
Abstract
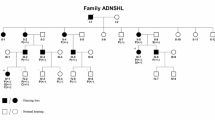
Autosomal dominant sensorineural hearing loss (ADSNHL) is extremely genetically heterogeneous, making it difficult to molecularly diagnose. We identified a multiplex (n=28 affected) family from the genetic isolate of Newfoundland, Canada with variable SNHL and used a targeted sequencing approach based on population-specific alleles in WFS1, TMPRSS3 and PCDH15; recurrent mutations in GJB2 and GJB6; and frequently mutated exons of KCNQ4, COCH and TECTA. We identified a novel, in-frame deletion (c.806_808delCCT: p.S269del) in the voltage-gated potassium channel KCNQ4 (DFNA2), which in silico modeling predicts to disrupt multimerization of KCNQ4 subunits. Surprisingly, 10/23 deaf relatives are non-carriers of p.S269del. Further molecular characterization of the DFNA2 locus in deletion carriers ruled out the possibility of a pathogenic mutation other than p.S269del at the DFNA2A/B locus and linkage analysis showed significant linkage to DFNA2 (maximum LOD=3.3). Further support of genetic heterogeneity in family 2071 was revealed by comparisons of audio profiles between p.S269del carriers and non-carriers suggesting additional and as yet unknown etiologies. We discuss the serious implications that genetic heterogeneity, in this case observed within a single family, has on molecular diagnostics and genetic counseling.
Similar content being viewed by others

Log in or create a free account to read this content
Gain free access to this article, as well as selected content from this journal and more on nature.com
or
References
Raviv D, Dror AA, Avraham KB : Hearing loss: a common disorder caused by many rare alleles. Ann N Y Acad Sci 2010; 1214: 168–179.
Young TL, Ives E, Lynch E et al: Non-syndromic progressive hearing loss DFNA38 is caused by heterozygous missense mutation in the Wolfram syndrome gene WFS1. Hum Mol Genet 2001; 10: 2509–2514.
Ahmed ZM, Li XC, Powell SD et al: Characterization of a new full length TMPRSS3 isoform and identification of mutant alleles responsible for nonsyndromic recessive deafness in Newfoundland and Pakistan. BMC Med Genet 2004; 5: 24.
Doucette L, Merner ND, Cooke S et al: Profound, prelingual nonsyndromic deafness maps to chromosome 10q21 and is caused by a novel missense mutation in the Usher syndrome type IF gene PCDH15. Eur J Hum Genet 2009; 17: 554–564.
Hilgert N, Smith RJ, Van Camp G : Forty-six genes causing nonsyndromic hearing impairment: which ones should be analyzed in DNA diagnostics? Mutat Res 2009; 681: 189–196.
Seary ER, Lynch SMP : Family Names of the Island of Newfoundland, Corrected edn St John’s, Newfoundland and Labrador, Canada: Memorial University of Newfoundland, 1998.
Miller SA, Dykes DD, Polesky HF : A simple salting out procedure for extracting DNA from human nucleated cells. Nucleic Acids Res 1988; 16: 1215.
Merner ND, Hodgkinson KA, Haywood AF et al: Arrhythmogenic right ventricular cardiomyopathy type 5 is a fully penetrant, lethal arrhythmic disorder caused by a missense mutation in the TMEM43 gene. Am J Hum Genet 2008; 82: 809–821.
Cottingham RW, Idury RM, Schaffer AA : Faster sequential genetic linkage computations. Am J Hum Genet 1993; 53: 252–263.
Ma D, Tillman TS, Tang P et al: NMR studies of a channel protein without membranes: structure and dynamics of water-solubilized KcsA. Proc Natl Acad Sci USA 2008; 105: 16537–16542.
Mencia A, Gonzalez-Nieto D, Modamio-Hoybjor S et al: A novel KCNQ4 pore-region mutation (p.G296S) causes deafness by impairing cell-surface channel expression. Hum Genet 2008; 123: 41–53.
Kubisch C, Schroeder BC, Friedrich T et al: KCNQ4, a novel potassium channel expressed in sensory outer hair cells, is mutated in dominant deafness. Cell 1999; 96: 437–446.
Coucke P, Van Camp G, Djoyodiharjo B et al: Linkage of autosomal dominant hearing loss to the short arm of chromosome 1 in two families. N Engl J Med 1994; 331: 425–431.
Kharkovets T, Dedek K, Maier H et al: Mice with altered KCNQ4 K+ channels implicate sensory outer hair cells in human progressive deafness. EMBO J 2006; 25: 642–652.
Hildebrand MS, Tack D, McMordie SJ et al: Audioprofile-directed screening identifies novel mutations in KCNQ4 causing hearing loss at the DFNA2 locus. Genet Med 2008; 10: 797–804.
Talebizadeh Z, Kelley PM, Askew JW, Beisel KW, Smith SD : Novel mutation in the KCNQ4 gene in a large kindred with dominant progressive hearing loss. Hum Mutat 1999; 14: 493–501.
Coucke PJ, Van Hauwe P, Kelley PM et al: Mutations in the KCNQ4 gene are responsible for autosomal dominant deafness in four DFNA2 families. Hum Mol Genet 1999; 8: 1321–1328.
Arnett J, Emery SB, Kim TB et al: Autosomal dominant progressive sensorineural hearing loss due to a novel mutation in the KCNQ4 gene. Arch Otolaryngol Head Neck Surg 2011; 137: 54–59.
Baek JI, Park HJ, Park K et al: Pathogenic effects of a novel mutation (c.664_681del) in KCNQ4 channels associated with auditory pathology. Biochim Biophys Acta 2010; 1812: 536–543.
Kamada F, Kure S, Kudo T et al: A novel KCNQ4 one-base deletion in a large pedigree with hearing loss: implication for the genotype-phenotype correlation. J Hum Genet 2006; 51: 455–460.
Su CC, Yang JJ, Shieh JC, Su MC, Li SY : Identification of novel mutations in the KCNQ4 gene of patients with nonsyndromic deafness from Taiwan. Audiol Neurootol 2007; 12: 20–26.
Topsakal V, Pennings RJ, te Brinke H et al: Phenotype determination guides swift genotyping of a DFNA2/KCNQ4 family with a hot spot mutation (W276S). Otol Neurotol 2005; 26: 52–58.
Van Hauwe P, Coucke PJ, Ensink RJ, Huygen P, Cremers CW, Van Camp G : Mutations in the KCNQ4 K+ channel gene, responsible for autosomal dominant hearing loss, cluster in the channel pore region. Am J Med Genet 2000; 93: 184–187.
del Castillo I, Villamar M, Moreno-Pelayo MA et al: A deletion involving the connexin 30 gene in nonsyndromic hearing impairment. N Engl J Med 2002; 346: 243–249.
Smith RJH, Hildebrand M : DFNA2 Nonsyndromic Hearing Loss, in Pagon RA, Bird TD, Dolan CR, Stephens K, Adam MP, (eds) Seattle, WA, USA: University of Washington, 2011.
Rahman P, Jones A, Curtis J et al: The Newfoundland population: a unique resource for genetic investigation of complex diseases. Hum Mol Genet 2003; 12: (Spec No 2): R167–R172.
Acknowledgements
We thank family members for participating and Drs S Kirby, T Batten and B Fernandez for help with clinical recruitment. This work was supported by grants from the Canadian Institutes of Health Research (CIHR MOP-66974), the Canadian Foundation for Innovation (New Investigator Award no. 9384; Leaders Opportunity Fund no.13120) and Genome Canada (Atlantic Medical Genetics and Genomics Initiative) to TLY. We gratefully acknowledge financial support from the Janeway Children’s Hospital Foundation, Memorial University IRIF and the Government of Newfoundland and Labrador. We also thank the Canadian Hard of Hearing Association-NL chapter for their continued support. TLY is a CIHR salary awardee; NA is a CIHR-RPP fellowship awardee.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Supplementary Information accompanies the paper on European Journal of Human Genetics website
Supplementary information
Rights and permissions
About this article
Cite this article
Abdelfatah, N., McComiskey, D., Doucette, L. et al. Identification of a novel in-frame deletion in KCNQ4 (DFNA2A) and evidence of multiple phenocopies of unknown origin in a family with ADSNHL. Eur J Hum Genet 21, 1112–1119 (2013). https://doi.org/10.1038/ejhg.2013.5
Received:
Revised:
Accepted:
Published:
Issue date:
DOI: https://doi.org/10.1038/ejhg.2013.5
Keywords
This article is cited by
-
Novel KCNQ4 variants in different functional domains confer genotype- and mechanism-based therapeutics in patients with nonsyndromic hearing loss
Experimental & Molecular Medicine (2021)
-
A novel pore-region mutation, c.887G > A (p.G296D) in KCNQ4, causing hearing loss in a Chinese family with autosomal dominant non-syndromic deafness 2
BMC Medical Genetics (2017)
-
A common variant in CLDN14 causes precipitous, prelingual sensorineural hearing loss in multiple families due to founder effect
Human Genetics (2017)
