
Abstract
The colonization history and present-day population structure of the European subspecies of the great tit Parus major major were studied using mitochondrial control region sequences. One major haplotype was found in all but one of the eight sampled populations from Spain to northern Finland. The other haplotypes differed from the common one by just a few substitutions; the overall nucleotide diversity was 0.00187 and haplotype diversity 0.8633. No population structuring was detected. The mismatch distribution followed the expected distribution of an expanding population. The estimated time to the most recent common ancestor coincides with the last glacial period. The results suggest that P. m. major survived the last glacial period in a single isolated refuge probably by the Mediterranean Sea. This was followed by rapid colonization of the European continent and population growth. The most recent range expansion northwards is still occurring. Gene flow between the sampled populations is extensive. It is aided by juvenile dispersal, long-distance movements of juvenile flocks and partial migration in the northern parts of the great tit’s range.
Similar content being viewed by others

Introduction
The races of the great tit Parus major have been classified into four subspecies groups, major, minor, cinereus and bokharensis, each consisting of several subspecies (Snow, 1954). These groups and subspecies form a classic example of a geographical ‘Rassenkreis’ among birds. The major group has the widest distribution area of these great tit allospecies, covering the whole of Europe and reaching the Pacific coast as a wide zone in northern China and southern Russia. The group consists of eight subspecies of which the nominate P. m. major occurs from the whole of Europe (excluding Britain, Ireland and some Mediterranean islands) eastwards to the Altai mountains. In the north, the distribution reaches the Fennoscandian Lapland and in the south it reaches the Mediterranean region, where five other allopatric subspecies of the major group have been described (Snow, 1954).
In Europe, the range of the great tit has expanded northwards during this century. The first records of breeding great tits in northern Norway (Finnmark and Troms; Haftorn, 1957) and northern Finland (Inari; Veistola et al., 1994) are from the 1950s. The great tit is able to survive and breed in many kinds of habitat as long as holes for nesting are provided. The range expansion northwards has therefore been aided by man by providing nest-boxes for the breeding season and food for winter (Haftorn, 1957; Orell, 1989). It has been suspected that the productivity of the northern areas is too low to maintain populations. Clutch size and number of fledglings are lower than in the mid-latitudes and a second breeding is attempted, but tends to fail, in the far north (Veistola et al., 1994). The northernmost birds breed almost exclusively in man-made nest boxes. In Kilpisjärvi, northern Finland, mortality among overwintering birds is very high (Järvinen, 1983). Such populations can be maintained only by immigrants from more productive areas, as suggested by source–sink dynamics (Pulliam, 1988).
In northern Europe the great tit is a partial resident migrating irregularly, unlike in the southern parts of Europe, where it is sedentary after the establishment of territories (Gosler, 1993). Irruptive movements in large numbers have been observed in the north during autumn (Linkola, 1961).
In the southern and south-eastern range of the distribution area P. m. major contacts five allospecifics. Racial differentiation here supports the idea that the Mediterranean region was a refuge during the last ice age. As the ice receded, P. m. major invaded north and east (Gosler, 1993). If this is true, molecular analyses should reveal signs of (i) population bottlenecks during the glacial period, and (ii) population growth connected to the vast range expansion after the bottleneck.
The mitochondrial DNA (mtDNA) allows us to trace recent dispersal events. Demographic events like population bottlenecks or rapid growth, and isolation into subpopulations, shape mtDNA variation (Baker & Marshall, 1997 and references therein). Here we present a wide-scale study of P. m. major using mitochondrial control region sequences to reveal the recent history and present day genetic structure of the population.
Materials and methods
Samples
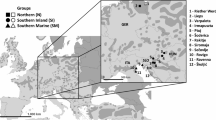
All populations of the study belong to the subspecies major. We received blood or egg samples from populations up in the northernmost parts (Kilpisjärvi, Finland) of the species’ range to those near to the southern limits of the subspecies’ range (Barcelona, Spain). Altogether eight populations (Fig. 1), each represented by six to ten individuals, were sampled (Table 1) during the years 1995–1997.

Map of the locations of the populations sampled and the distribution of the European great tits.
DNA isolation, PCR and sequencing
Isolation of total DNA from blood samples was performed according to standard procedures with proteinase K and SDS incubations and phenol-chloroform extractions. Mitochondrial DNA was isolated from embryonic plates, embryo and foetus as described in Kvist et al., 1998.
Primers for the polymerase chain reactions were designed at first from the homologous regions surrounding the mitochondrial control region. These regions were identified by comparing available bird sequences from GenBank. After successful amplification and sequencing of the first individuals, the primers were redesigned to be more specific for the great tit (L16700 tRNAGlu: 5′-ATCATAAATTCTCGCCGGGACTCT-3′ and H1471 12SRNA: 5′-AGTCAAGTTGCACTCATTGCTTAAT-3′). The PCR profile was the following: first denaturation at 94°C for 10 min, then 94°C for 1 min, 55°C for 1 min, 72°C for 2 min, repeated for 35 cycles and a final synthesis at 72°C for 10 min. The reactions were performed in 100 μL volume using Dynazyme (Finnzymes). The amplification product was purified from 0.8% agarose gel as in Kvist et al., 1998.
Direct sequencing of the double-stranded PCR product was performed using the dye terminator chemistry of Applied Biosystems and the ABI Prism 377 automatic sequencer according to the manufacturer’s instructions. The primers for the sequencing reactions were designed in such a way that the whole control region sequence was obtained in three parts, except the first eight nucleotides from the 5′ end (H422: 5′-CAATAAATAACCAGGCT-3′, L340: 5′-GCATCCACGAAGTCCAT-3′ and L689: 5′-TAAACC- CTTCCAGTGCG-3′). These were not sequenced from all the individuals because a stretch of cytosine bases following these nucleotides was causing ambiguities in sequencing reactions.
Sequence divergence analysis
Control region sequences were aligned using CLUSTAL W version 1.7 (Thompson et al., 1994). Pairwise genetic distances were calculated with MEGA (Kumar et al., 1993). The estimates for the nucleotide diversity π (Nei, 1987; eqns 10.5, 10.6 and 10.7) and the number of polymorphic nucleotides per nucleotide site, θ, (Nei, 1987; eqn 10.3) were calculated using DNASP version 2.52 (Rozas & Rozas, 1995).
Population genetic analyses
A parsimony network connecting the haplotypes was constructed manually from the aligned variable sites (Appendix). Most haplotypes differed from the common type by only one or two unique substitutions. Therefore it was easy to construct the network manually. On the other hand, there were not enough phylogenetically informative sites to perform parsimony analysis with phylogenetic programs. Four substitutions (at positions 439, 561, 1158 and 1120) were used in two different branches and one indel (at position 1118) was used in four different branches. This indel was located in a C-stretch consisting of seven or eight nucleotides. It is likely to have been generated by slippage, which occurs at higher rates than e.g. transitions or transversions, during replication (Berg et al., 1995). We considered that the phylogenetic signal of such an indel is weak, and felt it justified to spread this indel to separate branches of the network.
Because of small population sample sizes, we formed two groups; the Continental group consists of populations from Spain, the Netherlands, Germany and Estonia, and the Fennoscandian group populations from Gotland (Sweden) and Finland. Analysis of molecular variance (frequency-based AMOVA; Excoffier et al., 1992) was performed to compute the variance components within and between populations with the help of the ARLEQUIN program version 1.0b3. Tajima’s D testing for differences between nucleotide diversity π and θ, estimated from the number of segregating sites (Tajima, 1989), and the expected distribution of the pairwise genetic distances in population expansion (Rogers & Harpending, 1992) were calculated for the two groups and the total population using DNASP. The coalescence time for the haplotypes was approximated according to eqn 2 in Avise et al., (1988) using the conventional divergence rate of 2%/Myr between lineages for mitochondria (Klicka & Zink, 1997 and references therein). We compared the mean pairwise genetic distances obtained from mitochondrial RFLP (0.00186; Tegelström, 1987) and control region sequences (0.00187; this study) of the great tit. As these values were almost identical we felt comfortable in using the substitution rate also in control region sequence data, though the control region is often thought to evolve faster than the rest of the mtDNA. The same divergence rate was used to calculate the long-term effective population size for females according to Ne= (0.5 × 108p)/g (eqn 1, in Avise et al., 1988), where p is the average pairwise genetic distance and g is the generation time, which was assumed to be 1.972 years (calculated from Table 1 in Tinbergen & Daan, 1990).
Results
The length of the great tit control region was 1188–1189 nucleotides (GenBank accession no. AF059662). The nucleotide composition followed that of other avian control region sequences (e.g. Marshall & Baker, 1997); 13.6% G, 25.7% A, 31.2% T and 29.4% C. Comparison of the sequences obtained from the mitochondrial or total DNA templates showed no difference, confirming that we did not amplify nuclear pseudogenes. There were 52 polymorphic sites and two indels. Seventeen sites were phylogenetically informative (Appendix).
Twenty-four individuals shared the same haplotype, whereas other tits each had a unique haplotype, except that two German tits shared one, as did also a Spanish and an Estonian tit. Haplotype diversity 1 – ∑fi2 for the total sample was 0.8633. The most common haplotype was found in all populations, from the northernmost population in Kilpisjärvi, Finland to the southern population in Barcelona, Spain (except among the 10 birds from Harjavalta, Finland). Average nucleotide diversity π (Table 2) was 0.00187 varying from 0.00113 in the (combined) Dutch population to 0.00330 in the population from Harjavalta. There was a trend towards higher variability in the north than in the south in terms of nucleotide diversity, number of segregating sites per nucleotide and the haplotype diversity, though not significantly (Mann–Whitney for nucleotide diversity Z = 0.722, P = 0.471 and for haplotype diversity Z = 0.433, P = 0.067). This was partially because of the Harjavalta population but also the Continental populations because the common haplotype was present in higher frequencies in the south.
No geographical structuring was observed. AMOVA partitioned 98.3% of the total variance within populations, only 1.4% between populations within Fennoscandian and Continental groups and finally 0.3% between these groups. The lack of phylogeographical structure is evident from the minimum spanning network (Fig. 2), which seems to represent a model of an expanding population. This expansion of population size is further supported by the significantly negative Tajima’s D values (Table 2). The observed distribution of pairwise genetic distances (the mismatch distribution) followed the expected distribution of a growing population (Rogers & Harpending, 1992) in the total population, as well as in the Fennoscandian and Continental populations separately, though we lack statistical power to test this because of small sample sizes (Fig. 3).

The minimum spanning network obtained for eight populations of European great tits. Abbreviations: K, Kilpisjärvi, Finland; O, Oulu, Finland; H, Harjavalta, Finland; Go, Gotland, Sweden; E, Estonia; Ge, Germany; N, Netherlands; and S, Spain. The small black nodes refer to one substitution or indel. The alternative equally parsimonious locations of the haplotypes are shown with broken lines.

The observed and expected distribution of pairwise differences under the model for population expansion (Rogers & Harpending, 1992). The parameters used were for the Fennoscandian population (τ = 2.462, θ0= 0 and θ1= ∞), for the Continental (τ = 1.259, θ0= 0.655 and θ1= ∞) and for the total pooled population (τ = 2.001, θ0= 0.207 and θ1= ∞) of European great tits.
The coalescence time of the haplotypes was estimated to be 93 500 years based on the mean genetic pairwise distance of 0.00187. This leads to the long-term effective population size of 47 000 for the whole female population or 53 000 and 41 000 females estimated from the Fennoscandian and Continental samples, respectively.
Discussion
The last glaciation in Europe started 120 000 years ago and lasted some 100 000 years. The coldest period of this Weichselian ice age was 18–20 000 years ago when the ice covered northern Europe to approximately 50°N. The ice began retreating 13 000 years ago and finally receded 8000 years ago. During the coldest period, habitats suitable for great tits were present only in southern parts of Europe near the Mediterranean sea, which contained several refuges where great tits could have survived. This hypothesis is supported by the existence of six allospecies of the major group which possibly originate from isolated refuges of this area (Gosler, 1993).
If ancestral P. m. major was ‘trapped’ in a single refuge, the population size probably decreased drastically during the coldest period. When the glacial period ended, the species rapidly occupied new habitats and the population size increased (Gosler, 1993). The outcome of such a history is consistent with the mtDNA pattern. First, there is only one common haplotype. The other haplotypes are quite closely related to the most common one. Population expansion is supported by the mismatch distribution, the star-like parsimony network and the significantly negative Tajima’s D-value. Secondly, the wide distribution of the most common haplotype and lack of geographical structure support a recent range expansion. Thirdly, the approximated coalescence time coincides with the beginning of the glacial period.
A very similar history was suggested for the red-winged blackbird (Agelaius phoeniceus) by Ball et al., (1988). This species lacks phylogeographical structure and a single haplotype is widely distributed. The estimated coalescence times are 100 000 years and 93 500 years in blackbirds and great tits, respectively. The time of the glacial retreat is approximately the same in North America and Europe, and the blackbird possibly survived the glaciation in a single refuge, thereafter colonizing the continent rapidly (Ball et al., 1988).
Merilä et al., (1997) found that one haplotype predominated in greenfinches (Carduelis chloris) throughout Europe, being present in all ten sampled populations. The overall population structure resembled that of great tits, though some differentiation between the populations was detected. This was explained by the effect of several bottlenecks during and after the glacial period and the recent growth of the population. One difference in greenfinches compared to great tits is that there is a decrease of nucleotide diversity with the increasing latitude in greenfinches. The common chaffinch (Fringilla coelebs) populations (from Madeira, Norway, Italy and Tunisia) instead showed pronounced genetic differentiation between the island (Madeira) and continental populations and the existence of a divergent ancestral population in Tunisia (Marshall & Baker, 1997).
Life history and population dynamics
Closely connected to the historical range expansion is the possibility of present-day gene flow. Such dispersal would cause results similar to those we obtained by preventing the widely separated populations from differentiating.
Dispersal in the great tit has been estimated for many different populations in long-term studies. For example, the mean natal dispersal of female great tits (P. m. newtoni) has been estimated to be about 880 m per generation in the Wytham Wood population near Oxford, UK (Greenwood et al., 1979). Such a short dispersal combined with the site fidelity of the adults would mean that isolation by distance could cause differentiation in the genetic structure of the populations. However, no geographical structure was detected in this study. One explanation is the fact that measures of the dispersal distances are often underestimated because only the birds staying in or near the research area will be recorded and the probability of recovery is nonuniform (Barrowclough, 1978). Verhulst et al., (1997) recently reported that only 5% of the emigrant recruits from the same Wytham Wood population were found breeding in the surrounding area (within 2 km) and a considerable proportion of birds disperse further. Another explanation is that the long-distance movements of the juvenile flocks (hundreds of kilometres) are genetically effective, as suggested for Fennoscandian willow tit (Parus montanus) populations (Kvist et al., 1998).
Human influence on the northern populations, in the form of providing food for the winter and nest-boxes for breeding, has apparently had a large effect on the expansion of the range northwards and the permanent existence of the northern populations today (Haftorn, 1957; Orell, 1989). Thus far, the existence of the northern populations, like Kilpisjärvi, depends on immigration from a southern source population. This resembles what we found. Three individuals carried the common haplotype in Kilpisjärvi. The other five haplotypes were unique and differed from the common one by only one or two substitutions. Also these unique haplotypes may represent the source population, where they just remained undetected, but they may as well suggest a beginning of local differentiation. The immigration from the source to the northern parts of the great tit’s range is probably promoted by the partial seasonal migration which also enhances the gene flow.
The Harjavalta population showed a somewhat confusing pattern among the other populations in the sense that the common haplotype was not present in the sample. All the haplotypes were unique.
Island populations usually have lower levels of genetic variation than mainland populations (Frankham, 1997). This was not true in the great tit population on Gotland. The haplotype diversity was the same as in Kilpisjärvi and the nucleotide diversity was quite high. The immigration rate to another island population in Vlieland (the Netherlands) has been approximately estimated as 28% for females (Verhulst & van Eck, 1996). Though the island of Gotland is situated further away from the mainland continent than is Vlieland, the island is larger, and there is no reason to suppose that the immigration rate to Gotland would be low enough to facilitate differentiation.
References
Avise, J. C., Ball, M. and Arnold, J. (1988). Current versus historical population sizes in vertebrate species with high gene flow: A comparison based on mitochondrial DNA lineages and inbreeding theory for neutral mutations. Mol Biol Evol, 5: 331–344.
Baker, A. J. and Marshall, H. D. (1997). Mitochondrial control region sequences as tools for understanding evolution. In: Mindell, D. P. (ed.) Avian Molecular Evolution and Systematics. 51–82. Academic Press, San Diego, CA.
Ball, R. M., Freeman, S., James, F. C., Bermingham, E. and Avise, J. C. (1988). Phylogeographic population structure of Red-winged Blackbirds assessed by mitochondrial DNA. Proc Natl Acad Sci USA, 85: 1558–1562.
Barrowclough, G. F. (1978). Sampling bias in dispersal studies based on finite area. Bird-Banding, 49: 333–341.
Berg, T., Moum, T. and Johansen, S. (1995). Variable numbers of simple tandem repeats make birds of the order Ciconiiformes heteroplasmic in their mitochondrial genomes. Curr Genet, 27: 257–262.
Excoffier, L., Smouse, P. and Quattro, J. (1992). Analysis of molecular variance inferred from metric distances among DNA haplotypes: application to human mitochondrial DNA restriction data. Genetics, 131: 479–491.
Frankham, R. (1997). Do island populations have less genetic variation than mainland populations? Heredity, 78: 311–327.
Gosler, A. (1993). The Great Tit. Hamlyn, London.
Greenwood, P. J., Harvey, P. H. and Perrins, C. M. (1979). The role of dispersal in the great tit (Parus major): the causes, consequences and heritability of natal dispersal. J Anim Ecol, 48: 123–142.
Haftorn, S. (1957). Kjøttmeisas (Parus m. major L.) innvandring og nåværende utbredelse I Nord-Norge. D K N V S Forhandlinger, 30: 14–21.
Järvinen, A. (1983). Breeding strategies of hole nesting passerines in northern Lapland. Ann Zool Fenn, 20: 129–149.
Klicka, J. and Zink, R. M. (1997). The importance of recent Ice Ages in speciation: A failed paradigm. Science, 277: 1666–1669.
Kumar, S., Tamura, K. and Nei, M. (1993). MEGA:, Molecular Evolutionary Genetics Analysis. version 1.0. The Pennsylvania State University, University Park, PA 16802.
Kvist, L., Ruokonen, M., Thessing, A., Lumme, J. and Orell, M. (1998). Mitochondrial control region polymorphism reveals high amount of gene flow in Fennoscandian willow tits (Parus montanus borealis). Hereditas, 128: 133–143.
Linkola, P. (1961). Zur Kenntnis der Wanderungen finnischer Meisenvögel. Ornis Fennica, 38: 127–145.
Marshall, H. D. and Baker, A. J. (1997). Structural conservation and variation in the mitochondrial control region of fringilline finches (Fringilla spp.) and the greenfinch (Carduelis chloris). Mol Biol Evol, 14: 173–184.
Merilä, J., Björklund, M. and Baker, A. J. (1997). Historical demography and present day population structure of the greenfinch, Carduelis chloris – an analysis of mtDNA control-region sequences. Evolution, 51: 946–956.
Nei, M. (1987). Molecular Evolutionary Genetics. Columbia University Press, New York.
Orell, M. (1989). Population fluctuations and survival of great tits Parus major dependent on food supplied by man in winter. Ibis, 131: 112–127.
Pulliam, H. R. (1988). Sources, sinks and population regulation. Am Nat, 132: 652–661.
Rogers, A. J. and Harpending, H. (1992). Population growth makes waves in the distribution of pairwise genetic differences. Mol Biol Evol, 9: 552–569.
Rozas, J. and Rozas, R. (1997). DnaSP version 2.0: a novel software package for extensive molecular population genetics analysis. Comput Appl Biosci, 13: 307–311.
Snow, D. W. (1954). Trends in geographical variation in palaearctic members of the genus Parus. Evolution, 8: 19–28.
Tajima, F. (1989). Statistical method for testing the neutral mutation hypothesis by DNA polymorphism. Genetics, 123: 585–595.
Tegelström, H. (1987). Genetic variability in mitochondrial DNA in a regional population of the great tit (Parus major). Biochem Genet, 25: 95–110.
Thompson, J. D., Higgins, D. G. and Gibson, T. J. (1994). CLUSTAL W: improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucl Acids Res, 22: 4673–4680.
Tinbergen, J. M. and Daan, S. (1990). Family planning in the great tit (Parus major): Optimal clutch size as integration of parent and offspring fitness. Behaviour, 114: 161–190.
Veistola, S., Lehikoinen, E. and Iso-Iivari, L. (1994). Breeding biology of the great tit Parus major in a marginal population in northernmost Finland. Ardea, 83: 419–420.
Verhulst, S. and Vaneck, H. M. (1996). Gene flow and immigration rate in an island population of great tits. J Evol Biol, 9: 771–782.
Verhulst, S., Perrins, C. M. and Riddington, R. (1997). Natal dispersal of great tits in a patchy environment. Ecology, 78: 864–872.
Acknowledgements
We are grateful to all the following people for providing us with samples: Lluis Brotons (Barcelona, Spain), Tapio Eeva and Esa Lehikoinen (Harjavalta, Finland), Peeter Hõrak (Tartu, Estonia), Antero Järvinen (Kilpisjärvi, Finland), Katja Räsänen (Gotland, Sweden), Gernot Segelbacher (Tübingen, Germany), Marcel Visser and Christa Mateman (Veluwe and Speuld, the Netherlands) and Petteri Welling and Kari Koivula (Oulu, Finland). We thank also Dr Robert Zink for helpful comments to improve the manuscript. This work was funded by the Research Council for the Environment and Natural Resources of the Academy of Finland (to M.O.), the Thule Institute at the University of Oulu (to J.L. and M.O.) and the University of Oulu.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Kvist, L., Ruokonen, M., Lumme, J. et al. The colonization history and present-day population structure of the European great tit (Parus major major). Heredity 82, 495–502 (1999). https://doi.org/10.1038/sj.hdy.6885130
Received:
Accepted:
Published:
Issue date:
DOI: https://doi.org/10.1038/sj.hdy.6885130
Keywords
This article is cited by
-
Common patterns in the molecular phylogeography of western palearctic birds: a comprehensive review
Journal of Ornithology (2021)
-
Physiological condition and blood parasites of breeding Great Tits: a comparison of core and northernmost populations
Journal of Ornithology (2013)
-
Low genetic differentiation in a sedentary bird: house sparrow population genetics in a contiguous landscape
Heredity (2011)
-
Characterization of MHC-I in the blue tit (Cyanistes caeruleus) reveals low levels of genetic diversity and trans-population evolution across European populations
Immunogenetics (2011)
-
On the phylogenetics and population differentiation of the Great Tit and Blue Tit in Jordan
Journal of Ornithology (2007)
