
Abstract
The molecular divergence and phylogenetic relationships of the Iberian populations of Aphanius iberus were established using allozymes and the complete cytochrome b gene sequence. Congruent results were found with both nuclear and mitochondrial molecular markers. The Mediterranean and Atlantic populations are clearly differentiated into two independent lineages. Their high molecular divergence suggests an early isolation, and the absence of gene flow among the populations indicates their independent evolution. The nuclear and mitochondrial data reveal monophyletic clustering of the two geographical lineages, but provide weak support for the population relationships. However, the mitochondrial results differentiated the Villena population as a distinct mitochondrial unit within the Mediterranean group. Geographically broad studies across the distribution range of A. iberus have helped to elucidate the patterns of diversification of this species. The genetic divergence found between the Atlantic and the Mediterranean populations is of the same order as those found among recognized species of cyprinodontids. The identification of two discrete evolutionary lineages has important implications for the conservation of this species, since its recovery requires the recognition and preservation of natural diversity. The Mediterranean and Atlantic lineages should be managed separately to avoid loss of their genetic identity, and the genetic uniqueness of the populations should be preserved by using wild stocks as the source of genetic diversity in captive breeding programmes.
Similar content being viewed by others
Introduction
Maintenance of genetic diversity and population distinctiveness are two major goals in the conservation management plans of endangered species (Meffe, 1994). Genetic data and phylogeographic structure throughout the species’ range represent the basic information required to understand their evolution and biogeographic history. In recent years, the widespread recognition of a decline in cyprinodontiform populations in the Iberian waters has led to an increasing need to assess genetic variability in the wild populations for conservation management purposes. Allochthonous introductions and the deterioration of coastal brackish areas have been shown to be the main factors that threaten the Iberian cyprinodontiform populations and promote their fragmentation (Blanco & González, 1992; Vargas, 1993). Although genetic diversity was investigated at certain levels, the phylogeographic structure of some endangered Iberian freshwater fish remains unknown.
In an attempt to determine the genetic viability of the two unique endemic Iberian cyprinodontiforms, Aphanius iberus and Valencia hispanica, allozymic analyses were conducted on wild and captive populations of these species (Doadrio et al., 1996; Perdices et al., 1996; García-Marín & Pla-Zanuy, 1999). Genetic variation found among populations of both species was as high as is found in other non-endangered species and, apparently, enough to preserve their genetic viability if the same habitat conditions were maintained. Similar results were achieved using RFLP (restriction fragment length polymorphism) analysis of the mitochondrial DNA of A. iberus (Fernández-Pedrosa et al., 1995).
In particular, A. iberus has shown an interesting geographical pattern, with a high degree of isolation among populations. Aphanius iberus is restricted to the Spanish coastline and is found in marshes and brackish waters. The Mediterranean and Atlantic populations are disrupted by significant geographical gaps. These natural geographical barriers, and life-history traits which are similar to those observed in the closely related species A. fasciatus (e.g. benthic eggs and absence of larval stages) (Maltagliati & Camilli, 2000), should prevent gene flow between A. iberus populations. The allozymic variation and genetic divergences between A. iberus populations show two distinct genetic and geographical groups: the Mediterranean and the Atlantic lineages (Doadrio et al., 1996). Within the Mediterranean lineage, the unique inland population (Villena) was revealed as a well differentiated genetic population (Fernández-Pedrosa et al., 1995) and is considered an independent conservation unit (Doadrio et al., 1996). However, few Atlantic populations were analysed in previous studies and to further characterize these populations and to define any geographical subdivision, more populations need to be surveyed. Moreover, the strong genetic structure observed in the Mediterranean populations should be exhibited by the Atlantic populations of A. iberus (García-Marín et al., 1990; Fernández-Pedrosa et al., 1995). The results of such a study have further conservation implications since taxonomic accuracy is a prerequisite for conservation plans, since such programmes are only as effective as the taxonomy upon which they are based.
Thus, the objectivity of both nuclear and mitochondrial genetic information could significantly contribute to identifying evolutionarily important A. iberus lineages. In practice, the recognition of evolutionary lineages should be encouraged as a prerequisite in any sound conservation programme. These distinct lineages are revealed as the evolutionary framework for management plans and represent the essential units to delineate any effective conservation programme.
Given the lack of information on phylogeographic structure of A. iberus, our main purpose was to determine the A. iberus population structure using allozymic analysis. We collected data from additional populations to complement the previous, more geographically restricted studies (Doadrio et al., 1996), and determine patterns of gene flow among the wild populations of A. iberus. We further investigated the geographical differentiation of A. iberus across their entire range and determined whether data from the additional sites provided greater resolution of their relationships. We used mitochondrial DNA sequences to establish phylogenetic relationships. The mitochondrial lineages derived from this phylogeny permit inferences regarding the history of the A. iberus populations and provide information about the importance of taxonomy for effective conservation plans.
Materials and methods
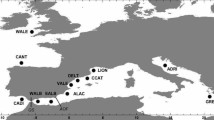
We have analysed a total of 20 A. iberus populations including the populations analysed in Doadrio et al. (1996) (with A* in Table 1) covering the entire range of the species (Fig. 1). Our analyses included the closely related species Aphanius fasciatus and Valencia hispanica, and the genus Poeciliopsis as taxonomic outgroup.

Map showing location of populations of Aphanius iberus analysed by allozymes and mtDNA sequences. 1–12 Mediterranean and 13–20 Atlantic populations. 1* Aïguamolls, 2* Salou, 3* Ebro Delta, 4* Peñíscola, 5* Albuixec, 6* Villena, 7* Santa Pola, 8* San Pedro Pinatar, 9* Carmolí, 10* Marchamalo, 11 Adra, 12 Albufera Adra, 13 Conil, 14 Roche, 15 Iro, 16 San Pedro, 17* Doñana, 18 Laguna Doñana, 19* Sevilla and 20 Salado. *Allozyme data from Doadrio et al. (1996).
Allozymic analyses and genetic diversity
A total of 319 individuals from eight populations of A. iberus were analysed together with 266 specimens retrieved from Doadrio et al. (1996) (Table 1). The loci surveyed, and the electrophoretic conditions, were the same as those described in Doadrio et al. (1996), except EST-2* and FH* which were not scored in the present study. Liver (L) and muscle (M) were dissected, homogenized with Tris-EDTA, NADP pH=6.8, and centrifuged; supernatants were stored at −70°C. Horizontal starch gel electrophoresis (12%) was performed and the products of the following 15 enzyme systems encoding 23 loci were scored: aspartate aminotransferase (mAAT-1*, L, M; sAAT-1*, M; sAAT-2*, L, M) E. C. 2.6.1.1, buffer system TC 6.7 (Tris-citrate pH=6.7) (after Pasteur et al., 1987); adenylate kinase (AK*, M) E. C. 2.7.4.3, TC 6.7; creatine kinase (CK*, M) E. C. 2.7.3.2, TC 6.7; esterase (EST-1*; EST-3*; EST-4*; M) (EST-1* and EST-3* substrate employed α and β-naphthyl-propionate 1% in acetone; EST-4* 4-methyl-umbelliferyl-acetate 0.5% in acetone) E. C. 3.1.1. -, TME 6.9 (Tris-maleate pH=6.9); glucose-6-phosphate isomerase (GPI-1*, M; GPI-2*, L) E. C. 5.3.1.9, Poulik; Isocitrate dehydrogenase (NADP +) (IDHP-1*, L; IDHP-2*, M) E. C. 1.1.1.42, TC 6.7; L-lactate dehydrogenase (LDH-1*, L, M; LDH-2*, L, M) E. C. 1.1.1.27, Poulik; malate dehydrogenase (mMDH-1*, L, M; sMDH-1*, L, M; sMDH-2*, L) E. C. 1.1.1.37, TC 6.7; malic enzyme (ME*, M) E. C. 1.1.1.38, Poulik; mannose 6-phosphate isomerase (MPI*, L, M), TC 6.7; phosphogluconate dehydrogenase (PGDH*, L) E. C. 1.1.1.44, TC 6.7; phosphoglucomutase (PGM-1*, L, M; PGM-2*, L) E. C. 5.4.2.2, Poulik; superoxide dismutase (SOD*, L, M) E. C. 1.15.1.1, Poulik. Enzyme nomenclature follows the recommendations of Shaklee et al. (1990). Alleles were designated by their mobility relative to the most frequent allele (100) in side-by-side comparisons and follow the nomenclature of Doadrio et al. (1996). We consider a locus to be diagnostic for a population (or a group of populations) when all the alleles at that locus are not present in any other population. A unique allele is an allele seen in only one population with a frequency <1.0.
Genetic variation was estimated as the percentage of polymorphic loci (P), mean observed heterozygosity (Ho) and expected heterozygosity (He), and mean number of alleles (A). Genotype frequencies were used to estimate gene frequencies, and pairwise genetic distances in A. iberus populations (Nei, 1972 and Rogers, 1972). Nei’s (1972) genetic distance, which facilitates comparison with previous studies, and Rogers’ (1972) genetic distance were used in the phenogram for their easily interpretable geometric basis (Swofford & Olsen, 1990). UPGMA phenograms were constructed to display major relationships among the A. iberus populations. All calculations were done using BIOSYS-2 (Swofford et al., 1997). Lévène’s corrections for small sample size was employed (Lévène, 1949). The Hardy–Weinberg equilibrium was assessed using an exact significance test in ARLEQUIN v. 2000 (Schneider et al., 2000). Support for the reliability of the branching pattern was evaluated by bootstrap analysis (100 replicates) (Felsenstein, 1985) in PHYLIP 3.5 (Felsenstein, 1993).
F-statistics were calculated to assess gene flow among the Mediterranean and Atlantic A. iberus populations. Pairwise FST significance was estimated using the analysis of molecular variance procedure (AMOVA) (Excoffier et al., 1992) in ARLEQUIN. A Mantel test was performed to examine the correlation between geographical and genetic distance matrices (Sokal et al., 1986). The test was done with 500 permutations using NTSYS v. 2.02 (Rohlf, 1998). Geographical distances between A. iberus populations were measured following the most direct routes along the main rivers and using the minimum distance across the littoral.
Mitochondrial DNA analyses and phylogenetic reconstruction
We characterized interpopulational variation within A. iberus by sequencing at least two individuals of a subset of eight different Mediterranean and Atlantic populations (Table 1). GenBank accession numbers for all individuals are listed in Table 1.
Total DNA was extracted from a small piece of dorsal muscle (1–3 cm3) using phenol/chloroform extraction and ethanol precipitation (Towner, 1991). The entire cytochrome b gene was PCR amplified using primers L14724 and H15915 (Schmidt & Gold, 1993). Double-stranded DNA was PCR synthesized in 25 μL reactions containing 67 mM Tris-HCl buffer pH=8.3, 1.5 mM MgCl2, 0.4 mM each dNTP, 2.5 μM each primer, 14.25 μL ddH20, 10–100 ng template DNA, and (1 U) Taq polymerase (Promega). Protocol for PCR amplifications was: denaturation at 92°C for 60 s, annealing at 48°C for 90 s, and extension at 72°C for 180 s, repeated for 35 cycles and a final extension for 240 s.
PCR products were cloned using the pGEM-T vector (Promega) and the E. coli JM109 strain, and sequenced using the FS-Taq Dye Deoxy Terminator cycle-sequencing kit (Applied Biosystems Inc.) on an automated DNA sequencer (Applied Biosystems 377) following the manufacturer’s instructions. DNA sequences of both strands were obtained using M13 universal (forward and reverse) sequencing primers.
Sequences were aligned by hand using as reference the cytochrome b of Poeciliopsis monacha (GenBank accession number AF047346). No ambiguous alignments were found and no gaps were postulated. No stop codons were found and all codon positions were included in the phylogenetic analyses. Using the matrix of HKY 85 distances, we generated in PAUP* v. 4.0B2 (Swofford, 1999) an initial Neighbour-Joining (NJ) tree which included eight A. iberus populations and A. fasciatus and P. monacha as outgroups. In Maximum Parsimony (MP) analysis, only minimal trees were retained and zero length branches were collapsed. Transversions (Tv) were given three times the weight of transitions (Ts), based on the empirically estimated Ts/Tv. The heuristic search with TBR branch swapping and 10 replicates of random addition of taxa was used. MP and NJ analysis were implemented in PAUP*. Bootstrap analysis (1000 replicates) was used to assess the relative robustness of inferred monophyletic groups.
For Maximum-likelihood (ML) analysis, we used the program MODEL TEST 2.1 (Posada & Crandall, 1998) to find the best model of evolution that fitted our data. The HKY model of evolution (Hasegawa et al., 1985) with gamma value was used for ML analyses. ML analyses were performed using the Quartet Puzzling method (Strimmer & Von Haeseler, 1996) as implemented in PAUP*.
Results
Population structure and molecular divergences
Among the 23 loci surveyed in the present study, three were monomorphic (LDH-1*, LDH-2* and SOD*) (P > 0.99) when all the Mediterranean and Atlantic populations of A. iberus were considered (Appendix 1 and table 3 in Doadrio et al., 1996). Allozyme loci frequencies observed in this study are shown in Appendix 1. Aphanius fasciatus and V. hispanica showed the highest proportion of diagnostic loci (13 and 14/23, respectively).
The eight Atlantic samples of A. iberus (including the populations of Sevilla and Doñana from Doadrio et al. (1996)) exhibited two diagnostic loci IDHP-2*110 and sMDH-2*110, and eight unique alleles (0.017 < frequencies < 0.333), when compared to the Mediterranean populations of A. iberus (Appendix 1). Atlantic populations showed 12/23 loci monomorphic (see Appendix 1).
Within all the Atlantic populations, the range of polymorphic loci was highly variable, ranging from 4.3% in the Conil sample to 30.4% in the Sevilla, Salado, Doñana, and Laguna de Doñana samples (P=20.6%). Observed heterozygosities (Ho) ranged from 0.0 in the Conil sample to 0.085 in the Salado sample (Ho=0.043) (Appendix 1). Among the Atlantic populations of A. iberus, in two out of 38 cases genotype frequencies were not in accordance with the Hardy–Weinberg equilibrium (***P < 0.001) due to heterozygote deficiency in the San Pedro and Iro samples at sAAT-2* and EST-1*, respectively.
All the Mediterranean populations of A. iberus (two in this study and 10 from Doadrio et al. (1996)) showed two diagnostic loci (IDHP-2* 100 and sMDH-2* 100). Five of the 23 loci analysed were monomorphic in these populations (see Appendix 1). The Adra and Albufera de Adra samples showed five unique alleles, similar to the number of unique alleles found previously in the Mediterranean populations (Appendix 1, and table 3 of Doadrio et al. (1996)). Deviations from the Hardy–Weinberg equilibrium, owing to heterozygote deficiencies, were found in both Adra samples at the following loci: EST-1*, EST-3* and MPI*.
Two main lineages within A. iberus can be distinguished using Nei distances: mean values greater than 0.15 which are observed between the Mediterranean and Atlantic populations (D=0.156) and mean values lower than 0.05 (D=0.034, Atlantic and D=0.037, Mediterranean) which are observed inside each lineage (one exception D=0.128 inside the Atlantic lineage) (Table 2). Within the Atlantic populations the lowest Nei distance (D=0.008) was observed between the Iro and Roche samples, and between the Doñana and Salado samples. The highest Nei value (D=0.128) was between the Conil and San Pedro samples. Genetic distances among both Adra populations and the rest of the Mediterranean populations were in the same range (0.005–0.062) as the genetic distances previously observed within the Mediterranean populations.
The UPGMA tree showed all the Atlantic and Mediterranean populations in two main groups (bootstrap value >78%), independently of the genetic distance used (Fig. 2a). Significant pairwise FST values were exhibited among most populations (FST=0.8). High FST values were also found within the Atlantic populations (FST=0.41) and the Mediterranean populations (FST=0.33). According to the AMOVA analysis, nearly 68% of the variation detected was due to differences among the Atlantic and Mediterranean A. iberus lineages, whereas 17.2% of the variation was due to among-population differences within each group. The Mantel test showed that the patterns of allozyme variation exhibited by the A. iberus groups could not be explained by geographical distances (r=0.63, t=8.754, P=1.0).

Phylogenetic relationships of Aphanius iberus populations based on nuclear and mitochondrial data. (a) UPGMA dendrogram based on Rogers’ (1972) allozymic distances with bootstraps values (100 replicates) above branches (> 50%). (b) Phylogenetic relationships based on cytochrome b sequences. Bootstrap values at each node (1000 replicates) correspond to: above branches MP (1 Ts: 3 Tv), below branches NJ (HKY85) and lowest value ML. Branches not recovered in all analyses were forced to collapse.
Mitochondrial DNA analysis
The complete (1140 bp) cytochrome b sequence was obtained for eight selected populations representing the Mediterranean and Atlantic A. iberus lineages. A total of 358 variable characters, 143 of which were phylogenetically informative for MP analysis, were found when including the outgroups A. fasciatus and P. monacha, and 112 variable and 70 informative characters for A. iberus alone.
Plots of the numbers of transitions and transversions against distances at all positions revealed no evidence of saturation (plots not shown). All codon positions were included in the phylogenetic analysis. The estimated transition: transversion ratio among all taxa analysed was 2.87.
Congruent phylogenies recovered with MP, NJ and ML using A. fasciatus and P. monacha which were the outgroups. Maximum parsimony analysis of the cytochrome b sequence data resulted in one most-parsimonious tree of 729 steps and a consistency index of 0.923 when weighting was 3 Ts:1 Tv. Congruent trees were found when different weighting schemes (2:1, 4:1, 6:1) or when equal weight was applied (trees not shown).
All analyses recovered two groups (>95% bootstrap) that represent the Mediterranean and Atlantic lineages (Fig. 2b). The Mediterranean lineage includes the Aïguamolls sample, which represents the northernmost A. iberus population, and the Adra samples, with the Villena population well differentiated. All the Atlantic populations belong to the second Aphanius lineage without well-resolved phylogenetic relationships.
Sequence divergence (HKY85 distances) of haplotypes supported a differentiation pattern among populations similar to that detected with Nei’s genetic distances. Low sequence divergence was found within the Atlantic samples (range 0.3–1.6%) and the Mediterranean samples (range 0.2–2.9%). The Villena Lagoon sample (Mediterranean lineage) differed from the rest of the Mediterranean samples by a mean of 2.4% (range 2.1–2.9%). Nevertheless, the highest differences were detected between the samples from the Atlantic and the Mediterranean slopes with a mean of 5.5% (range 4.7–6.4%).
Discussion
Molecular divergences found within Aphanius iberus populations clearly differentiated two Iberian groups: the Mediterranean and the Atlantic lineages (Fig. 2a,b). Similar divergences and identical groups were recovered using nuclear (allozymes) and mitochondrial genes (cytochrome b). The degree of genetic divergence found between the Mediterranean and Atlantic lineages suggests that both A. iberus groups have evolved independently, resulting in the presence of diagnostic (IDHP-2* and sMDH-2*) and unique alleles.
Both the Atlantic and the Mediterranean A. iberus groups exhibited genetic variation values in the same range as the congeneric species A. fasciatus (Maltagliati, 1998) and A. dispar (Kornfield & Nevo, 1976). However, the Aphanius cyprinodontids show lower levels of genetic heterozygosity than the average heterozygosity reported for freshwater fish (He=0.062) (Ward et al., 1994). Some studies have shown that organisms which inhabit brackish environments have low levels of genetic polymorphism (Battaglia et al., 1978). In particular, low genetic variation in A. fasciatus and in the Mediterranean A. iberus populations was associated with habitat conditions, e.g. individuals inhabiting high salinity waters exhibited lower genetic variation (Doadrio et al., 1996; Maltagliati, 1998). In the present study, the Atlantic populations of A. iberus exhibited the lowest genetic variation values. Although the observed reduction in allozymic variation in the Atlantic populations may be attributed to inbreeding or bottleneck processes, the decrease of genetic variation could also be related to the environmental variability of their brackish habitat.
Most of the genetic variation was found between the Mediterranean and Atlantic A. iberus lineages. Although the observed patterns of the A. iberus lineage subdivision, and their fixed genetic variation, suggest that discontinuous distributions might have played an important role in their divergence, presumably other external factors have widely contributed to their current extensive genetic differentiation.
A long-term interruption of gene flow (FST=0.8) was also detected, reflecting the strong genetic discontinuity found between the lineages and suggesting that current lineages undergo no genetic exchange. Previous genetic studies have found significant spatial heterogeneity among the Mediterranean populations of A. iberus (García-Marín et al., 1990; Fernández- Pedrosa et al., 1995; García-Marín & Pla-Zanuy, 1999). Allozymic and mitochondrial cytochrome b gene sequence results confirmed this genetic spatial differentiation in the Atlantic lineage of A. iberus. Similar genetic heterogeneity was also detected among populations of A. fasciatus (Maltagliati, 1998) and other cyprinodontids (Dunham & Minckley, 1998). It is likely that cyprinodontid species that inhabit naturally fragmented areas, e.g. A. fasciatus and A. iberus, are genetically structured according to a geographical pattern. Population genetic structure in cyprinodontid species distributed in larger, continuous areas seems to be avoided by gene flow among populations (Ashbaugh et al., 1994; Dunham & Minckley, 1998); however, the naturally disrupted habitat of A. iberus has facilitated population fragmentation. Disrupted A. iberus ranges are reflected in unique alleles in many of the populations analysed; however, the presence of diagnostic alleles indicates a complete interruption of gene flow between the Mediterranean and Atlantic lineages. This evidence favours the presence of historical geological barriers that prevented gene flow between the Mediterranean and Atlantic lineages.
All molecular evidence found in the present study (diagnostic loci, allozymic distances, FST and mitochondrial divergences) indicate the possibility that the observed interruption of gene flow between the Atlantic and Mediterranean lineages was not recent. This hypothesis has been suggested, based on the biogeographical areas currently inhabited by A. iberus and their coincidence with palaeogeographical data (Doadrio et al., 1996). Our analyses indicate that the Mediterranean and Atlantic groups represent two divergent lineages of Aphanius; both lineages inhabit isolated areas with no genetic exchange. Their molecular distinctiveness suggests early isolation of the two lineages. The opening of the Strait of Gibraltar in the Late Pliocene (5.5 Ma) (Krijgsman et al., 1999) and the absence of current connections between both Aphanius lineages, suggest a Pliocene origin to these two lineages. The effectiveness of the Strait of Gibraltar as a barrier for fish dispersion has been documented for Fundulus heteroclitus in south-west Spain (Moreno-Amich et al., 1999). The current genetic independence of the two Aphanius lineages was also corroborated by diagnostic morphological characters, suggesting that the Mediterranean and Atlantic lineages should not be considered the same species (Doadrio et al., in press). Recent breeding experiments have shown that Aphanius species hybridize in captivity and produce fertile offspring (Sienknecht, 1999). However, those experiments do not invalidate our conclusions since there is no evidence of their hybridization in nature. Sienknecht (1999) argued that both Iberian lineages are the same species because of an absence of reproductive isolation in captivity. However, the biological species concept applies poorly to fish which even hybridize between genera (Arnold, 1997) and in such cases the phylogenetic species concept may be more applicable (Kullander, 1999). The Mediterranean and Atlantic lineages represent two genetically and geographically isolated monophyletic groups, whose diagnostic characters (synapomorphies) permit their unambiguous identification.
The establishment of two differentiated taxonomic groups is particularly important for an endangered species such as A. iberus for which management is strongly promoted. In the present case, correct conservation plans should recognize the two A. iberus lineages as evolutionarily independent groups that should be managed separately. Crucial to preservation of genetic variability within each Aphanius lineage is the consideration of locally adapted populations, e.g. Villena, as unique entities. Recovery programmes have heavily promoted the use of natural stocks as sources of genetic diversity (Leberg et al., 1994), whereas mixed populations are discouraged to avoid loss of locally adapted genotypes (Vrijenhoek, 1994). The use of wild stocks to initiate captive breeding programmes, e.g. Doñana National Park, and genetic monitoring of those derived populations, seem to be effective tools in preserving the natural diversity of Aphanius populations (Schönhuth, et al. submitted). The existence of high genetic variation inside the Mediterranean and Atlantic A. iberus lineages should prevent the decrease of initial diversity, if enough individuals are selected to establish captive stocks.
References
Arnold, M. L. (1997). Natural Hybridization and Evolution. Oxford University Press, New York.
Ashbaugh, N. A., Echelle, A. A. and Echelle, A. F. (1994). Genetic diversity in Red River pupfish Cyprinodon rubrofluviatilis (Atheriniformes, Cyprinodontidae) and its implications for the conservation genetics of the species. J Fish Biol, 45: 291–302
Battaglia, B., Bisol, P. M. and Fava, G. (1978). Genetic variability in relation to the environment in some marine invertebrates. In: Battaglia, B. and Beardmore, J. (eds) Marine Organism: Genetics, Ecology and Evolution, pp. 53–70. Plenum Press, New York.
Blanco, J. C., González, J. L. eds. (1992). Libro Rojo de los Vertebrados de España. Colección Técnica, ICONA, Madrid.
Doadrio, I., Perdices, A. and Machordom, A. (1996). Allozymic variation of the endangered killifish Aphanius iberus and its application to conservation. Environ Biol Fishes, 45: 259–271.
Doadrio, I., Carmona, J. A. and Fernández-Delgado, C. In pressMorphometric study of the Iberian Aphanius (Actinopterigii, Cyprinodontiformes) with description of a new species. Folia Zool
Dunham, J. B. and Minckley, W. L. (1998). Allozymic variation in desert pupfish from natural and artificial habitats: genetic conservation in fluctuating populations. Biol Conserv, 84: 7–15.
Excoffier, L., Smouse, P. and Quattro, J. (1992). Analysis of molecular variance inferred from metric distances among DNA haplotypes: application to human mitochondrial DNA restriction data. Genetics, 131: 479–491.
Felsenstein, J. (1985). Confidence limits on phylogenies: an approach using the bootstrap. Evolution, 39: 783–791.
Felsenstein, J. (1993). PHYLIP (Phylogenetic inference package) vers. 3.5c. Computer program and manual distributed by the author, Seattle.
Fernández-Pedrosa, V., González, A., Planelles, M. and Moya, A. et al. (1995). Mitochondrial DNA variability in three Mediterranean populations of Aphanius iberus. Biol Conserv, 72: 251–256
García-Marín, J. L. and Pla-Zanuy, C. (1999). Conservación de la diversidad genética en el fartet, L. ibera. In: Planelles-Gomis, M. (Coord.) Peces Ciprinodóntidos Ibéricos Fartet Y Samaruc, pp. 169–187. Generalitat Valenciana, Conselleria de Medio Ambiente, Valencia, Spain.
García-Marín, J. L., Vila, A. and Pla, C. (1990). Genetic variation in the Iberian toothcarp Aphanius iberus (Cuvier & Valenciennes). J Fish Biol, 37 (Suppl. A), 1–2.
Hasegawa, M., Kishino, H. and Yano, T. (1985). Dating of the human–ape splitting by a molecular clock of mitochondrial DNA. J Mol Evol, 22: 160–174.
Kornfield, J. L. and Nevo, E. (1976). Likely pre-Suez occurrence of a Red Sea fish Aphanius dispar in the Mediterranean. Nature, 264: 289–291.
Krijgsman, W., Hilgen, F. J., Raffi, I. and Sierro, F. J. et al. (1999). Chronology, causes and progression of the Messinian salinity crisis. Nature, 400: 652–655
Kullander, S. O. (1999). Fish species – how and why. Rev Fish Biol Fisher, 9: 325–352.
Leberg, P. L., Stangel, P. W., Hillestad, H. O. and Marchinton, R. L. et al. (1994). Genetic structure of reintroduced wild turkey and white-tailed deer populations. J Wildlife Manage, 58: 698–711.
Lévène, H. (1949). On a matching problem arising in genetics. Ann Math Stat, 20: 91–94.
Maltagliati, F. (1998). A preliminary investigation of allozyme genetic variation and population geographical structure in Aphanius fasciatus from Italian brackish-water habitats. J Fish Biol, 52: 1130–1140
Maltagliati, F. and Camilli, L. (2000). Temporal genetic variation in a population of Aphanius fasciatus (Cyprinodontidae) from a brackish-water habitat at Elba Island (Italy). Environ Biol Fish, 57: 107–112.
Meffe, G. K. (1994). Genetics: Conservation of diversity within species. In: Meffe, G. K. and Carroll, C. R. (eds) Principles of Conservation Biology, pp. 161–201. Sinauer Associates, Sunderland, MA.
Moreno-Amich, R., Planelles-Gomis, M., Fernández-Delgado, C. and García-Berthou, E. (1999). Distribución geográfica de los ciprinodontiformes en la península ibérica. In: Planelles-Gomis, M. (Coord.) Peces Ciprinodóntidos Ibéricos Fartet Y Samaruc, pp. 33–57. Generalitat Valenciana, Conselleria de Medio Ambiente, Valencia, Spain.
Nei, M. (1972). Genetic distance between populations. Am Nat, 106: 183–192.
Pasteur, N., Pasteur, G., Bonhomme, F. and Catalan, J. et al. (1987). Manuel Technique de Génétique par Électrophorèse des Protéines. Collection Technique et Documentation, Lavoisier, Paris, France.
Perdices, A., Machordom, A. and Doadrio, I. (1996). Allozymic variation and relationships of the endangered cyprinodontid genus Valencia and its implications for conservation. J Fish Biol, 49: 1112–1127
Posada, D. and Crandall, K. A. (1998). MODELTEST: testing the model of DNA substitution. Bioinformatics, 14: 817–818.
Rogers, J. S. (1972). Measures of genetic similarity and genetic distance. Studies in Genetics University of Texas Publication, 7213: 145–153.
Rohlf, F. J. (1998). NTSYS-Pc, version 2.02i. Numerical taxonomy and multivariate analysis system. State University of New York, Stony Brook, USA.
Schmidt, T. R. and Gold, J. R. (1993). Complete sequence of the mitochondrial cytochrome b gene in the cherryfin shiner, Lythrurus roseipinnis (Teleostei: Cyprinidae) Copeia, 3: 880–883.
Schneider, S., Roessli, D. and Excoffier, L. (2000). ARLEQUIN, version 2000. A software for population genetics data analysis. Genetics and Biometry Laboratory, University of Geneva, Switzerland.
Schönhuth, S., Luikart, G. and Doadrio, I. Submitted. Effects of a founder event and supplementary introductions on genetic variation in a captive breeding population of the endangered Spanish killifish (Aphanius iberus). Biol Conserv
Shaklee, J. B., Allendorf, F. W., Morizot, D. G. and Whitt, G. G. (1990). Gene nomenclature for protein-coding loci in fish. Trans Am Fish Soc, 119: 2–15.
Sienknecht, U. (1999). Diferencias genéticas a nivel de población de Lebias ibera (Valenciennes 1846) (Teleostei: Cyprinodontidae). In: Planelles-Gomis, M. (Coord.) Peces Ciprinodóntidos Ibéricos Fartet Y Samaruc, pp. 213–223. Generalitat Valenciana, Conselleria de Medio Ambiente, Valencia, Spain.
Sokal, R. R., Smouse, P. E. and Neel, N. J. (1986). The genetic structure of a tribal population, the Yanomama Indians. XV. Patterns inferred by autocorrelation analysis. Genetics, 114: 259–287.
Strimmer, K. and Von Haeseler, A. (1996). Quartet puzzling: a quartet maximum-likelihood for reconstructing tree topologies. Mol Biol Evol, 13: 964–969.
Swofford, D. L. (1999). PAUP*: Phylogenetic analysis using parsimony (*and other methods), version 4.0b2. Sinauer Associates, Sunderland, MA.
Swofford, D. L. and Olsen, G. J. (1990). Phylogeny reconstruction. In: Hillis, D. M. and Moritz, C. (eds) Molecular Systematics, pp. 411–500. Sinauer Associates, Sunderland, MA.
Swofford, D. L., Selander, R. B. and Black, W. C. (1997). BIOSYS-2: A computer program for the analysis of allelic variation in populations genetics and biochemical systematics. Illinois Natural History Survey, IL, USA.
Towner, P. (1991). Purification of DNA. In: Brown, T. A. (ed.) Essential Molecular Biology. A Practical Approach, pp. 47–68. Oxford University Press, Oxford.
Vargas, M. J. (1993). Interacción entre Aphanius iberus y Gambusia holbrooki en el Delta del Ebro: Sus Ciclos Biológicos y Ecologías Tróficas. Tesis Dostoral, Universidad Central de Barcelona.
Vrijenhoek, R. C. (1994). Genetic diversity and fitness in small populations. In: Loeschcke, V., Tomiuk, J. and Jain, S. K. (eds) Conservation Genetics, pp. 37–53. Switzerland, Birkhäuser Basel.
Ward, R. D., Woodwark, M. and Skibinski, D. O. F. (1994). A comparison of genetic diversity levels in marine, freshwater, and anadromous fishes. J Fish Biol, 44: 213–232.
Acknowledgements
We would like to thank L. Domínguez, P. Economidis, P. Garzón, F. Gómez-Caruana, F. Lerma, R. Moreno Amich, J.C. Nevado, M. Planelles, S. Peiró, and P. Risueño for their help in collecting material for this study. Thanks to A. Machordom, O. Sanjur, S. Schönhuth and S. Williams for providing helpful comments and assistance. Partial financial support was provided by the ICONA-CSIC project ‘Caracterización poblacional y estudio de la variabilidad genética de especies en Peligro de Extinción’ and Junta de Andalucía-Córdoba University project ‘Plan de recuperación del Fartet en Andalucía’.
Author information
Authors and Affiliations
Corresponding author
Appendix 1
Appendix 1
(See Table3 below)
Rights and permissions
About this article
Cite this article
Perdices, A., Carmona, J., Fernández-Delgado, C. et al. Nuclear and mitochondrial data reveal high genetic divergence among Atlantic and Mediterranean populations of the Iberian killifish Aphanius iberus (Teleostei: Cyprinodontidae). Heredity 87, 314–324 (2001). https://doi.org/10.1046/j.1365-2540.2001.00888.x
Received:
Accepted:
Published:
Issue date:
DOI: https://doi.org/10.1046/j.1365-2540.2001.00888.x
Keywords
This article is cited by
-
Complete mitochondrial genome of the Spanish toothcarp, Aphanius iberus (Valenciennes, 1846) (Actinopterygii, Aphaniidae) and its phylogenetic position within the Cyprinodontiformes order
Molecular Biology Reports (2023)
-
Historical biogeography reveals new independent evolutionary lineages in the Pantosteus plebeius-nebuliferus species-group (Actinopterygii: Catostomidae)
BMC Evolutionary Biology (2018)
-
Diversity, mitochondrial phylogeny, and ichthyogeography of the Capoeta capoeta complex (Teleostei: Cyprinidae)
Hydrobiologia (2018)
-
Mitochondrial phylogeography of the killifish Aphanius fasciatus (Teleostei, Cyprinodontidae) reveals highly divergent Mediterranean populations
Marine Biology (2013)
-
Phylogeography and genetic structure of the Mediterranean killifish Aphanius fasciatus (Cyprinodontidae)
Marine Biology (2007)
