
Abstract
Ductal carcinoma in situ (DCIS) is a non-obligate precursor of invasive breast cancer and a frequent mammographic finding requiring treatment. Up to 25% of DCIS can recur and half of recurrences are invasive, but there are no reliable biomarkers for recurrence. We hypothesised that copy number aberrations could predict likelihood of recurrence. We analysed a cohort of pure DCIS cases treated only with wide local excision for genome-wide copy number and loss of heterozygosity using Affymetrix OncoScan MIP arrays. Cases included those without recurrence within 7 years (n=25) and with recurrence between 1 and 5 years after diagnosis (n=15). Pure DCIS were broadly similar in copy number changes compared with invasive breast cancer, with the consistent exception of a greater frequency of ERBB2 amplification in DCIS. There were no significant differences in age or ER status between the cases with a recurrence vs those without. Overall, the DCIS cases with recurrence had more copy number events than the DCIS without recurrence. The increased copy number appeared non-random with several genomic regions showing an increase in frequency in recurrent cases, including 20q gain, ERBB2 amplification and 15q loss. Copy number changes may provide prognostic information for DCIS recurrence, but validation in additional cohorts is required.
Similar content being viewed by others


Main
Ductal carcinoma in situ (DCIS) is a proliferation of malignant cells within breast ducts without invasion through the basement membrane. A number of lines of evidence demonstrate that DCIS is a non-obligate precursor of invasive breast cancer, including the natural history of untreated DCIS,1 the high risk of ipsilateral invasive breast cancer after a diagnosis of DCIS and the common clonal genetic events found in DCIS and synchronous or recurrent invasive breast cancer.2 Prior to population-based screening mammography, DCIS was an unusual finding comprising only 3–5% of all breast neoplasms. Currently, ~20% of cases of screen-detected breast neoplasia are DCIS.3 Standard treatment has become wide local excision of DCIS with or without adjuvant radiotherapy.4
Recent clinical trials have found that ~25% of DCIS treated with local excision only will recur within 15 years, with the greatest risk of recurrence in the first 5 years.5 Approximately half of the recurrences will be invasive carcinoma, requiring more extensive treatment, and with a possible decrease in disease-specific survival.6 Radiotherapy reduces the recurrence rate by about 50%, so despite the fact that only small a small proportion of patients will benefit, many clinicians recommend radiotherapy because of the implications of invasive recurrence. The most commonly used predictive models to estimate the recurrence risk with or without radiotherapy include DCIS grade, tumour size, width of the margin of normal tissue and patient age.4 The evidence base for the selection criteria is inadequate as it is not possible to reliably identify the group of DCIS patients at minimal risk of recurrence. Consequently, clinical management of DCIS is problematic with many women being over-treated, whereas others potentially receive inadequate therapy. Genomic factors have not been widely used in the management of DCIS but have the potential to predict the outcome, in the way gene signatures are used to provide prognostic and predictive information for ER-positive and HER2-negative invasive breast cancer.7
Breast cancer develops with the accumulation of somatic genetic alterations, including point mutations, copy number alterations (gains and losses), copy number neutral loss of heterozygosity and structural chromosome alterations, such as translocations. Breast cancer is considered to be a ‘copy number’ class tumour, whereby the predominant driver events are copy number alterations.8 Based on copy number similarities, DCIS synchronous with invasive breast cancer appears to already be an advanced tumour;9, 10, 11 however, ‘pure’ DCIS (i.e., DCIS in the absence of an invasive carcinoma) may have substantial biological differences to invasive breast cancer. For example, ERBB2 overexpression is significantly more frequent in pure DCIS (up to 50%) than invasive breast cancer (5–15%).12, 13 The limited existing genetic data for pure DCIS9, 14 suggests that copy number events are common and likely to reflect biological behaviour, such as likelihood of progression to invasive carcinoma.
In this study, we hypothesise that copy number profiles may be predictive of whether a case of DCIS is likely to recur or progress to invasive carcinoma. We compare pure DCIS cases with and without a subsequent recurrence using genome-wide SNP arrays.
Materials and methods
Tumour Samples, DNA Extraction and Copy Number Analysis
This study was approved by the Peter MacCallum Cancer Centre Human Research Ethics Committee (06/24). Cases were recruited as described previously through NorthWestern BreastScreen Victoria, a population-based mammographic screening service, between 1994 and 2005.15 All women were treated with wide local excision but without radiotherapy. Non-recurrent cases were defined as those with no incidence of ipsilateral or contralateral DCIS or invasive breast cancer within 7 years of the primary DCIS diagnosis (n=46). Recurrent cases were defined as those cases with recurrent ipsilateral DCIS (n=11) or invasive breast cancer (n=6) between 1 and 5 years after primary DCIS diagnosis. Cases with a recurrence within 12 months were excluded to reduce the likelihood that these were due to sub-optimal surgery, although in all cases margins were reported as clear. Exclusion of cases with recurrence after 5 years was implemented to reduce the chance of new independent primaries (i.e., non-clonal with the initial DCIS) being included.
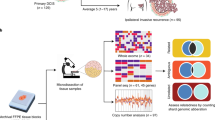
Archived formalin-fixed, paraffin-embedded pathology blocks were obtained, and H&E sections were reviewed by two pathologists to identify areas of DCIS for microdissection. Tumour and normal cells were needle microdissected from 10 to 40 10-micron sections, and DNA was extracted using the Qiagen DNeasy Kit as described previously.10 Of the 46 non-recurrent cases, 15 were excluded owing to insufficient DNA. At least 35 ng of DNA from each DCIS (31 non–recurrent cases and 17 primary recurrent cases, including 8 cases with both the primary and recurrence tumour tissue) and matching normal cells extracted from the same tissue block (n=26) were analysed for genomic copy number using the Affymetrix Molecular Inversion Probe 330 K array by Affymetrix OncoScan FFPE Express Services.16
Data Processing and Analysis
Data preprocessing was performed by Affymetrix OncoScan FFPE Express Services as described,16 where all tumour samples were batch normalised against the normal patient DNA as well as internal Affymetrix controls. Data are available through GEO (Accession no. GSE66746). Genome build hg18 (March 2006) was used for probe locations. Samples were imported into Nexus (BioDiscovery, Hawthorne, CA, USA) and segmented using a circular binary segmentation algorithm. Gains were called if the log2 ratio was >0.3 and losses at <−0.3. High copy number gains were >1.2 and homozygous deletions <−1.2. Segmented copy number data were also obtained from published studies17, 18, 19 and imported into Nexus. Copy number thresholds of ±0.2 were used for the invasive cohorts to take into account the higher level of normal cell contamination.
Comparisons between groups were made using Nexus applying thresholds of P<0.05 and at least 25% frequency difference unless otherwise stated. Significantly different regions of copy number alteration were merged into a single interval if they were within 0.5 Mb and filtered to exclude small regions (<0.5 Mb) if another similar region was not present within 10 Mb. The lowest P-value (from a segment of at least 50 kb in length) from within merged regions was used to annotate the region.
Droplet Digital PCR (ddPCR)
PCR validation of clonal copy number status was performed for ARID4B (Hs05760195_cn), ZNF217 (Hs01203936_cn) and ERBB2 (Hs00641606_cn) compared with the reference gene RNAseP (Hs01203936_cn) using Taqman probes (Life Technologies) and the QX200 ddPCR system (Bio-Rad). The results were analysed using the QuantaSoft analysis software (Bio-Rad).
Immunohistochemistry
If available, ER status was taken from the pathology report (n=8). ER and Ki67 immunohistochemistry was performed by the Peter MacCallum Cancer Centre Anatomical Pathology department using standard protocols. ER was scored using the Allred system.20 Ki67 was scored using the ImageScope software (Leica Microsystems) and the Nuclear v9 algorithm to estimate the number of positively staining nuclei. Ki67 was scored as low if <12% of cells were positive, high if >19% of cells were positive and equivocal if between 12 and 19% of cells were positive.
Results
DCIS cases (n=48) were ascertained from a population-based mammographic screening cohort with clinical follow-up available15 (Table 1, Supplementary Table S1). All cases were treated by surgical wide excision only and were selected to enrich for high-grade cases (77% of cases compared with 58% in the full cohort15). DNA was extracted and copy number was obtained by using SNP arrays (Figure 1). For comparisons, we used our previously published analysis of DCIS synchronous with invasive breast cancer analysed on the same platform10 and invasive breast cancer cases from the META-BRIC study,19 TCGA18 and from a study of early-stage breast cancer also using the OncoScan assay (Brewster cohort17).

Copy number analysis of pure DCIS. (a) Frequency plots of copy number gain (blue) and loss (red) in pure DCIS, DCIS synchronous with invasive breast cancer and invasive breast cancer.17 Below are difference plots comparing pure DCIS with invasive breast cancer and pure DCIS with DCIS synchronous to invasive breast cancer. Blue line is the difference in gain frequency; red is the difference in loss frequency. Regions of gain that are more frequent in pure DCIS are below the line, and regions of loss more frequent in pure DCIS are above the line; vice versa for the comparator data set. Bars underneath show significant regions of difference at P<0.05 (uncorrected), with a difference of >25% for the synchronous comparison and >15% for the invasive breast cancer comparisons. Copy number spikes in the TCGA data are copy number polymorphisms generally not detectable on the lower resolution Oncoscan array. (b) Fraction of the genome altered by copy number. Open circles (non-recurrent) and open triangles (synchronous) represent low-grade DCIS. Open squares (recurrent) indicate non-clonal cases. Error bars indicate mean and s.d. (c) Difference plot for chromosome 17 showing the increase in frequency of gain in pure DCIS compared with invasive breast cancer at ERBB2 (arrow) and also more distally.
Comparison of Pure DCIS to Synchronous DCIS and Invasive Breast Cancer
In pure DCIS, the fraction of the genome affected by copy number alterations ranged from 0.03 to 55% with a diverse range of recurrent gains and losses observed (Figure 1). When compared to CN data from three published cohorts of invasive breast cancer, the pure DCIS cases were broadly similar in the frequency of copy number alteration at specific loci (Figure 1) and the overall fraction of the genome altered (Table 1). The exception was the META-BRIC cohort, which had a lower fraction altered than all other groups; however, examination of this cohort using the ASCAT algorithm21 suggests that many cases had a high level of normal cell contamination. The overall pattern of gains and losses was similar between pure DCIS and invasive breast cancer with commonalities such as frequent gains on 8q and 16p and losses on 8p and 17p. The most consistent differences were lower frequency of pure DCIS cases with 1q gain and more DCIS cases with 17q gain, including ERBB2 as well as more distal regions (Figure 1, Supplementary Table S2).
As we have previously reported, synchronous DCIS generally appears to be an advanced lesion with most of the same copy number changes as the synchronous invasive cancer;9, 10, 11 this likely represents an entity with invasive capacity that is potentially quite distinct from pure DCIS. The synchronous DCIS cases (n=18) had a higher fraction genome altered with an average of 29% of the genome affected compared with 17.5% of the pure DCIS cases (Figure 1b; P=0.004, Mann–Whitney two-tailed). Compared with the synchronous DCIS, the pure DCIS had fewer 1q, 5p and 8q gains, and 1p, 6q, 9q and 13q losses, but a higher frequency of 20q gains. However, the synchronous cases had a higher proportion of ER-negative cases (61 vs 21%, P<0.01 Fisher’s exact test) and a slightly different distribution of grade (P=0.05, 3 × 2 Fisher’s exact test), which may have confounded these genomic comparisons.
Clonal Analysis of Primary and Recurrent Tumours
Within the cohort were included patients who had an ipsilateral recurrence of DCIS (n=9), DCIS with a microinvasive component (n=2), invasive breast cancer with DCIS (n=5) or invasive breast cancer alone (n=1) within 5 years of the primary cancer diagnosis (n=17, Supplementary Table S3). We were able to obtain copy number data from eight of the recurrent tumours; six DCIS, one invasive breast cancer and one mixed DCIS and invasive breast cancer. Compared with the matched primary tumours, there was no significant difference in the fraction genome altered (Wilcoxon matched-paired test, Supplementary Figure S1). However, there was a large variation in the copy number altered base pairs showing the same event (gain or loss) between the matched pairs, ranging from 11 to 58%. Six cases showed clear clonal copy number events, including shared breakpoints (Supplementary Figure S2). The case with mixed DCIS/invasive breast cancer recurrence showed a clonal relationship of the primary with the invasive breast cancer but not the DCIS part of the recurrence, suggesting that the latter was a new primary (Supplementary Figure S1). One primary case with a DCIS recurrence (T12664/T38458) only shared a gain of 16p with a similar peri-centromeric breakpoint (but did not share imbalance of the same alleles), and both tumours had multiple distinct copy number events (only 11% shared). A final case with invasive breast cancer only recurrence (P15661/P17661) shared 16% of copy number changes but none of these had shared breakpoints and may comprise random overlap of common events.
For the remaining 9 cases with no recurrence array data, 6/8 pairs shared the same grade (one case lacked information on the recurrence). For 5 cases with immunohistochemistry available, 3/3 pairs shared profiles (ER, Ki67 and HER2) consistent with a clonal relationship, and a further 2/2 pairs shared the same ER status. We also obtained ddPCR data for four cases: three showed concordant gains, while one showed one concordant and one discordant gain. Thus the majority of cases (15/17) lacked evidence to exclude a clonal relationship between primary and recurrence. Two cases (12%) appeared not to share a clonal relationship by copy number array and were excluded from subsequent analyses.
Identification of Copy Number Events Associated with Recurrence
We selected a subset of intermediate or high-grade cases of DCIS with and without recurrence (n=15 and 25, respectively) to identify copy number changes associated with recurrence. Six low-grade non-recurrent cases were excluded on the basis that no low-grade tumours were present in the recurrent cohort and copy number differences between high- and low-grade DCIS have been reported.22, 23, 24 There were no differences in grade, ER status, Ki67 staining and patient age between the two groups, although tumour size was slightly larger in the recurrence cohort (P=0.04, Table 2). The fraction genome altered was significantly different (P=0.03, Mann–Whitney two-tailed) between the non-recurrent (15.7%) and recurrent cases (23.7%), and it was notable that only 2/15 of the recurrent cases had a fraction genome altered <10% compared with 12/25 of the non-recurrent cases (P=0.04, Fisher’s exact test).
Comparison of the copy number events between the recurrence and non-recurrence groups identified a number of differences (Figure 2, Table 3,Supplementary Table S4). The most significant (P<0.001) were gain of 20q (Figure 2b) and loss on 15q and 10q. These differences were also reflected in the allelic imbalance, with significantly different regions identified on 10, 15q and 20q. Allelic imbalance on chromosome 10 affected most of the chromosome but was unusual in that it was not associated strongly with copy number status (Figure 2c). Copy number gain was somewhat more common on 10p (9/15 gain vs 6/15 loss or copy number neutral allelic imbalance), while copy number loss was more common on 10q (8/14 loss vs 6/14 gain or neutral).

Comparison of non-recurrent with recurrent DCIS. (a) Frequency plots of copy number gain (blue) and loss (red) in DCIS without recurrence and DCIS with recurrence within 5 years. Below is the difference plot comparing the two groups. Blue line is the difference in gain frequency; red is the difference in loss frequency. Regions of gain that are more frequent in recurrent DCIS are below the line, and regions of loss more frequent in pure DCIS are above the line; vice versa for the non-recurrent cases. Bars underneath show significant regions of difference at P<0.05 (uncorrected), with a difference of >25%. (b) Difference plot for chromosome 20 showing the increase in frequency of gain in recurrent DCIS. (c) Difference plot for chromosome 10 showing the increase in frequency of allelic imbalance (purple) in recurrent DCIS.
We considered whether there were any copy number differences between DCIS that recurred as DCIS (n=8) or as invasive/microinvasive disease (n=7) (Supplementary Figure S3). There were no striking differences although gain of 20p12 and 20q11 were slightly higher in DCIS recurring as invasive breast cancer (P<0.01), while 2p24–25 gain and 17p loss were more frequent in DCIS recurring as DCIS (P<0.05).
ERBB2 Status: Differences in Recurrence Cases and Copy Number Events
It has previously been shown that HER2 status is prognostic for DCIS,25, 26 thus we assessed this locus specifically in our cohort. Copy number gain (including low-level gain) at ERBB2 was significantly different between recurrence and non-recurrence cases (P=0.02, Fisher's exact test), with 12/15 (80%) recurrence and 10/25 (40%) non-recurrence cases showing gain. When considering high-level gain only, 9/15 (60%) recurrence and 8/25 (32%) non-recurrence cases were highly amplified at ERBB2 (P=0.1, Fisher's exact test). In this data set, therefore, copy number amplification at ERBB2 is not strongly associated with recurrence overall. However, previous studies have found a stronger association of ERBB2 amplification with DCIS that recurred as DCIS compared with DCIS recurring as invasive breast cancer, and indeed when we only consider purely DCIS recurrences, 6/8 cases were highly amplified for ERBB2 compared with 8/25 non-recurrences (P=0.047, Fisher's exact test). This result is consistent with the fewer invasive cases with ERBB2 amplification (13% overall) compared with pure DCIS (35%) (all invasive breast cancer cohorts P<0.001, Fisher’s exact test).
We compared DCIS cases with and without recurrence after stratifying by ERBB2 copy number amplification status, identifying a number of differences distinct from the overall comparison (Supplementary Figures S4 and S5, Supplementary Table S2). For example, within ERBB2+ cases, those with recurrence had more frequent gain of 11q, including CCND1 (P=0.01), while for the ERBB2− recurrence cases 7p11 gain was more frequent (at EGFR, P=0.008). Intriguingly, 13/15 (87%) DCIS with recurrence had either ERBB2 high-level amplification or EGFR gain, compared with 9/25 (36%) non-recurrences (P=0.003). We evaluated EGFR staining by immunohistochemistry in three recurrent cases with chromosome 7 gain. Two cases with low-level gain were EGFR negative; however, one case with a high-level gain of EGFR showed some protein positivity in a low percentage of cells (<10%). These results suggest that either EGFR is not the target of the chromosome 7 gain in these cases and/or that the gains were sub-clonal and not represented in the sections used for immunohistochemistry.
If gain of 11q or 7p was a marker of DCIS aggressiveness, we might predict a higher percentage of cases with gain of these regions in invasive cohorts compared with DCIS. However, when stratifying the invasive cases by ERBB2 copy number status, the frequency of 11q or 7p copy number gain was not increased in any invasive breast cancer cohort compared with DCIS (Supplementary Figures S6 and S7).
ER Status is Associated with Different Copy Number Changes in DCIS Recurrence Cases
We stratified the pure DCIS cohort by ER status and repeated the comparison between recurrent and non-recurrent cases. The strongest differences within ER− cases were loss on 15q25.3 and allelic imbalance on 10p15.1 (P=0.005 for both, Supplementary Figure S10), but other differences were not as strong. Inversely, in ER+ cases most differences were maintained; however, losses at 15q and allelic imbalance on chromosome 10 were less significant. Thus the changes on chromosomes 10 and 15 may be more specific to ER− cases. Consistent with this idea, 10p gain and 15q loss are more frequently observed in ER− invasive breast cancer compared with ER+ invasive breast cancer and also more frequently in ER− invasive breast cancer compared with ER− DCIS (Supplementary Figures S8 and S9). Thus genes on 10p and 15q could be important for aggressiveness and/or invasive potential in ER− DCIS cases.
Subgroup Comparisons of DCIS Copy Number
We performed sub-analyses on the pure DCIS cohort, comparing copy number differences by grade, ER status and ERBB2 copy number amplification status and looking for consistencies with published invasive breast cancer copy number data (Supplementary Figure S11, Supplementary Table S5). The differences between groups within DCIS were often also observed in the invasive breast cancer data, for example increased 16p gain in ER+ cases and more frequent 8q gains and 5q, 14q and 15q losses in ER− cases. High-grade cases had more frequent 8q gain but less 16q loss compared with low-grade cases, similar to previous reports.22, 23, 24 The ERBB2+ DCIS cases shared increased gains on 17q and losses of 8p with the ERBB2+ invasive breast cancer cases, but the consistent increase in 8q gain in the ERBB2+ invasive breast cancer cases was not statistically significant in the DCIS cases. This might suggest that gain of this chromosome arm differentiates the type of ERBB2+ cases that have become invasive.
Discussion
Our study has been performed on a carefully selected cohort, comprising tumours treated only with wide local excision but all with clear margins, no radiotherapy and either with at least 7 years of follow-up or with a recurrence at least 12 months but <5 years after initial surgery. The cohort has enabled us to undertake a comparison of copy number between DCIS that have recurred and those that have not. This is the first such study to be undertaken and only the second study to report copy number changes in DCIS with follow-up data. The only previous study used cytogenetic CGH to compare 18 DCIS cases with their recurrences and found 1/18 cases to be non-clonal.27 However, they did not compare their data to non-recurrent cases, and in addition, their cohort included a range of grades and, most critically, cases with involved margins. The presence of DCIS in the margins is a prognostic factor (reviewed by Benson and Wishart4) that we have removed in our current study. Nonetheless, the high proportion (6/8) of DCIS sharing clonal genetic changes with the recurrences in our study implies that some tumorigenic cells must be left behind after surgery and are able to re-establish a tumour within 5 years. Identification of DCIS cases with or without the capacity for such regeneration from a small population of remaining cells would enable stratification for radiotherapy. The cases analysed here were not treated with radiotherapy; therefore, it is not certain whether the regions we identified would be predictive of a good outcome under radiotherapy conditions. This possibility will be important to test in the future.
In this study, DCIS with recurrence had significantly more copy number changes overall and specifically had a higher frequency of 20q gain, 15q loss, chr10 allelic imbalance and, to a lesser extent, ERBB2 amplification. There are many plausible cancer-driver genes within these regions, such as ZNF217,28, 29 NCOA3,30 GNAS31 and AURKA32 on 20q, GATA3 on 10p18, 33 and NTRK3 on 15q.34 Sub-analyses identified CCND1 amplification to be enriched in ERBB2-amplified recurrence cases. All of these genes have previously been implicated in breast cancer pathogenesis, and NCOA3 has recently been shown to be a critical gene in the development and maintenance of DCIS.35
The allelic imbalance identified on chromosome 10 was somewhat complex. The signal was strongest on the p arm, where cases had gains (3 non-recurrent, 7 recurrent), losses (1 non-recurrent, 3 recurrent) and copy number neutral allelic imbalance (3 recurrent). On the q arm, recurrent cases with allelic imbalance were more likely to also have copy number loss (n=6) or no copy number change (n=2), while non-recurrent cases had a mixture of allelic imbalance with gain (n=2), loss (n=2) and neutrality (n=2). Thus it is not clear what gene(s) are being targeted. Gain of 10p is most commonly seen in Basal-type invasive breast cancer, but the underlying gene(s) have not been identified.19 It is possible that the allelic imbalance signal observed is a combination of gain of an unknown oncogene on 10p, loss of some tumour-suppressor gene such as PTEN (10q) and possibly selection for a sequence variation on either 10p or 10q, such as in GATA3 or FGFR2. Regions of allelic imbalance, gain or loss were often non-overlapping from case to case, suggesting that more than one gene may be the target across multiple samples. This complex pattern requires many more samples to be analysed with a technique such as massively parallel sequencing that would enable all possibilities to be examined simultaneously.
We found that DCIS cases with recurrence had a higher level of genomic instability than those without recurrence. In a recent aCGH study, large (>40 mm) high-grade DCIS without invasion were compared with small (<15 mm) high-grade DCIS.36 The smaller tumours had a slightly higher overall level of genomic aberrations and non-significant increases in 8q and 20q gain frequency, the latter present in 27% of large and 48% of small cases. This result suggested that large tumours without progression, and thus presumably with a less aggressive phenotype, had fewer copy number changes, similar to that observed in our study for non-recurrent cases.
Levels of genome instability have previously been associated with both better and worse clinical outcome in a variety of tumour types. On the one hand, ongoing genetic instability may predispose to better outcome due to sensitivity to genotoxic chemotherapeutic agents, as suggested for BRCA1/BRCA2-deficient ovarian carcinomas.37 In contrast, clonal evolution theory supports the concept of a high level of preexisting genetic variations and tumour heterogeneity providing multiple options for clonal selection under stress. In breast cancer, this dichotomy is supported by a chromosomal instability expression signature that showed a better prognosis for the fourth quartile (most genetically unstable profile) but a worse prognosis for the third quartile.38 In other studies, image cytometry has found breast cancers with high and more heterogeneous DNA content to have worse outcome,39, 40 and specific patterns of copy number variation identified by array CGH have also been correlated with survival.41 In the context of DCIS untreated by radiotherapy or chemotherapy, a higher level of genomic instability may provide greater genetic diversity to promote survival of disseminated cells after surgery. This possibility is supported by the highly divergent copy number profiles of some of the later recurrences compared with the primary tumours, where, despite a clear clonal origin, multiple unique copy number events in both tumours are indicative of high tumour heterogeneity.
The principal objective of this study was to identify regions of genomic aberration in DCIS that might predict recurrence following surgery. No previous studies have attempted to find such regions; however, immunohistochemical markers and gene expression profiles have been evaluated. In some of the largest studies to date, ERBB2, COX2 and p16 have emerged as the most promising candidates.25, 26, 42 As in our study, ERBB2 positivity appears to be more predictive for recurrence as DCIS rather than invasive breast cancer. A recent study using immunohistochemistry as a surrogate for intrinsic subtyping was unable to find a prognostic signature for DCIS, despite its relatively large size.43 The difficulty in implementing predictive markers for DCIS remains the availability of large cohorts with consistent treatment for validation, and our study is no exception. Despite starting with a database of >400 screen-detected patients, we were only able to retrospectively identify <20 suitable DCIS cases that had recurred in under 5 years, had tissue available, clear margins and high grade. A concerted prospective international effort will be required to validate the regions we have identified and evaluate their usefulness in a clinical setting. Validation would be facilitated by the development of in situ assays to extend the number of cases beyond those able to be microdissected for DNA.
References
Sanders ME, Schuyler PA, Dupont WD et al. The natural history of low-grade ductal carcinoma in situ of the breast in women treated by biopsy only revealed over 30 years of long-term follow-up. Cancer 2005;103:2481–2484.
Lopez-Garcia MA, Geyer FC, Lacroix-Triki M et al. Breast cancer precursors revisited: molecular features and progression pathways. Histopathology 2010;57:171–192.
Leonard GD, Swain SM . Ductal carcinoma in situ, complexities and challenges. J Natl Cancer Inst 2004;96:906–920.
Benson JR, Wishart GC . Predictors of recurrence for ductal carcinoma in situ after breast-conserving surgery. Lancet Oncol 2013;14:e348–e357.
Boxer MM, Delaney GP, Chua BH . A review of the management of ductal carcinoma in situ following breast conserving surgery. Breast 2013;22:1019–1025.
Donker M, Litiere S, Werutsky G et al. Breast-conserving treatment with or without radiotherapy in ductal carcinoma In Situ: 15-year recurrence rates and outcome after a recurrence, from the EORTC 10853 randomized phase III trial. J Clin Oncol 2013;31:4054–4059.
Andre F, Pusztai L . Molecular classification of breast cancer: implications for selection of adjuvant chemotherapy. Nat Clin Pract Oncol 2006;3:621–632.
Ciriello G, Miller ML, Aksoy BA et al. Emerging landscape of oncogenic signatures across human cancers. Nat Genet 2013;45:1127–1133.
Liao S, Desouki MM, Gaile DP et al. Differential copy number aberrations in novel candidate genes associated with progression from in situ to invasive ductal carcinoma of the breast. Genes Chromosomes Cancer 2012;51:1067–1078.
Johnson CE, Gorringe KL, Thompson ER et al. Identification of copy number alterations associated with the progression of DCIS to invasive ductal carcinoma. Breast Cancer Res Treat 2012;133:889–898.
Hernandez L, Wilkerson PM, Lambros MB et al. Genomic and mutational profiling of ductal carcinomas in situ and matched adjacent invasive breast cancers reveals intra-tumour genetic heterogeneity and clonal selection. J Pathol 2012;227:42–52.
Allred DC, Clark GM, Molina R et al. Overexpression of HER-2/neu and its relationship with other prognostic factors change during the progression of in situ to invasive breast cancer. Hum Pathol 1992;23:974–979.
Tamimi RM, Baer HJ, Marotti J et al. Comparison of molecular phenotypes of ductal carcinoma in situ and invasive breast cancer. Breast Cancer Res 2008;10:R67.
Gao Y, Niu Y, Wang X et al. Genetic changes at specific stages of breast cancer progression detected by comparative genomic hybridization. J Mol Med 2009;87:145–152.
Fong J, Kurniawan ED, Rose AK et al. Outcomes of screening-detected ductal carcinoma in situ treated with wide excision alone. Ann Surg Oncol 2011;18:3778–3784.
Wang Y, Moorhead M, Karlin-Neumann G et al. Analysis of molecular inversion probe performance for allele copy number determination. Genome Biol 2007;8:R246.
Brewster AM, Thompson P, Sahin AA et al. Copy number imbalances between screen- and symptom-detected breast cancers and impact on disease-free survival. Cancer Prev Res (Phila) 2011;4:1609–1616.
Cancer Genome Atlas Network. Comprehensive molecular portraits of human breast tumours. Nature 2012;490:61–70.
Curtis C, Shah SP, Chin SF et al. The genomic and transcriptomic architecture of 2,000 breast tumours reveals novel subgroups. Nature 2012;486:346–352.
Harvey JM, Clark GM, Osborne CK et al. Estrogen receptor status by immunohistochemistry is superior to the ligand-binding assay for predicting response to adjuvant endocrine therapy in breast cancer. J Clin Oncol 1999;17:1474–1481.
Van Loo P, Nordgard SH, Lingjaerde OC et al. Allele-specific copy number analysis of tumors. Proc Natl Acad Sci USA 2010;107:16910–16915.
Buerger H, Otterbach F, Simon R et al. Comparative genomic hybridization of ductal carcinoma in situ of the breast-evidence of multiple genetic pathways. J Pathol 1999;187:396–402.
Hwang ES, DeVries S, Chew KL et al. Patterns of chromosomal alterations in breast ductal carcinoma in situ. Clin Cancer Res 2004;10:5160–5167.
Vos CB, ter Haar NT, Rosenberg C et al. Genetic alterations on chromosome 16 and 17 are important features of ductal carcinoma in situ of the breast and are associated with histologic type. Br J Cancer 1999;81:1410–1418.
Kerlikowske K, Molinaro AM, Gauthier ML et al. Biomarker expression and risk of subsequent tumors after initial ductal carcinoma in situ diagnosis. J Natl Cancer Inst 2010;102:627–637.
Rakovitch E, Nofech-Mozes S, Hanna W et al. HER2/neu and Ki-67 expression predict non-invasive recurrence following breast-conserving therapy for ductal carcinoma in situ. Br J Cancer 2012;106:1160–1165.
Waldman FM, DeVries S, Chew KL et al. Chromosomal alterations in ductal carcinomas in situ and their in situ recurrences. J Natl Cancer Inst 2000;92:313–320.
Littlepage LE, Adler AS, Kouros-Mehr H et al. The transcription factor ZNF217 is a prognostic biomarker and therapeutic target during breast cancer progression. Cancer Discov 2012;2:638–651.
Nonet GH, Stampfer MR, Chin K et al. The ZNF217 gene amplified in breast cancers promotes immortalization of human mammary epithelial cells. Cancer Res 2001;61:1250–1254.
Anzick SL, Kononen J, Walker RL et al. AIB1, a steroid receptor coactivator amplified in breast and ovarian cancer. Science 1997;277:965–968.
Garcia-Murillas I, Sharpe R, Pearson A et al. An siRNA screen identifies the GNAS locus as a driver in 20q amplified breast cancer. Oncogene 2014;33:2478–2486.
Zhou H, Kuang J, Zhong L et al. Tumour amplified kinase STK15/BTAK induces centrosome amplification, aneuploidy and transformation. Nat Genet 1998;20:189–193.
Usary J, Llaca V, Karaca G et al. Mutation of GATA3 in human breast tumors. Oncogene 2004;23:7669–7678.
Jin W, Kim GM, Kim MS et al. TrkC plays an essential role in breast tumor growth and metastasis. Carcinogenesis 2010;31:1939–1947.
Ory V, Tassi E, Cavalli LR et al. The nuclear coactivator amplified in breast cancer 1 maintains tumor-initiating cells during development of ductal carcinoma in situ. Oncogene 2014;33:3033–3042.
Hwang ES, Lal A, Chen YY et al. Genomic alterations and phenotype of large compared to small high-grade ductal carcinoma in situ. Hum Pathol 2011;42:1467–1475.
Wang ZC, Birkbak NJ, Culhane AC et al. Profiles of genomic instability in high-grade serous ovarian cancer predict treatment outcome. Clin Cancer Res 2012;18:5806–5815.
Birkbak NJ, Eklund AC, Li Q et al. Paradoxical relationship between chromosomal instability and survival outcome in cancer. Cancer Res 2011;71:3447–3452.
Auer GU, Caspersson TO, Wallgren AS . DNA content and survival in mammary carcinoma. Anal Quant Cytol 1980;2:161–165.
Kronenwett U, Ploner A, Zetterberg A et al. Genomic instability and prognosis in breast carcinomas. Cancer Epidemiol Biomarkers Prev 2006;15:1630–1635.
Hicks J, Krasnitz A, Lakshmi B et al. Novel patterns of genome rearrangement and their association with survival in breast cancer. Genome Res 2006;16:1465–1479.
Generali D, Buffa FM, Deb S et al. COX-2 expression is predictive for early relapse and aromatase inhibitor resistance in patients with ductal carcinoma in situ of the breast, and is a target for treatment. Br J Cancer 2014;111:46–54.
Zhou W, Jirstrom K, Amini RM et al. Molecular subtypes in ductal carcinoma in situ of the breast and their relation to prognosis: a population-based cohort study. BMC Cancer 2013;13:512.
Acknowledgements
This work was supported by the Victorian Breast Cancer Research Consortium and the Australian National Health and Medical Research Council (NHMRC, No. 509050).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Supplementary Information accompanies the paper on Modern Pathology website
Supplementary information
Rights and permissions
About this article

Cite this article
Gorringe, K., Hunter, S., Pang, JM. et al. Copy number analysis of ductal carcinoma in situ with and without recurrence. Mod Pathol 28, 1174–1184 (2015). https://doi.org/10.1038/modpathol.2015.75
Received:
Revised:
Accepted:
Published:
Issue date:
DOI: https://doi.org/10.1038/modpathol.2015.75
This article is cited by
-
Genomic analysis defines clonal relationships of ductal carcinoma in situ and recurrent invasive breast cancer
Nature Genetics (2022)
-
The prognostic significance of wild-type isocitrate dehydrogenase 2 (IDH2) in breast cancer
Breast Cancer Research and Treatment (2020)
-
Thioredoxin-interacting protein is an independent risk stratifier for breast ductal carcinoma in situ
Modern Pathology (2018)
-
Utilization of the oncoscan microarray assay in cancer diagnostics
Applied Cancer Research (2017)
-
Copy number analysis by low coverage whole genome sequencing using ultra low-input DNA from formalin-fixed paraffin embedded tumor tissue
Genome Medicine (2016)

{kind=link}
{kind=link}