
Abstract
Deletion 20q is a common chromosomal abnormality in myeloid neoplasms. Detection of del(20q) in patients following cytotoxic therapies raises concerns for an emerging therapy-related myeloid neoplasm. In this study, we identified 92 patients who acquired isolated del(20q) in their bone marrow following cytotoxic therapies for malignant neoplasms. Seventy-six patients showed interstitial and sixteen patients showed terminal 20q deletion. The median interval from prior cytotoxic therapies to detection of del(20q) was 58 months (range, 5–213 months). With a median follow-up of 23 months (range, 1–183 months), 21 (23%) patients developed therapy-related myeloid neoplasm and 71 (77%) patients did not. In patients who developed therapy-related myeloid neoplasm, del(20q) presented in a higher percentage of metaphases (60 vs 25%, P<0.0001); persisted for a longer period of time (24 vs 10 months, P=0.0487); and was more often a terminal deletion (33 vs 13%, P=0.0006) compared with patients who did not develop therapy-related myeloid neoplasm. Clonal evolution was only detected in patients with therapy-related myeloid neoplasm (4 patients, 19%). We conclude that del(20q) emerging after cytotoxic therapy represents an innocuous finding in more than two-thirds of patients. In patients who develop a therapy-related myeloid neoplasm, del(20q) often involves a higher percentage of metaphases, persists longer and more frequently is a terminal rather than an interstitial deletion.
Similar content being viewed by others
Main
Deletion of the long arm of chromosome 20, del (20q), occurs in a variety of myeloid neoplasms, including myelodysplastic syndromes, acute myeloid leukemia, and Philadelphia chromosome-negative myeloproliferative neoplasms, seen in approximately 5–8%, 2%, and 2–10% of cases, respectively.1, 2, 3, 4 The presence of del(20q) appears to be associated with a favorable prognosis and low risk for acute myeloid leukemia transformation in patients with Philadelphia chromosome-negative myeloproliferative neoplasms and myelodysplastic syndromes.5, 6, 7, 8 However, in patients with de novo acute myeloid leukemia, del(20q) has been associated with a poor response to chemotherapy and reduced overall survival, and has been classified as intermediate II risk category.8, 9
Isolated del(20q) has been well studied in de novo myelodysplastic syndromes.1 Patients with de novo myelodysplastic syndromes and isolated del(20q) often have an indolent clinical course, frequent thrombocytopenia, low blast counts, and high reticulocyte counts.6, 10, 11 The occurrence of isolated del(20q) is not considered as a definitive evidence for myelodysplastic syndromes in the absence of morphological evidence according to the 2008 WHO classification,1 whereas some studies reported that del(20q) in de novo myelodysplastic syndromes may only be associated with minimal morphological dysplasia.6, 12
Therapy-related myeloid neoplasm is a late complication of cytotoxic chemotherapy and/or radiotherapy for a prior neoplastic or non-neoplastic disorders.13 Patients with therapy-related myeloid neoplasm usually have a dismal clinical outcome, with a median overall survival of 6–8 months.14, 15 Cytogenetic abnormalities can be detected in over 80% of these patients with more than half having a complex karyotype.13, 16 Isolated del(20q) is uncommon in therapy-related myelodysplastic syndromes, and only five cases in one study have been reported with detailed clinicopathologic features.17 In this study, patients with therapy-related myelodysplastic syndromes and isolated del(20q) were often associated with morphological dysplasia and had a variable clinical course.17
Del(20q) as a sole clonal cytogenetic abnormality is not uncommon in patients following cytotoxic therapies; however, the clinical significance of emergence of isolated del(20q) in this clinical setting is largely unknown. In our experience, the question of whether this finding should be concerning for a therapy-related myeloid neoplasm17 or represents a 'silent' clonal cytogenetic abnormality as we recently described18 is often raised. To address these questions, we retrospectively reviewed the archives of the cytogenetics laboratory at our hospital and identified 92 patients who acquired isolated del(20q) following cytotoxic therapies and analyzed its clinical implications with emphasis on the incidence of therapy-related myeloid neoplasm of this cohort.
Materials and methods
Case Selection
We searched the database of Clinical Cytogenetic Laboratory at the University of Texas MD Anderson Cancer Center for cases with isolated del(20q) between 1 January 2003 and 30 June 2014. We then narrowed down to the patients who had received prior cytotoxic therapies. Cases with heavy (≥50%) bone marrow involvements by primary malignancy, which may compromise the morphological evaluation of hematopoietic cells for myelodysplasia, were excluded from the study. A detailed chart review was conducted in all patients, including prior history of malignancies and cytotoxic therapies, cytogenetic features, and outcomes. The diagnoses of therapy-related myeloid neoplasms, including therapy-related myeloproliferative neoplasms, therapy-related myelodysplastic syndromes, therapy-related myelodysplastic syndromes/myeloproliferative neoplasms, and therapy-related acute myeloid leukemia, were based on clinical, morphologic, immunophenotypic, and molecular genetic criteria as specified in the WHO classification.1 All samples were collected following institutional guidelines with informed consent in accordance with the Declaration of Helsinki.
Peripheral Blood and Bone Marrow Assessment
Complete blood cell counts at the time of del(20q) detection and during follow-up were collected. Peripheral blood smears, bone marrow core biopsy and clot sections, as well as aspirate smears and touch imprints were reviewed. A 500-cell differential count on bone marrow aspirate smears or touch imprints was performed. Particular attention was paid to the cytological features of the myeloid cells, erythrocytes, and megakaryocytes for morphologic evidence of dysplasia. Involvement of bone marrow by primary malignancy was evaluated by morphology, immunohistochemistry, and/or flow-cytometry immunophenotyping analysis.
Conventional Cytogenetic and Fluorescence In Situ Hybridization Analyses
Conventional chromosomal analysis was performed on G-banded metaphase cells prepared from unstimulated 24-h and 48-h bone marrow aspirate cultures (for primary diagnosis of myeloid neoplasms or solid tumors), or unstimulated 24-h and mitogens stimulated 72-h bone marrow aspirate cultures (for primary diagnosis of lymphoid neoplasms) using standard techniques. Mitogens included 12-O-TetradecanoylphorboM 3-acetate, oligonucleotides, and lipopolysaccharide. Twenty metaphases were analyzed and the results were reported using the International System for Human Cytogenetic Nomenclature (ISCN 2013).19 The presence of del(20q) was further confirmed by fluorescence in situ hybridization analysis using a fluorescently labeled LSI D20S108 probe (Vysis/Abbott Laboratories, Des Plaines, IL, USA) according to the manufacturer’s instructions. Two hundred interphases were counted and the percentage of del(20q)-positive cells was calculated. The cutoff for del(20q) positivity established in our laboratory is 5%.
Combined Morphologic and Fluorescence In Situ Hybridization Analysis
Combined morphologic and fluorescence in situ hybridization analysis was performed on a subset of cases with bone marrow involvement by primary tumors according to the methods described previously20 with minor modifications. Briefly, morphologic evaluation and image capture were performed on Wright-Giemsa stained bone marrow aspirate smears; smears were then destained using 1% acid alcohol, followed by protease II (Vysis/Abbott Laboratories) treatment, and hybridization with LSI D20S108 probe. Target cell populations were captured under the fluorescence microscope.
Statistical Analysis
The unpaired t-test was used for numerical comparisons. Chi-square and Fisher’s exact tests were applied for categorical variables. Overall survival was estimated by the Kaplan–Meier method from the date of del(20q) detection until death from any cause or censored at last follow-up for alive patients.
Results
Clinical Findings
We identified 356 patients with del(20q) as a sole cytogenetic abnormality in the bone marrow at our institution during the study interval. Of them, 106 patients acquired del(20q) in the bone marrow following cytotoxic therapies for various malignant tumors. Fourteen cases were found to have bone marrow involvement by primary cancer >50% and were excluded from this study. The remaining 92 patients formed our study group. There were 78 men and 14 women with a median age of 64 years (range, 42–89 years). Primary malignancies included lymphoid neoplasms (n=82), solid tumors (n=6), and composite tumors (n=4) (Table 1). Sixty-three (68%) patients had bone marrow involvement by the primary malignancy at the time of diagnosis of the initial neoplasm.
Ninety patients received chemotherapies. The major chemotherapeutic regimens included: fludarabine, cyclophosphamide, and rituximab (n=30); cyclophosphamide, doxorubicin, vincristine, and prednisone with or without rituximab (n=25); hyperfractionated cyclophosphamide, vincristine, doxorubicin, dexamethasone alternating with high-dose methotrexate and cytarabine (n=7); and common therapeutic regimens for plasma cell myeloma (bortezomib, revlimid, dexamethasone, thalidomide, n=15). In all, 16 patients received combined chemotherapy and radiotherapy; 38 patients received chemotherapy and stem-cell transplantation; and 10 patients received chemotherapy, radiotherapy and stem-cell transplantation. Two patients received radiotherapy only (Table 2).
Peripheral Blood and Bone Marrow Findings
At the time of del(20q) detection, 6 patients had pancytopenia; 10 had anemia and thrombocytopenia; 22 had isolated thrombocytopenia; 7 had isolated anemia; and 1 had marked leukocytosis and thrombocytosis. The remaining 46 patients had a normal blood cell count. The median of white-blood cell count was 4.6 × 109 /l (range, 1.3–39.2 × 109 /l), hemoglobin 12.6 g/dl (range, 8.7–17.4 g/dl), and platelet count 114 × 109 /l (range, 24–765 × 109 /l). One patient had circulating blasts (2%).
Bone marrow was involved by primary tumors in 34 patients at the time of del(20q) detection, including chronic lymphocytic leukemia/small lymphocytic lymphoma (n=23), plasma cell myeloma (n=6), hairy cell leukemia (n=2), mantle cell lymphoma (n=1), marginal zone lymphoma (n=1), and lymphoplasmacytic lymphoma (n=1); 14 of 34 patients had minimal involvement (<10%). The remaining 58 patients had no evidence of bone marrow involvement by their primary malignancies.
At the time of detection of del(20q), morphological assessment was assessed without knowledge of cytogenetic findings. Twelve patients showed marked dysplasia, and/or increased blasts in the bone marrow meeting the criteria for therapy-related myeloid neoplasms, including 10 therapy-related myelodysplastic syndromes, 1 therapy-related acute myeloid leukemia, and 1 therapy-related myelodysplastic syndrome/myeloproliferative neoplasm, unclassifiable. Of the remaining 80 patients, 16 patients had mild dysplasia and there was no evidence of dysplasia in 64 patients. Ninety patients had bone marrow blasts <5%, and two patients had bone marrow blasts of 10 and 21%, respectively.
During the follow-up interval, nine patients showed hematologic and morphologic evidence of myeloid neoplasms after a median of 18 months (range, 4–68 months) after del(20q) detection, including eight therapy-related myelodysplastic syndromes and one chronic myelomonocytic leukemia. One of these nine patients developed therapy-related myelodysplastic syndromes after disappearance of del(20q) and the emergence of a new hypodiploid clone with monosomy 5 and add(6p).
Cytogenetic Findings
Baseline bone marrow cytogenetic data at the time of diagnosis of primary tumors (before chemotherapy and/or radiotherapy) were available in 49 (53%) patients (Table 3) and unknown in 43 patients. In all, thirty-eight patients had a diploid karyotype and 11 had an abnormal karyotype, including 4 patients with cytogenetic abnormalities associated with chronic lymphocytic leukemia, 4 patients with hyperdiploid clones associated with plasma cell myeloma, 2 patients with -Y, and 1 patient (Burkitt lymphoma) with t(8;14)(q24.1;q32). No patients showed del(20q) before cytotoxic therapies. The median interval from the first dose of chemotherapy/radiotherapy to the detection of del(20q) was 58 months (5–213 months) (Table 3).
The number of metaphases showing del(20q) by conventional cytogenetic analysis ranged from 2 to 20 (10–100%), with a median of 6 (30%). The percentage of metaphases with del(20q) was significantly higher in patients who developed therapy-related myeloid neoplasm than those who did not (60 vs 25%; P<0.0001). Seventy-six patients showed interstitial 20q deletion and sixteen had terminal 20q deletion. The most common breakpoints for interstitial deletion were at 20q11.2 (centromeric) and 20q13.1 or 20q13.3 (telomeric). The breakpoint for terminal deletion was at 20q11.2. Compared with patients who did not develop therapy-related myeloid neoplasm, patients with therapy-related myeloid neoplasm showed a higher frequency of terminal deletion (33 vs 13%); lower frequency of interstitial deletion at 20q11.2q13.1 (5 vs 24%) (P=0.0004), and comparable frequency of interstitial deletion at 20q11.2q13.3 (Table 3).
Interphase fluorescence in situ hybridization for del(20q) was performed on 49 patients. The clonal size was significantly larger in patients who developed therapy-related myeloid neoplasms (median, 46%; range, 11–93%) compared with patients who did not develop therapy-related myeloid neoplasms (median, 15%; range, 6–78%) (P=0.0055). The clonal size detected by interphase fluorescence in situ hybridization was slightly smaller but proportional to the percentage of metaphases detected by conventional chromosomal analysis (r=0.9285).
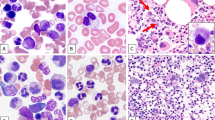
Combined morphologic and fluorescence in situ hybridization analysis was performed in 13 patients who had 11–50% primary tumor involvement in bone marrow and who had Wright-Giemsa stained bone marrow aspirate smears available. These included 12 patients with chronic lymphocytic leukemia and 1 with plasma cell myeloma. Del(20q) was detected exclusively in myeloid and erythroid cells, but not in lymphocytes or plasma cells (Figure 1).

Combine morphologic and fluorescence in situ hybridization analysis with LSI D20S108 probe ( × 100 objective) in two patients without therapy-related myeloid neoplasm. No morphological dysplasia was noted in myeloid or erythroid lineages. Del(20q) was detected in myeloid and erythroid lineages (red arrows), but not in lymphocytes or plasma cells (green arrows). (a and c) Bone marrow with involvement of plasma cell myeloma; (b and d) Bone marrow with involvement of chronic lymphocytic leukemia. (a and b) Bone marrow smear stained with Wright-Giemsa; (c and d) fluorescence in situ hybridization analysis with probe of D20S108.
Sixty-two (67%) patients had one or more follow-up cytogenetic studies (median 2; range 1–9 times). Patients who developed therapy-related myeloid neoplasms were significantly more likely to have persistence of del(20q) on follow-up bone marrow samples (median 6 samples; range 1–9 samples) compared with patients who did not develop therapy-related myeloid neoplasms (median 2 samples; range 1–5 samples) (P<0.0001). In addition, the duration of del(20q) persistence was significantly longer in patients who developed therapy-related myeloid neoplasms than those who did not (24 vs 10 months; P=0.0487). At the time of last follow-up, 15 (71%) patients who developed therapy-related myeloid neoplasms and 31 (44%) patients who did not develop therapy-related myeloid neoplasms had persistent del(20q) (P=0.0006) (Table 3).
Clonal evolution was detected in four (19%) patients who developed therapy-related myeloid neoplasms after 2–48 months of having del(20q). Acquired additional clonal abnormalities were as follows (1 patient each): 47,XY,+13,del(20)(q11.2); 46,X,der(Y)t(Y;1)(q12;q12),del(20)(q11.2q13.3); 47,del(20)(q11.2q13.3),+mar; and 45,XY,add(2)(q37),-7,del(20)(q11.2q13.3). In the group of patients who did not develop therapy-related myeloid neoplasms, no patients showed clonal evolution.
Last Clinical Follow-up and Outcome
The median follow-up interval was 23 months (range, 1–183 months) after detection of del(20q). Among the 71 patients who did not meet the criteria for therapy-related myeloid neoplasm, 46 patients had completely normal blood cell counts and never showed dysplasia in bone marrow or peripheral blood at the time of del(20q) detection or during follow-up. For the remaining 25 patients, 3 patients had anemia and thrombocytopenia with hypoplastic bone marrow, no dysplasia was detected; 3 patients had persistent but stable thrombocytopenia, and mild or occasional dysplasia in one or two hematopoietic lineages that did not meet the criteria for therapy-related myelodysplastic syndromes; 12 patients had cytopenia and/or mild dysplasia but were either receiving active chemotherapy or had substantial bone marrow involvement by primary tumor (or both); 7 patients had cytopenia without dysplasia at the time of del(20q) detection, but did not have adequate follow-up (<3 months) due to death, stem-cell transplantation, or being lost to follow-up.
Among the 21 patients who developed therapy-related myeloid neoplasms, 1 patient was diagnosed with therapy-related acute myeloid leukemia at the time of del(20q) detection; and 4 patients were transformed from myelodysplastic syndromes into acute myeloid leukemia 6–12 months after diagnosis of therapy-related myelodysplastic syndromes. At last follow-up, 18 (86%) patients had died of acute myeloid leukemia/myelodysplastic syndromes (n=12), complications of stem-cell transplantation (n=2), peripheral T-cell lymphoma (n=1), or unknown causes (n=3); 3 (14%) patients were alive with persistent myelodysplastic syndromes and concurrent chronic lymphocytic leukemia (n=2) or follicular lymphoma (n=1). The median overall survival was 24 months after detection of del(20q) and 13 months after diagnosis of therapy-related myeloid neoplasms.
Of the group of patients who never developed therapy-related myeloid neoplasm, 25 (35%) patients died, 18 (25%) patients were alive with persistent primary malignancy, and 28 (40%) patients were alive and in clinical remission. The cause of death included primary malignancy (n=21), complications of stem-cell transplantation (n=1), or unknown causes (n=3). The median overall survival was 86 months after detection of del(20q), which was significantly longer than 24 months in patients who developed therapy-related myeloid neoplasms (P=0.0036).
Discussion
In this study, we present detailed clinicopathologic follow up of 92 patients who developed isolated del(20q) following cytotoxic chemotherapy/radiotherapy. Twenty-one (23%) patients developed therapy-related myeloid neoplasms at the time of del(20q) detection or during the follow-up period, but the majority (71, 77%) of patients did not develop therapy-related myeloid neoplasm after a median of 23 months follow-up. We also found three factors that are associated with a high risk of developing therapy-related myeloid neoplasm: clone size of del(20q), the deleted region of 20q, and persistence of del(20q).
In this study, the size of del(20q) clone was significantly higher in patients who developed therapy-related myeloid neoplasm than those who did not. Among the 21 patients who developed therapy-related myeloid neoplasms, 15 (71%) had del(20q) as the major clone (in ≥50% cells); 3 showed del(20q) in 20–40% metaphases at the first time detection, but the clone became a major clone in the follow-up bone marrow; and 2 had persistent del(20q) as the minor clone (20–30%). One patient showed del(20q) in 10% of metaphases with no cytopenia or dysplasia; however, 17 months later, del(20q) was disappeared and a new hypodiploid clone of 45,XX,-5,add(6)(p23) emerged, accompanied with cytopenia and overt dysplasia. Therefore, it seems likely that the therapy-related myeloid neoplasm in this patient was associated with the hypodiploid clone instead of del(20q). Among the 71 patients who did not develop therapy-related myeloid neoplasm, 55 (77%) patients had del(20q) as a minor clone (<50% cells) and 16 patients had del(20q) as a major clone. Among these 16 patients with major clone, 6 had cytopenia but no overt dysplasia, the other 10 patients showed no evidence of cytopenia or dysplasia. Although longer follow-up time may be helpful in determining whether these patients will eventually develop therapy-related myeloid neoplasm, our data clearly show that patients with a major del(20q) clone are more likely to develop therapy-related myeloid neoplasm compared with those patients with a minor clone. When del(20q) presents as a minor clone in the bone marrow, normal hematopoietic elements are usually adequate to compensate and maintain the normal hematopoietic function, patients are therefore unlikely show cytopenia or overt dysplasia. This may also explain why patients with a minor del(20q) clone have a superior overall survival compared with patients with a major clone.21, 22
Most patients in our cohort and those reported in the literatures have interstitial deletion of 20q. The breakpoints of 20q deletion reported have been heterogeneous. The most common breakpoints were at 20q11.2 (for both interstitial and terminal deletion), 20q13.1 or 20q13.3 (for interstitial deletion).4, 23, 24, 25 Two common deleted regions (CDRs) have been identified, one is located within 20q12 and spans ~8 Mb;24, 26 the other is located within 20q13.2 and spans ~1.8 Mb.27 In this study, 20q12 was deleted in all patients; however, 20q13.2 was preserved in 17 (24%) patients who did not develop therapy-related myeloid neoplasm and only 1 (5%) patient with therapy-related myeloid neoplasm. In addition, a higher percentage of patients with therapy-related myeloid neoplasm showed terminal deletion. Loss of genetic material from 20q may confer a proliferative advantage to myeloid cells, possibly through loss of function of a tumor suppressor gene. Although no specific gene thought to be responsible for the pathogenesis of myeloid neoplasms has been identified on chromosome 20q, L3MBTL1 is considered as a candidate gene. L3MBTL1 is a human homolog of the Drosophila polycomb L(3)MBT tumor suppressor gene which is located at 20q12. By using a loss-of-function approach, Gurvich et al28 found that deletion of the L3MBTL1 gene causes replicative stress, DNA breaks, activation of the DNA damage response, and genomic instability, which could contribute to the development of myeloid neoplasms.
The results in this study also show that del(20q) persists longer (24 vs 10 months, P=0.0487) and is detected more often (6 vs 2 times, P<0.0001) in patients who developed therapy-related myeloid neoplasm compared with those who did not. Among the 21 patients who developed therapy-related myeloid neoplasm, only 1 patient showed disappearance of del(20q) with replacement by a hypodiploid clone as mentioned above. Among the patients who did not develop therapy-related myeloid neoplasm, 13 (30%) patients showed transient and 31 showed persistent del(20q) detection; the other 27 patients had no further cytogenetic testing, which is mainly due to a lack of clinical indication (primary disease in remission and no cytopenia). An abnormal clone could be a transient finding, especially when it presents as a small clone,29 or a clone developing during or immediately following chemotherapy30 or stem-cell transplant.31 Bone marrow repair capacity and body immunosurveillance may have key roles in suppressing abnormal clones and clearing abnormal clones from the bone marrow.
Among the 21 patients who developed therapy-related myeloid neoplasm, 12 patients were diagnosed with therapy-related myeloid neoplasm at the time when del(20q) was first detected, whereas the other 9 patients were diagnosed 4–68 months after detection of del(20q). Of these 9 patients, 2 had cytopenia but no dysplasia; 2 showed mild/occasional dysplasia but no cytopenia, and 5 had no dysplasia or cytopenia at the first time of del(20q) detection. Three of these patients showed del(20q) as major clone and six as minor clone at the time of initial detection. Interestingly, other karyotypic alternations occurred in six of these nine patients at the time when therapy-related myeloid neoplasm was diagnosed: a minor clone evolved to a major clone in bone marrow (n=3); clonal evolution (n=2); or a new hypodiploid clone emerged (n=1). These findings suggest that (1) detection of del(20q) clone can occur before the detection of cytopenia and/or dysplasia, which is consistent with the findings by Kanagal-Shamanna et al;17 (2) clonal size is a very important risk factor for developing therapy-related myeloid neoplasm as mentioned above; (3) del(20q) may not be sufficient to cause therapy-related myeloid neoplasm in some patients and additional genetic changes are required to further potentiate the leukemic clone.
Among the 71 patients who did not meet the criteria for therapy-related myeloid neoplasm, 25 patients showed different degrees of cytopenia at the time of del(20q) detection or during the follow-up. Although the cytopenia could be explained by hypoplastic bone marrow (n=3) or the undergoing chemotherapy and/or bone marrow involvement by primary malignancy (n=12), we cannot exclude the possibility of therapy-related myeloid neoplasm in these 15 patients with certainty. This is also true for the seven patients who had cytopenia but had inadequate follow-up (<3 months), as we know that the detection of del(20q) may precede the appearance of overt dysplasia as discussed above. It is debatable whether the remaining three patients who had persistent but stable thrombocytopenia, and mild or occasional dysplasia, should be diagnosed with therapy-related myeloid neoplasm. These three patients would not meet the WHO criteria for therapy-related myeloid neoplasm due to lack of overt dysplasia;1 however, these three patients might have a low-grade myelodysplastic syndromes under the observations that myelodysplastic syndromes with isolated del(20q) may only show minimal/mild dysplasia.6, 12
In summary, isolated del(20q) developing in patients with prior cytotoxic therapies is not always associated with progression to a therapy-related myeloid neoplasm, only a subset of the patients will develop therapy-related myeloid neoplasm. In patients who develop therapy-related myeloid neoplasm, del(20q) usually presents in a higher percentage of metaphases, persists for a longer time period, and is more often a terminal deletion or manifestation of clonal evolution. Close follow-up and surveillance for clonal evolution rather than an immediate therapeutic intervention seems most appropriate for patients with a minor del(20q) clone and a morphologically normal bone marrow.
References
Brunning RD, Orazi A, Germing U et al. Myelodysplastic syndromes/neoplasms, overview. In: Swerdlow S, Campo E, Harris N et al(eds) WHO Classification of Tumors of Hematopoietic and Lymphoid Tissues. Stylus Publishing, LLC: Sterling, VA, 2008, pp 88–93.
Bacher U, Schnittger S, Kern W et al. Distribution of cytogenetic abnormalities in myelodysplastic syndromes, Philadelphia negative myeloproliferative neoplasms, and the overlap MDS/MPN category. Ann Hematol 2009;88:1207–1213.
Heim S, Mitelman F . Cytogenetic analysis in the diagnosis of acute leukemia. Cancer 1992;70:1701–1709.
Bench AJ, Aldred MA, Humphray SJ et al. A detailed physical and transcriptional map of the region of chromosome 20 that is deleted in myeloproliferative disorders and refinement of the common deleted region. Genomics 1998;49:351–362.
Greenberg PL, Tuechler H, Schanz J et al. Revised international prognostic scoring system for myelodysplastic syndromes. Blood 2012;120:2454–2465.
Gupta R, Soupir CP, Johari V et al. Myelodysplastic syndrome with isolated deletion of chromosome 20q: an indolent disease with minimal morphological dysplasia and frequent thrombocytopenic presentation. Br J Haematol 2007;139:265–268.
Caramazza D, Begna KH, Gangat N et al. Refined cytogenetic-risk categorization for overall and leukemia-free survival in primary myelofibrosis: a single center study of 433 patients. Leukemia 2011;25:82–88.
Campbell LJ, Garson OM . The prognostic significance of deletion of the long arm of chromosome 20 in myeloid disorders. Leukemia 1994;8:67–71.
Dohner H, Estey EH, Amadori S et al. Diagnosis and management of acute myeloid leukemia in adults: recommendations from an international expert panel, on behalf of the European Leukemia Net. Blood 2010;115:453–474.
Braun T, de Botton S, Taksin AL et al. Characteristics and outcome of myelodysplastic syndromes(MDS) with isolated 20q deletion: a report on 62 cases. Leuk Res 2011;35:863–867.
Sashida G, Takaku TI, Shoji N et al. Clinico-hematologic features of myelodysplastic syndrome presenting as isolated thrombocytopenia: an entity with a relatively favorable prognosis. Leuk Lymphoma 2003;44:653–658.
Steensma DP, Dewald GW, Hodnefield JM et al. Clonal cytogenetic abnormalities in bone marrow specimens without clear morphologic evidence of dysplasia: a form fruste of myelodysplasia? Leuk Res 2003;27:235–242.
Vardiman J, Arber D, Brunning R et alTherapy-related myeloid neoplasms. In: Swerdlow S, Campo E, Harris N et al (eds). WHO Classification of Tumors of Hematopoietic and Lymphoid Tissues. IARC: Lyon, 2008, pp 127–129.
Ok CY, Hasserjian RP, Fox PS et al. Application of the international prognostic scoring system-revised in therapy-related myelodysplastic syndromesand oligoblastic acute myeloid leukemia. Leukemia 2014;28:185–189.
Klimek VM . Recent advances in the management of therapy-related myelodysplastic syndromesand acute myeloid leukemia. Curr Opin Hematol 2013;20:137–143.
Pedersen-Bjergaard J, Pedersen M, Roulston D et al. Different genetic pathways in leukemogenesis for patients presenting with therapy-related myelodysplasia and therapy-related acute myeloid leukemia. Blood 1995;86:3542–3552.
Kanagal-Shamanna R, Yin CC, Miranda RN et al. Therapy-related myeloid neoplasms with isolated del(20q): comparison with cases of de novo myelodysplastic syndrome with del(20q). Cancer Genet 2013;206:42–46.
Tang G, Wang SA, Lu V et al. Clinically silent clonal cytogenetic abnormalities arising in patients treated for lymphoid neoplasms. Leuk Res 2014;38:896–900.
Shaffer L, McGowan-Jordan J, Schmid M International system for human cytogenetic nomenclature (ISCN 2013) Recommendations of the International Standing Commitee on Human Cytogenetic Nomenclature. Basel: Karger, 2013;39–83.
Wang SA, Hutchinson L, Tang G et al. Systemic mastocytosis with associated clonal hematological non-mast cell lineage disease: clinical significance and comparison of chomosomal abnormalities in SM and AHNMD components. Am J Hematol 2013;88:219–224.
Pan J, Wu C, Xue Y et al. The characteristics and prognostic analysis in 213 myeloid malignancy patients with del(20q): a report of a single-center case series. Cancer Genet 2014;207:51–56.
Mallo M, Luno E, Sanzo C et al. Clinical impact of the clone size in MDS cases with monosomy 7 or 7q deletion, trisomy 8, 20q deletion and loss of Y chromosome. Leuk Res 2011;35:834–836.
Okada M, Suto Y, Hirai M et al. Microarray CGH analyses of chromosomal 20q deletions in patients with hematopoietic malignancies. Cancer Genet 2012;205:18–24.
Wang PW, Iannantuoni K, Davis EM et al. Refinement of the commonly deleted segment in myeloid leukemias with a del(20q). Genes Chromosomes Cancer 1998;21:75–81.
Nacheva E, Holloway T, Carter N et al. Characterization of 20q deletions in patients with myeloproliferative disorders or myelodysplastic syndromes. Cancer Genet Cytogenet 1995;80:87–94.
Bench AJ, Nacheva EP, Hood TL et al. Chromosome 20 deletions in myeloid malignancies: reduction of the common deleted region, generation of a PAC/BAC contig and identification of candidate genes. UK Cancer Cytogenetics Group (UKCCG). Oncogene 2000;19:3902–3913.
Huh J, Tiu RV, Gondek LP et al. Characterization of chromosome arm 20q abnormalities in myeloid malignancies using genome-wide single nucleotide polymorphism array analysis. Genes Chromosomes Cancer 2010;49:390–399.
Gurvich N, Perna F, Farina A et al. L3MBTL1 polycomb protein, a candidate tumor suppressor in del(20q12) myeloid disorders, is essential for genome stability. Proc Natl Acad Sci USA 2010;107:22552–22557.
Goswami RS, Liang CS, Bueso-Ramos CE et al. Isolated +15 in bone marrow: Disease-associated or a benign finding? Leuk Res 2015;39:72–76.
Basecke J, Podleschny M, Becker A et al. Therapy-associated genetic aberrations in patients treated for non-Hodgkin lymphoma. Br J Haematol 2008;141:52–59.
Lopez de Mesa R, Sierrasesumaga L, Calasanz MJ et al. Nonclonal chromosomal aberrations induced by anti-tumoral regimens in childhood cancer: relationship with cancer-related genes and fragile sites. Cancer Genet Cytogenet 2000;121:78–85.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Rights and permissions
About this article

Cite this article
Yin, C., Peng, J., Li, Y. et al. Clinical significance of newly emerged isolated del(20q) in patients following cytotoxic therapies. Mod Pathol 28, 1014–1022 (2015). https://doi.org/10.1038/modpathol.2015.66
Received:
Revised:
Accepted:
Published:
Issue date:
DOI: https://doi.org/10.1038/modpathol.2015.66
This article is cited by
-
The significance of genetic mutations and their prognostic impact on patients with incidental finding of isolated del(20q) in bone marrow without morphologic evidence of a myeloid neoplasm
Blood Cancer Journal (2020)
-
Myelodysplastic Syndromes: Laboratory Workup in the Context of New Concepts and Classification Criteria
Current Hematologic Malignancy Reports (2018)
-
Marginal zone lymphoma-derived interfollicular diffuse large B-cell lymphoma harboring 20q12 chromosomal deletion and missense mutation of BIRC3 gene: a case report
Diagnostic Pathology (2016)
-
Targeted next-generation sequencing identifies a subset of idiopathic hypereosinophilic syndrome with features similar to chronic eosinophilic leukemia, not otherwise specified
Modern Pathology (2016)
