
Abstract
The population genetic structure of the Macaronesian planktonic-developing periwinkle Littorina striata was analysed, using random amplified polymorphic DNA analysis (RAPD). Two primers, yielding six polymorphic loci, were surveyed to infer the population genetic structure of five geographically separated populations (i.e. 10–2000 km). Biased and unbiased allele frequency and heterozygosity levels were estimated and were found to be highly similar. As in previous allozyme studies, our results suggest that populations of L. striata display (i) only little amounts of genetic heterogeneity and population differentiation, (ii) high levels of gene flow, between geographically separated populations and within a single population, between two distinct shell morphs (i.e. nodulose and smooth shells) and (iii) a tendency for northern populations to be less heterozygous than southern populations. The current results reveal a high congruence between allozyme and RAPD data, suggesting that geographically separated populations and different shell morphs share a common gene pool. Given the fact that our RAPD loci might be considered as neutral markers, we conclude that the previously analysed allozymes were representative for L. striata's entire genome.
Similar content being viewed by others

Introduction
Littorina striata King & Broderip 1832 is a strictly Macaronesian (Azores, Madeira, Canary Islands and Cape Verde Islands) periwinkle (Littorinidae) inhabiting the upper eulittoral and littoral fringes of rocky shores (Reid, 1996). Despite the species' planktonic development, it displays a number of shell polymorphisms that do not appear to be randomly distributed at either micro- or macrogeographical scales (e.g. De Wolf et al., 1997, 1998a). This variation mainly involves sculpture (nodulose vs. smooth), shell and aperture size, and shell weight (i.e. thickness) (Reid, 1996; De Wolf et al., 1997). Formerly, the sculpture polymorphism was even interpreted as a taxonomic difference, with nodulose specimens being assigned to a separate species, namely L. affinis d'Orbigny 1839 (in Reid, 1996). Because of this taxonomic uncertainty and because species with planktonic development are not expected to show extensive population differentiation and structuring (Chapman, 1995 and references therein; but see Palumbi, 1996), it has been questioned whether L. striata represents a single, homogeneous gene pool and if so, which factors cause the nonrandom distribution of its shell variation. In order to address these issues, allozyme data have been used to assess the degree of genetic exchange (i.e. gene flow) between L. striata populations and morphotypes at various geographical scales (De Wolf et al., 1997, 1998b). These studies revealed no significant genetic structuring, even though there were signs of a macrogeographical N–S differentiation with respect to numbers of alleles, heterozygosity estimates and esterase band counts (De Wolf et al., 1998c). In addition, there was a tendency for isolation by distance, but this pattern was not statistically significant (De Wolf et al. unpubl. data).
However, these results were based on only four to five polymorphic enzyme loci and nonspecific esterases. Moreover, allozymes may be a biased sample of a genome (e.g. Karl & Avise, 1992; Lu & Rank, 1996) and/or may be subject to selection (e.g. Hilbisch & Koehn, 1985; Johannesson et al., 1995). Hence the allozyme-based population genetic data of L. striata needed to be verified by other, supposedly unbiased and neutral, markers. Random Amplified Polymorphic DNA (RAPD) (Welsh & McClelland, 1990; Williams et al., 1990) may provide such data. Although this technique has many weaknesses (e.g. Clark & Lanigan, 1993; Lynch & Milligan, 1994; Backeljau et al., 1995; Fritsch & Rieseberg, 1996), it also offers some important advantages such as its relative ease and speed, the high degree of (artificially simple) polymorphisms and the virtually inexhaustible pool of possible genetic markers (e.g. Clark & Lanigan, 1993; Lynch & Milligan, 1994; Fritsch & Rieseberg, 1996). Nevertheless, RAPDs have hitherto only marginally been applied in littorinid (Crossland et al., 1993, 1996) or gastropod population genetics (e.g. Jacobsen et al., 1996; Larson et al., 1996; Stothard & Rollinson, 1996).
In the present study we used RAPDs to assess population genetic differentiation in L. striata, i.e. differentiation between nodulose and smooth specimens and between five macrogeographical populations which had been previously analysed by allozyme electrophoresis.
Materials and methods
Sample collection and DNA extraction
A total of 320 specimens of L. striata were collected at wave-exposed shores in Macaronesia, including two populations from the Azores (AZ1: Vila de Nordeste, São Miguel; AZ2: Faial da Terra, São Miguel), two populations from the Cape Verde Islands (CV1: Baia das Gatas, São Vicente; CV2: Mindelo, São Vicente) and one population from the Canary Islands (CA1: Playa de America, Tenerife). Populations were separated by distances ranging from 10 km (over land) between CV1 and CV2 to 2000 km between CV1,2 and AZ1,2.
Genomic DNA was extracted from individual columellar muscles following the CTAB protocol of Winnepenninckx et al. (1996), modified so that tissues were not ground under liquid nitrogen, but instead, directly incubated in CTAB buffer.
RAPD amplification
Prior to PCR, DNA concentrations were adjusted to 50 ng/μL. PCR reactions were performed in a final volume of 25 μL, containing 25 pmol RAPD primer, 50 ng template DNA and a standard quantity ‘Ready To Go’ RAPD analysis mixture (Pharmacia Biotech). Amplifications were performed with the Hybaid Omn-E thermocycler (Hybaid Ltd, Teddington, Middlesex, UK). PCR conditions were: 5 min at 95°C followed by 45 cycles, each of 1 min at 95°C, 1 min at 36°C and 2 min at 72°C. After the last cycle, a final step of 5 min at 72°C was added to allow complete extension of all amplified fragments. Amplification products (10 μL) were separated for 90 min at 120 V on 0.8% agarose TBE gels (1.78 mM Tris; 1.78 mM Boric Acid; 0.4 mM EDTA). These were stained with 10 μL ethidium bromide added to the gel and 0.25 μL ethidium bromide/mL TBE buffer to the TBE running buffer. Gels were photographed under UV light.
Forty random, decamer primers were screened (Kits H and G of Operon Technologies), two of which, OPH-12 (5§-ACGCGCATGT-3§) and OPG-08 (5§-TCACGTCCAC-3§) were retained for further analysis. RAPD bands were scored as present/absent (p/a=1/0) and only well-resolved profiles were considered. The reproducibility of RAPD profiles was tested for each primer, by reanalysing 20 individuals and comparing both profiles.
Data analysis
Assuming that each RAPD band represents a single diallelic locus in Hardy–Weinberg (H–W) equilibrium, the p/a data set can be converted into allele frequencies (e.g. Apostol et al., 1996; Lu & Rank, 1996). However, because RAPDs behave as dominant markers (i.e. 1=AA or Aa and 0=aa), allele frequency estimates are less accurate than with codominant markers (Lynch & Milligan, 1994). Using the fraction of double-recessive genotypes (x) (i.e. band absence at a given locus), the biased frequency of the null allele is q=x1/2 and its unbiased estimate is given by eqn (2a) in Lynch & Milligan (1994).
Unbiased allele frequencies were calculated by hand and were subsequently entered into the program BIOSYS-1 (Swofford & Selander, 1989) to compute heterozygosity levels (H). Unbiased H-values for the ith locus and jth population were obtained according to Lynch & Milligan (1994) (i.e. eqns 4a and 2b). However, because unbiased and biased allele frequencies and H estimates only differed very slightly (Table 1) and as the available software [RAPDFST (Apostol et al., 1996) and POPGENE v. 1.1 (Yeh & Boyle, 1996)] did not allow entering unbiased allele frequencies, all further analyses used biased allele frequency and H estimates.
The homogeneity of allele frequencies between (i) the five populations, (ii) both Azorean populations, (iii) both Cape Verde populations, and (iv) nodulose and smooth Canarian specimens, was tested with the contingency chi-squared analysis option of POPGENE. FST-values and FST-based gene flow (Nm) estimates, for these comparison levels, were estimated with the RAPDFST program. The comparison of nodulose and smooth specimens was restricted to the Canarian population because that was the only one in which both morphotypes were sufficiently represented (nodulose=19; smooth= 21). The numbers of nodulose specimens in the other populations were too small (<7).
The BIOSYS-1 program was used to calculate Prevosti distances between the five populations. For comparative purposes, we did the same for the available, unpublished, allozyme data of these populations. Both distance matrices were subjected to UPGMA clustering and matrix correlations were tested with Mantel's statistic (permutation level=1000) as implemented by the NTSYS v. 1.8 program (Rohlf, 1993).
Results
The two primers retained for analysis yielded six polymorphic, reproducible and consistently scorable RAPD-bands (i.e. six loci: four with OPH-12 and two with OPG-08), whose size ranged from 220 bp to 1636 bp (Fig. 1). None of these RAPD bands was diagnostic for either nodulose or smooth specimens.

RAPD profiles of Littorina striata, using the (a) OPG08 and (b) OPH12 primers of the Operon kits H and G (Operon Technologies). [White arrow represents first locus, respectively OPG08-1 in (a) and OPH12-1 in (b); white and black arrows represent remaining loci in numerical order, respectively OPG08-2 in (a) and OPH12-2, OPH12-3 and OPH12-4 in (b); left lane represents a kb DNA ladder in both (a) and (b) ].
Although mean H estimates were more or less comparable among populations, the two lowest values were observed in the Azores (Table 1).
There were six cases of significant genetic heterogeneity. However, after sequential Bonferroni correction only one locus remained significant (locus OPG08–02, all populations compared) (Table 2). In contrast, no allele frequency heterogeneities were observed between nodulose and smooth specimens (Table 2).
FST-values were small, ranging between 0.001 and 0.076 (Table 3). The highest mean FST-value was observed among the five populations (mean FST=0.044), although it was more or less halved among Cape Verdian or Azorean populations and decreased even further between the two morphotypes (Table 3). Consequently, FST-based gene flow estimates were high, ranging from Nm=5.475 among the five populations to Nm=16.412 between both morphotypes.
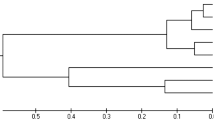
The UPGMA trees based on Prevosti distances calculated from, respectively, RAPD and allozyme data (Table 4) were comparable in that both placed the Cape Verdian populations at the base of the tree and both showed the same cluster of Azorean and Canary Island populations (Fig. 2 and 3). Yet, in the RAPD tree the Cape Verdian populations appeared paraphyletic with CV2 as the first branch (Fig. 3), whereas in the allozyme tree the Cape Verdian populations appeared as a monophyletic sister group of the Azorean–Canary Islands cluster (Fig. 2). Hence, both distance matrices were not significantly correlated (Mantel test: r=0.267, P=0.202).

Prevosti genetic-based UPGMA tree, based on allozyme data, for populations of Littorina striata.

Prevosti genetic-based UPGMA tree, based on RAPD data, for populations of Littorina striata.
Discussion
Previous surveys of four to five enzyme loci in 66 populations (including the present ones) of L. striata from all over Macaronesia, revealed no consistent and, after Bonferroni correction, significant deviations from H–W equilibrium conditions (De Wolf et al. unpubl. data). This implies that the assumption of H–W frequencies may also be acceptable for the RAPD data. In contrast, none of the enzyme loci was diallelic, but instead carried at least five to seven alleles (De Wolf et al. unpubl. data). Therefore, we feel less confident about the assumption of RAPD diallelism. This issue cannot be resolved without further, labour-intensive work (e.g. Fritsch & Rieseberg, 1996). Yet, imposing a diallelic structure on RAPD data can be considered as a pooling of genotypes, making statistical tests more conservative.
Although eqn (2a) in Lynch & Milligan (1994) provides an unbiased estimate of the null allele frequency (q) by considering small sample sizes, it still may be biased downwards if the null allele is rare (Lynch & Milligan, 1994). Therefore, RAPD analyses should be restricted to amplified bands that are not too common, i.e. those with a frequency of less than 1−(3/N) in each population (Lynch & Milligan, 1994). This was the case in the present study. Anyway, as already noted in Materials and methods, biased and unbiased allele frequencies were very similar (Table 2) because the correction term in eqn (2a) of Lynch & Milligan (1994) differed only slightly from 1 (range: 1.00007–1.005837). Hence the outcome of our analyses is not expected to be affected by whether or not our allele frequency estimates were biased. The same applies to our heterozygosity estimates.
The RAPD heterozygosity values for L. striata (0.244–0.306) were slightly higher than those observed using allozyme loci (0.151–0.216), but tentatively suggested the same N–S differentiation, viz. Hnorth<Hsouth (De Wolf et al. unpubl. data). However, testing the significance of this difference will require more data. Likewise, the RAPD FST values of L. striata (0.002–0.076) were of comparable magnitude to the allozyme-based FST-values (0.001–0.032 De Wolf et al. unpubl. data) and indicated only very limited differentiation between populations. Hence, Nm estimates were high (Nm>≲1). Both the RAPD H- and FST-values in L. striata were comparable to allozyme-based figures in other mollusc species with planktonic development (e.g. Johannesson, 1992; Dayan & Dillon, 1995).
Because none of the RAPD markers studied was diagnostic for shell morphotype, and because there was no significant genetic differentiation between nodulose and smooth specimens, we conclude that only one biological species is involved. This result is consistent with previous allozyme data (De Wolf et al., 1998b) and suggests that the sculpture polymorphism is indeed intraspecific.
In conclusion, the present RAPD data nearly perfectly fit previous allozyme data on L. striata (except perhaps for the basal branches in the UPGMA trees, which produced a nonsignificant correlation between the distance matrices involved), even to the point that tendencies suggested by allozymes remain tendencies with RAPDs (i.e. the N–S difference of H). This congruence between our allozyme and RAPD data strongly contrasts with previous studies, which reported that allozymes underestimate genetic diversity compared to RAPDs (e.g. Apostol et al., 1996; Crossland et al., 1993, 1996). As pointed out by Schaal et al. (1991) and by Karl & Avise (1992), the differences between RAPD- and allozyme-based estimates of genetic diversity suggest that allozymes may not be a representative sampling of the entire genome. Yet, this does not follow from the present work. Indeed, given that there was no reason to assume that our data were non-neutral, we regard the numerical congruence between our allozyme and RAPD data as an indication that in the case of L. striata the allozyme data provide a fairly good picture of the population genetic structure of this species.
References
Apostol, B. L., Black, W. C., IV, Reiter, P. and Miller, B. R. (1996). Population genetics with RAPD–PCR markers: the breeding structure of Aedes aegypti in Puerto Rico. Heredity, 76: 325–334.
Backeljau, T., de Wolf, H., Jordaens, K., van Dongen, S., Verhagen, R. and Winnepenninckx, B. (1995). Random Amplified Polymorphic DNA (RAPD) and parsimony methods. Cladistics, 11: 119–130.
Chapman, M. G. (1995). Spatial patterns of shell shape of three species with co-existing littorinid snails in New South Wales, Australia. J Moll Stud, 61: 141–162.
Clark, A. G. and Lanigan, C. M. S. (1993). Prospects for estimating nucleotide divergence with RAPDs. Mol Biol Evol, 10: 1096–1111.
Crossland, S., Coates, D., Grahame, J. and Mill, P. J. (1993). Use of random amplified polymorphic DNA's (RAPDs) in separating two sibling species of Littorina. Mar Ecol Prog Ser, 96: 301–305.
Crossland, S., Coates, D., Grahame, J. and Mill, P. J. (1996). The Littorina saxatilis species complex — interpretation using random amplified polymorphic DNAs. In: Taylor, J. T. (ed.) Origin and Evolutionary Radiation of the Mollusca. pp. 205–210. Oxford University Press, Oxford.
Dayan, S. N., Dillon, T. R., Jr (1995). Florida as a biogeographic boundary: Evidence from population genetics of Littorina irrorata. The Nautilus, 108: 49–54.
de Wolf, H., Backeljau, T., Medeiros, R. and Verhagen, R. (1997). Microgeographical shell variation in Littorina striata a planktonic developing periwinkle. Mar Biol, 129: 331–342.
de Wolf, H., Backeljau, T. and Verhagen, R. (1998a). Large scale patterns of shell variation in Littorina striata a planktonic developing periwinkle from Macaronesia (Gastropoda, Prosobranchia). Mar Biol, 131: 309–317.
de Wolf, H., Backeljau, T. and Verhagen, R. (1998b). Spatio-temporal genetic structure and gene flow between two distinct shell morphs of the planktonic developing periwinkle, Littorina striata (Mollusca, Prosobranchia). Mar Ecol Prog Ser, 163: 155–163.
de Wolf, H., Backeljau, T. and Verhagen, R. (1998c). Lack of significant esterase and myoglobin differentiation in the periwinkle, Littorina striata (Gastropoda, Prosobranchia). Adv Hydrobiol. in press.
Fritsch, P. and Rieseberg, L. H. (1996). The use of random amplified polymorphic DNA (RAPD) in conservation genetics. In: Smither, T. B. and Wayne, R. K. (eds) Molecular Genetic Approaches in Conservation. pp. 54–73. Oxford University Press, Oxford.
Hilbisch, T. J. and Koehn, R. K. (1985). The physiological basis of natural selection at the LAP locus. Evolution, 39: 1302–1317.
Jacobsen, R., Forbes, V. E. and Skovgaard, O. (1996). Genetic population structure of the prosobranch snail Potamopyrgus antipodarum (Gray) in Denmark using PCR-RAPD fingerprints. Proc Roy Soc B, 263: 1065–1070.
Johannesson, K. (1992). Genetic variability and large scale differentiation in two species of littorinid gastropods with planktotrophic development, Littorina littorea (L.) and Melarhaphe (Littorina) neritoides (L.). Biol J Linn Soc, 47: 285–299.
Johannesson, K., Johannesson, B. and Lundgren, U. (1995). Strong natural selection causes microscale allozyme variation in a marine snail. Proc Natl Acad Sci USA, 92: 2602–2606.
Karl, S. A. and Avise, J. C. (1992). Balancing selection at allozyme loci in oysters: implications from nuclear RFLPs. Science, 256: 99–102.
Larson, S. E., Andersen, P. L., Miller, A. N., Cousin, C. E., Richards, C. S., Lewis, F. A. and Knight, M. (1996). Use of RAPD-PCR to differentiate genetically defined lines of an intermediate host of Schistosoma mansoni, Biomphalaria glabrata. J Parasitol, 82: 237–244.
Lu, R. and Rank, G. H. (1996). Use of RAPD analyses to estimate population genetic parameters in the alfalfa leaf-cutting bee, Megachile rotundata. Genome, 39: 655–663.
Lynch, M. and Milligan, B. G. (1994). Analysis of population genetic structure with RAPD markers. Mol Ecol, 3: 91–99.
Palumbi, S. R. (1996). Macrospatial genetic structure and speciation in marine taxa with high dispersal abilities. In: Ferraris, J. D. and Palumbi, S. R. (eds) Molecular Zoology: Advances, Strategies and Protocols. pp. 101–117. Wiley-Liss, New York.
Reid, D. G. (1996). Systematics and Evolution of Littorina. The Ray Society, London.
Rohlf, F. J. (1993). NTSYS-PC. Numerical taxonomy and multivariate analysis system. Version 1.80. Applied Biostatistics Inc., Setauket, NY.
Schaal, B. A., O'Kane, S. L., Jr and Rogstad, S. H. (1991). DNA variation in plant populations. Trends Ecol Evol, 6: 329–333.
Stothard, J. R. and Rollinson, D. (1996). An evaluation of random amplified polymorphic DNA (RAPD) for the identification and phylogeny of freshwater snails of the genus Bulinus (Gastropoda: Planorbidae). J Moll Stud, 62: 165–176.
Swofford, D. L. and Selander, R. B. (1989). BIOSYS-1. A computer program for the analysis of allelic variation in population genetics and biochemical systematics. Illinois Natural History Survey, Champaign, IL.
Welsh, J. and McClelland, M. (1990). Fingerprinting genomes using PCR with arbitrary primers. Nucl Acids Res, 18: 7213–7218.
Williams, J. G. K., Kubelik, A. R., Livak, K. J., Rafalski, J. A. and Tingey, S. V. (1990). DNA polymorphisms amplified by arbitrary primers are useful as genetic markers. Nucl Acid Res, 18: 6531–6335.
Winnepenninckx, B., Backeljau, T. and de Wachter, R. (1996). Investigation of molluscan phylogeny on the basis of 18S rRNA sequences. Mol Biol Evol, 13: 1306–1313.
Yeh, F. C. and Boyle, T. (1996). POPGENE v1.1. Microsoft Windows-based software for population genetic analysis. University of Alberta, Edmonton.
Acknowledgements
The authors would like to thank J. Grahame, C. Wilding, B. Winnepenninckx and R. De Wachter. This research was supported by the MAST 3 programme of the European Commission under contract number MAS3-CT95–0042 (AMBIOS) and PRAXIS XXI 2/2.1/169/94 (Portugal). H. De Wolf holds an IWT scholarship.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
de Wolf, H., Backeljau, T. & Verhagen, R. Congruence between allozyme and RAPD data in assessing macrogeographical genetic variation in the periwinkle Littorina striata (Mollusca, Gastropoda). Heredity 81, 486–492 (1998). https://doi.org/10.1046/j.1365-2540.1998.00433.x
Received:
Accepted:
Published:
Issue date:
DOI: https://doi.org/10.1046/j.1365-2540.1998.00433.x
Keywords
This article is cited by
-
Sharp decrease of genetic variation in two Spanish localities of razor clam Ensis siliqua: natural fluctuation or Prestige oil spill effects?
Ecotoxicology (2012)
-
Strong genetic differentiation among east Atlantic populations of the sword razor shell (Ensis siliqua) assessed with mtDNA and RAPD markers
Helgoland Marine Research (2011)
-
No evidence for genetic differentiation between Antarctic limpet Nacella concinna morphotypes
Marine Biology (2010)
-
Completely disjunct mitochondrial DNA haplotype distribution without a phylogeographic break in a planktonic developing gastropod
Marine Biology (2008)
-
The genetic structure of endangered populations in the Cranberry Fritillary, Boloria aquilonaris (Lepidoptera, Nymphalidae): RAPDs vs allozymes
Heredity (2002)
