
Abstract
Purpose: Designating mutations as recessive or dominant is a function of the effect of the mutant allele on the phenotype. Genes in which both classes of mutations are known to exist are particularly interesting to study because these mutations typically define distinct pathogenic mechanisms at the molecular level.
Methods: We studied two consanguineous families with different eye phenotypes and used a combination of candidate gene analysis and homozygosity mapping to identify the underlying genetic defects.
Results: In one family, a novel BFSP2 mutation causes autosomal recessive diffuse cortical cataract with scattered lens opacities, and in another, a novel PITX3 mutation causes an autosomal recessive severe form of anterior segment dysgenesis and microphthalmia.
Conclusion: We show that BFSP2 and PITX3, hitherto known to cause eye defects only in a dominant fashion, can also present recessively. The likely null nature of both mutations and lack of manifestation in heterozygotes strongly argues for a mechanism other than loss of function in the previously reported dominant mutations in these two genes. Thus, study of consanguineous populations has the additional advantage of not only identifying novel recessive genes but also defining the mutational mechanism of dominant disorders.
Similar content being viewed by others


Main
The gene and disease paradigm has benefited greatly from the expanding compendium of mutations and their effect on health. Key to the mechanistic understanding of this link between mutations and the diseases they cause is the study of the molecular consequences of these mutations. Although an overall pattern can be appreciated in the mutational behavior of many genes, e.g., mutations in enzyme-coding genes tend to behave recessively whereas multimeric structural protein-coding genes tend to behave dominantly, there are instances in which individual genes can harbor both classes of mutations. These genes are particularly interesting to study because the distinct pathogenic mechanisms for recessive and dominant mutations often provide new insights into the molecular pathogenesis of the mutation and the associated phenotype.1
We have shown that for genetically heterogeneous disorders that can be caused by both dominant and recessive mutations, recessive mutations are typically overrepresented in consanguineous populations.2 However, we share here an emerging theme wherein even for genes in which only dominant mutations are known to exist, consanguineous populations can reveal the presence of recessively acting mutations which adds a new dimension to the mechanistic characterization of the molecular phenotype associated with these genes.
MATERIALS AND METHODS
Patients were evaluated ophthalmologically by one of the authors (AOK). Written informed consent was obtained to recruit patients (RAC #2070023). Sequencing of candidate genes with or without homozygosity mapping was done as dictated by the family history. Homozygosity mapping was performed by Axiom CEU Human Array (Affymetrix) genotyping according to the manufacturer's protocol followed by analysis for runs of homozygosity using autoSNPa as described before.3
RESULTS
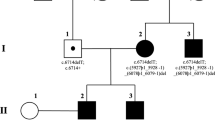
The pedigrees of both study families are shown in Figure 1. Family 1 consists of healthy first cousin parents and one daughter with bilateral severe congenital microphthalmia and a very severe form of anterior segment dysgenesis (ASD) best described as sclerocornea (Fig. 2). An affected cousin was unavailable. Family 2 consists of healthy first cousin parents and three daughters with juvenile-onset diffuse cortical cataract with scattered lens opacities (age of symptoms approximately 12 years of age for each; Fig. 1). The father had had bilateral cataract surgery for posterior subcapsular cataract at 44 years of age. The 50-year-old mother and six other children were unaffected.

Pedigrees of Family 1 and Family 2. Affected cousins (by history) in both families were unavailable.

A, Orbital CT shows microphthalmia in patient in Family 1. B, Slit-lamp examination of one of the patients in Family 2 showing cortical cataract with scattered lens opacities. C and D, Sequence chromatogram showing mutations in PITX3 and BFSP2, respectively.
For Family 1, we sequenced a panel of genes known to cause ASD (CYP1B1, PAX6, PITX2, and PITX3) and identified a homozygous mutation in PITX3 c.640_656del (p.(Ala214ArgfsX42)) (Fig. 1). For Family 2, we performed homozygosity scan and identified a single run of a homozygosity shared between all affected patients. This is a known dominant cataract locus that is linked to BFSP2 mutations. Surprisingly, we identified a homozygous BFSP2 mutation c.598_599dup (p.(Ala201ArgfsX19)) (Fig. 1). Both PITX3 and BFSP2 mutations were present in a heterozygous state in the unaffected parents. The BFSP2 mutation was also identified in three of the six healthy children.
DISCUSSION
We report the identification of the first recessive mutations in two key regulators of lens development in humans: BFSP2 and PITX3. BFSP2 encodes CP49, an intermediate filament (IF) protein that is lens-specific in its expression.4 Like other IF proteins in the lens, CP49 plays a critical role in the highly orchestrated organization of the cytoskeleton of the differentiated fiber cells.5 The hexagonal architecture of these fiber cells provides a remarkably efficient packaging mechanism that reduces intercellular spaces which is highly suited for the strict optical properties of the lens.6 CP49 is an atypical IF protein, however, in that it lack the C-tail seen in other IF.7 It does not form homopolymers but rather heterodimerizes with BFSP1, another lens-specific IF protein.8 Mutations of both BFSP1 and BFSP2 are known to cause cataract in humans.
Three dominant mutations have been reported to date in BFSP2, all involving single amino acids (two missense and one in-frame deletion).9–11 The resulting phenotype was a cataract phenotype (juvenile and congenital) that is comparable with the one reported here, but the pathogenic mechanism of those dominant mutations remains unknown. The involvement of single amino acids makes it tantalizingly plausible that the intact mutant protein acts in a dominant negative fashion by interfering with the assembly of the wild-type protein, particularly when one considers that all three mutations affect the rod domain which is required for the beaded filament assembly. The mutation we report here is almost certainly null because the mutant protein lacks most of the Coil1b domain and the entire rod domain and the downstream Coil-2 domain.12 Therefore, we hypothesize that this mutant protein that completely lacks the ability to assemble or polymerize is unlikely to interfere with the wild-type protein. This is likely tolerated by the lens fiber cells and may explain the lack of phenotypic consequences in the heterozygotes in this family (mother, grandparents, and three healthy children); however, we cannot exclude that early onset senile cataract in the father, although different in morphology, may have been influenced by his carrier status. Our hypothesis is also supported by the lack of phenotype in the mice that are heterozygous for complete null alleles.13 Furthermore, the only human mutation to date in the closest relative of BFSP2, BFSP1, is also a null mutation that causes cataract only recessively.14
Similarly, PITX3, which is known for its pivotal role in the normal formation of the lens vesicle and separation from the lens ectoderm, is only known to cause ASD dominantly in humans. Three mutations have been reported to date in this transcription factor, but the pathogenic mechanism of the mutation is still unclear.15,16 For instance, one recent study of two of the three dominant human mutations showed that the mutant PITX3 retains the capacity to localize to the nucleus and bind to the consensus promoter sequence.17 However, it seems that mutant PITX3 was unable to form homo or heterodimers. This, in addition to the documented reduced transcriptional activation by these mutant proteins (40–70%), makes it tempting to speculate a dominant negative mechanism in which a PITX3 protein with reduced activation/transactivation capacity occupies the site of action of the normal counterpart.17 The novel PITX3 mutation we report here involves deletion of the same 17 bp that are duplicated in a recurrent dominant mutation. It seems that both our deletion and the previously reported duplication are mediated by the presence of an 11 bp repeat.15 We note here that our recessive mutation results in a more severe phenotype (sclerocornea and microphthalmia) compared with the cataract/ASD phenotype described with dominant mutations. Why deletion should act recessively while a duplication should act dominantly is unclear but we note the two frameshifting events create different novel amino acids for varying lengths at the C-terminus. PITX3 encodes a 302 aa protein and comprises four exons with the third exon being the last coding exon. The previously reported 17 bp duplication results in a frameshift in codon 220 and produces an aberrant protein consisting of 94 additional residues, i.e., the novel stop codon is introduced downstream to the original stop codon so nonsense-mediated decay is unlikely to be triggered. Similarly, our 17 bp deletion results in a frameshift in codon 220, introduces 40 novel residues followed by a stop. Because this premature stop codon is also in exon 3 which is the last coding exon, NMD would also be unlikely. Lack of any phenotype in the carrier parents of this patient makes it highly unlikely that previously reported mutations are simple loss of function. This may also explain why mice heterozygous for null Pitx3 were phenotypically normal compared with homozygotes.18 We posit, therefore, that at least some instances of dominant human diseases which can only be recapitulated in homozygous mouse models can be explained by different mutational mechanisms between human and the mouse model rather than by difference in redundancy level between species.
Consanguineous populations have been invaluable to the identification of recessively acting disease genes. In this study, and in another where we described the first recessively acting CRYAB mutation,19 we demonstrate the potential of these populations in improving our understanding the molecular mechanism of dominant disorders, an important step toward the design of molecular therapeutics.
REFERENCES
Antonarakis SE, Cooper DN . Human gene mutations: mechanisms and consequences, 4th ed. Berlin: Springer, 2010.
Aldahmesh MA, Safieh LA, Alkuraya H, et al. Molecular characterization of retinitis pigmentosa in Saudi Arabia. Mol Vis 2009; 15: 2464–2469.
Carr IM, Flintoff KJ, Taylor GR, Markham AF, Bonthron DT . Interactive visual analysis of SNP data for rapid autozygosity mapping in consanguineous families. Hum Mutat 2006; 27: 1041–1046.
Georgatos SD, Gounari F, Remington S . The beaded intermediate filaments and their potential functions in eye lens. Bioessays 1994; 16: 413–418.
Perng MD, Zhang Q, Quinlan RA . Insights into the beaded filament of the eye lens. Exp Cell Res 2007; 313: 2180–2188.
Song S, Landsbury A, Dahm R, Liu Y, Zhang Q, Quinlan RA . Functions of the intermediate filament cytoskeleton in the eye lens. J Clin Invest 2009; 119: 1837–1848.
Perng MD, Quinlan RA . Seeing is believing! The optical properties of the eye lens are dependent upon a functional intermediate filament cytoskeleton. Exp Cell Res 2005; 305: 1–9.
Carter JM, Hutcheson AM, Quinlan RA . In vitro studies on the assembly properties of the lens proteins CP49, CP115: coassembly with alpha-crystallin but not with vimentin. Exp Eye Res 1995; 60: 181–192.
Conley YP, Erturk D, Keverline A, et al. A juvenile-onset, progressive cataract locus on chromosome 3q21-q22 is associated with a missense mutation in the beaded filament structural protein-2. Am J Hum Genet 2000; 66: 1426–1431.
Ma X, Li FF, Wang SZ, Gao C, Zhang M, Zhu SQ . A new mutation in BFSP2 (G1091A) causes autosomal dominant congenital lamellar cataracts. Mol Vis 2008; 14: 1906–1911.
Jakobs PM, Hess JF, FitzGerald PG, Kramer P, Weleber RG, Litt M . Autosomal-dominant congenital cataract associated with a deletion mutation in the human beaded filament protein gene BFSP2. Am J Hum Genet 2000; 66: 1432–1436.
Merdes A, Gounari F, Georgatos SD . The 47-kD lens-specific protein phakinin is a tailless intermediate filament protein and an assembly partner of filensin. J Cell Biol 1993; 123: 1507–1516.
Sandilands A, Prescott AR, Wegener A, et al. Knockout of the intermediate filament protein CP49 destabilises the lens fibre cell cytoskeleton and decreases lens optical quality, but does not induce cataract. Exp Eye Res 2003; 76: 385–391.
Ramachandran RD, Perumalsamy V, Hejtmancik JF . Autosomal recessive juvenile onset cataract associated with mutation in BFSP1. Hum Genet 2007; 121: 475–482.
Semina EV, Ferrell RE, Mintz-Hittner HA, et al. A novel homeobox gene PITX3 is mutated in families with autosomal-dominant cataracts and ASMD. Nat Genet 1998; 19: 167–170.
Berry V, Yang Z, Addison PK, et al. Recurrent 17 bp duplication in PITX3 is primarily associated with posterior polar cataract (CPP4). J Med Genet 2004; 41: e109.
Sakazume S, Sorokina E, Iwamoto Y, Semina EV . Functional analysis of human mutations in homeodomain transcription factor PITX3. BMC Mol Biol 2007; 8: 84.
Ho HY, Chang KH, Nichols J, Li M . Homeodomain protein Pitx3 maintains the mitotic activity of lens epithelial cells. Mech Dev 2009; 126: 18–29.
Safieh LA, Khan AO, Alkuraya FS . Identification of a novel CRYAB mutation associated with autosomal recessive juvenile cataract in a Saudi family. Mol Vis 2009; 15: 980–984.
Acknowledgements
This study was funded by a grant from KACST (08-MED497-20) and a Collaborative Research Grant from DHFMR to FSA. We sincerely thank the patients and their families for their enthusiastic participation.
Author information
Authors and Affiliations
Corresponding author
Additional information
Disclosure: The authors declare no conflict of interest.
Rights and permissions
About this article
Cite this article
Aldahmesh, M., Khan, A., Mohamed, J. et al. Novel recessive BFSP2 and PITX3 mutations: Insights into mutational mechanisms from consanguineous populations. Genet Med 13, 978–981 (2011). https://doi.org/10.1097/GIM.0b013e31822623d5
Received:
Accepted:
Published:
Issue date:
DOI: https://doi.org/10.1097/GIM.0b013e31822623d5
Keywords
This article is cited by
-
Unilateral buphthalmos, corneal staphyloma and corneal fistula caused by pathogenic variant in the PITX3 gene: a case report
BMC Ophthalmology (2022)
-
Variants in PAX6, PITX3 and HSF4 causing autosomal dominant congenital cataracts
Eye (2022)
-
Genetics of anophthalmia and microphthalmia. Part 1: Non-syndromic anophthalmia/microphthalmia
Human Genetics (2019)
-
Novel phenotypes and loci identified through clinical genomics approaches to pediatric cataract
Human Genetics (2017)
-
Expanding the genetic heterogeneity of intellectual disability
Human Genetics (2017)
