earlier (McCauley et al, 1995). In the context of the present report, the 1994 and 1999 population-specific samples were compared in order to evaluate changes in population size, within-population heterozygosity, and allele identity and number. The genetic structure across populations was also estimated for the two samples by calculating Wright's Fst for the 1994 and 1999 data, respectively. DNA extractions were taken from individuals in eight of the 11 populations. These were used as templates for the PCR reactions needed to evaluate cpDNA haplotype diversity (McCauley, 1994). Correlations between demographic and genetic change were also investigated. In order to scale populations to a common demographic currency, population growth rate was calculated for each as (ln (size in 1999/size in 1994)). Correlations of this quantity were tested with relative changes in expected heterozygosity ((heterozygosity in 1999−heterozygosity in 1994)/heterozygosity in 1994) and number of alleles ((value in 1999−value in 1994)/value in 1994). Ratios were used to account for the fact that individual populations varied in their initial genetic diversity (other measures, such as (value in 1999−value in 1994), were also explored with similar results).
Results
The average population size in 1999 (12.7) was not significantly different from 1994 (11.8). However, some individual populations more than doubled in size during this time period, while others declined by up to five-fold (Table 1). The mean absolute change in population size was 6.72 individuals.
The number and identity of alleles pooled across all six loci and all 11 populations did not change between 1994 and 1999. That is, the same 27 alleles were detected in each sample. The mean number of alleles/population was 17.2 in 1994 and 16.7 in 1999, not a significant difference (Table 1). However, there was considerable change within individual populations, with some populations gaining as many as six alleles, and others losing as many as eight alleles. The average absolute net change was 3.18 alleles (Table 1). Moreover, alleles that were globally rare were more likely to disappear from some populations, and appear for the first time in others, between 1994 and 1999 (Figure 1). The relative distribution of genetic diversity within and among populations, as measured by Wright's Fst, also remained more or less constant between years (Table 1). Comparison of the cpDNA haplotype composition of the eight sampled populations in 1994 and 1997 using PCR length polymorphisms reported earlier (McCauley, 1994) showed that Wright's Fst increased significantly (from 0.68 in 1994 to 0.98 in 1997) because of the loss of individual haplotypes during the sampling interval (Table 2).

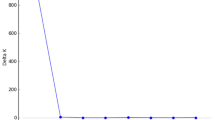
Turnover of allozyme alleles as a function of their frequency pooled across all S. alba populations that were sampled in 1994. Turnover is defined as the fraction of all populations carrying a given allele in the 1999 that did not carry it in 1994. Note: all alleles seen in the 1999-pooled sample were seen in the 1994-pooled sample.
The mean within-population heterozygosities in 1994 and 1999 (0.50 in each case) were not significantly different (Table 1). However, heterozygosity increased considerably in some populations and decreased considerably in others with a mean absolute change of 0.07 (Table 1).
Figures 2a and b present the correlations between the changes over the sampling interval in population-specific demographic and genetic properties. There was a statistically significant positive product-moment correlation between change in population size and change in both heterozygosity (Figure 2a; r=0.64, df=9, P<0.03) and allele number (Figure 2b; r=0.81, df=9, P<0.002). Populations that grew between 1994 and 1999 also experienced a net increase in these two measures of genetic diversity.

Relation between demographic and genetic changes in 11 study populations of S. alba that were sampled in 1994 and 1999. Panel (a) presents the relation between population-specific changes in expected heterozygosity and a measure of population growth. Panel (b) presents the relation between population-specific changes in allele number and a measure of population growth. See the text in Material and methods for details on calculations of the variables.
Discussion
In all species, individual population histories are crucial in determining the levels of genetic diversity. The unique behaviour of individual populations, each with a different size, population history, and genetic diversity, should give a powerful assessment of how genetics and demography influence population fitness. It is interesting to note the contrast in our results between the dynamical behaviour of individual populations and the apparent stasis evident when the set of populations is described by summary statistics, such as mean population size, mean heterozygosity, or Fst.
It is well known that the genetic properties of small populations can be influenced by the joint action of gene flow and genetic drift (reviewed by Ellstrand and Elam, 1993). What appears to be happening in the S. latifolia study populations is that the relative strength of these two processes differs from population to population, such that some populations increase in genetic diversity between successive samples, whereas others decrease in diversity. Nevertheless, Fst remained remarkably constant over the sampling period. The trajectories that each new colony took during the sampling period do not appear to be because of deterministic features of the colony founding event. There were no statistical relations between the number of individuals, the number of alleles present and the heterozygosity (expected or observed) of the colony in 1994 and its relative growth rate measured in 1999.
A comparison of the cpDNA haplotype composition of eight (of the initial eleven populations) sampled in 1994 and 1997 gives a different view of the dispersal dynamics after colonization. The chloroplast markers are matrilineally inherited and therefore describe the pattern of seed dispersal (McCauley, 1994). As shown in Table 2, there was a clear decline in haplotype diversity in this 3-year period. No population added a haplotype not seen in 1994, but several populations lost haplotypes.
The data suggest that many of the 11 populations were subjected to severe genetic drift. The sharp overall decline in within-population cpDNA haplotype diversity and the more moderate and population-specific loss of allozyme alleles suggest that gene flow among these populations was primarily through pollen movement and not through seed. New alleles could also appear through seed immigration, but it would be unlikely that seeds could contribute to allozyme diversity without jointly contributing to cpDNA diversity. The extent of ongoing pollen-mediated gene flow is also detected through the turnover rate among rare alleles. Alleles that were globally rare experienced the highest rate of local extinction and recolonization (Figure 1). These vagrant alleles persist in the metapopulation only because gene flow, presumably through pollen, transports them to new localities at approximately the same rate that they are lost through local extinction.
Theory predicts that smaller populations should have lower genetic diversity, and lose it faster than larger populations. Not surprisingly, our results show that genetics and demography are not independent of one another in S. latifolia. What is interesting in this study, however, is that a significant correlation was shown between change in the genetic properties of populations and change in population size. Populations that increased in size increased in genetic diversity, whereas populations that decreased in size lost genetic diversity. That is, the relative influence of gene flow and genetic drift varied according to whether the population was growing or shrinking.
Of course, the directionality of cause and effect could be such that demography influences genetics, rather than genetics influencing demography. For example, larger populations could be more attractive to pollinators, which would result in a higher rate of gene flow. Thus, growing populations would increase in genetic diversity. It would be tempting to ask which particular attribute of the colony site was critical in determining the extent of subsequent gene flow, but because of the limited number of colonies that were followed in this study, we did not have the statistical power to identify any one particular factor. It is known from previous work (Richards et al, 1999) that population size influences the rate of gene flow into artificial populations of S. latifolia. However, this relation is complex because it is influenced by the joint effects of population size and degree of isolation. Whatever the causal mechanism for the observed correlation between genetic and demographic dynamics, it is clear that the individual study populations are not interchangeable entities.
While the detrimental effects of inbreeding in many domesticated and naturally occurring species have been widely documented over the last century (reviewed by Charlesworth and Charlesworth, 1987; Keller and Waller, 2002), the issue of how genetic variation and, most importantly, the change of genetic variation influence the viability of natural populations has only been studied in a small number of taxa (Jimenez et al, 1994; Saccheri et al, 1998; Westemeier et al, 1998; Madsen et al, 1999; reviewed by Hedrick and Kalinowski, 2000 and also Dudash and Fenster, 2000). There have been several studies of how population size and degrees of isolation have negative affects on survival and fecundity in many plant taxa, but few longitudinal studies have tracked population size and measures of genetic variation in natural populations through time (Heschel and Paige, 1995). For this reason, the correlation between measures of genetic diversity and population fitness, as measured through survival and fecundity, has been complex. For example, in a series of studies of the genetic diversity and population measures of growth rate and fecundity of the threatened perennial plant Scabiosa columbaria in the Netherlands, allozyme diversity decreased in the smaller populations. However, early fitness estimates based on seed set, seed size, and seedling size were mostly influenced by nongenetic maternal effects rather than by population origin (van Treuren et al, 1991). Similarly, in populations in the long-lived perennial Gentiana pneumonanthe, measures of early fitness were positively correlated with population size; however, correlations between population sizes with fitness traits later in the life cycle were not significant (Oostermeijer et al, 1994; Raijman et al, 1994). In contrast, studies of the short-lived plants, Gentianella germaninca and Ipomopsis aggregata, showed reduced seed production and flower production in smaller populations and the authors attributed these responses to genetic factors (Heschel and Paige, 1995; Fischer and Matthies, 1998).
The most important question raised by these results would be to ask what cause-and-effect mechanism underlies the observed correlation. It is often hypothesized that there can be a positive feedback between dwindling population size and dwindling genetic diversity: the so-called extinction vortex (Gilpin and Soulé, 1986). In fact, another set of newly established small populations of S. latifolia has been shown to suffer from inbreeding depression, so that could be the case here (Richards, 2000b). Similarly, it has been hypothesized that gene flow can rescue small populations from extinction if an increase in population fecundity because of heterosis accompanies the import of new alleles (Thrall et al, 1998; Madsen et al, 1999; Richards, 2000a; Ingvarsson, 2001; Ebert et al, 2002). Thus, it could be postulated that the correlation observed here is because of inbreeding depression reducing the size of some populations even as gene flow permits others to grow.
References
Altizer SM, Thrall PH, Antonovics J (1998). The role of pollinators as vectors for anther-smut infection in Silene alba. Am Midland Nat 139: 147–163.
Amos W, Balmford A (2001). When does conservation genetics matter? Heredity 87: 257–265.
Antonovics J, Thrall P, Jarosz A, Stratton D (1994). Ecological genetics of metapopulations: the Silene–Ustilago plant–pathogen system. In: Real L (ed) Ecological Genetics, Princeton University Press: Princeton, NJ. pp 129–145.
Brown JH, Kodric-Brown A (1977). Turnover rates in insular biogeography: effect of immigration and extinction. Ecology 58: 445–449.
Charlesworth D, Charlesworth B (1987). Inbreeding depression and its evolutionary consequences. Ann Rev Ecol Syst 18: 237–268.
Dudash MR, Fenster CB (2000). Inbreeding and outbreeding depression in fragmented populations. In: Young AC and Clarke GM (eds) Genetics, Demography and Viability of Fragmented Populations, Cambridge University Press: Cambridge, UK. pp 35–54.
Ebert D, Haag C, Kirkpatrick M, Riek M, Hottinger J, Pajunen VI (2002). A selective advantage to immigrant genes in a Daphnia metapopulation. Science 295: 485–488.
Ellstrand N, Elam D (1993). Population genetic consequences of small population size: implications for plant conservation. Ann Rev Ecol Syst 24: 217–242.
Fischer M, Matthies D (1998). Effects of population size on performance in the rare plant Gentianella germanica. J Ecol 86: 195–203.
Frankham R (1995). Conservation genetics. Ann Rev Genet 29: 305–327.
Gilpin M, Soulé M (1986). Minimum viable populations: the process of species extinction. In: Soulé ME (ed) Conservation Biology; the Science of Scarcity and Diversity, Sinauer Associates: Sunderland, MA. pp 19–34.
Goodman D (1987). The demography of chance extinction. In: Soulé ME (ed) Viable Populations for Conservation. Cambridge University Press: Cambridge, UK. pp 11–34.
Hanski I, Ovaskainen O (2000). The metapopulation capacity of a fragmented landscape. Nature 404: 755–758.
Hedrick PW, Kalinowski ST (2000). Inbreeding depression in conservation biology. Ann Rev Ecol Syst 31: 139–163.
Heschel M, Paige K (1995). Inbreeding depression, environmental stress, and population size variation in Scarlet gilia (Ipomopsis aggregata). Conservation Biol 9: 126–133.
Higgins K, Lynch M (2001). Metapopulation extinction caused by mutation accumulation. Proc Nat Acad Sci USA 98: 2928–2933.
Ingvarsson PK (2001). Restoration of genetic variation lost – the genetic rescue hypothesis. Trends Ecol Evol 16: 62–63.
Ingvarsson PK, Giles BE (1999). Kin-: structured colonization and small-scale genetic differentiation in Silene dioica. Evolution 53: 605–611.
Jimenez JA, Hughes KA, Alaks G, Graham L, Lacy RC (1994). An experimental- study of inbreeding depression in a natural habitat. Science 266: 271–273.
Keller LF, Waller DM (2002). Inbreeding effects in wild populations. Trends Ecol Evol 17: 230–241.
Lynch M, Conery J, Bürger R (1995). Mutational meltdown in sexual populations. Evolution 49: 1067–1080.
Madsen T, Shine R, Olsson M, Wittzell H (1999). Conservation biology – restoration of an inbred adder population. Nature 402: 34–35.
McCauley DE (1994). Contrasting the distribution of chloroplast DNA and allozyme polymorphism among local populations of Silene alba: implications for studies of gene flow in plants. Proc Nat Acad Sci USA 91: 8127–8131.
McCauley DE . (1997). The relative contributions of seed and pollen movement to local genetic structure of Silene alba. J Heredity 88: 257–263.
McCauley D, Raveill J, Antonovics J (1995). Local founding events as determinants of genetic structure in a plant metapopulation. Heredity 75: 630–636.
McNeill J (1977). The biology of Canadian weeds. 25. Silene alba (Miller) E. H. L. Krause. Can J Plant Sci 57: 1103–1114.
Menges ES (1991). Seed germination percentage increases with population size in a fragmented prairie species. Conservation Biol 5: 158–164.
Newman D, Pilson D (1997). Increased probability of extinction due to decreased genetic population size: experimental populations of Clarkia pulchella. Evolution 51: 354–362.
Ouborg J, van Treuren R (1995). The significance of genetic erosion in the process of extinction. IV. Inbreeding load and heterosis in relation to population size in the mint Salvia pratensis. Evolution 48: 996–1008.
Oostermeijer JGB, Van Eijck MW, Den Nijs HCM (1994). Offspring fitness in relation to population size and genetic variation in the rare perennial plant species Gentiana pneumonanthe (Gentianaceae). Oecologia 97: 289–296.
Raijman LEL, van Leewen NC, Kersten R, Oostermeijer JGB, den Nijs HCM, Menken SBJ (1994). Genetic variation and outcrossing rate in relation to population size in Gentiana pneumonanthe L. Conservation Biol 8: 1014–1026.
Richards CM (1997). The ecological genetics of population establishment in Silene alba. Ph.D. Dissertation, Duke University, Durham, NC. USA.
Richards CM (2000a). Genetic and demographic influence on population persistence: gene flow and genetic rescue in Silene alba. In: Young AC, Clarke GM (eds) Genetics, Demography and Viability of Fragmented Populations, Cambridge University Press: Cambridge, UK. pp 271–292.
Richards CM (2000b). Inbreeding depression and genetic rescue in a plant metapopulation. Am Nat 155: 383–394.
Richards CM, Church S, McCauley DE (1999). The influence of population size and isolation on gene flow by pollen. Evolution 53: 63–73.
Saccheri I, Kuusaari M, Kankare M, Vikman P, Fortelius W, Hanski I (1998). Inbreeding and extinction in a butterfly metapopulation. Nature 392: 491–494.
Thrall P, Richards C, McCauley D, Antonovics J (1998). Metapopulation collapse: consequences of limited gene flow in spatially structured populations. In: Bascompte J, Solé RV (eds) Modeling Spatiotemporal Dynamics in Ecology, Springer-Verlag: Berlin. pp 83–104.
van Treuren R, Bijlsma N, Ouborg J, van Delden W (1991). The significance of genetic erosion in the process of extinction. I. Genetic differentiation in Salvia pratensis and Scabiosa columbaria in relation to population size. Heredity 48: 996–1008.
Werth C (1985). Implementing an isozyme laboratory in a field station. Virginia J Sci 36: 53–76.
Westemeier RL, Brawn JD, Simpson SA, Esker TL, Jansen RW, Walk JW, Kershner EL, Bouzat JL, Paige KN (1998). Tracking the long-term decline and recovery of an isolated population. Science 282: 1695–1698.
Young AG, Clarke GM (2000). Genetics, Demography and Viability of Fragmented Populations. Cambridge University Press: Cambridge, UK.
McCauley DE, Richards CM, Emery SN, Smith RA, McGlothlin JW (2001). The interaction of genetic and demographic processes in plant metapopulations: A case study in Silence Alba. In: Silvertown J and Antonovics J (eds) Integrating Ecology and Evolution in a Spatial Context, Blackwell Scientific: London. pp. 177–196.
Acknowledgements
We thank the University of Virginia's Mountain Lake Biological Station for material and logistical support and an anonymous reviewer who greatly improved the text. This work was supported by a grant from the National Science Foundation (DEB-9610496) to DE McCauley.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Richards, C., Emery, S. & McCauley, D. Genetic and demographic dynamics of small populations of Silene latifolia. Heredity 90, 181–186 (2003). https://doi.org/10.1038/sj.hdy.6800214
Received:
Accepted:
Published:
Issue date:
DOI: https://doi.org/10.1038/sj.hdy.6800214
Keywords
This article is cited by
-
Cryptic molecular-geographical divergence among Iranian Silene aucheriana populations inferred from ISSR and climatic data
Biologia (2023)
-
Annual climatic fluctuations and short-term genetic variation in the eastern spadefoot toad
Scientific Reports (2021)
-
Assessment of spatial–temporal variation in natural populations of Brassica incana in south Italy: implications for conservation
Plant Systematics and Evolution (2018)
-
Phenotypic divergence and inter-specific trait correlation in a plant-pollinator/seed predator mutualism
Evolutionary Ecology (2014)
-
Population genetics and fitness in fragmented populations of the dioecious and endangered Silene otites (Caryophyllaceae)
Plant Systematics and Evolution (2012)
