
Abstract
Melanoma may be difficult to identify histologically and relatively high rates of misdiagnosis leads to many malpractice claims. Currently separation of melanomas from nevi is based primarily on light microscopic interpretation of hematoxylin and eosin stained sections with limited assistance from immunohistology. To increase the accuracy of discrimination of benign and malignant melanocytic lesions we identified DNA microarray-derived gene expression profiles of different melanocytic lesions and evaluated the performance of these gene signatures as molecular diagnostic tools in the molecular classification and separation of melanomas and nevi. Melanocyte-derived cells were isolated by laser capture microdissection from 165 formalin-fixed and paraffin-embedded melanocytic nevi and melanoma tissue sections. RNA was isolated, amplified, labeled, and hybridized to a custom DNA microarray. In all 120 samples were used to identify differentially expressed genes and generate a gene expression classifier capable of distinguishing between melanomas and nevi. These classifiers were tested by the leave-one-out method and in a blinded study. RT-PCR verified the results. Unsupervised hierarchical clustering identified two distinct lesional groups that closely correlated with the histopathologically identified melanomas and nevi. Analysis of gene expression levels identified 36 significant differentially expressed genes. In comparison with nevi, melanomas expressed higher levels of genes promoting signal transduction, transcription, and cell growth. In contrast, expression of L1CAM (homolog) was reduced in melanomas relative to nevi. Genes differentially expressed in melanomas and nevi, on the basis of molecular signal, sub classified a group of unknown melanocytic lesions as melanomas or nevi and had high concordance rates with histopathology. Gene signatures established using DNA microarray gene expression profiling can distinguish melanomas from nevi, indicating the feasibility of using molecular classification as a supplement to standard histology. Our successful use of a standard formalin-fixed and paraffin-embedded tissue further supports the practicability of combining molecular diagnostic testing with histopathology in evaluation of difficult melanocytic lesions.
Similar content being viewed by others


Main
Melanoma is the most fatal cutaneous neoplasm. Surgical resection is curative early in the disease, but chemotherapy, biotherapy and immunotherapy remain ineffective for more advanced melanoma, mean survival from first detection of metastases being only 6–9 months.1 Accurate pathologic identification of the primary melanoma is critical for optimal clinical management. Unfortunately, diagnosis by standard histologic criteria can be very difficult and distinction of a melanoma from a benign nevus may be impossible.
Misdiagnosis of melanoma, mainly under-diagnosis, constitutes 13% of total medical malpractice lawsuits; the second largest group of malpractice claims. Recurrent problems include melanoma misdiagnosed as a melanocytic nevus (without disclosure of diagnostic doubt), chronically inflamed nevus, spitz's nevus, and dysplastic nevus/atypical mole.2 These are lesions that are often ‘difficult to diagnose’ and interpretation is subjective leading to a range of diagnoses from different pathologists. In a study where 11 expert pathologists reviewed 37 ‘classic’ melanocytic lesions there was total agreement in only 30% of cases.3 These high rates of misdiagnosis and the possibility that they may contribute to an unfavorable outcome are of great concern to dermatologists and dermatopathologists. With advances in biomedical technology and the completion of the human genome project, the possibility of genome-based ancillary testing is of great interest.
Functional genomics also known as transcriptomics (permitting simultaneous analysis of the expression of thousands of genes) exploded with the development of DNA microarray systems in the mid 1990s.4, 5 These systems have been used to find disease-associated biomarkers, classify tumors, distinguish tumor subclasses, and predict outcome and response to chemotherapy.6 They provide high specificity based on the binding of DNA and RNA to complimentary sequences 7 and it is appropriate to consider their role in the future practice of diagnostic dermatopathology. The DNA microarray technique has been used to study molecular changes in breast carcinoma, lung carcinoma, rhabdomyosarcoma, glioblastoma, and melanoma. Initial DNA microarray studies of melanoma evaluated tumor cell lines and frozen tissues from melanocytic lesions.8 Bittner et al.9 were the first group to use molecular classification of frozen tissues to distinguish the metastatic and non-metastatic phenotypes. The classification strategy is to develop a DNA microarray classifier and then generalize to predict the class of members of a new and unknown sample by comparing it to the classifier.8 Becker et al.10 used cDNA array hybridization of frozen tissue specimens to classify and distinguish melanomas and nevi. Their specimens included lesional cells of interest isolated by laser capture microdissection; drastically minimizing contaminating of non-melanocytic cells, such as inflammatory cells, keratinocytes, histiocytes, and vascular endothelial cells. Additional studies, using DNA microarrays prepared from frozen tissues have also reported that melanomas, nevi, and normal melanocytes have separate gene profiles.11, 12, 13
Studies of cell lines and fresh frozen tissues suggest that it will be possible to distinguish the molecular changes associated with melanoma cells from those seen in nevocytes and melanocytes. Molecular classifiers may also be able to predict metastatic potential and clinical outcome. We do not know of any reports of DNA microarray studies that used formalin-fixed and paraffin-embedded tissues. Quantitative gene expression studies have, however, been successfully performed using laser-captured formalin-fixed and paraffin-embedded tissue in real time RT-PCR studies.14, 15 Given that RNA quantitative studies have been successfully undertaken using formalin-fixed and paraffin-embedded tissue, their use for quantitative analysis of the DNA microarray system is of great interest, as tissue archives can provide a vast resource of desirable samples.
This report discusses our use of the DNA microarray expression profiling system in studies of primary cutaneous melanomas and benign nevi using formalin-fixed and paraffin-embedded tissues with the eventual aim of applying these approaches to an analysis of difficult melanocytic lesions. Class comparison, development of gene signatures, and the predictive molecular classification of melanomas and nevi were investigated. In addition, genes that were differentially expressed between melanomas and nevi were analyzed. Results were verified by quantitative RT-PCR. Our results indicate that DNA microarray technology is a potentially valuable molecular diagnostic tool that may be of assistance in determining the true nature of melanocytic lesions that are difficult to diagnose by light microscopy.
Materials and methods
Case Selection Method
Cases were selected from two independent archival sources (UCLA Department of Pathology and Laboratory Medicine and Pathology Inc., Torrance). Formalin-fixed and paraffin-embedded tissue was selected from procedures performed within 3 years of our study with an average storage time of 1 year. Each case was independently reviewed by 2–4 different pathologists from two separate institutions. Morphologic diagnostic criteria for nevi were architectural symmetry, nested proliferation, maturation, small nuclei, no distinct eosinophilic nucleoli, and lack of architectural and cytologic atypia. Features of melanoma were asymmetry, pagetoid spread, radial growth, lack of maturation, lentiginous growth, clefting, loss of nested growth, ulceration, regression, sun damage, necrosis, mitoses, and atypical mitoses, prominent eosinophilic nucleoli, and dusty cytoplasm. Cases were divided into two separate groups of 136 and 50. From the 136 set group, an initial 86-sample subset was used for unsupervised hierarchical clustering. Subsequently 120 samples from the same 136 set group: 62 primary melanomas (superficial spreading, desmoplastic, nodular, acral-lentiginous, and metastatic) and 58 nevi (common acquired and congenital type, no Spitz nevi were included), were selected for classification analysis. Sixteen cases were omitted from the classification study because they had insufficient tissue available for analysis. Of the 50 set group, 26 primary melanomas and 19 nevi (common acquired and congenital type nevi) were selected for blinded validation studies. Five cases were omitted because they had an insufficient tissue for the planned analysis.
Laser Capture Microdissection and RNA Isolation
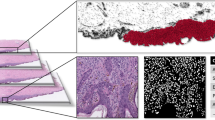
Sections of each sample (8 μm thick) were mounted on both positively charged glass microscope slides (Richard Allen Bond-Rite) for pathological examination and membrane slides (Molecular Machines Industries) for laser capture microdissection respectively (Figure 1). Glass and membrane slides were hematoxylin- and eosin-stained utilizing standard protocols. Laser capture microdissection was performed on either a Molecular Machines Industries or Palm system. The number of nevus cells or tumor cells varied with sample type size and cellularity.

Overall sample processing scheme. Sections (8 μm) of each sample were mounted on the microscope and membrane slides. Laser-captured microdissection was performed and melanocytes isolated. RNA was extracted and samples with greater than 1 ng of RNA were amplified. Patient sample RNA was labeled with Cy-5 and Universal Human Reference RNA was labeled with Cy-3. DNA microarray hybridizations were conducted and data were analyzed.
RNA was extracted from laser capture microdissection captured samples by incubating in 100 μl Proteinase K buffer plus 10 μl Proteinase K (Sigma) at 55°C for 16 h. The remaining steps of RNA purification were conducted by following the Qiagen RNeasy Mini Kit protocol. RNA yield was assessed by measurement of the ACTB gene by quantitative RT-PCR (see Quantitative PCR section). Samples with >1 ng of RNA were selected for amplification.
RNA Amplification
Between 1 ng and 100 ng of total RNA was used as a template for amplification with ‘RampUP™ ‘(Genisphere). This method utilizes two rounds of in vitro transcription with the T7 and T3 RNA polymerases. In the second round of amplification, UTP was substituted with 5-(3-aminoallyl-) UTP (Epicentre) for indirect labeling. Following amplification cleanup, RNA was re-suspended in 10 μl 0.1 M NaHCO3. Here, 10μl of DMSO reconstituted Cy-3 or Cy-5 dye (Amersham) was added to each RNA sample and incubated at 37°C in the dark for 60 min. Labeled samples were purified by RNeasy Mini Kit. Spectrophotometric absorbance readings were obtained at 260, 280, 532, and 650 nm.
DNA Microarray Synthesis
Melanoma Gene Expression Microarrays were developed and synthesized utilizing the Combimatrix 12 000 feature CustomArray platform. These chips utilize silicon chip technology and are instantly customizable utilizing in situ synthesis technology. Each array was designed with over 12 000 individual features representing over 1100 unique genes identified to be associated with melanoma, other tumors, and control genes. Probes were designed utilizing the proprietary Combimatrix probe design software and designed with an average length of 35 bases.
DNA Microarray Hybridizations
DNA microarray hybridizations utilized a two color methodology in which the patient sample was labeled with Cy-5 and Universal Human Reference RNA (Stratagene CAT no. 740 000) was labeled with Cy-3. To normalize for variations in sample to sample labeling efficiencies, DNA microarray hybridizations were conducted by mixing the appropriate volume of Cy-5-labeled sample and Cy-3-labeled Universal RNA control such that vol=200/A650 or vol=200/A550, respectively. This mix was dried and fragmented to an average size of 50–100 bases by incubating in 10 μl fragmentation buffer (10 mM ZnCl2, 10 mM Tris-HCl pH 7) at 60°C for 30 min. DNA microarray hybridizations and washes were then conducted according to the manufacturer's instructions with the exception that hybridizations were conducted at 45°C.
Data Analysis
Hybridized arrays were imaged using an Axon Genepix 4200AL scanner. Feature data was extracted utilizing the Combimatrix software ‘Microarray Imager’. Data was imported into ‘GeneMaths’ software for normalization and analysis. For each probe, the Cy3 (reference) and Cy5 (sample) signal intensities were log2 transformed and from these data a (Cy5/Cy3) ratio was calculated. These ratios were then median and s.d. normalized such that the median signal intensity of all probes was equal to zero and the s.d. of all probe signal intensities was equal to one.
For classifier generation, normalized ratio data for all arrays (120) was subjected to a 2-tailed t-test to identify the top 100 probes which displayed the most significant gene expression differences between malignant melanoma and nevi specimens. Normalized data for the top 100 probes, representing 36 unique genes, were utilized to generate a classifier based on a Support Vector Machine Training algorithm embedded in the ‘GeneMaths’ software.
Unknown samples were classified utilizing the classifier described above. Samples were normalized as described above, compared to the previously described classifier, and categorized as a ‘Best Match’ to the melanoma or nevus gene expression signatures.
Quantitative PCR
Taqman q-PCR probes were designed with the aid of ‘Primer 3’ software specific for the mRNA sequences for FABP7, PCNA, L1CAM homolog (NCBI accession number BC104918), and ACTB. To minimize the effect of variability in RNA degradation, PCR amplicons for each gene were kept constant at 71 to 74 bases. A one step RT-qPCR was performed with the addition of reverse transcriptase (Stratagene Catalog no. 600085) to Taqman 2 × Universal PCR mix (Applied Biosystems Catalog no. 4324018), gene specific primers, probe, and sample. The reaction was performed on an ABI 7500 instrument according to the manufacturer's instructions. For each sample, the gene expression levels of FABP7, PCNA, and L1CAM homolog were normalized relative to the expression level of ACTB (GeneX/ACTB). These normalized expression levels were then converted to a percentage of the highest expression level observed among the six patient samples tested.
For comparison to quantitative RT-PCR data, DNA microarray data was normalized according to the previously described method for microarray data. The normalized expression levels for the three genes of interest were then converted to a percentage of the highest expression level observed among the six patient samples tested.
Results
Unsupervised Hierarchical Clustering of Melanocytic Lesions Identified Distinctive Molecular Phenotypes that Distinguish Melanomas and Benign Nevi
To demonstrate distinctive genetic phenotypes and generate gene expression signature profiles for primary cutaneous melanoma and benign nevi, we utilized a two-channel hybridization oligonucleotide array representing 1100 unique genes of interest with high relevance to cancer biology. Isolated specimen RNA and target preparations were derived from formalin-fixed and paraffin-embedded tissue. The first 86 melanocytic lesions sampled comprised 38 primary cutaneous melanomas and 48 nevi. Following image analysis, summarization and data normalization, we selected 100 top probes on the basis of their demonstrated significance and these were retained for comparative analysis (see next section and Figure 3a). Unsupervised hierarchical clustering resulted in distinctive groups determined by molecular phenotype respectively comprised of melanomas and benign nevi (Figure 2). The cluster on the left contain samples that are virtually all (36/38–95%) from histopathologically diagnosed primary cutaneous melanomas and that on the right contain a high preponderance (47/48–98%) of samples from histopathologically diagnosed benign nevi. Of the 38 samples that clustered left, that is, the gene expression profile-determined melanoma cluster, the two discrepant samples corresponded to lesions diagnosed respectively on histology as a ‘dysplastic’ nevus (with mild atypia) and a compound nevus. The single discrepant sample in the gene expression profile-determined nevic cluster was diagnosed as a melanoma on histologic examination. This represents gene microarray/histopathology concordance of 95% for melanomas and 98% for nevi. Although correlation was not quite 100% these data strongly support the existence of distinctive and separate gene expression profiles in melanomas and nevi, suggesting that these two classes of melanocytic lesions, even if fixed and mounted in paraffin blocks can be classified and separated on the basis of their genetic phenotypes.

Unsupervised hierarchical clustering. Initial 86 melanocytic lesions resulted in two distinct tight clusters of 38 specimens (left) and 48 specimens (right), delineated by a black vertical line. Each row represents a single probe and each column represents a melanocytic lesion. The colored bar just above the heat map represents each melanocytic lesion with a histopathologic diagnosis of melanoma (red box) or nevus (green box). Relative expressions of mRNA in the lesion are color represented: red is higher level expression, green is lower level expression, and black is no change.
Differentially Expressed Genes that Separately and Reliably Identify Melanoma and Nevi: Validation of DNA Microarray Expression Profiling by Quantitative RT-PCR
Significance of the microarray results using t-test and data analysis permitted the identification of the 100 top probes representing 36 unique genes that we retained for subsequent comparative analysis and development of distinctive gene signatures for melanoma and nevi. Each selected probe had a P-value at least P<2.42 × 10−6 The 14 most statistically significant genes (at least P<8.2 × 10−8) demonstrating significant differential expression in melanoma relative to nevi are represented in Figure 3a. Relative to gene expression in nevi, melanomas had higher expression of PHACTR1, HLA-B, PRAME, STAT1, LCP2, and HLA-A. In contrast, melanoma had a lower expression of PTN, L1CAM homolog, GPX3, FABP7, RPL12, DLC1, NACA, and GSTM2. A review of the literature demonstrated prior reports of similar findings; increased relative expression levels of PHACTR1, PRAME, and STAT1; and decreased expression of GPX3, FABP7 and DLC1 in melanomas relative to nevi. A few discrepancies were observed (far right column) in which previous reports12, 13 have shown an increased expression of L1CAM and no change in levels of the HLA MHC class I expression whereas our findings show a decreased expression of L1CAM (homolog) and an increased expression of HLA MHC class I in melanoma relative nevi.

Identification of genes predictive of melanoma. (a) Within the 100 probe/36 genes selected for gene array analysis, 14 individual gene expression results comparing levels in melanoma relative to nevi are represented. The 14 represented genes are those with the most significant P-values. Comparative expression of some genes has been previously performed by various molecular biology techniques (last column) and is consistent with the results from the DNA microarray. ‘Up’ represents higher level of gene expression and ‘down’ represents lower level of gene expression in melanoma compared with nevi. L1CAM probe in this study was derived from a L1CAM homolog. L1CAM expression results from literature tabulated on the right column represent L1CAM and not the L1CAM homolog. (b) Quantitative polymerase chain reaction (Q-PCR) was performed from three random melanoma (M1-M3) and nevi (N1-N3) samples with primers specific for FABP7, L1CAM homolog, and PCNA to verify the DNA microarray results. Relative signals indicating gene expression levels are measured on the y axis. Expression level tested by DNA microarray (left column) was comparable to quantitative RT-PCR (right column).
To verify the DNA microarray measurements, three representative genes subjected to expression profiling were further evaluated using quantitative RT-PCR. Expression of genes for FABP7, PCNA, and L1CAM homolog in three representative melanomas and nevi are presented as DNA microarray-derived relative signals (Figure 3b). Results from the same samples evaluated by quantitative RT-PCR are presented in parallel. From the microarray and Q-PCR results the three melanoma samples expressed less FABP7 and L1CAM homolog than the three nevus samples. However, the melanoma samples expressed more PCNA, a DNA polymerase cofactor involved in cell replication. These two independent methods showed a high degree of correlation. The results from the quantitative RT-PCR studies thus confirm the accuracy of the DNA microarray measurements.
Validation: Assessment of Assay Concordance
Ideally a large set of samples (N) with a high power value is required to develop and validate a classification. In studies where there is a high power value, an independent test set can be used for validation. In practice the number of samples available is usually limited, requiring the use of alternative methods to form independent test sets that may be used for building and training an algorithm. Cross-validation using the ‘leave-one-out’ approach is currently regarded as the simplest approach for studies of limited sample size.6 In this investigation, we undertook a ‘leave-one-out’ cross-validation study of 120 samples of melanomas and nevi (Figure 4a). A total of 120 samples (N) was analyzed (62 histologically identified primary cutaneous melanomas and 58 benign nevi). Genes that were differentially expressed at a statistically significantly level (at least P<2.42 × 10−6) were selected as a 100 probe classifier and predictor for melanoma or nevi. For the cross-validation study, a classifier was developed when one sample (k) was omitted from the 120 total samples (N-k=119). This 119 sample classifier (N-k) was applied to the single omitted sample (k) as a test set and predicted the molecular class of k as melanoma or nevus. This process was repeated omitting a new sample (k) and a further new classification utilized. The concordance rate between gene classification and histopathologic diagnosis for all samples was 89%. Concordance rates for melanoma and nevi were 90 and 86%, respectively (Figure 4a).

Validation and assessment of assay concordance. (a) Results of ‘leave-one-out’ cross-validation studies are tabulated. One sample (k) of a collection of 120 samples (N) was left out and a classifier was developed using the remaining (N-k) samples. After each sample was analyzed through the cross-validation study, percent concordance with histopathologic diagnosis was calculated for all samples and also subdivided into melanoma and nevi. (b) Blinded validation results showing percent concordance of 45 new samples applied to the 120 sample classifier are as tabulated.
To further assess concordance, 45 histologically diagnosed primary cutaneous melanoma and benign nevus samples were applied ‘blind’ to the 120 case-derived classifier. These 45 samples were separate from the cases used to develop the 120-sample classifier. Comparison of classification by gene array with histopathologic diagnosis showed overall concordance of 89% for all blinded samples, 96% for melanomas, and 79% for nevi (Figure 4b).
Discussion
We believe that this is the first study to utilize laser capture microdissected formalin-fixed and paraffin-embedded tissues to analyze melanocytic lesions by DNA microarray. Previous DNA microarray studies of melanocytic lesions used frozen tissue and did not employ laser capture microdissection.9, 10, 11, 12, 13, 16 In this study, arrays were processed in two groups; the initial 120 cases and a ‘blind’ group of 45. As slight variations in array data between different lots can artificially bias the results of an unsupervised clustering study, tissues from primary cutaneous melanomas and nevi from the first 86-sample subset were subjected to unsupervised hierarchical clustering which identified two separate and distinctive groups of lesions. Many genes were significantly and differentially expressed either in primary melanomas or nevi. Distinct molecular signatures derived from gene expression profiling identified melanocytic lesions as melanomas or nevi with high accuracy. Signatures were originally created on the basis of a 120 sample pool of primary melanomas and nevi. A ‘leave-one-out’ validation study showed an 89% overall concordance with expert histopathologic diagnosis made independently by 2–4 reviewing pathologists from two separate institutions. Classification of an additional 45 ‘blinded’ samples also demonstrated an overall concordance rate of 89%. These promising results suggest that our classification rule may be generalizable and useful in the assignment of new ‘unknown’ samples to melanoma or nevus categories. There may be an adjunctive role for molecular diagnostic tools in diagnostic dermatopathology (and potentially in other areas of surgical pathology). The capacity to use formalin-fixed and paraffin-embedded tissues certainly facilitates movement of these novel approaches towards practical application.
Molecular testing may not be necessary for the many melanocytic lesions that are diagnosed with relative ease by microscopic examination of standard hematoxylin- and eosin-stained sections. The availability of a confirmatory adjunctive test is, however, extremely appealing in assessment of difficult melanocytic lesions where interpretation of morphology is challenging. Unfortunately, the level of interpretative difficulty of a lesion is not usually apparent until it is examined microscopically, by which time fresh tissue is not available. Freezing or reserving fresh portions of melanocytic lesions prior to histopathologic analysis is neither customary nor particularly practical, would add significant cost and poses administrative problems. Thus most tumors will not be evaluable by molecular testing unless such testing can use formalin-fixed and paraffin-embedded material. Because of variations in the quality and quantity of extracted RNA, previous attempts to evaluate formalin-fixed and paraffin-embedded tissues have not produced acceptable results. The development of techniques to extract adequate RNA from formalin-fixed and paraffin-embedded tissue as a basis for production of quality DNA microarray data would eliminate the need for fresh/frozen specimens and make adjunctive molecular testing a more practical diagnostic option. However, our study is limited to formalin-fixed and paraffin-embedded tissue evaluated within 3 years of its removal from the patient, with an average ‘delay’ of 1 year. Advances in biotechnology, have lead to recent studies that yielded high quality data from formalin-fixed and paraffin-embedded tissues, data that are reproducible and precise, and comparable to data obtained from frozen specimens.14 These advances derive from determination of optimized reagent conditions and (particularly) the use of random hexamers (rather than traditional poly-T oligomers) to prime the reverse transcription step (as performed in RampUP). The rationale for utilizing random hexamers comes from observations showing loss of the poly-T site due to increased susceptibility to degradation of RNA of the poly-A tail in extracted formalin-fixed and paraffin-embedded tissue.15 Failure of reverse transcription and diminished successful recovery of RNA is regarded as secondary to loss of the priming site for poly-T. Random hexamers appear to facilitate successful RNA recovery both qualitatively and quantitatively. Adequate RNA extraction from formalin-fixed and paraffin-embedded tissue that facilitates successful subsequent gene expression analysis (including DNA microarray studies) is critical in our successful analysis of the DNA expression profiles of melanocytic lesions from formalin-fixed and paraffin-embedded tissue. To confirm the accuracy of our DNA microarray studies and the quality of the RNA we extracted, an independent molecular biology technique (quantitative RT-PCR) was performed and generated similar observations. Formalin-fixed and paraffin-embedded tissue can thus be employed as source material for an analysis by a DNA microarray system, providing practicality and ‘ease of use’. This makes the use of molecular classification assays feasible, as potential ancillary diagnostic tests and possibly guides to prognosis and treatment planning. The formalin-fixed and paraffin-embedded tissue in this study had an average storage of 1 year; however, there are limitations to its use due to the fragility of RNA with increasing time in storage. Unfortunately this also limits our ability to conduct retrospective ‘prospective’ studies on older archival blocks. Study of primary melanomas excised in the past would be ideal, allowing comparisons between microarray results and clinical outcome in patients followed for an extended period. The incorporation of clinical outcome would greatly improve the confidence to be placed on a given diagnosis and would provide a ‘gold standard’ for this type of study. Inevitably, in the absence of prolonged follow-up even the best histopathology-based diagnostic opinion remains unconfirmed by clinical outcome. The study supports the possibility that molecular methods may be developed to become a part of routine diagnostics. Additional studies of this exciting possibility, correlating the clinical outcome of patients with melanocytic lesions (the most precise index of malignancy or benignancy) and their gene expression profiles will be required to determine the confidence that can be placed on the capacity of genetic analysis to distinguishing melanomas from nevi with clinically acceptable accuracy.
We have identified by statistical analysis 100 top probes representing 36 genes with substantially different expression levels in specimens that clustered as melanomas or nevi. Of the 36 genes, the 14 most statistically significant were tabulated as to their expression levels in melanoma relative to nevi and some confirmed observations in the recent literature (Figure 3a). These previous reports however, reflected expression levels determined by DNA microarray studies using frozen tissues. Some of these genes are characterized, L1CAM homolog, cell adhesion molecule; GPX3, glutathione peroxidase; FABP7, fatty acid-binding protein; PHACTR1, phosphatase inhibitor and actin regulator; DLC, dynein light chain; HLA-A and HLA-B, MHC class I proteins; PRAME, retinoic acid receptor repressor and cell growth promoter; and STAT1, signal transducer and transcription activator. Overall, the relative gene expression levels in melanomas are consistent with the current understanding of molecular biology. For example, compared to nevi, melanoma had increased expressions of PHACTR1, PRAME, STAT1, and LCP2, gene products involved in promoting signal transduction, transcription, and cell growth. Malignancy is associated with decreased adhesive states and our observation of decreased L1CAM homolog expression in melanomas is in line with that observation. The L1CAM homolog gene expression pattern in our study was inconsistent with a previous study of melanomas and nevi that used frozen tissue on DNA microarray and reported increased L1CAM expression in melanoma compared to nevi.12 The discrepancy may relate to our study of a L1CAM homolog, rather than the L1CAM examined in the prior study. Previous studies reported relatively similar expression levels of HLA (MHC class I) genes in melanoma and nevi11 whereas we found higher expression of HLA-A and HLA-B (MHC class I) genes in melanoma relative to nevi (Figure 3a).
The significance of differences between studies is uncertain. Different studies have used cell lines, fresh frozen tissue, FFPE, differing proportions, of primary melanomas, metastatic melanomas and different types of nevi, various approaches to tissue collection (eg, laser capture microdissection), a range of sample sizes, different microarray platforms and a range of techniques of data analysis. Discrepancies between DNA microarray data from melanoma studies are highlighted by diversity between gene expression profiles reported in a meta-analysis of nine published DNA microarray gene expression profile studies.17 Such lack of comparability and reproducibility between studies is unsatisfactory. Gyorffy et al,17 suggests potential solutions, including sufficient sample size, clinical standards, standard microarray data processing and exchange of samples for quality control. Despite these discrepancies data from early research using melanoma DNA microarrays have yielded promising results and raise the possibility that standardized expression profile approaches may be developed for clinical use in the future.
In summary, diagnosis of melanocytic tumors based on histopathology alone may lead to a minority of clinically significant incorrect diagnoses. Reduction of the frequency of misdiagnosis and the possibility of improper management may require the development of new adjunctive non-morphological techniques that can be applied to difficult melanocytic lesions. We here report the successful use of laser capture microdissected formalin-fixed and paraffin-embedded tissue in an analysis of melanocytic lesions by DNA microarray technology. Our data showed that statistically significant differential expression levels of some genes could be used to separate malignant melanomas and nevi. We applied these gene signatures to unknown melanocytic lesions to predict whether they were melanomas or nevi and achieved good concordance with histopathology. Molecular based ancillary approaches to diagnosis of difficult melanocytic tumors are of high interest though their development is at an early stage. This study demonstrates the possibility of achieving such goals on the basis of additional studies. Here, gene expression profiles accurately separated lesions considered as melanomas or nevi on the basis of expert histopathologic evaluation. Clinical outcome is the ultimate proof that a lesion is either a melanoma or a nevus and future studies will need to correlate gene expression profiling with outcome. We believe that for the foreseeable future histopathology will remain the primary diagnostic method with molecular testing, as it becomes more readily available it will serve as an ancillary approach to diagnosis.
This is the first in a proposed series of studies that will incorporate larger study sets that include diagnostically difficult melanocytic lesions (eg, dysplastic/atypical nevi, spitz tumors), to determine specific genetic expression differences between such lesions. Potentially, such studies may allow us to identify key gene(s) to distinguish melanocytic lesions at different stages of evolution and of widely differing morphology. Such studies will elucidate critical basic features of melanocyte pathobiology and may enhance the accuracy of the diagnosis.
Accession codes
References
Whitaker S . Adjuvant therapy in melanoma. Clin Exp Dermatol 2000;25:497–502.
Troxel DB . Pitfalls in the diagnosis of malignant melanoma. Am J Surg Pathol 2003;27:1278–1283.
Ackerman AB . Discordance among expert pathologists in diagnosis of melanocytic neoplasms. Human Pathol 1996;27:1115–1166.
Liphshutz RJ, Morris D, Chee M, et al. Using oligonucleotide probe arrays to access genetic diversity. Biotechniques 1995;19:442–447.
Schena M, Shalon D, Davis RW, et al. Quantitative monitoring of gene expression patterns with a complementary DNA microarray. Science 1995;270:467–470.
Quackenbush J . Microarray Analysis and Tumor Classification. N Engl J Med 2006;354:2463–2472.
Southern E, Mir K, Shchepinov M . Molecular interactions on microarrays. Nat Genet 1999;21:5–9.
Kunz M, Ibrahim SM, Koczan D, et al. DNA microarray technology and its applications in dermatology. Exp Dermatol 2004;13:593–606.
Bittner M, Meltzner P, Chen Y, et al. A. molecular classification of cutaneous malignant melanoma by gene expression profiling. Nature 2000;406:536–540.
Becker B, Roesch A, Hafner C, et al. Discrimination of melanocytic tumors by cDNA array hybridization of tissues prepared by laser pressure catapulting. J Invest Dermatol 2004;122:361–368.
Seykora JT, Jih D, Elenitsas R, et al. Gene Expression Profiling of Melanocytic Lesions. Am J Dermatopathol 2003;25:6–11.
Talantov D, Mazumder A, Yu JX, et al. Novel genes associated with malignant melanoma but not benign melanocytic lesions. Clin Cancer Res 2005;11:7234–7242.
Haqq C, Nosrati M, Sudilovsky D, et al. The gene expression signatures of melanoma progression. Proc Natl Acad Sci USA 2005;102:6092–6097.
Specht K, Richter T, Muller U, et al. Quantitative gene expression analysis in microdissected archival formalin-fixed and paraffin-embedded tumor tissue. Am J Pathol 2001;158:419–429.
Lewis F, Maughan NJ, Smith V, et al. Unlocking the archive—gene expression in paraffin-embedded tissue. J Pathol 2001;195:66–71.
Winnepenninckx V, Lazar V, Michiels S, et al. Gene expression profiling of primary cutaneous melanoma and clinical outcome. J Natl Cancer Inst 2006;98:472–482.
Gyorffy B, Lage H . A Web-Based Data Warehouse on Gene Expression in Human Malignant Melanoma. J Invest Dermatol 2007;127:394–399.
Author information
Authors and Affiliations
Corresponding author
Additional information
Disclosure/conflicts of interest
There are no conflicts of interest or any disclosures necessary.
Rights and permissions
About this article
Cite this article
Koh, S., Opel, M., Wei, JP. et al. Molecular classification of melanomas and nevi using gene expression microarray signatures and formalin-fixed and paraffin-embedded tissue. Mod Pathol 22, 538–546 (2009). https://doi.org/10.1038/modpathol.2009.8
Received:
Revised:
Accepted:
Published:
Issue date:
DOI: https://doi.org/10.1038/modpathol.2009.8
Keywords
This article is cited by
-
Virulence determinants associated with the Asian community-associated methicillin-resistant Staphylococcus aureus lineage ST59
Scientific Reports (2016)
-
The role of osteopontin expression in melanoma progression
Tumor Biology (2015)
-
Comparison of Gene Expression Profile Between Tumor Tissue and Adjacent Non-tumor Tissue in Patients with Gastric Gastrointestinal Stromal Tumor (GIST)
Cell Biochemistry and Biophysics (2015)
-
Future perspectives in melanoma research: meeting report from the "Melanoma Bridge", Napoli, December 5th-8th 2013
Journal of Translational Medicine (2014)
-
MRSA epidemic linked to a quickly spreading colonization and virulence determinant
Nature Medicine (2012)
