
Abstract
The diagnosis of malignant peripheral nerve sheath tumor remains challenging, especially in the sporadic setting. Malignant peripheral nerve sheath tumor is a rare malignancy, and owing to the lack of specific histological criteria, immunohistochemical and molecular diagnostic markers, several differential diagnoses must be considered, in particular melanoma. Recently, inactivation of the polycomb repressive complex 2 (PRC2), induced by inactivating mutations in two of its critical constituents SUZ12 and EED, was reported in a large subset of malignant peripheral nerve sheath tumors. Homozygous PRC2 inactivation induces complete loss of trimethylation at lysine 27 of histone 3 (H3K27me3). Recent studies suggest that complete loss of H3K27me3 is highly specific for malignant peripheral nerve sheath tumor and may be a useful immunohistochemical diagnostic marker. Therefore, to determine the specificity of the complete loss of H3K27me3 expression in the context of the differential diagnosis of malignant peripheral nerve sheath tumor from melanoma (its major potential mimic), we performed H3K27me3 immunohistochemistry in a pathologically and genetically well-characterized cohort of primary (neurofibromatosis type 1 (NF1), radiation-associated and sporadic context) malignant peripheral nerve sheath tumors (n=122) and in a cohort or primary (desmoplastic) and metastatic melanomas (n=265). In total, 88 (72%) malignant peripheral nerve sheath tumors, including 46 (71%) NF1-associated, 4 (100%) radiation-associated, and 38 (72%) sporadic tumors, showed complete loss of H3K27me3. We observed increased loss of H3K27me3 with increasing histological grade. Interestingly, we found complete loss of H3K27me3 in 37% (n=98) of all melanomas, including 25% (n=9) of primary desmoplastic melanomas. Moreover, partial loss ('mosaic' pattern) was observed in 23 (19%) of all malignant peripheral nerve sheath tumors and in 136 (51%) of all melanomas. Complete loss of H3K27me3 detected by immunohistochemistry is not specific for malignant peripheral nerve sheath tumor and cannot be used safely when distinguishing malignant peripheral nerve sheath tumor from melanoma.
Similar content being viewed by others


Main
Malignant peripheral nerve sheath tumors, which account for ~5% of soft tissue sarcomas,1 are spindle cell sarcomas that are rare, aggressive, and locally invasive with poor prognosis. Up to 50% of malignant peripheral nerve sheath tumors arise in patients with neurofibromatosis type 1 (NF1), fewer than 10% of malignant peripheral nerve sheath tumors are radiation associated, and the remainder are without a known genetic predisposition ('sporadic' form).2 Establishing the correct diagnosis of malignant peripheral nerve sheath tumor remains a challenge not only due to morphological and immunohistochemical complexity (particularly outside the NF1 context) but also because a number of other sarcomas, benign soft tissue tumors, and non-epithelial tumors (especially melanoma) can appear similar to malignant peripheral nerve sheath tumors. Recently, studies have identified loss of trimethylation at lysine 27 of histone H3 (H3K27me3) in a subset of malignant peripheral nerve sheath tumors reflecting inactivation of the polycomb repressive complex 2 (PRC2), resulting from inactivating mutations of its constituents SUZ12 or EED.3, 4, 5 Mutations in EED or SUZ12 were mutually exclusive and identified in 23–70% of NF1-associated, 90% of radiation-associated, and 36–92% of sporadic malignant peripheral nerve sheath tumors.4, 5 Therefore, recent studies suggest that loss of H3K27me3 detected by immunohistochemistry may be helpful in the distinction of malignant peripheral nerve sheath tumors from other malignant spindle cell neoplasms.6, 7, 8 However, it is unclear whether H3K27me3 loss is specific for malignant peripheral nerve sheath tumor, as loss of this protein has also been reported recently in some other sarcomas, especially synovial sarcoma and dermatofibrosarcoma protuberans, and in other malignant spindle cell tumors (rare subset of spindle cell melanoma).9, 10
In the current study, we evaluated the potential role of H3K27me3 immunohistochemical expression in daily routine practice to distinguish malignant peripheral nerve sheath tumor from its main morphological mimicker, namely melanoma. H3K27me3 immunohistochemistry was performed in a large series of 122 primary malignant peripheral nerve sheath tumors and 265 melanomas of different variants and stages.
Materials and methods
Malignant Peripheral Nerve Sheath Tumor Patients and Sample Selection
Our cohort consisted of a total of 122 samples from 122 malignant peripheral nerve sheath tumor patients. All samples originated from primary malignant peripheral nerve sheath tumors. Eighty-nine patients were previously included in the study by Le Guellec et al.11 As described in that study, the diagnosis of malignant peripheral nerve sheath tumor is difficult and has historically suffered from a lack of entirely specific histological criteria. It is therefore likely that a variety of non-nerve sheath tumors have been placed incorrectly in this category over the years. To obtain a homogeneous, robust and relevant cohort of definitive primary malignant peripheral nerve sheath tumors, we systematically performed a histologic review, immunohistochemistry, and molecular analysis, if relevant. The precise strategy is described in the 'Materials and methods' of that study.11 Thirty-three additional patients with primary malignant peripheral nerve sheath tumor were included between 1 January 2014 and 15 September 2016 with the same diagnostic strategy.11 Patients were treated at 11 participating cancer centers and entered either into the Conticabase (European retrospective clinicopathologic database on sarcomas, https://conticabase.sarcomabcb.org) or the NetSarc (National prospective database, https://netsarc.sarcomabcb.org). Ethics approval from the appropriate committees was obtained.
For the purpose of this study, we classified sporadic malignant peripheral nerve sheath tumor cases as 'certain' when the diagnosis of malignant peripheral nerve sheath tumor was certain (tumor originating in a peripheral nerve or in a pre-existing nerve sheath tumor, usually a neurofibroma) or focal S100 protein/SOX10 positivity in the absence of other markers and absence of specific simple genomic alteration), or as 'probable' when histologic features was typical but supportive immunohistochemical and clinical data were lacking (not originating from a nerve or benign nerve sheath tumor, absence of S100 protein/SOX10 expression, absence of other immunohistochemical markers, and absence of specific simple genomic alteration). Data regarding patients’ characteristics and tumor description were obtained by a retrospective review of the medical records. These records and histologic data were entered into a centralized database (https://conticabase.sarcomabcb.org). The following variables were compared among the three cohorts (NF1-associated, radiation-associated, and sporadic malignant peripheral nerve sheath tumor): NF1 status, age at diagnosis, gender, tumor location, tumor size, depth location, histologic subtype, and tumor grade (FNCLCC). Epithelioid malignant peripheral nerve sheath tumors were excluded from this study.
Five tissue microarrays were constructed with a 1.5-mm diameter tissue core from each formalin-fixed paraffin-embedded tumor tissue sample of the 122 malignant peripheral nerve sheath tumors using a semiautomatic tissue arrayer (Alphelys, Plaisir, France). At least three cores from each tumor were sampled to outweigh intratumoral heterogeneity. Each tissue microarrays contained additional cores of variable tissue type (tonsil, kidney), both as internal control for immunohistochemistry and for the purpose of orientation.
Melanoma Sample Selection
To test the specificity of this marker, we analyzed different variants of melanomas (desmoplastic/spindle cells melanoma, superficial spreading melanoma (SSM) and nodular melanoma), melanomas at different stages (primary and metastatic), and tested tissue microarray slides, as well as whole-tissue sections (WTS). WTS of 19 primary desmoplastic/spindle cell melanomas, a tissue microarrays containing a total of 8 primary melanomas (6 SSM and 2 nodular), and a tissue microarrays of 25 metastatic melanomas (16 lymph node and 9 skin) from the same number of patients were collected from the Department of Pathology at IUCT-Oncopole (Toulouse, France). Samples were analyzed on at least three representative 1.5-mm cores taken from each donor block following the selection of regions of interest by a pathologist. WTS of nine primary desmoplastic/spindle cell melanomas were collected from the Department of Pathology at Curie Institut (Paris, France). A tissue microarrays containing a total of 204 metastatic melanomas (71 lymph nodes, 55 skin, 33 brain, 15 lung, 14 small bowel, 6 liver, 4 parotid, 2 bone, 2 nasal sinus, 1 breast, and 1 spleen) from the same number of patients were collected from the Department of Pathology at La Timone Hospital (Marseille, France). Samples were analyzed on at least four representative 0.6-mm cores taken from each donor block following the selection of regions of interest by a pathologist.
Immunohistochemistry
Five tissue microarrays slides of the 122 cases of malignant peripheral nerve sheath tumor, WTS slides of the 40 cases of malignant peripheral nerve sheath tumor (matched with those in tissue microarrays of malignant peripheral nerve sheath tumor), WTS slides of the 28 cases of primary desmoplastic/spindle cell melanomas, tissue microarrays slides of the 8 cases of primary melanomas and tissue microarrays slides of 229 cases of metastatic melanomas were deparaffinized in xylene, hydrated in alcohol, and baked in a microwave (30 min in Tris-buffer, pH 9). Endogenous peroxidase was blocked. H3K27me3 rabbit monoclonal antibody (clone C36B11, dilution 1:200; Cell Signaling Technology, Danvers, MA) was used and staining was performed on the Benchmark Ultra-automated Stainer (Ventana) using diaminobenzidine as the chromogen. Immunohistochemical expression was quantitatively scored according to the percentage of nuclear-positive tumor cells. The proportion of tumor cells showing nuclear staining was semiquantitatively recorded as 'complete loss' (<5%), 'partial loss' (≥5 and <90%), and 'intact/retained' (≥90%). The presence of positive internal controls (inflammatory or endothelial cells) was strictly required to assess the immunohistochemical signal. Samples not presenting these positive internal controls were excluded. A positive control was tested on each run of immunohistochemical analysis.
In the 28 cases of primary desmoplastic/spindle cell melanomas, additional antibody against SOX10 (clone EP268, dilution 1:250; Epitomics, Burlingame, CA, USA) was used.
Statistical Analysis
Data were described as median (range) for quantitative and by frequencies and percentages for qualitative. Comparisons between groups were performed using the Kruskall–Wallis test for continuous data and the χ2 test (or Fisher's exact) for qualitative data. All P-values reported were two-sided. For all statistical tests, differences were considered significant at the 5% level. Statistical analyses were performed using the STATA 11.0 Software.
Results
Clinicopathological Characteristics of Malignant Peripheral Nerve Sheath Tumors
Patient characteristics of 122 malignant peripheral nerve sheath tumors are presented in Table 1. The mean and median age at diagnosis was 44 and 41.5 years, respectively (range, 7–94 years). The average age at diagnosis varied depending on the clinical setting of malignant peripheral nerve sheath tumors. Patients with NF1 syndrome were younger compared with those with sporadic malignant peripheral nerve sheath tumors (median age, 37 vs 52.5 years). In radiation-related malignant peripheral nerve sheath tumor patients, tumors presented at an older age compared with NF1-associated malignant peripheral nerve sheath tumors, with a mean of 41 years (range, 20–68 years).
H3K27me3 Immunohistochemical Expression in Malignant Peripheral Nerve Sheath Tumors
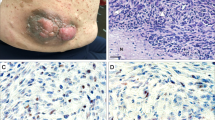
The results of the immunohistochemical analysis are summarized in Tables 1 and 2. Seventy-two percent (n=88) of all malignant peripheral nerve sheath tumors demonstrated complete loss of staining with the anti-H3K27me3 monoclonal antibody (Figures 1b,2b, 2c and 3b, 3c, 3e, 3f). Whereas sporadic and NF1-associated malignant peripheral nerve sheath tumors showed a comparable frequency of loss of H3K27 trimethylation (72% vs 71% respectively), all radiation-associated malignant peripheral nerve sheath tumors (n=4) exhibited loss of H3K27me3 expression. In addition, a heterogeneous staining pattern with a subset of tumor cells (≥ 5 and <90%) showing loss of staining for H3K27me3 ('partial loss') was observed in 12 of 65 (18%) NF1-associated (Figures 1d and e) and in 11 of 53 (20%) sporadic malignant peripheral nerve sheath tumors (Figures 2d and e). Nine percent (11 out of 122) of malignant peripheral nerve sheath tumors showed retention of H3K27 trimethylation, as they demonstrated nuclear staining in ≥90% of the tumor cells (Figures 1f, g and 2f, 2g). Among the sporadic tumors, 10 of 15 (67%) 'certain' (Figures 3b and c) and 28 of 38 (74%) 'probable' (Figures 2b and c) malignant peripheral nerve sheath tumors showed complete loss of H3K27 trimethylation.

Neurofibromatosis type 1-associated malignant peripheral nerve sheath tumor. (a) Hematoxylin and eosin (H&E × 100). (b) Complete loss of expression of trimethylation at lysine 27 of histone 3 (H3K27me3) on whole-tissue section. (c) Complete loss of expression of H3K27me3 on tissue microarrays. Note the internal positive controls with vessels and inflammatory cells retaining staining. (d) Partial loss of expression of H3K27me3 on whole-tissue section. (e) Partial loss of expression of H3K27me3 on tissue microarrays. (f) Strong and diffuse expression of H3K27me3 on tissue microarrays. (g) Strong and diffuse expression of H3K27me3 on tissue microarrays.

Sporadic ('probable') malignant peripheral nerve sheath tumor. (a) Hematoxylin and eosin (H&E × 100). (b) Complete loss of expression of trimethylation at lysine 27 of histone 3 (H3K27me3) on whole-tissue section. (c) Complete loss of expression of H3K27me3 on tissue microarrays. Note the internal positive controls with vessels and inflammatory cells retaining staining. (d) Partial loss of expression of H3K27me3 on whole-tissue section. (e) Partial loss of expression of H3K27me3 on tissue microarrays. (f) Strong and diffuse expression of H3K27me3 on tissue microarrays. (g) Strong and diffuse expression of H3K27me3 on tissue microarrays.

Sporadic ('certain') malignant peripheral nerve sheath tumor. (a) Hematoxylin and eosin (H&E × 100). (b) Complete loss of expression of trimethylation at lysine 27 of histone 3 (H3K27me3) on whole-tissue section. (c) Complete loss of expression of H3K27me3 on tissue microarrays. Note the internal positive controls with vessels and inflammatory cells retaining staining. (d) Radiation exposure-malignant peripheral nerve sheath tumor (MPNST). Hematoxylin and eosin (H&E × 100). (e), Complete loss of expression of H3K27me3 on whole-tissue section of radiation exposure-MPNST. (f) Complete loss of expression of H3K27me3 on tissue microarrays of radiation exposure-MPNST.
When comparing immunoreactivity observed on tissue microarrays slides with that of the WTS slides, only 1/40 (3%) MPNST tumor was found to demonstrate discordant (status of 'partial loss' with 20% of loss of staining on TMA and status of 'intact/retained' with 5/10% of loss of staining on the WTS slides).
In the cohort of the 122 malignant peripheral nerve sheath tumors, complete loss of H3K27 trimethylation was observed in 50% of low-grade, 55% of intermediate-grade, and 80% of high-grade tumors. Loss of H3K27 trimethylation was less frequent in low- and intermediate-grade (20 of 37; 54%) compared with high-grade (68 of 85; 80%) malignant peripheral nerve sheath tumors (P=0.0066). Moreover, complete loss of H3K27 trimethylation was observed in 67% of spindle and pleomorphic cell subtype and in 95% of Triton subtype malignant peripheral nerve sheath tumors. Loss of H3K27 trimethylation was more frequent in Triton subtype (21 of 22; 95%) compared with spindle and pleomorphic cell subtype (64 of 96; 67%) malignant peripheral nerve sheath tumors (P=0.0369). Finally, complete loss of H3K27 trimethylation was more frequent in male patients (64 of 79; 81%) than in female patients (24 of 43; 56%) (P=0.0001).
H3K27me3 Immunohistochemical Expression in Melanomas
The results of the immunohistochemical analysis are summarized in Table 2. Thirty-seven percent (98 out of 265) of melanomas demonstrated complete loss of H3K27 trimethylation. Among the primary tumors, 9 of 36 (25%) desmoplastic/fusiform cells (Figures 4b and c) and 2 of 8 (25%) nodular/SSM melanomas showed complete loss of expression of H3K27 trimethylation. Complete loss of H3K27me3 immunoreactivity was observed in 89 of 229 (39%) melanoma metastases (Figure 5b). Interestingly, 136 of 265 (51%) of melanomas showed a heterogeneous (mosaic) staining pattern with a subset of tumor cells (≥5 and <90%) showing loss of staining for H3K27me3 ('partial loss'). Forty-three percent (19 of 36) of primary melanomas (Figures 4d and e) and 51% (117 of 229) of melanoma metastases (Figure 5c) exhibited partial loss of H3K27 trimethylation. Finally, intact H3K27 trimethylation was seen in 31 of 265 (12%) melanomas, with 8 of 36 (22%) primary desmoplastic/fusiform melanomas (Figures 4f and g) and 23 of 229 (10%) melanoma metastases (Figure 5d).

(a) Primary desmoplastic/spindle cell melanoma. Hematoxylin and eosin (H&E × 100). (b) Complete loss of expression of trimethylation at lysine 27 of histone 3 (H3K27me3) on whole-tissue section. (c) Complete loss of expression of H3K27me3 on tissue microarrays. Note the internal positive controls with vessels and inflammatory cells retaining staining. (d) Partial loss of expression of H3K27me3 on whole-tissue section. (e) Partial loss of expression of H3K27me3 on tissue microarrays. (f) Strong and diffuse expression of H3K27me3 on tissue microarrays. (g) Strong and diffuse expression of H3K27me3 on tissue microarrays.

(a) Melanoma metastasis. Hematoxylin and eosin (H&E × 100). (b) Strong and diffuse expression of trimethylation at lysine 27 of histone 3 (H3K27me3) on tissue microarrays. (c) Partial loss of expression of H3K27me3 on tissue microarrays. (d), Complete loss of expression of H3K27me3 on tissue microarrays. Note the internal positive controls with vessels and inflammatory cells retaining staining.
Discussion
The diagnosis of malignant peripheral nerve sheath tumor is difficult and has historically suffered from a lack of entirely specific histological criteria, ancillary immunohistochemical and molecular diagnostic markers, especially outside the NF1 context. Recent studies have suggested that loss of H3K27me3 may serve as a useful diagnostic marker in the distinction of malignant peripheral nerve sheath tumor from other malignant cell neoplasms (and notably melanoma). In the current study, we first show in a large multicenter cohort of primary malignant peripheral nerve sheath tumors and second in a series of primary and metastatic melanomas that complete loss of H3K27 trimethylation is not a reliable marker to distinguish malignant peripheral nerve sheath tumor from melanoma. Complete loss of expression of H3K27me3 was observed in 72% of malignant peripheral nerve sheath tumors compared with 37% of loss found in melanomas, including primary desmoplastic/fusiform cell melanomas, indicating that complete loss of H3K27me3 is not specific for malignant peripheral nerve sheath tumor in the differential diagnosis, especially with desmoplastic/fusiform cell melanomas.
Proteins of the polycomb group are transcriptional repressors that modify chromatin and promote cancer processing by epigenetic modifications.12, 13 The PRC2 is composed of SUZ12, one EED isoform and EZH1 or EZH2. These chromatin-modifying proteins establish and maintain the dimethylation at lysine 27 of histone H3 (H3K27me2) and H3K27me3.4 Depending on the cellular context, the PRC2 proteins are able to exert different functions in human cancers. PRC2 was initially thought to be oncogenic. EZH2 is overexpressed in a variety of cancers (breast, lymphoma, glioblastoma, and synovial sarcoma) in which this upregulation is associated with aggressive behavior and unfavorable outcome.14, 15, 16 However, recent work suggests that PRC2 can act as tumor suppressor with loss of function induced by somatic genetic alterations (majority in EZH2 gene) found in several myeloid disorders and acute lymphoblastic leukemias.17 Whereas EZH2 can present both oncogenic and tumor suppressor functions, EED and SUZ12 appear to act predominantly as tumor suppressors.3, 5, 12 Recently, several studies in malignant peripheral nerve sheath tumors identified loss of function of the PRC2, resulting from the inactivation of mutually exclusive mutations of two of its constituents SUZ12 or EED. Mutations in EED or SUZ12 were detected in 36–92% of sporadic, 23–70% of NF1-associated, and 90% of radiotherapy-associated malignant peripheral nerve sheath tumors.4, 5 In malignant peripheral nerve sheath tumor, inactivation of PRC2 due to somatic mutations in EED and SUZ12 induces loss of H3K27me3, and promotes cell proliferation and tumor growth.4
When considering only NF1-associated, radiation-exposed, and sporadic malignant peripheral nerve sheath tumors, previous studies showed complete loss of H3K27 trimethylation in variable proportions: 29% (12/42),5 34% (55/162),10 56% (30/54),8 57% (51/90),6 68% (17/25),9 73% (45/62),4 and 75% (47/63).7 Epithelioid malignant peripheral nerve sheath tumors were excluded because this rare malignant peripheral nerve sheath tumor variant arises in a different biologic context, most of the time accompanied by loss of INI1 (SMARCB1) expression. Several explanations have been proposed to clarify these differences such as different protocols (antibodies, dilutions, antigen retrieval, and cutoff) and the variable frequency of NF1-associated vs sporadic and radiation-associated malignant peripheral nerve sheath tumors between these cohorts. However, only two of these studies related PRC2 functional status with the mutational status (of SUZ12 and EED) and immunohistochemical expression of H3K27me3.4, 7 In particular, Lee et al4 reported inactivation of PRC2 in 88% (46/52) of malignant peripheral nerve sheath tumors because of SUZ12 (n=27) or EED (n=19) heterozygous and homozygous inactivating mutations.4 However, they showed that DNA sequencing alone cannot predict PRC2 functional status in all malignant peripheral nerve sheath tumors and that the positive and negative immunostaining for H3K27me3 is highly concordant with the PRC2 wild-type (no mutation) and the homozygous loss of genetic status, respectively. However, the latter was not predictive of H3K27me3 immunohistochemical staining, so H3K27me3 expression may be more accurate in this context. To have the optimal and most realistic conditions, we applied the same technical criteria (same antibody, same dilution, and same cutoff) as in the study of Prieto-Granada et al.7 On the one hand, they showed a good correlation between the H3K27me3 expression pattern (C36B11 immunostaining) and the PRC2 component genotype (EED and/or SUZ12) and, on the other hand, tested a new monoclonal H3K27me3 antibody (clone C36B11), which demonstrated a better signal-to-noise ratio with a crisper appearance and less background staining than that used by Lee et al,4 that is, polyclonal antibody 07-449. In the study by Lee et al,4 biallelic inactivation of SUZ12 or EED was identified in 69% (34/49) of malignant peripheral nerve sheath tumors (epithelioid variant excluded), which is in accordance with the rate of H3K27me3 complete loss (72%) observed in our study.
In malignant peripheral nerve sheath tumors, these inactivations of PRC2 components significantly co-occurred with somatic alterations of CDKN2A (81% of all MPNST) and NF1 (72% of non-NF1-associated MPNST).4 In the pathogenesis of malignant peripheral nerve sheath tumor, loss of PRC2 is thought to be the additional, that is, third hit that could explain progression of neurofibromas towards malignant peripheral nerve sheath tumor.4, 5 PRC2 inactivation may occur at some point during progression to higher grade malignant peripheral nerve sheath tumor, which is in accordance with the observation that in our study H3K27me3 expression was lost significantly more frequently in high-grade than in low/intermediate-grade tumors. Moreover, complete loss of H3K27 trimethylation was significantly more frequent in the Triton subtype compared with spindle and pleomorphic cell subtype malignant peripheral nerve sheath tumors, which is in accordance with literature data showing that MPNSTs demonstrating rhabdomyoblastic differentiation are particularly aggressive and associated with poor prognosis.11, 18, 19 Among the sporadic tumors, no significant difference in complete loss of H3K27me3 was observed between 'certain' (67%) and 'probable' (74%) malignant peripheral nerve sheath tumors. Therefore, our cohort is homogeneous and the diagnostic strategy used to build this cohort (systematical histologic review, immunohistochemistry, and molecular analysis) is relevant.
The differential diagnosis of malignant peripheral nerve sheath tumor from melanoma can also be very challenging, especially as these melanoma variants, particularly desmoplastic/spindle cell melanomas, show no or very little expression of melanocytic markers such as MelanA and HMB45. Although the majority of desmoplastic/spindle cell melanomas demonstrate S100 protein and/or Sox10 immunoreactivity, partial/or complete loss of expression of these markers such as that observed in malignant peripheral nerve sheath tumors can be found,20, 21 especially in metastatic cases.22, 23 Indeed, in our series of 28 desmoplastic/spindle cell melanomas, although 24 tumors showed a diffuse expression of SOX10, 3 exhibited a partial expression of SOX10 and 1 showed a complete loss of SOX10 expression. Interestingly, the desmoplastic/spindle cell melanoma with complete loss of SOX10 expression showed complete loss of H3K27me3 expression; 2 of the desmoplastic/spindle cell melanomas with partial expression of SOX10 presented a complete loss of H3K27me3 expression and finally the desmoplastic/spindle cell melanoma with a partial expression of SOX10 exhibited a partial loss of H3K27me3 expression. These data show that additional melanocyte markers (such as SOX10) are useful when faced within this differential diagnosis (melanoma vs malignant peripheral nerve sheath tumor), but may be insufficient in some situations (particularly in desmoplastic melanomas). Recently, altered PRC2 components were also found to have a putative role in the progression of melanoma. Activating mutations in EZH2 were found in a subset of melanoma that contributed to disease progression by inactivating tumor suppressor genes.24 A recent study demonstrated overexpression of H3K27me3, particularly at the tumor front region, in superficial spreading, as well as in nodular and acral melanoma.25 However, no case of desmoplastic/spindle cell melanomas was included in that publication and somatic mutations in EZH2 were reported in only 3% of melanoma. Moreover, a recent study reported a significant loss of H3K27me3 expression in four of five childhood melanomas.26 Conversely, two studies reported a retained expression of H3K27me3 in 20 cases (20/20)6 and in 53 cases (53/53)7 of spindle cell and/or desmoplastic melanomas, with different antibodies and technical conditions, whereas a third study reported the complete loss of H3K27me3 in 11% (1/9) of spindle cell melanomas10 and a fourth study reported the partial loss of H3K27me3 in 12% (1/8) of spindle cell melanomas.8 In our study, complete loss of expression of H3K27me3 was observed in 37% of melanomas, with 25% of primary desmoplastic/fusiform cells melanomas. De Raedt et al.3 suggested that SUZ12 inactivation potentiates the effects of NF1 mutations by amplifying RAS signaling. The co-occurrence of genomic NF1 alterations and PRC2 complex inactivating mutations has been observed in melanoma and glioblastoma. Thus, genomic human tumor data (TCGA data (cBioPortal)) demonstrate that at least one allele of NF1 and SUZ12 are codeleted/mutated in 24% of melanomas. Moreover, EED deletions/mutations are present in 44% of NF1-mutant melanomas, most of which also lack one SUZ12 allele.3 These data suggest that a number of melanomas present an alteration of the PRC2 complex, which leads to a loss of H3K27me3 expression and could explain the complete loss of H3K27me3 immunohistochemical expression observed in our study. Moreover, a recent study reported loss of H3K27me3 in several sarcomas (synovial sarcoma, fibrosarcomatous dermatofibrosarcoma protuberans), which may present overlapping histological and immunohistochemical features with MPNST.8 All these data indicate that complete loss of H3K27me3 occurs frequently in several malignant peripheral nerve sheath tumor mimics and cannot be used as a specific diagnostic marker for malignant peripheral nerve sheath tumor.
Partial loss ('mosaic' pattern) of H3K27me3 was observed in 23 (19%) of all malignant peripheral nerve sheath tumors and in 136 (51%) of all melanomas, corresponding with loss of expression in a subset of tumor cells (≥5 and <90%). The precise explanation for this heterogeneous staining pattern was proposed by Schaefer et al.6 for malignant peripheral nerve sheath tumors. They hypothesized that a subset of tumor cells with heterozygous PRC2 subunit mutations has acquired an additional genomic alteration that inactivates the second allele. This loss of heterozygosity occurs during tumor progression. On the other hand, further studies are needed for melanoma since no data have been published to explain this observation.
In summary, the complete loss of H3K27me3 detected by immunohistochemistry is not a robust diagnostic marker for differentiating malignant peripheral nerve sheath tumor from melanoma and other malignant peripheral nerve sheath tumor mimics. This suggests that PRC2 inactivation in malignant peripheral nerve sheath tumor may occur during progression to higher grade. Further investigations are needed to determine the value of H3K27me3 as a predictive marker for establishing sensitivity to epigenetic-based therapies (EZH2 inhibitors, BRD4 targeted therapies) in tumors such as malignant peripheral nerve sheath tumor and melanoma.
References
Lewis JJ, Brennan MF . Soft tissue sarcomas. Curr Probl Surg 1996;33:817–872.
Ducatman BS, Scheithauer BW, Piepgras DG et al. Malignant peripheral nerve sheath tumors. A clinicopathologic study of 120 cases. Cancer 1986;57:2006–2021.
De Raedt T, Beert E, Pasmant E et al. PRC2 loss amplifies Ras-driven transcription and confers sensitivity to BRD4-based therapies. Nature 2014;514:247–251.
Lee W, Teckie S, Wiesner T et al. PRC2 is recurrently inactivated through EED or SUZ12 loss in malignant peripheral nerve sheath tumors. Nat Genet 2014;46:1227–1232.
Zhang M, Wang Y, Jones S et al. Somatic mutations of SUZ12 in malignant peripheral nerve sheath tumors. Nat Genet 2014;46:1170–1172.
Schaefer IM, Fletcher CD, Hornick JL . Loss of H3K27 trimethylation distinguishes malignant peripheral nerve sheath tumors from histologic mimics. Mod Pathol 2016;29:4–13.
Prieto-Granada CN, Wiesner T, Messina JL et al. Loss of H3K27me3 Expression Is a Highly Sensitive Marker for Sporadic and Radiation-induced MPNST. Am J Surg Pathol 2016;40:479–489.
Asano N, Yoshida A, Ichikawa H et al. Immunohistochemistry for trimethylated H3K27 in the diagnosis of malignant peripheral nerve sheath tumours. Histopathology 2017;70:385–393.
Röhrich M, Koelsche C, Schrimpf D et al. Methylation-based classification of benign and malignant peripheral nerve sheath tumors. Acta Neuropathol 2016;131:877–887.
Cleven AH, Al Sannaa GA, Briaire-de Bruijn I et al. Loss of H3K27 tri-methylation is a diagnostic marker for malignant peripheral nerve sheath tumors and an indicator for an inferior survival. Mod Pathol 2016;29:1113.
Le Guellec S, Decouvelaere AV, Filleron T et al. Malignant peripheral nerve sheath tumor is a challenging diagnosis: a systematic pathology review, immunohistochemistry, and molecular analysis in 160 patients from the French Sarcoma Group Database. Am J Surg Pathol 2016;40:896–908.
Sauvageau M, Sauvageau G . Polycomb group proteins: multi-faceted regulators of somatic stem cells and cancer. Cell Stem Cell 2010;299–313.
Lee SC, Miller S, Hyland C et al. Polycomb repressive complex 2 component Suz12 is required for hematopoietic stem cell function and lymphopoiesis. Blood 2015;126:167–175.
Kleer CG, Cao Q, Varambally S et al. EZH2 is a marker of aggressive breast cancer and promotes neoplastic transformation of breast epithelial cells. Proc Natl Acad Sci USA 2003;100:11606–11611.
Changchien YC, Tátrai P, Papp G et al. Poorly differentiated synovial sarcoma is associated with high expression of enhancer of zeste homologue 2 (EZH2). J Transl Med 2012;10:216.
Zhang P, Yang X, Ma X et al. Antitumor effects of pharmacological EZH2 inhibition on malignant peripheral nerve sheath tumor through the miR-30a and KPNB1 pathway. Mol Cancer 2015;14:55.
Nikoloski G, Langemeijer SM, Kuiper RP et al. Somatic mutations of the histone methyltransferase gene EZH2 in myelodysplastic syndrome. Nat Genet 2010;42:665–667.
Woodruff JM, Chernik NL, Smith MC et al. Peripheral nerve tumors with rhabdomyosarcomatous differentiation (malignant 'Triton' tumors). Cancer 1973;32:426–439.
Brooks JS, Freeman M, Enterline HT . Malignant 'Triton' tumors. Natural history and immunohistochemistry of nine new cases with literature review. Cancer 1985;55:2543–2549.
Nonaka D, Chiriboga L, Rubin BP . Differential expression of S100 protein subtypes in malignant melanoma, and benign and malignant peripheral nerve sheathtumors. J Cutan Pathol 2008;35:1014–1019.
Miller DD, Emley A, Yang S et al. Mixed versus pure variants of desmoplastic melanoma: a genetic and immunohistochemical appraisal. Mod Pathol 2012;25:505–515.
Kacerovska D, Michal M, Kutzner H et al. Metastatic desmoplastic malignant melanoma associated with low-grade myofibroblastic sarcoma. Am J Dermatopathol 2009;31:490–494.
Kiuru M, McDermott G, Berger M et al. Desmoplastic melanoma with sarcomatoid dedifferentiation. Am J Surg Pathol 2014;38:864–870.
Zingg D, Debbache J, Schaefer SM et al. The epigenetic modifier EZH2 controls melanoma growth and metastasis through silencing of distinct tumour suppressors. Nat Commun. Nat Commun 2015;6 6051.17.
Kampilafkos P, Melachrinou M, Kefalopoulou Z et al. Epigenetic modifications in cutaneous malignant melanoma: EZH2, H3K4me2, and H3K27me3 immunohistochemical expression is enhanced at the invasion front of the tumor. Am J Dermatopathol 2015;37:138–144.
Busam KJ, Shah KN, Gerami P et al. Reduced H3K27me3 expression is common in nodular melanomas of childhood associated with congenital melanocytic nevi but not in proliferative nodules. Am J Surg Pathol 2017;41:396–404.
Acknowledgements
We thank L M’Hamdi and L Puydenus from SBEC-BEC (Support Biopathologique aux Etudes Cliniques - Bureau des Etudes Cliniques) from Institut Claudius Regaud-IUCT-Oncopole for TMAs and technical support. We also thank the 'Assistance Publique Hopitaux de Marseille Biobank' (authorization number: AC2013-1786; BIOBANQUES BB-0033-00097) for retrieving samples. The data used in this publication was provided by the Conticanet (Connective Tissue Cancer Network) database (https://conticabase.sarcomabcb.org) and the French sarcoma network RRePS (https://rreps.sarcomabcb.org). These databases are financially supported by Conticanet and INCa (Institut National du Cancer). The following centers participated in the study: Paul Papin Center, Angers; Bergonié Institute, Bordeaux; Jean Perrin Center, Clermont-Ferrand; Georges-Francois Leclerc Center, Dijon; Oscar Lambret Center, Lille; Léon-Bérard Center, Lyon; Hôtel-Dieu CHU, Nantes; Antoine Lacassagne Center, Nice; CHU La Timone, Marseille, CHU-Guy de Chauliac, Montpellier; Institut Gustave Roussy, Paris; Paul Strauss Center, Strasbourg; CHU Toulouse, Toulouse; Claudius Regaud Institute, Toulouse and CHU Bretonneau, Tours.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Rights and permissions
About this article

Cite this article
Le Guellec, S., Macagno, N., Velasco, V. et al. Loss of H3K27 trimethylation is not suitable for distinguishing malignant peripheral nerve sheath tumor from melanoma: a study of 387 cases including mimicking lesions. Mod Pathol 30, 1677–1687 (2017). https://doi.org/10.1038/modpathol.2017.91
Received:
Revised:
Accepted:
Published:
Issue date:
DOI: https://doi.org/10.1038/modpathol.2017.91
This article is cited by
-
Histologische und molekularpathologische Besonderheiten von Tumoren des peripheren Nervensystems
Die Pathologie (2023)
-
Update from the 5th Edition of the World Health Organization Classification of Head and Neck Tumors: Soft Tissue Tumors
Head and Neck Pathology (2022)
-
Assessment of H3K27me3 immunohistochemistry and combination of NF1 and p16 deletions by fluorescence in situ hybridization in the differential diagnosis of malignant peripheral nerve sheath tumor and its histological mimics
Diagnostic Pathology (2021)
-
Clinicopathological and prognostic significance of H3K27 methylation status in malignant peripheral nerve sheath tumor: correlation with skeletal muscle differentiation
Virchows Archiv (2021)
-
Mesenchymal Neoplasms of Salivary Glands: A Clinicopathologic Study of 68 Cases
Head and Neck Pathology (2021)
