
Abstract
Backgroud:
Glioblastoma is a kind of highly malignant and aggressive tumours in the central nervous system. Previously, we found that neurotensin (NTS) and its high-affinity receptor 1 (NTSR1) had essential roles in cell proliferation and invasiveness of glioblastoma. Unexpectedly, cell death also appeared by inhibition of NTSR1 except for cell cycle arrest. However, the mechanisms were remained to be further explored.
Methods:
Cells treated with SR48692, a selective antagonist of NTSR1, or NTSR1 shRNA were stained with Annexin V-FITC/PI and the apoptosis was assessed by flow cytometry. Cytochrome c release was detected by using immunofluorescence. Mitochondrial membrane potential (MMP, ΔΨm) loss was stained by JC-1 and detected by immunofluorescence or flow cytometry. Apoptosis antibody array and microRNA microarray were performed to seek the potential regulators of NTSR1 inhibition-induced apoptosis. Interaction between let-7a-3p and Bcl-w 3′UTR was evaluated by using luciferase assay.
Results:
SR48692 induced massive apoptosis, which was related to mitochondrial cytochrome c release and MMP loss. Knockdown of NTSR1 induced slight apoptosis and significant MMP loss. In addition, NTSR1 inhibition sensitised glioblastoma cells to actinomycin D or doxorubicin-induced apoptosis. Consistently, NTSR1 inhibition-induced mitochondrial apoptosis was accompanied by downregulation of Bcl-w and Bcl-2. Restoration of Bcl-w partly rescued NTSR1 deficiency-induced apoptosis. In addition, NTSR1 deficiency promoted higher let-7a-3p expression and inhibition let-7a-3p partly rescued NTSR1 inhibition-induced apoptosis. In addition, let-7a-3p inhibition promoted 3′UTR activities of Bcl-w and the expression of c-Myc and LIN28, which were the upstream of let-7a-3p, decreased after NTSR1 inhibition.
Conclusions:
NTSR1 had an important role in protecting glioblastoma from intrinsic apoptosis via c-Myc/LIN28/let-7a-3p/Bcl-w axis.
Similar content being viewed by others


Main
Glioblastoma (GBM) is the most commonly diagnosed primary malignant brain tumour in adults. It recurs in almost all patients, and there is no commonly accepted standard treatment, so the prognosis is extremely poor. The median survival of patients with newly diagnosed GBM is nearly 12 months (Mischel and Cloughesy, 2003). In addition, the median overall survival time is only about 15–17 months even with the current gold-standard first-line therapeutic method—maximal safe resection followed by radiotherapy combined with temozolomide chemotherapy (Gilbert et al, 2014; Preusser et al, 2015). The excessive proliferation nature of GBM is due to frequent genetic alterations and subsequent stimulation of abnormal signal transduction pathways (Holland et al, 2000; Sturm et al, 2014). However, it remains unknown which of these pathways is essential to GBM initiation and progression.
Receptors for some regulatory peptides were reported to be highly expressed in tumours and associated with tumorigenesis, such as aberrant expression and activity of G-protein-coupled receptors (GPCRs; O’Hayre et al, 2013; Venkatakrishnan et al, 2013). As one member of GPCRs, neurotensin receptors (NTSRs)-induced signaling pathways were considered to contribute to malignance of multiple tumours and were suggested to be potential targets for cancer treatment. Neurotensin (NTS) is a short oligopeptide chain containing 13 amino-acid residues and works as a neuromodulator in the central nervous system and as an endocrine agent in the periphery nervous system (Carraway and Leeman, 1973). The biological functions of NTS were mediated via specific receptors, including two GPCRs, NTSR1 and NTSR2, as well as a sortilin receptor NTSR3 (Dal Farra et al, 2001). Among them, NTSR1 was the most affinitive receptor and had important roles during tumorigenesis and cancer development. We previously demonstrated that activation of NTS-NTSR1 signaling impacted the poor prognosis of glioma patients and promoted cell proliferation and invasion of GBM cells (Ouyang et al, 2015). In addition, we found that NTS-induced IL-8/CXCR1/STAT3 pathway was crucial for the maintenance of stem-like traits in glioma stem cells (Zhou et al, 2014). However, the underlying mechanisms were not fully elucidated.
In this study, we further showed an antiapoptotic property of NTSR1 in GBM. We found that inhibition of NTSR1 induced intrinsic apoptosis and sensitised GBM cells to stress-induced apoptosis by downregulation of Bcl-w. In addition, we showed that microRNA let-7a-3p targeted 3′UTR of Bcl-w and participated in this process. Our results provide clues for targeted therapy in GBM treatment.
Materials and methods
Reagents
SR48692 (SML0278), actinomycin D (1162400) and Doxorubicin (D1515) were synthesised at Sigma (Sigma-Aldrich Corp., Castle Hill, NSW, Australia). ABT-199 (GDC-0199) was purchased from MCE (Medchem Express Corp., Monmouth Junction, NJ, USA). All of them were dissolved with dimethyl sulphoxide (DMSO, Sigma-Aldrich Corp.).
Cell lines
Human GBM cell lines U251, LN229, A172 and U87 and mouse GBM cell line GL261 (ATCC, Manassas, VA, USA) were maintained in Dulbecco’s modified Eagle’s medium (DMEM, Life Technologies, NY, USA) supplemented with 10% foetal bovine serum (Invitrogen, Burlington, ON, Canada). Primary GBM tissue named 0807T and para-carcinoma tissue from the same tumour named 0807P were procured from Daping Hospital, the second affiliated hospital of the Third Military Medical School.
Real-time quantitative PCR
RNA was extracted using Trizol (Takara, Dalian, China) according to the manufacturer’s protocol and real-time quantitative PCR was performed for gene expression by using the SYBER Green PCR Master mix (Takara) and LightCycler96 real-time PCR system (Roche, Indianapolis, IN, USA). The forward and reverse primers are listed in Supplementary Table S1. Results were calculated using the ΔΔCt method with β-actin as a control.
Western blotting analysis
Western blottings on total cell lysates were performed using the following antibodies: Anti-NTSR1, anti-c-Myc (ab39688) and anti-LIN28 (ab46020) were purchased from Abcam (Abcam plc. Cambridge, MA, USA). Anti-caspase 3 (9665), anti-GAPDH (5174), anti-cleaved-caspase 3 (9664) and Bcl-2 (15071) were purchased from CST (Cell Signaling Technology Inc., Danvers, MA, USA). Anti-Bcl-w (G154) antibody was purchased from Bioworld (Bioworld Technology Inc., MN, USA).
Cell viability assay
3-(4,5-Dimethyl-2-thiazolyl)-2,5-diphenyl-2-H-tetrazolium bromide (MTT, Sigma) at 5 mg ml−1 was diluted in growth media and incubated for 4 h, then the precipitate was dissolved with DMSO (Sigma) and absorption values were obtained at 570 nm using an MULTISCAN GO multilabel plate reader (Thermo scientific, Vantaa, Finland).
Apoptosis assay
Cell apoptosis were detected by using double staining with Annexin V-FITC/PI (APOAF, Sigma) according to the manufacturer’s instructions. Briefly, cells were harvested by trypsin digestion and washed twice with precooled PBS. The cell pellets were suspended in 1 × binding buffer (10 mM HEPES/NaOH, 140 mM NaCl and 2.5 mM CaCl2, pH 7.4) at a concentration of 5 × 106 cells ml−1. Then the cells were incubated with AnnexinV-FITC and propidium iodide (PI) for 15 min (22–25 °C) in dark. The stained cells were immediately analysed by a BD Acurri C6 flow cytometry (BD, Franklin Lakes, NJ, USA). All data were analysed with Flowjo 7.6. Each measurement was carried out at least in triplicate.
Sub-G1 analysis assay
Treated cells, including medium, were collected and centrifuged at 600 g for 5 min and the supernatant was removed. Cells were washed twice with PBS and fixed with 70% ice-cold ethanol for 24 h. Then the cells were centrifuged and washed again and adjusted to a final concentration of 1 × 106 cells ml−1. And 50 mg l−1 PI and 0.25 ml RNase (1 mg ml−1 in PBS) was added to a 0.25 ml cell sample. Cells were incubated in the dark at 4 °C for 30 min before flow cytometric analysis (BD Accurri C6 cytometer and Modifit LT3.3). In the DNA histogram, the amplitude of the sub-G1 DNA peak represents the number of apoptotic cells. The experiment was repeated at least three times.
Caspase 3 activity assay
Cells were harvested with lysis buffer and added in Ac-DEVD-AMC (caspase-3 tetrapeptide substrate, Beyotime, Haimen, Jiangsu, China) and then incubated at 37 °C for 1 h. Then the suspension was placed in an influorescence spectrometer (Promega, Madison, WI, USA) to analyse influorescent intensity (excitation wavelength: 380 nm, emission wavelength: 430–460 nm). Ac-DEVD-CHO (Beyotime) was used as a caspase 3 inhibitor.
JC-1 staining
To evaluate the mitochondrial membrane potential (MMP), a lipophilic cationic probe 5,5′,6,6′-tetrachloro-1,1′,3,3′-tetraethylbenzimidazol-carbocyanine iodide (JC-1, 5 mg l−1, Biotime) was added to cell cultures for 30 min at 37 °C and fluorescence was measured by using a fluorescent microscope (Olympus, Tokyo, Japan) or a BD Accurri C6 Flow Cytometer. In all, 10 μ M CCCP (carbonyl cyanide 3-chlorophenylhydrazone, Beyotime)-pretreated groups were used as positive control. The experiment was repeated at least three times.
Cytochrome c release assay
Cells treated with 10 μ M SR48692 for 24 h and then were fixed and permeabilised as previously described. Cells were stained with anti-cytochrome c antibodies. Nuclei were stained with DAPI (C1002, Beyotime). Immunofluorescence was analysed by a Nikon microscope (80i, Nikon, Tokyo, Japan).
Vector construction and virus infection
The RNAi target site was designed by using the website of GeneSil (http://www.genesil.com/business/products/order2.htm), and the target sequences are listed at Supplementary Table S2. The hairpin oligonucleotides were synthesised in Beijing Genomics Institute (BGI, Beijing, China) and cloned into the pLKO.1 lentivirus vector. Human full-length Bcl-w (GenBank: NM_004050.4) cDNA was obtained by using PCR (primers were shown in Supplementary Table S3), and the DNA fragments were then inserted into PCDH-CMV-MCS-EF1-puro vectors to generate the recombinant plasmids. Vectors were transfected into 293FT cells to generate lentiviruses by using the Lipofectamine 2000 reagent (Invitrogen, Carlsbad, CA, USA). Subsequently, the lentiviruses were infected into cells according to the manufacturer’s protocol from Invitrogen and the cells were selected with puromysin (5 μg ml−1, Sigma) for at least 72 h.
Soft agar assay
Colony-formation ability was determined by soft agar assay on GBM cells. Briefly, 1.5 ml DMEM medium containing 0.6% agarose were added to each well of a six-well culture plates and allowed to solidify (base agar). One thousand cells were then mixed with 1 ml DMEM medium containing 0.3% agarose and added to the top of base agar (top agar). The cells were cultured in 5% CO2 incubator at 37 °C for 15–20 days. Colonies were imaged and counted by an Olympus inverted microscope (Olympus, Tokyo, Japan). Then cells were stained with MTT and imaged using a scanner (Epson Co., Ltd., Beijing, China).
TaqMan miRNA assay
Expression of mature miRNAs (let-7a-3p and let-7a-5p) was analysed by using the Bulge-Loop miRNA qRT–PCR assay (Guangzhou RiboBio Co., Ltd., Guangzhou, China). Briefly, single-stranded cDNA was gained from 5 ng total RNA in a 10 μl reaction volume, using the system provided by Bulge-Loop miRNA qRT–PCR Starter Kit (Guangzhou RiboBio, 1 μg RNA Template, 1 μl 5 μ M Bulge-Loop miRNA RT Primer, 2 μl 5 × Reverse Transcription Buffer, 2 μl RTase Mix). The reactions were incubated at 42 °C for 60 min and then inactivated by incubation at 70 °C for 10 min. For miRNA qPCR reations, each 20 μl PCR included 10 μl 2 × SYBR Green Mix, 2 μl RT Product, 0.8 μl 5 μ M Bulge-Loop miRNA Forward Primer and 0.8 μl 5 μ M Bulge-Loop U6 miRNA Reverse Primer. The reactions were incubated in a 96-well plate at 95 °C for 10 min, followed by 40 cycles of 95 °C for 2 s, 60 °C for 20 s and 70 °C for 10 s.
Let-7a-3p microRNA inhibition
micrOFF let-7a-3p inhibitors was purchased from Guangzhou RiboBio Co., Ltd. and applied at a final concentration 100 nM with the help of riboFECT CP (Guangzhou RiboBio). Briefly, 5 × 105 cells were seeded in six-well plates and cultured for 24 h. Then we diluted 2.5 μl miRNA inhibitor with 60 μl 1 × riboFECT CP Buffer and gently mixed with 6 μl riboFECT CP Reagent and incubated at room temperature for 15 min. The mixture was added to the six-well plates, gently blended and then cultured for 48 h.
Tumour xenografts
Four-week-old female nude mice (BALA/c; Beijing Laboratory Animal Research Center, Beijing, China) were purchased and housed in the SPF room to acclimate for a week. These mouse were divided into two groups. In one group, A172 cells (11 × 106) in 100 μl DMEM were transplanted subcutaneously into female BALA/c nude mice. Then the mice were injected with 10 mg kg−1 SR49692 every 3 days for 7 times after the tumour plumped. In another group, scramble or NTSR1 knockdown A172 cells (1 × 106) in 100 μl DMEM were subcutaneously injected into both flanks of each mouse, respectively. Then the mice were injected with 5 μg kg−1 actinomycin D every day for 7 times after the tumour plumped. Tumour growth was measured by caliper measurement daily, and tumour volume was then calculated with the formula (volume=tumour length × width2 × 0.5236) after tumour plumped. At the termination of the experiment, tumours were removed and weighed. All animal experiments were preapproved by the Institutional Animal Care and Use Committees of the Southwest University.
Luciferase assay
Part-length of the 3′UTR of Bcl-w as shown in Supplementary Table S4 was synthesised in Beijing Genomics Institute and cloned into the pGL3 basic vector. After that, vectors were transfected into A172 and U251 cells. Firefly luciferase gene in the vector pGL3-control (Promega) was used as a control for transfection efficiency. Luciferase assays were performed using the Dual-Luciferase Reporter Assay System Kit (Promega) according to the manufacturer’s instructions. Luciferase expression was analysed by Modulus single-tube multimode reader (Promega). The relative luciferase expression equals the expression of Renilla luciferase (pRL-TK) divided by the expression of firefly luciferase.
Apoptosis protein array
In all, 500–600 μg ml−1 protein lysate preparation was performed on biological triplicate samples. Then the apoptosis protein array was applied by using RayBio Human Apoptosis Antibody Array (Cat# AAH-APO-G1, RayBiotech Inc., Guangzhou, China) as the providers described. The signals were imaged by using an Axon GenePix laser scanner via cy3 channel. Supplementary Table S5 reports detailed information concerning the antibodies used and signal values. Then the heatmap was drawn by using the Heml: Heatmap Illustrator v.1.0.3.3 software (Deng et al, 2014).
microRNA array
Total RNAs from scramble or shNTSR1 U251 cells treated with DMSO or actinomycin D were extracted using Trizol (Takara) according to the manufacturer’s protocol. The quality of RNA was detected by using a NanoDrop ND-1000 Spectrophotometer (NanoDrop Technologies, Wilmington, DE, USA) and an Agilent 2100 Bioanalyzer (Agilent Technologies, Foster, CA, USA). RNA samples with RNA integrity number >7, 28S/18S ratio>0.7 and RIN⩾6.0 were used for the Agilent microRNA Chip. The candidate miRNAs expressed differently between scramble and NTSR1-silenced groups were performed via microRNA microarray profiling by using Agilent miRNA array 18.0 (ShanghaiBio Corporation, Shanghai, China). RNA labelling and hybridisation were performed by using a Human miRNA Microarray and a miRNA Complete Labeling and Hyb Kit (Agilent Technologies, Santa Clara, CA, USA). After washing twice with Gene Expression Wash Buffer, the slides were scanned by using an Agilent Microarray Scanner. The signal values were normalised by values of scramble groups and were converted into base-e logarithmic values. The heatmap was drawn by using the Heml: Heatmap Illustrator v.1.0.3.3 software (Deng et al, 2014). Relative signal values of microRNA array are shown in Supplementary Table S6.
Statistical analysis
The statistical significance of the data was performed using Graphpad Prism 5 (GraphPad Software, La Jolla, CA, USA) and conducted by a two-tailed, unpaired Student’s t-test.
Results
NTSR1 antagonist SR48692 induced mitochondrial apoptosis in GBM cells
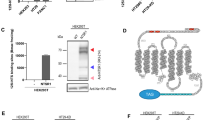
First, we analysed NTSR1 expression of different GBM cell lines. The results showed that NTSR1 expressed in these cell lines and a primary GBM in a higher level than in para-carcinoma tissue (Figure 1A and Supplementary Figure S1A). Among them, the expression levels of NTSR1 in U251, A172 and a mouse GBM cell line GL261 GBM cells were higher than in other cells (Figure 1A and Supplementary Figure S1A). Meanwhile, we conducted an MTT assay to detect IC50 of SR48692 in these cell lines. The results showed that IC50 of SR48692 is 25.74 μ M in GL261 cells, while 47.13 μ M in A172 cells and 17.07 μ M in U251 cells (Supplementary Figure S1B). These results showed that sensitiveness of these cell lines were roughly associated with NTSR1 expression. So during the next experiments, we used these cell lines as models in vitro. By treatment of 5 or 10 μ M SR48692, a potent NTSR1 antagonist, cell viabilities were decreased in U251, A172 and GL261 by a concentration-dependent manner, compared with DMSO-treated cells (Figure 1B). Meanwhile, sub-G1 analysis showed a higher sub-G1 phrase appeared in GBM cells by exposure to SR48692 treatment for 48 h (Figure 1C). By using an Annexin V-FITC/PI dual staining apoptosis kit, we confirmed that NTSR1 inhibition by SR48692 treatment induced apoptosis in GL261, U251 and A172 GBM cells (Figure 1D). In addition, caspase 3 activities were also increased in SR48692-treated cells in a dose-dependent manner and the induced caspase 3 activities could be rescued by a caspase inhibitor, Ac-DEVD-CHO (Figure 1E). Collectively, these evidences indicated that NTSR1 inhibition by SR48692 induced apoptosis in GBM cells.

SR48692 induced mitochondrial apoptosis in GBM cells. (A) Representative western blotting of NTSR1 expression in mouse GBM GL261 cells, human GBM U87, LN229, A172 and U251 cells, and a primary GBM tissue 0807T as well as a counterpart para-carcinoma tissue 0807P. GAPDH expression levels served as a loading control. (B) Cell viability assay in GL261, A172 and U251 cells after treatment of 5 or 10 μ M SR48692 or DMSO at 0, 2, 4 and 6 days. (C) Image and quantification of Sub-G1 phrase analysis by flow cytometry in GL261, A172 and U251 GBM cells after treatment of 10 μ M SR48692 or DMSO for 48 h. (D) The effect of Annexin V-FITC/PI dual staining apoptosis assays analysed by flow cytometry in GL261, A172 and U251 GBM cells after treatment of 10 μ M SR48692 or DMSO for 48 h. (E) The effect of relative caspase 3 activities assays in GL261, A172 and U251 GBM cells after treatment of 10 μ M SR48692 or DMSO for 48 h. In all, 20 μ M Ac-DEVD-CHO, a caspase 3 inhibitor, was used to perform a negative control. (F) Image of cytochrome c release assay in GL261, A172 and U251 GBM cells after treatment of 10 μ M SR48692 or DMSO for 48 h. Scale bars represent 200 μm. (G) Flow cytometric analysis of JC-1 staining in GL261, A172 and U251 GBM cells after treatment of 10 μ M SR48692 or DMSO for 48 h. Scale bars represent 100 μm. All data were shown as mean±s.d. Student’s t-test was carried out. *P<0.05, **P<0.01, ***P<0.001.
Mitochondria are important participants in apoptosis by releasing cytochrome c into the cytoplasm (Green and Kroemer, 2004). Cytochrome c in cytoplasm recruits Apaf1 and procaspase 9 to form apoptosome, which activates caspase 3 activity (Green et al, 2014). To determine whether SR48692-induced apoptosis was related to mitochondria, we used cytochrome c-releasing apoptosis assay in SR48692-treated GBM cells. Surprisingly, 24 h SR48692-treated cells exhibited mostly diffuse cytoplasmic cytochrome c staining, while DMSO-treated cells displayed punctate cytoplasmic staining (Figure 1F). These evidences showed that cytochrome c was released from mitochondria to cytoplasm. Meanwhile, a decline in MMP (ΔΨm) was observed both by fluorescence microscopy or flow cytometry at 12 h in GBM cells after SR48692 treatment by using JC-1 staining (Figure 1G and Supplementary Figure S1C). These results indicated that NTSR1 antagonist SR48692 induced intrinsic apoptosis pathway in GBM cells.
NTSR1 knockdown induced slight apoptosis in GBM cells
To elucidate whether NTSR1 was responsible for its antiapoptotic effect, we knocked down NTSR1 in U251 and A172 GBM cells by lentivirus infection. RT–PCR and western blotting analysis showed that NTSR1 expression was decreased in NTSR1-silenced cells compared with control groups (Supplementary Figures S2A and B). Consistently, cell viabilities were decreased in NTSR1 knockdown U251 and A172 GBM cells, compared with scramble-infected cells (Supplementary Figure 2C). Meanwhile, Annexin V-FITC/PI dual staining assay also showed that the apoptotic rate was slightly increased after NTSR1 knockdown (Figure 2A). Meanwhile, caspase 3 activities were also slightly elevated and ΔΨm was significantly lost after NTSR1 knockdown (Figure 2B and C). These evidences showed that NTSR1 knockdown induced slight mitochondrial apoptosis in GBM cells.

NTSR1 knockdown induced slight mitochondrial apoptosis in GBM cells. (A) The effect of Annexin V-FITC/PI dual staining apoptosis assays analysed by flow cytometry in A172 and U251 GBM cells after NTSR1 knockdown. (B) The effect of relative caspase 3 activities assays in A172 and U251 GBM cells after NTSR1 knockdown. In all, 20 μ M Ac-DEVD-CHO, a caspase 3 inhibitor, was used to perform a negative control. (C) Flow cytometric analysis of JC-1 staining in A172 and U251 GBM cells after NTSR1 knockdown. All data were shown as mean±s.d. Student’s t-test was carried out. *P<0.05, **P<0.01, ***P<0.001, NS, no sense.
NTSR1 inhibition sensitised drug-induced apoptosis in GBM cells
We hypothesised that the abundant NTSR1 contribute to chemoresistance, while NTSR1 reduction may sensitise GBM cells to stress-induced apoptosis. Actinomycin D, known to inhibit both RNA pol I and RNA pol II (Perry and Kelley, 1970), would trigger apoptosis of tumour cells, especially GBM at appropriate concentration (Narita et al, 2000). To further elucidate whether NTSR1 inhibition could sensitise actinomycin D-induced apoptosis, we used a low-dose actinomycin D (10 nM) during the cell culture. Annexin V-FITC/PI dual staining assay showed that the apoptotic rate was massively increased after SR48692 treatment compared with DMSO treatment in the presence of actinomycin D (Figure 3A). In addition, caspase 3 activities were also increased after SR48692 treatment compared with DMSO treatment in the presence of actinomycin D (Figure 3B). What is more, NTSR1 knockdown showed similar results in the presence of actinomycin D compared with scramble groups (Figure 3C and D). In addition, we also found that NTSR1 knockdown also promoted U251 and A172 cells to apoptosis induced by doxorubicin (0.5 μ M), a DNA damage drug (Supplementary Figures S3A and B). These results indicated that NTSR1 inhibition sensitised drug-induced apoptosis in GBM cells.

NTSR1 knockdown sensitised GBM cells to drug-induced apoptosis. (A) The effect of Annexin V-FITC/PI dual staining apoptosis assays analysed by flow cytometry in GL261, A172 and U251 GBM cells after treatment of 10 μ M SR48692 or DMSO for 48 h in the presence or absence of 5 nM actinomycin D in cultural medium. (B) The effect of relative caspase 3 activities assays in GL261, A172 and U251 GBM cells after treatment of 10 μ M SR48692 or DMSO for 48 h in the presence or absence of 5 nM actinomycin D in cultural medium. (C) The effect of Annexin V-FITC/PI dual staining apoptosis assays analysed by flow cytometry in A172 and U251 GBM cells after NTSR1 knockdown in the presence of 5 nM actinomycin D in cultural medium for 24 h. (D) The effect of relative caspase 3 activities assays in A172 and U251 GBM cells after NTSR1 knockdown in the presence of 5 nM actinomycin D in cultural medium for 24 h. In all, 20 μ M Ac-DEVD-CHO, a caspase 3 inhibitor, was used to perform a negative control. All data were shown as mean±s.d. Student’s t-test was carried out. *P<0.05, **P<0.01, ***P<0.001, NS, no sense.
NTSR1 inhibition downregulated Bcl-w and Bcl-2 in GBM cells
To determine which factor of apoptosis pathway was responsible for the cell death of GBM cells after NTSR1 inhibition, we set up a RayBio Human Apoptosis Antibody Array (AAH-APO-G1-4) analysis of 43 proteins, which was tightly related to apoptosis pathway, in U251 GBM cells after NTSR1 knockdown in the absence or presence of actinomycin D. The heatmap showed that Bcl-w was downregulated after NTSR1 knockdown in the absence or presence of actinomycin D (Figure 4A). Meanwhile, Bcl-2 and caspase 3 were significantly decreased after NTSR1 knockdown compared with scramble groups only in the presence of actinomycin D (Figure 4A). Furthermore, we conducted western blotting experiments and the results showed that, after SR48692 treatment, Bcl-w and Bcl-2 was downregulated, while cleaved-caspase 3 was elevated (Figure 4B). Bcl-w was also significantly reduced in NTSR1 knockdown cells, but Bcl-2 and caspase 3 was not significantly affected (Figure 4C). However, when actinomycin D was added into the medium, Bcl-2 was significantly downregulated and cleaved-caspase 3 was increased after NTSR1 knockdown (Figure 4D). To explore whether Bcl-w was essential during the process of NTSR1 inhibition-induced apoptosis, we restored Bcl-w in SR48692-treated and NTSR1 knockdown cells. The results showed that Bcl-w could rescue NTSR1 inhibition or deficiency-induced Bcl-2 downregulation and subsequent caspase 3 activation (Figure 4E and F), as well as apoptosis (Supplementary Figures S4A and B). These results indicated that NTSR1 inhibition induced intrinsic apoptosis via downregulation of Bcl-w and Bcl-2 in GBM cells.

NTSR1 inhibition induced downregulation of Bcl-w and Bcl-2 in GBM cells. (A) Heatmap of apoptosis-related protein array in U251 cells after NTSR1 knockdown in the absence or presence of 5 nM actinomycin D in cultural medium for 24 h. (B) Representative western blotting of Bcl-w, Bcl-2, caspase 3 and cleaved-caspase 3 expression in GL261, A172 and U251 cells after 10 μm SR48692 treatment in the absence or presence of 5 nM actinomycin D in cultural medium for 24 h. (C) Representative western blot of NTSR1, Bcl-w, Bcl-2, caspase 3 and cleaved-caspase 3 expression in A172 and U251 cells after NTSR1 knockdown. (D) Representative western blotting of NTSR1, Bcl-w, Bcl-2, caspase 3 and cleaved-caspase 3 expression in A172 and U251 cells after NTSR1 knockdown in the absence or presence of 5 nM actinomycin D in cultural medium for 24 h. (E) Representative western blotting of Bcl-w, Bcl-2, caspase 3 and cleaved-caspase 3 expression in Bcl-w or vector transient-transfected A172 and U251 cells after 10 μm SR48692 treatment in cultural medium for 24 h. (F) Representative western blot of Bcl-w, Bcl-2, caspase 3 and cleaved-caspase 3 expression in scramble or NTSR1-silenced A172 and U251 cells after transient transfected with Bcl-w for 48 h in the presence of 5 nM actinomycin D. GAPDH expression levels served as a loading control.
NTSR1 inhibition-induced apoptosis was rescued by let-7a-3p suppression in GBM cells
MicroRNAs are critical regulators in the modulation of signaling pathways, such as apoptosis (Su et al, 2015). To explore the essential microRNA during the NTSR1 inhibition induced apoptosis pathway, we conducted a microRNA array. The results showed that let-7a-3p significantly upregulated after NTSR1 knockdown in the absence or presence of actinomycin D (Supplementary Figure 5), which was also subsequently confirmed by qRT–PCR (Figure 5A). And both knockdown of NTSR1 or pharmaceutical inhibition of NTSR1 by SR48692 promoted let-7a-3p expression (Supplementary Figures S6A and B). However, another mature microRNA let-7a-5p was not affected by these alterations (Supplementary Figures S6C and D). Meanwhile, let-7a-3p inhibition could not affect the expression of NTSR1 (Figure 5B). Furthermore, we found that the seed sequence of let-7a-3p had a complementary sequence with 3′UTR of Bcl-w, companied with several G:U wobbles nearby (Figure 5C). Next, wild and mutant 3′UTR of Bcl-w (Figure 5C and Supplementary Table S4) was synthesised and linked up to pGL3-basic vector. Luciferase assay showed that luciferase activities were increased in pGL3-Bcl-w 3′UTR wild but not in pGL3-Bcl-w 3′UTR mutant transfected U251 and A172 cells after let-7a-3p inhibitor treatment (Figure 5D and E). These results showed that let-7a-3p was the downstream of NTSR1 inhibition and could be an important regulator in NTSR1 inhibition-induced apoptosis.

NTSR1 inhibition-induced apoptosis was rescued by let-7a-3p suppression in GBM cells. (A) Relative expression of let-7a-3p in A172 and U251cells after NTSR1 knockdown in the absence or presence of actinomycin D in cultural medium for 24 h. (B) Representative western blotting of NTSR1 in A172 and U251cells after let-7a-3p inhibitor treatment. GAPDH expression levels served as a loading control. (C) Schematic diagram of the binding of let-7a-3p to the 3′UTR region of Bcl-w mRNA. (D and E) Relative luciferase activity (FFL/RL) was assessed by using luciferase assay in A172 and U251 cells after negative control or let-7a-3p inhibitor and wild or mutant pGL3-Bcl-w 3′UTR transfection. (F) The effect of Annexin V-FITC/PI dual staining apoptosis assays analysed by flow cytometry in DMSO or 10 μm SR48692-treated A172 and U251 cells after negative control or let-7a-3p inhibitor transfection. (G) The effect of Annexin V-FITC/PI dual staining apoptosis assays analysed by flow cytometry in NTSR1-silenced A172 and U251 cells after negative control or let-7a-3p inhibitor transfection. (H) The effect of Annexin V-FITC/PI dual staining apoptosis assays analysed by flow cytometry in DMSO or 10 μm SR48692-treated A172 and U251 cells after negative control or let-7a-3p inhibitor transfection in the presence of 5 nM actinomycin D in cultural medium for 24 h. (I) The effect of Annexin V-FITC/PI dual staining apoptosis assays analysed by flow cytometry in NTSR1-silenced A172 and U251 cells after negative control or let-7a-3p inhibitor transfection in the presence of 5 nM actinomycin D in cultural medium for 24 h. (J) Representative western blotting of NTSR1, Bcl-w and Bcl-2 in NTSR1-silenced A172 and U251 cells after negative control or let-7a-3p inhibitor transfection. (K) Relative expression of c-Myc and LIN28 in U251 and A172 cells after NTSR1 knockdown. (L) Representative western blotting of c-Myc and LIN28 in NTSR1-silenced A172 and U251 cells. GAPDH expression levels served as a loading control. All data were shown as mean±s.d. Student’s t-test was carried out. *P<0.05, **P<0.01, ***P<0.001, NS, no sense.
To determine whether let-7a-3p was essential during apoptosis induced by NTSR1 inhibition, we used a let-7a-3p inhibitor to transfected into U251 cells after NTSR1 inhibition. The results showed that let-7a-3p inhibition partly rescued SR48692-induced apoptosis and almost completely retrieved NTSR1 knockdown-induced apoptosis (Figure 5F and G). In addition, let-7a-3p inhibition could partly rescue SR48692 or NTSR1 knockdown-sensitised actinomycin D-induced apoptosis (Figure 5H and I). Importantly, let-7a-3p inhibition also abrogated NTSR1 knockdown-induced Bcl-w and Bcl-2 reduction both in A172 and U251 cells (Figure 5J). These results showed that microRNA let-7a-3p at least partly recovered NTSR1 inhibition-induced apoptosis and might be an important regulator during GBM survival. It was reported that the expression of let-7 family was regulated by LIN28, which was transcriptionally upregulated by c-Myc in multiple advanced human malignancies (Viswanathan et al, 2008; Chang et al, 2009). Our previous data also showed that NTS/NTSR1 could transcriptionally upregulate the expression of c-Myc in neuroblastoma cells (Ouyang et al, 2016). So we thought that NTS/NTSR1/c-Myc/LIN28 might be an upstream pathway of let-7a-3p/Bcl-w-induced apoptosis. As shown in Figure 5K and L as well as Supplementary Figure S6E, NTSR1 deficiency induced decrease of c-Myc and LIN28 expression. In conclusion, our data revealed the molecular mechanism of NTSR1 inhibition-induced apoptosis.
NTSR1 inhibition reduced tumour growth in vivo
Before animal experiments, we measured the capacity of tumour colony formation in A172 GBM cells by treatment of SR49692 or knockdown of NTSR1. The result showed that formatted colony number of cells treated with SR49692 or knockdown of NTSR1 was significantly less than the control groups, and colony size was also smaller, too (Figure 6A and B). GBM A172 cells were transplanted subcutaneously into female BALA/c nude mice. Then the mice were injected with 10 mg kg−1 SR49692 every 3 days for 7 times after the tumour plumped. The mice were killed at the termination of the experiment, and the formed tumours were excised. The results showed that SR49692 treatment significantly blocked tumour growth both in weight and in size (Figure 6C–E). Treatment with SR49692 did not affect the weight of the mice (Supplementary Figure S7A). In addition, scramble or NTSR1 knockdown A172 cells were also transplanted subcutaneously into female BALA/c nude mice. Then the mice were injected with 5 μg kg−1 actinomycin D every day for 7 times after the tumour plumped. The mice were killed at the termination of the experiment, and the formed tumours were excised. Similar to the above, NTSR1 knockdown remarkably inhibited the tumour growth both in weight and in size (Figure 6F–H). To test whether NTSR1 inhibition was also associated with apoptosis in vivo, the expression of Bcl-w and Bcl-2 was investigated in the xenograft tumour tissues. In consistent with our previous results, these proteins were all markedly downregulated in SR49692-treated or NTSR1 knockdown tumours compared with their controls (Figure 6I and J). Treatment with actinomycin D did not affect the weight of the mice (Supplementary Figure S7B). In addition, expression of microRNA let-7a-3p was also detected in the xenograft tumour tissues. As expected, the expression of let-7a-3p was upregulated in the NTSR1 inhibition groups compared with their controls (Figure 6K and L). Conclusively, these results indicated that NTSR1 inhibition reduced tumour growth in vivo.

NTSR1 inhibition reduced tumour growth of GBM cells in vivo. (A) The colony formation was examined by soft agar assay (500 cells per well) in DMSO or 10 μ M SR48692-treated A172 cells. Scale bars represent 500 μm. (B) The colony formation was examined by soft agar assay (500 cells per well) in scramble or NTSR1-silenced A172 cells (scale bar, 100 μm). (C–E) A172 cells were injected into the flank of BALA/c nude mice and treated with DMSO or 10 mg kg−1 SR48692 by subcutaneous injection when tumours were palpable. Tumour volume (D) was measured. When tumours reached large enough, mice were killed, tumour was imaged and tumour was weighed (C and E). (F–H) scramble or NTSR1-silenced A172 cells were injected into the flank of BALA/c nude mice and treated with 5 μg kg−1 actinomycin D by subcutaneous injection when tumours were palpable. Tumour volume (G) was measured. When tumours reached large enough, mice were killed, tumour was imaged and weighed (F and H). (I) Representative western blotting of NTSR1, Bcl-w and Bcl-2 in xenograft tumours treated with DMSO or 10 mg kg−1 SR48692. GAPDH expression levels served as a loading control. (J) Representative western blotting of NTSR1, Bcl-w and Bcl-2 in xenograft tumours after scramble or NTSR1 knockdown. GAPDH expression levels served as a loading control. (K) Relative expression of let-7a-3p in xenograft tumours treated with DMSO or 10 mg kg−1 SR48692. (L) Relative expression of let-7a-3p in xenograft tumours after scramble or NTSR1 knockdown. All data were shown as mean±s.d. Student’s t-test was carried out. **P<0.01, ***P<0.001.
Discussion
GBM is one of the most causes of tumour-related death in the world and has an extreme poor prognosis about 1 year (DeSantis et al, 2014; Siegel et al, 2014). So far, multiple biologicals, including anti-invasive cilengitide and antiangiogenic cediranib and bevacizumab or specific inhibitors of some oncogenic signaling pathways such as enzastaurin, everolimus, erlotinib, imatinib, gefitinib and temsirolimus, have failed to be demonstrated to cure GBM (Chinot et al, 2014). Therefore, identifying novel biomarkers and exploring novel therapeutic targets for the treatment of GBM are urgent tasks for scientists.
As one member of GPCRs, NTSR1 is a seven-span transmembrane protein in the plasma membrane and is the most affinitive receptor of neurotensin. Recently, NTSR1 was reported to have a negative role in the initiation and development of various tumours, including prostate cancer (Taylor et al, 2012), colorectal cancer (Bakirtzi et al, 2011), lung cancer (Wakabayashi-Nakao et al, 2012), breast cancer (Heakal and Kester, 2009), squamous cell carcinoma (Shimizu et al, 2008), gastrointestinal cancer (Gromova et al, 2011), neuroblastoma (Gustafson et al, 2005), neuroendocrine tumour (Kim et al, 2014), malignant melanoma (Zhang et al, 2014), lymphoma (Saada et al, 2012) and liver cancer (Tang et al, 2012). Previously, we found that activation of NTS/NTSR1 signaling was tightly related to the poor prognosis of glioma patients and increased proliferative and invasive characteristics of GBM cells (Ouyang et al, 2015, 2016). However, the mechanism underlying was remained to be further elucidated.
In this study, we confirmed that NTSR1 was highly expressed in GBM cell lines, and the expression of NTSR1 in a primary tumour was higher than that of para-carcinoma tissue from the same sample (Figure 1A). This results confirmed our previous conclusions that the expression levels of NTSR1 was related to the progression of GBM (Ouyang et al, 2015). To explore the function of NTSR1 in GBM cells, we used an inhibitor of NTSR1, SR-48692, also known as meclinertant. SR48692 was a drug acting as a selective, non-peptide antagonist at NTSR1 (Gully et al, 1993). It was used in scientific research to explore the physical or pathological functions, especially antitumour activities of NTS/NTSR1 signaling. In addition, SR48692 showed antitumour activities in multiple tumours, including melanoma (Zhang et al, 2014), prostate cancer (Baxendale et al, 2013), gastric cancer (Akter et al, 2015), pancreatic carcinoma (Iwase et al, 1997) and so on. These evidences indicated that SR48692 might be a potent drug for cancer treatments. Similarly, we showed that inhibition of NTSR1 via its antagonist SR48692 decreased cell viabilities and induced apoptosis in mouse GBM GL261 cells and human GBM A172 and U251 cells (Figure 1B–D). In addition, SR48692-treated cells also showed increased caspase 3 activities, which could be rescued by a specific caspase 3 inhibitor, Ac-DEVD-CHO (Figure 1E). Meanwhile, NTSR1 knockdown in A172 and U251 GBM also induced slight apoptosis (Figure 2A and B). In addition, soft agar assay revealed decreased colony number and size in NTSR1 inhibition or knockdown groups compared with control groups (Figure 6A and B). In vivo studies also showed that SR48692 treatment reduced tumour size, weight and volume (Figure 6C–E) and NTSR1 knockdown reduced tumour size, weight and volume induced by actinomycin D (Figure 6F–H). These evidences demonstrated that NTSR1 inhibition induced apoptosis in GBM cells and SR48692 might be a candidate drug for GBM treatment.
The intrinsic apoptosis pathway, which is regulated by mitochondria, is an important signaling pathway for cancer survival and treatment (Reed, 2011). In this pathway, caspase activation is closely linked to mitochondrial outer membrane permeabilisation (MOMP), which is mainly controlled by the Bcl-2 families (Green and Kroemer, 2004). Increased MOMP results in the release of proteins, including cytochrome c, thus initiating a caspase cascade and subsequent cell death (Kluck et al, 1997; Wei et al, 2001). In addition, loss of MMP (ΔΨm) has been recognised as an early feature of apoptosis (Green and Reed, 1998). Our data showed that SR48692 treatment induced cytochrome c release and decline of ΔΨm (Figure 1F and G). Meanwhile, NTSR1 knockdown also induced significant decline of ΔΨm (Figure 2C). These data indicated that NTSR1 inhibition-induced apoptosis was tightly related to mitochondrial activities.
Inherent complexity and multiple mechanisms of drug resistance make it difficult to treat GBM (Haar et al, 2012). Currently used chemotherapy drug and radiotherapy have done little in prolonging the life expectancies of GBM patients. To determine whether NTSR1 inhibition increased GBM cells to drug-induced apoptosis, we added low dose of actinomycin D and doxorubicin in the medium. Actinomycin D was the first antibiotic that has been demonstrated to have anticancer activity (Tacar et al, 2013). And doxorubicin has been used in cancer treatment for decades but causes toxicity to normal tissues, which makes the treatment dose limiting (Tacar et al, 2013). Surprisingly, the results showed that both SR48692 treatment and NTSR1 knockdown increased apoptosis induced by low dose of actinomycin D or doxorubicin (Figure 3A–D and Supplementary Figures S3A–B). This result indicated that NTSR1 protected GBM cells from apoptosis and inhibition of NTSR1 sensitised GBM cells to drug-induced apoptosis. Chemotherapy combined with NTSR1 inhibition might be a new strategy for GBM treatment.
Bcl-2 family members are central regulators in mitochondrial apoptosis pathway by governing MOMP and can be either proapoptotic (Bax, BAD, Bak and Bok) or antiapoptotic (Bcl-2, Bcl-xL and Bcl-w) (Green and Kroemer, 2004). Bcl-w, also known as BCL2L2, can inhibit the intrinsic pathway of apoptosis in some tumours (Oltersdorf et al, 2005). Recently, Bcl-w was reported to have an important role in some solid tumours. For example, Bcl-w also induced matrix metalloproteinase-2 expression through activating phosphoinositide 3-kinase, Akt and specificity protein 1 to promote invasion of gastric cancer (Bae et al, 2006). Meanwhile, Bcl-w could enhance mesenchymal changes and invasiveness of GBM cells by inducing nuclear accumulation of β-catenin (Lee et al, 2013). In our results, apoptosis-related protein array showed that Bcl-w was downregulated after NTSR1 knockdown both in the absence and in the presence of actinomycin D, while Bcl-2 and caspase 3 significantly decreased after NTSR1 knockdown only in the presence of actinomycin D (Figure 4A). This data indicated that massive apoptosis might emerge only when Bcl-2 was inhibited. Inhibition of NTSR1 by SR48692 could decrease both Bcl-w and Bcl-2, thus inducing significant apoptosis (Figure 4B). However, NTSR1 knockdown could significantly downregulate the expression of Bcl-w but not sharply alter the expression of Bcl-2 (Figure 4C). It was reported that actinomycin D and doxorubicin could downregulate Bcl-2 expression (Merkel et al, 2012; Florou et al, 2013). So we added low concentration of actinomycin D and doxorubicin in NTSR1-downregulated cells and the results showed that both Bcl-w and Bcl-2 was inhibited, thus sensitising actinomycin D-induced apoptosis (Figure 3D and F, Supplementary Figures S4A–B). Consistently, restoration of Bcl-w could partly rescue decrease of Bcl-2 and increase of cleaved-caspase 3 in NTSR1-inhibited or NTSR1-deficient cells (Figure 4E and F). In vivo studies also showed decreased Bcl-w and Bcl-2 expression in NTSR1-inhibited or NTSR1-deficient xenograft tumours (Figure 6I and J). These evidences indicated that Bcl-w was an essential modulator in NTSR1-inhibition- or -deficiency-induced mitochondrial apoptosis.
miRNAs are a class of non-coding RNAs composed of 18–24 bp nucleotides that suppress gene expression through directly binding to the complementary sequences in the 3′UTR of target mRNAs (Rottiers et al, 2011). They have been reported to participate in regulating tumorigenesis and cancer progression by acting as tumour promotors or suppressors (Lin and Gregory, 2015). miRNAs are suggested to be potential therapeutic targets for tumour treatment (Seton-Rogers, 2012). As the first known human miRNA, let-7 family has been demonstrated to involve in cell differentiation and proliferation during tumour development and usually act as tumour suppressor. There are 12 different let-7 family members (let-7a-1, -7a-2, -7a-3, -7b, -7c, -7d, -7e, -7f-1, -7f-2, -7g, -7i and mir-98) expressed in humans (Roush and Slack, 2008). let-7a-3p is a less abundant mature microRNA that comes from the left arm of let-7a-1 or let-7a-3 stem-loop; another higher expressed synonymous mature microRNA is let-7a-5p, which comes from the right arm of let-7a-1 or let-7a-3 stem-loop, according to miRBase website (http://www.mirbase.org/) (Ambros et al, 2003). In addition, let-7a-5p is also one of the mature microRNA of let-7a-2; another synonymous mature microRNA is let-7a-2-3p. It is reported that pre-let-7a was further processed by Dicer and turned into a mature let-7a duplex, which subsequently yielded let-7a-3p mediated by Ago3 (Winter and Diederichs, 2013). However, this process might be affected by LIN28, which bound to pre-let-7a and recruited a UTUase to irreversibly transform pre-let-7a into uridylated pre-miRNA (up-miRNA), and the later one might be swiftly degraded by nucleases afterward (Heo et al, 2008). Previous studies showed that let-7a (including let-7a-1, -7a-2, -7a-3) functioned as a tumour repressor via regulating of cell proliferation, differentiation, apoptosis and metabolism in various cancers (Johnson et al, 2007; Zhu et al, 2011; Lu et al, 2016; Tang et al, 2016). In addition, let-7a transcriptionally suppressed K-Ras and inhibited glioma malignancy independent of PTEN expression (Wang et al, 2013). However, the functions of let-7a mature microRNAs was less known.
Herein, we reported that let-7a-3p was significantly upregulated after NTSR1 inhibition, while let-7a-5p expression was not changed (Figure 5A, Supplementary Figures S5 and S6A–D). In addition, the expression of let-7a-3p was also increased in NTSR1-inhibited or NTSR1-deficient xenograft tumours (Figure 6K and L). And let-7a-3p inhibition could not affect the expression of NTSR1 (Figure 5B). Importantly, the seed sequence of let-7a-3p was found to have a complementary sequence with 3′UTR of Bcl-w, companied with several G:U wobbles nearby (Figure 5C). So we synthesised wild and mutant 3′UTR of Bcl-w (Figure 5C and Supplementary Table S3) and linked them up to pGL3-basic vector. We conducted a luciferase assay and the result showed that luciferase activities were increased in pGL3-Bcl-w 3′UTR wild while not in pGL3-Bcl-w 3′UTR mutant-transfected U251 and A172 cells after let-7a-3p inhibitor treatment (Figure 5D and E). Besides, inhibition of microRNA let-7a-3p could partly recover NTSR1 inhibition-induced apoptosis and downregulation of Bcl-w and Bcl-2 both in the presence and absence of actinomycin D (Figure 5F–I). c-Myc/LIN28/let-7 axis was a well-known pathway in multiple tumours (Viswanathan et al, 2008; Chang et al, 2009), and previous reports showed that NTS/NTSR1 could transcriptionally activiate c-Myc in GBM cells and colorectal cancers (Wang et al, 2010; Ouyang et al, 2016). Thus we measured the expression of c-Myc and LIN28 in NTSR1 knockdown cells and the result showed that NTSR1 knockdown induced decrease of c-Myc and LIN28 expression (Figures 5K and L). In conclusion, as shown in Figure 5N, our data indicated that NTS/NTSR1/c-Myc/LIN28/let-7a-3p axis had an important role in the regulation of apoptosis in GBM cells.
In summary, our data showed that NTSR1 inhibition induced mitochondrial apoptosis in GBM cells and sensitised them to drug-induced apoptosis. And NTSR1 inhibition induced downregulation of Bcl-w and Bcl-2, which had pivotal roles in regulating intrinsic apoptosis. In addition, NTSR1 inhibition also induced upregulation of let-7a-3p, which had an essential function in Bcl-w- and Bcl-2-induced mitochondrial apoptosis. Our results depicted the molecular mechanisms of NTSR1 inhibition-mediated apoptosis and provided clues for GBM treatment.
Change history
06 June 2017
This paper was modified 12 months after initial publication to switch to Creative Commons licence terms, as noted at publication
References
Akter H, Park M, Kwon OS, Song EJ, Park WS, Kang MJ (2015) Activation of matrix metalloproteinase-9 (MMP-9) by neurotensin promotes cell invasion and migration through ERK pathway in gastric cancer. Tumour Biol 36 (8): 6053–6062.
Ambros V, Bartel B, Bartel DP, Burge CB, Carrington JC, Chen X, Dreyfuss G, Eddy SR, Griffiths-Jones SAM, Marshall M, Matzke M, Ruvkun G, Tuschl T (2003) A uniform system for microRNA annotation. RNA 9 (3): 277–279.
Bae IH, Park MJ, Yoon SH, Kang SW, Lee SS, Choi KM, Um HD (2006) Bcl-w promotes gastric cancer cell invasion by inducing matrix metalloproteinase-2 expression via phosphoinositide 3-kinase, Akt, and Sp1. Cancer Res 66 (10): 4991–4995.
Bakirtzi K, Hatziapostolou M, Karagiannides I, Polytarchou C, Jaeger S, Iliopoulos D, Pothoulakis C (2011) Neurotensin signaling activates microRNAs-21 and -155 and Akt, promotes tumor growth in mice, and is increased in human colon tumors. Gastroenterology 141 (5): 1749–1761, e1.
Baxendale IR, Cheung S, Kitching MO, Ley SV, Shearman JW (2013) The synthesis of neurotensin antagonist SR 48692 for prostate cancer research. Bioorg Med Chem 21 (14): 4378–4387.
Carraway R, Leeman SE (1973) The isolation of a new hypotensive peptide, neurotensin, from bovine hypothalami. J Biol Chem 248 (19): 6854–6861.
Chang TC, Zeitels LR, Hwang HW, Chivukula RR, Wentzel EA, Dews M, Jung J, Gao P, Dang CV, Beer MA, Thomas-Tikhonenko A, Mendell JT (2009) Lin-28B transactivation is necessary for Myc-mediated let-7 repression and proliferation. Proc Natl Acad Sci USA 106 (9): 3384–3389.
Chinot OL, Wick W, Mason W, Henriksson R, Saran F, Nishikawa R, Carpentier AF, Hoang-Xuan K, Kavan P, Cernea D, Brandes AA, Hilton M, Abrey L, Cloughesy T (2014) Bevacizumab plus radiotherapy-temozolomide for newly diagnosed glioblastoma. N Engl J Med 370 (8): 709–722.
Dal Farra C, Sarret P, Navarro V, Botto JM, Mazella J, Vincent JP (2001) Involvement of the neurotensin receptor subtype NTR3 in the growth effect of neurotensin on cancer cell lines. Int J Cancer 92 (4): 503–509.
Deng W, Wang Y, Liu Z, Cheng H, Xue Y (2014) HemI: a toolkit for illustrating heatmaps. PLoS One 9 (11): e111988.
DeSantis CE, Lin CC, Mariotto AB, Siegel RL, Stein KD, Kramer JL, Alteri R, Robbins AS, Jemal A (2014) Cancer treatment and survivorship statistics, 2014. CA Cancer J Clin 64 (4): 252–271.
Florou D, Patsis C, Ardavanis A, Scorilas A (2013) Effect of doxorubicin, oxaliplatin, and methotrexate administration on the transcriptional activity of BCL-2 family gene members in stomach cancer cells. Cancer Biol Ther 14 (7): 587–596.
Gilbert MR, Dignam JJ, Armstrong TS, Wefel JS, Blumenthal DT, Vogelbaum MA, Colman H, Chakravarti A, Pugh S, Won M, Jeraj R, Brown PD, Jaeckle KA, Schiff D, Stieber VW, Brachman DG, Werner-Wasik M, Tremont-Lukats IW, Sulman EP, Aldape KD, Curran WJJ, Mehta MP (2014) A randomized trial of bevacizumab for newly diagnosed glioblastoma. N Engl J Med 370 (8): 699–708.
Green DR, Galluzzi L, Kroemer G (2014) Cell biology. Metabolic control of cell death. Science 345 (6203): 1250256.
Green DR, Kroemer G (2004) The pathophysiology of mitochondrial cell death. Science 305 (5684): 626–629.
Green DR, Reed JC (1998) Mitochondria and apoptosis. Science 281 (5381): 1309–1312.
Gromova P, Rubin BP, Thys A, Erneux C, Vanderwinden JM (2011) Neurotensin receptor 1 is expressed in gastrointestinal stromal tumors but not in interstitial cells of Cajal. PLoS One 6 (2): e14710.
Gully D, Canton M, Boigegrain R, Jeanjean F, Molimard JC, Poncelet M, Gueudet C, Heaulme M, Leyris R, Brouard A, Pelaprat D, Labbe- Jullie, Mazelia J, Soubrie P, Maffrand JP, Rostene W, Kitabgi P, Fur GL (1993) Biochemical and pharmacological profile of a potent and selective nonpeptide antagonist of the neurotensin receptor. Proc Natl Acad Sci USA 90 (1): 65–69.
Gustafson WC, De Berry BB, Evers BM, Chung DH (2005) Role of gastrointestinal hormones in neuroblastoma. World J Surg 29 (3): 281–286.
Haar CP, Hebbar P, Wallace GC, Das A, Vandergrift WA, Smith JA, Giglio P, Patel SJ, Ray SK, Banik NL (2012) Drug resistance in glioblastoma: a mini review. Neurochem Res 37 (6): 1192–1200.
Heakal Y, Kester M (2009) Nanoliposomal short-chain ceramide inhibits agonist-dependent translocation of neurotensin receptor 1 to structured membrane microdomains in breast cancer cells. Mol Cancer Res 7 (5): 724–734.
Heo I, Joo C, Cho J, Ha M, Han J, Kim VN (2008) Lin28 mediates the terminal uridylation of let-7 precursor microRNA. Mol Cell 32 (2): 276–284.
Holland EC, Celestino J, Dai C, Schaefer L, Sawaya RE, Fuller GN (2000) Combined activation of Ras and Akt in neural progenitors induces glioblastoma formation in mice. Nat Genet 25 (1): 55–57.
Iwase K, Evers BM, Hellmich MR, Kim HJ, Higashide S, Gully D, Thompson JC, Townsend CM Jr. (1997) Inhibition of neurotensin-induced pancreatic carcinoma growth by a nonpeptide neurotensin receptor antagonist, SR48692. Cancer 79 (9): 1787–1793.
Johnson CD, Esquela-Kerscher A, Stefani G, Byrom M, Kelnar K, Ovcharenko D, Wilson M, Wang X, Shelton J, Shingara J, Chin L, Brown D, Slack FJ (2007) The let-7 microRNA represses cell proliferation pathways in human cells. Cancer Res 67 (16): 7713–7722.
Kim JT, Liu C, Zaytseva YY, Weiss HL, Townsend CM Jr., Evers BM (2014) Neurotensin, a novel target of Wnt/beta-catenin pathway, promotes growth of neuroendocrine tumor cells. Int J Cancer 136 (6): 1475–1481.
Kluck RM, Bossy-Wetzel E, Green DR, Newmeyer DD (1997) The release of cytochrome c from mitochondria: a primary site for Bcl-2 regulation of apoptosis. Science 275 (5303): 1132–1136.
Lee WS, Woo EY, Kwon J, Park MJ, Lee JS, Han YH, Bae IH (2013) Bcl-w enhances mesenchymal changes and invasiveness of glioblastoma cells by inducing nuclear accumulation of beta-catenin. PLoS One 8 (6): e68030.
Lin S, Gregory RI (2015) MicroRNA biogenesis pathways in cancer. Nat Rev Cancer 15 (6): 321–333.
Lu L, Katsaros D, Risch HA, Canuto EM, Biglia N, Yu H (2016) MicroRNA let-7a modifies the effect of self-renewal gene HIWI on patient survival of epithelial ovarian cancer. Mol Carcinog 55 (4): 357–365.
Merkel O, Wacht N, Sifft E, Melchardt T, Hamacher F, Kocher T, Denk U, Hofbauer JP, Egle A, Scheideler M, Schlederer M, Steurer M, Kenner L, Greil R (2012) Actinomycin D induces p53-independent cell death and prolongs survival in high-risk chronic lymphocytic leukemia. Leukemia 26 (12): 2508–2516.
Mischel PS, Cloughesy TF (2003) Targeted molecular therapy of GBM. Brain Pathol 13 (1): 52–61.
Narita Y, Asai A, Kuchino Y, Kirino T (2000) Actinomycin D and staurosporine, potent apoptosis inducers in vitro, are potentially effective chemotherapeutic agents against glioblastoma multiforme. Cancer Chemother Pharmacol 45 (2): 149–156.
O’Hayre M, Vazquez-Prado J, Kufareva I, Stawiski EW, Handel TM, Seshagiri S, Gutkind JS (2013) The emerging mutational landscape of G proteins and G-protein-coupled receptors in cancer. Nat Rev Cancer 13 (6): 412–424.
Oltersdorf T, Elmore SW, Shoemaker AR, Armstrong RC, Augeri DJ, Belli BA, Bruncko M, Deckwerth TL, Dinges J, Hajduk PJ, Joseph MK, Kitada S, Korsmeyer SJ, Kunzer AR, Letai A, Li C, Mitten MJ, Nettesheim DG, Ng S, Nimmer PM, O’Connor JM, Oleksijew A, Petros AM, Reed JC, Shen W, Tahir SK, Thompson CB, Tomaselli KJ, Wang B, Wendt MD, Zhang H, Fesik SW, Rosenberg SH (2005) An inhibitor of Bcl-2 family proteins induces regression of solid tumours. Nature 435 (7042): 677–681.
Ouyang Q, Chen G, Zhou J, Li L, Dong Z, Yang R, Xu L, Cui H, Xu M, Yi L (2016) Neurotensin signaling stimulates glioblastoma cell proliferation by upregulating c-Myc and inhibiting miR-29b-1 and miR-129-3p. Neuro Oncol 18 (2): 216–226.
Ouyang Q, Gong X, Xiao H, Zhou J, Xu M, Dai Y, Xu L, Feng H, Cui H, Yi L (2015) Neurotensin promotes the progression of malignant glioma through NTSR1 and impacts the prognosis of glioma patients. Mol Cancer 14 (1): 1–13.
Perry RP, Kelley DE (1970) Inhibition of RNA synthesis by actinomycin D: characteristic dose-response of different RNA species. J Cell Physiol 76 (2): 127–139.
Preusser M, Lim M, Hafler DA, Reardon DA, Sampson JH (2015) Prospects of immune checkpoint modulators in the treatment of glioblastoma. Nat Rev Neurol 11 (9): 504–514.
Reed JC (2011) Cancer. Priming cancer cells for death. Science 334 (6059): 1075–1076.
Rottiers V, Najafi-Shoushtari SH, Kristo F, Gurumurthy S, Zhong L, Li Y, Cohen DE, Gerszten RE, Bardeesy N, Mostoslavsky R, Naar AM (2011) MicroRNAs in metabolism and metabolic diseases. Cold Spring Harb Symp Quant Biol 76: 225–233.
Roush S, Slack FJ (2008) The let-7 family of microRNAs. Trends in cell biology 18 (10): 505–516.
Saada S, Marget P, Fauchais AL, Lise MC, Chemin G, Sindou P, Martel C, Delpy L, Vidal E, Jaccard A, Troutaud D, Lalloue F, Jauberteau MO (2012) Differential expression of neurotensin and specific receptors, NTSR1 and NTSR2, in normal and malignant human B lymphocytes. J Immunol 189 (11): 5293–5303.
Seton-Rogers S (2012) MicroRNAs: editing changes the meaning. Nat Rev Cancer 12 (12): 797.
Shimizu S, Tsukada J, Sugimoto T, Kikkawa N, Sasaki K, Chazono H, Hanazawa T, Okamoto Y, Seki N (2008) Identification of a novel therapeutic target for head and neck squamous cell carcinomas: a role for the neurotensin-neurotensin receptor 1 oncogenic signaling pathway. Int J Cancer 123 (8): 1816–1823.
Siegel R, Ma J, Zou Z, Jemal A (2014) Cancer statistics, 2014. CA Cancer J Clin 64 (1): 9–29.
Sturm D, Bender S, Jones DTW, Lichter P, Grill J, Becher O, Hawkins C, Majewski J, Jones C, Costello JF, Iavarone A, Aldape K, Brennan CW, Jabado N, Pfister SM (2014) Paediatric and adult glioblastoma: multiform (epi)genomic culprits emerge. Nat Rev Cancer 14 (2): 92–107.
Su Z, Yang Z, Xu Y, Chen Y, Yu Q (2015) MicroRNAs in apoptosis, autophagy and necroptosis. Oncotarget 6 (11): 8474–8490.
Tacar O, Sriamornsak P, Dass CR (2013) Doxorubicin: an update on anticancer molecular action, toxicity and novel drug delivery systems. J Pharm Pharmacol 65 (2): 157–170.
Tang KH, Ma S, Lee TK, Chan YP, Kwan PS, Tong CM, Ng IO, Man K, To KF, Lai PB, Lo CM, Guan XY, Chan KW (2012) CD133(+) liver tumor-initiating cells promote tumor angiogenesis, growth, and self-renewal through neurotensin/interleukin-8/CXCL1 signaling. Hepatology 55 (3): 807–820.
Tang R, Yang C, Ma X, Wang Y, Luo D, Huang C, Xu Z, Liu P, Yang L (2016) MiR-let-7a inhibits cell proliferation, migration, and invasion by down-regulating PKM2 in gastric cancer. Oncotarget 7 (5): 5972–5984.
Taylor RM, Severns V, Brown DC, Bisoffi M, Sillerud LO (2012) Prostate cancer targeting motifs: expression of alphanu beta3, neurotensin receptor 1, prostate specific membrane antigen, and prostate stem cell antigen in human prostate cancer cell lines and xenografts. Prostate 72 (5): 523–532.
Venkatakrishnan AJ, Deupi X, Lebon G, Tate CG, Schertler GF, Babu MM (2013) Molecular signatures of G-protein-coupled receptors. Nature 494 (7436): 185–194.
Viswanathan SR, Daley GQ, Gregory RI (2008) Selective blockade of microRNA processing by Lin28. Science 320 (5872): 97–100.
Wakabayashi-Nakao K, Maruyama K, Ishii H, Muramatsu K, Hatakeyama K, Ohshima K, Ogura S, Nakajima T, Yamaguchi K, Mochizuki T (2012) Investigation of proNT/NMN secretion from small cell lung carcinoma cells using a mouse xenograft model. Oncol Rep 28 (4): 1181–1186.
Wang X, Jackson LN, Johnson SM, Wang Q, Evers BM (2010) Suppression of neurotensin receptor type 1 expression and function by histone deacetylase inhibitors in human colorectal cancers. Mol Cancer Ther 9 (8): 2389–2398.
Wang XR, Luo H, Li HL, Cao L, Wang XF, Yan W, Wang YY, Zhang JX, Jiang T, Kang CS, Liu N, You YP (2013) Overexpressed let-7a inhibits glioma cell malignancy by directly targeting K-ras, independently of PTEN. Neuro Oncol 15 (11): 1491–1501.
Wei MC, Zong W-X, EH-Y Cheng, Lindsten T, Panoutsakopoulou V, Ross AJ, Roth KA, MacGregor GR, Thompson CB, Korsmeyer SJ (2001) Proapoptotic BAX and BAK: a requisite gateway to mitochondrial dysfunction and death. Science 292 (5517): 727–730.
Winter J, Diederichs S (2013) Argonaute-3 activates the let-7a passenger strand microRNA. RNA Biol 10 (10): 1631–1643.
Zhang Y, Zhu S, Yi L, Liu Y, Cui H (2014) Neurotensin receptor1 antagonist SR48692 reduces proliferation by inducing apoptosis and cell cycle arrest in melanoma cells. Mol Cell Biochem 389 (1-2): 1–8.
Zhou J, Yi L, Ouyang Q, Xu L, Cui H, Xu M (2014) Neurotensin signaling regulates stem-like traits of glioblastoma stem cells through activation of IL-8/CXCR1/STAT3 pathway. Cell Signal 26 (12): 2896–2902.
Zhu H, Shyh-Chang N, Segrè AV, Shinoda G, Shah SP, Einhorn WS, Takeuchi A, Engreitz JM, Hagan JP, Kharas MG (2011) The Lin28/let-7 axis regulates glucose metabolism. Cell 147 (1): 81–94.
Acknowledgements
This work is supported by the National Key Research and Development Program of China (2016YFC1302204), the National Natural Science Foundation of China (81672502, 81602479, 31501100), Chongqing Postdoctoral Science Foundation (No. xm2015026, No. xm2016005) and Fundamental Research Funds for the Central Universities (XDJK2016E015).
Author Contributions
ZD, HC and LY designed the study. DZ, QL, RY, SZ and X-XK performed the experiments and acquired the data. DZ, QL and YL analysed and interpreted the acquired data. ZD, HC and LY participated in discussion and writing of the manuscript.
This work is published under the standard license to publish agreement. After 12 months the work will become freely available and the license terms will switch to a Creative Commons Attribution-NonCommercial-Share Alike 4.0 Unported License.
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Supplementary Information accompanies this paper on British Journal of Cancer website
Supplementary information
Rights and permissions
From twelve months after its original publication, this work is licensed under the Creative Commons Attribution-NonCommercial-Share Alike 4.0 Unported License. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-sa/4.0/
About this article

Cite this article
Dong, Z., Lei, Q., Yang, R. et al. Inhibition of neurotensin receptor 1 induces intrinsic apoptosis via let-7a-3p/Bcl-w axis in glioblastoma. Br J Cancer 116, 1572–1584 (2017). https://doi.org/10.1038/bjc.2017.126
Received:
Revised:
Accepted:
Published:
Issue date:
DOI: https://doi.org/10.1038/bjc.2017.126
Keywords
This article is cited by
-
Neurotensin receptor1 signaling promotes progression of epithelial–mesenchymal transition in mice with chronic rhinosinusitis through Wnt3a/β-catenin pathway
Journal of Molecular Histology (2025)
-
TRIP13 regulates progression of gastric cancer through stabilising the expression of DDX21
Cell Death & Disease (2024)
-
Let-7a-3p overexpression increases chemosensitivity to carmustine and synergistically promotes autophagy and suppresses cell survival in U87MG glioblastoma cancer cells
Naunyn-Schmiedeberg's Archives of Pharmacology (2024)
-
Modulatory effects of miRNAs in doxorubicin resistance: A mechanistic view
Functional & Integrative Genomics (2024)
-
HECTD3 regulates the tumourigenesis of glioblastoma by polyubiquitinating PARP1 and activating EGFR signalling pathway
British Journal of Cancer (2022)
