
Abstract
Background
Citrin deficiency (CD), a disorder caused by mutations in the SLC25A13 gene, may result in neonatal intrahepatic cholestasis. This study was purposely to explore the mutation spectrum of SLC25A13 gene in Vietnamese CD patients.
Methods
The 292 unrelated CD patients were first screened for four high-frequency mutations by PCR/PCR-RFLP. Then, Sanger sequencing was performed directly for heterozygous or undetected patients. Novel mutations identified would need to be confirmed by their parents.
Results
12 pathogenic SLC25A13 mutations were identified in all probands, including three deletions c.851_854del (p.R284Rfs*3), c.70-63_133del (p.Y24_72Ifs*10), and c.[1956C>A;1962del] (p.[N652K;F654Lfs*45]), two splice-site mutations (IVS6+5G>A and IVS11+1G>A), one nonsense mutations c.1399C>T (p.R467*), one duplication mutation c.1638_1660dup (p.A554fs*570), one insertion IVSl6ins3kb (p.A584fs*585), and four missense mutation c.2T>C (p.M1T), c.1231G>A (p.V411M), c.1763G>A (p.R588Q), and c.135G>C (p.L45F). Among them, c.851_854del (mut I) was the most identified mutant allele (91.78%) with a total of 247 homozygous and 42 heterozygous genotypes of carriers. Interestingly, two novel mutations were identified: c.70-63_133del (p.Y24_72Ifs*10) and c.[1956C>A;1962del] (p.[N652K;F654Lfs*45]).
Conclusion
The SLC25A13 mutation spectrum related to intrahepatic cholestasis infants in Vietnam revealed a quite similar pattern to Asian countries’ reports. This finding supports the use of targeted SLC25A13 mutation for CD screening in Vietnam and contributed to the SLC25A13 mutation spectra worldwide. It also helps emphasize the role of DNA analysis in treatment, genetic counseling, and prenatal diagnosis.
Similar content being viewed by others
Introduction
Neonatal intrahepatic cholestasis caused by citrin deficiency (NICCD) is an autosomal recessive disorder caused by a mutation in SLC25A13 gene that leads to the deficiency of a trans-membrane transport protein, called citrin [1]. Citrin is a Ca2+ dependent mitochondrial solute carrier [1], which is expressed not only in the liver but also in the kidneys and heart, however, it is most abundant in the hepatic mitochondria and is a component of the malate-aspartate reduced nicotinamide adenine dinucleotide shuttle [1,2,3,4]. Citrin forms an essential shuttle for hepatic glycolysis, and the condition of deficiency is basically characterized by impairment of hepatic glycolysis [4].
The disorder was first described in Japanese and East Asians but is now considered a pan-ethnic disease [4], which is quite common among the East Asian population [5]. The frequency of homozygotes with SLC25A13 reported in related papers is 1 in 19,000 [6, 7]. In Vietnam, the frequency of homozygotes or compound heterozygotes for SLC25A13 pathogenic variants was estimated at 1:3800 based on the carrier frequency of 1:31, which was expected to be several times higher than other East Asia [8]. Together with other inherited metabolic disorders, many newborn patients with citrin deficiencies have missed diagnosis or lately recognized based on clinical features alone. This is extremely concerning because these patients could become phenotype of recurrent hyperammonemia with neuropsychiatric symptoms in Citrullinemia type II (CTLN2) in their adulthood with many complications and severe prognoses.
Citrin deficiency (CD) has three phenotypes based on the age of onset: (1) NICCD (MIM # 605814) in newborns, (2) failure to thrive and dyslipidemia caused by CD (FTTDCD) in older children, and (3) recurrent hyperammonemia with neuropsychiatric symptoms in citrullinemia type II (CTLN2; MIM # 603471) in adults. Among these conditions, NICCD presents in the first few weeks of life with history of low birthweight and growth retardation [9, 10]. The common clinical manifestations include transient intrahepatic cholestasis and varying symptoms of hepatic dysfunctional conditions [11] such as jaundice, hepatomegaly, diffuse fatty liver, elevation of plasma protein (hypoproteinemia), transient elevation of multiple amino acid in blood (citrulline, methionine, tyrosine, and/or fetoprotein), decreased coagulation factors, hemolytic anemia [12], and/or hypoglycemia/hypergalactosemia [13].
In an early infant on-set, CD symptoms might overlap with other cholestatic liver disorders (hepatitis, biliary atresia), which makes it difficult for physicians to obtain an accurate diagnosis [14]. NICCD in general is not a severe disease, which is believed to be resolved in the first year of life with appropriate treatment. However, in some cases, patients may develop other chronic conditions as well as liver disorders that require liver transplantation, and furthermore, fatal cases have been reported somewhere [9].
Previously, CD was thought to be restricted to the Japanese population when it was first reported in Japan [15]. To date, almost all reported patients were from East Asia and only few cases from Caucasian origin have been described [13]. The pathogenic gene of CD is localized on the long arm of chromosome 7 (7q21.3) and is about 200 kb in size, which composes of 18 exons and encodes for 675 amino acids (aa).
This study purposely investigated the mutation spectrum of the SLC25A13 gene of NICCD by Polymerase Chain Reaction—Restriction Fragment Length Polymorphism (PCR-RFLP) analysis in Vietnam. Then, direct DNA sequencing of the entire coding region and adjacent splice sites of the SLC25A13 gene was applied for the case of one or unknown mutation after the conventional genetic analysis. Information about mutations in the SLC25A13 gene specified for Vietnamese population would be helpful for diagnosing CD disease, providing early treatment, and screening targeted mutations for their siblings.
Patients and method
Editorial policies and ethical considerations
All the methods were obtained in accordance with relevant guidelines and regulations and approved by the Research Institute for Child Health (RICH) - Vietnam National Children’s Hospital (VNCH) and Hanoi University of Public Health (HUPH), Ministry of Health, Hanoi, Vietnam. Informed consent for genetic analysis was obtained from the child’s parent or guardian prior.
Patients
From 2010 to 2021, all infants admitted to the VNCH, who had suspicious clinical characteristics of neonatal hepatitis or cholestasis, were included in the study at the baseline. After that, cases of jaundice due to thalassemia or other confirmed hereditary metabolic diseases were excluded. Eligible patients, who presented features of prolonged jaundice, acholic stool, and poor weight gain then had their peripheral blood collected. All patients were confirmed with no consanguineous relationship.
Genetic analysis
Genomic DNA was isolated from peripheral blood samples (including samples from patients and their families) using a Qiagen DNA blood mini kit (QIAamp DNA Blood Mini preparation kits, German) following the manufacturer’s guidelines. The DNA concentration was determined using a Thermo Scientific NanoDrop spectrophotometer (Waltham, MA, USA).
PCR/PCR-RFLP analysis
Oligonucleotide primers were synthesized and purchased from Invitrogen (USA) (Supplementary Table S1). The four known SLC25A13 mutations (I, III, X, XIX) were detected by PCR/PCR-RFLP as described previously [1]. Mutation III, XIX were direct electrophoresis on 3% Amplisize gel; The restriction endonuclease for RFLP analysis was Tail I and Tas I (New England Biolabs Inc., USA). Mutation I and X were treated by restriction enzymes Tail I and Tas I at 65 °C, then electrophoresis on 3% agarose gel.
Sequencing analysis
To identify the genotype of patients with one or unknown mutation detected by PCR-RFLP, sequencing analysis of the DNA fragment amplified by genomic DNA-PCR. Eighteen exons and their intron-flanking region of the SLC25A13 gene were successfully polymerase chain reaction (PCR) amplified by using 18 primer pairs synthesized and purchased from Introgen (USA) (Supplementary Table S2) and addition primer pairs for exon 3 designed by Primer3 (https://primer3.ut.ee/): 5′-CAGTAGCACCCTTCCTCACA-3′ and 5′-AGAAAATGGTCCTAAGAGATGGG-3′ (using Reference sequence NG_012247.1) (Supplementary Table S3). Fifty nanograms of genomic DNA was subjected to 32 cycles of PCR amplification in a 25 μL volume consisting of 10× PCR buffer (Invitrogen, USA), 10 μM concentration of each primer, 20 mM MgCl2, 10 μM dNTPs, and 5U Taq DNA polymerase (Invitrogen, USA). DNA was denatured at 95 °C for 5 min followed by 35 cycles of denaturation for 1 min at 95 °C, annealing for 1 min at 54–62 °C (Supplementary Table S2), and extension for 2 min at 72 °C, and a final extension for 7 min at 72 °C. PCR amplification was carried out on an ABI 9700 GeneAmp PCR system (USA). Direct sequencing with both forward and reverse primers was performed by an initial PCR reaction. Singer strands with a BigDye terminator v3.1 cycle sequencing kit (Applied Biosystems, USA) were purified by Bigdye X-terminator purification kit (Applied Biosystems, USA). The sequence analyses were carried out with an ABI PRISM 3500 Genetic Analyzer machine (Applied Biosystems, USA).
Gene analysis used Chromas software, or equivalent software, comparing SLC25A13 gene of the patients with the standard sequence on Genbank NG_012247.1. Reference gene were obtained from NCBI (https://www.ncbi.nlm.nih.gov/) and variants are written according to international conventions [12].
Result
Patient characteristics
This study is the largest Vietnamese CD cohort described in official references to date. It was composed of 182 males and 110 females from 292 families in the North of Vietnam. The ratio of males and females is about 2:1. The mean age of onset for the patients was 3.05 months (Standard deviation [SD]:1.61).
Identification of SLC25A13 mutation and genotype
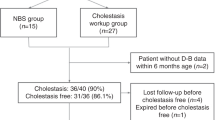
The PCR/PCR-RFLP method to detect four targeted mutations (I, III, X, and XIX) was used for the first step in DNA diagnosis of the CD patients, then the heterozygous patients would be directly sequenced 18 exons of SLC25A13 gene. In total of 292 CD patients, two hundred and seventy-eight patients were defined by PCR/PCR-RFLP, including 247 homozygotes of I/I, 2 homozygotes of XIX/XIX, and the remaining 29 of heterozygotes of mut I. Fourteen patients were defined as carrying only one or none of these 4 common mutations (undetected patients), then these patients continued to be DNA diagnosed by direct Sanger sequencing for whole 18 exons and their adjacent introns of the SLC25A13 gene (Fig. 1).

Flow diagram for genetic analysis of SLC25A13 in citrin deficiency patient cohort
249 homozygous patients and 43 compound heterozygous patients were defined. Homozygous genotypes were detected only in patients with homozygous mut I (247/249) and mut XIX (2/249), in which the homozygous genotype for mut I dominated the most considerable portion with 84.59% of all patients. Interestingly, most of the remaining genotypes are compound heterozygous with mut I (42/43). Overall, participants carrying at least one of 4 target mutations (I, III, X, and XIX) accounted for 99.7% (Table 1).
SLC25A13 mutation spectrum
In total, 12 different mutations were detected in 292 patients, including 4 missenses, 3 deletions, 2 splice sites, 1 nonsense, 1 duplication, and 1 insertion. Of which, two novel mutations were first discovered, c.70–63_133del (p.Y24_72Ifs*10) and c.[1956C > A;1962del] (p.[N652K;F654Lfs*45]), while the remaining mutations have been previously published in the literatures. Totally, 97.43% of detected alleles belonged to the group of four common mutations I, III, X, and XIX, with 91.78% was mut I, 3.6% was mut XIX, and 1.37% was mut X. (Table 2)
Pedigree analysis of two patients with novel missense mutation combined a frameshift mutation
Of the reported twelve mutations, two novel mutations c.[1956C>A;1962del] and c.70-63_133del, which have not been reported to be pathogenic, were identified in two compound heterozygous patients. Mutation c.[1956C>A;1962del] was found in a 1.5-month-old patient that had compound heterozygous of c.851-854del/c.[1956C>A;1962del] (Fig. 2). In another case, mutation c.70-63_133del was found in 27 days old patient whose genotype was c.851-854del/c.70-63_133del (Fig. 3). These novel mutations were also confirmed on peripheral blood of their parents. As below, two cases with novel mutations were detailed, from their admission to current recovered conditions by laboratory test performance.

Pedigree, agarose electrophoresis of mutation c.851_854del and chromatogram of novel mutation c.[1956C>A;1962del]. a Pedigree of the patient WBCT170102:  affected female;
affected female;  carrier female;
carrier female;  carrier male;
carrier male;  the indication case. b Electrophoresis of mutation 851_854del. M: Marker 100 bp (Invitrogen); 1: Normal control; 2: Homozygous control; 3: Heterozygous control; 4: Father (heterozygous); 5 and 6: indication case (Patient, heterozygous) and her older sister (heterozygous); 7: Mother (Normal); BC (blank control). c Chromatogram of mutation c.[1956C>A;1962del]. This mutation is a combination of c.1956C>A and c.1962del which is a deletion nucleotide T (1 bp deletion) at the nucleotide 1962 in exon 18 of SLC25A13 leading to a new stop codon at position 690; d Schematic diagram of the mutant models of mutation c.[1956C>A;1962del] [1]. Full-length blot of b was shown in Supplementary Fig. S1
the indication case. b Electrophoresis of mutation 851_854del. M: Marker 100 bp (Invitrogen); 1: Normal control; 2: Homozygous control; 3: Heterozygous control; 4: Father (heterozygous); 5 and 6: indication case (Patient, heterozygous) and her older sister (heterozygous); 7: Mother (Normal); BC (blank control). c Chromatogram of mutation c.[1956C>A;1962del]. This mutation is a combination of c.1956C>A and c.1962del which is a deletion nucleotide T (1 bp deletion) at the nucleotide 1962 in exon 18 of SLC25A13 leading to a new stop codon at position 690; d Schematic diagram of the mutant models of mutation c.[1956C>A;1962del] [1]. Full-length blot of b was shown in Supplementary Fig. S1

Pedigree, agarose electrophoresis of mutation c.851_854del and chromatogram of novel mutation c.70-63_133del. a Pedigree of the patient WBCT210304:  affected male;
affected male;  carrier female;
carrier female;  carrier male;
carrier male;  the indication case. b Electrophoresis of mutation c.851_854del: M: Marker 100 bp; 1: Normal control; 2: Heterozygous control; 3: Homozygous control; 4: Mother (normal); 5: Father (heterozygous); 6: Patient (heterozygous); BC: (blank control). (c1, c2): Electrophoresis of mutation c.70-63_133del (p.Y24_72Ifs*10) by routine primer and additional primer for exon 3 and intron adjacent. d Chromatogram of sequences of a novel mutation. A novel mutation, c.70-63_133del (p.Y24_72Ifs*10) is a deletion of 127 bp with 64 bp in exon 3 and 63 bp in intron 2 of SLC25A13 gene that results in a frameshift at codon 24 and a novel stop codon is introduced at position 34. e Schematic diagram of the mutant models of mutation c.70-63_133del. Full-length blots of b, c were shown in Supplementary Figs. S2, S3
the indication case. b Electrophoresis of mutation c.851_854del: M: Marker 100 bp; 1: Normal control; 2: Heterozygous control; 3: Homozygous control; 4: Mother (normal); 5: Father (heterozygous); 6: Patient (heterozygous); BC: (blank control). (c1, c2): Electrophoresis of mutation c.70-63_133del (p.Y24_72Ifs*10) by routine primer and additional primer for exon 3 and intron adjacent. d Chromatogram of sequences of a novel mutation. A novel mutation, c.70-63_133del (p.Y24_72Ifs*10) is a deletion of 127 bp with 64 bp in exon 3 and 63 bp in intron 2 of SLC25A13 gene that results in a frameshift at codon 24 and a novel stop codon is introduced at position 34. e Schematic diagram of the mutant models of mutation c.70-63_133del. Full-length blots of b, c were shown in Supplementary Figs. S2, S3
Case 1 (WBCT170102)
The first patient (WBCT170102) was a female 1.5-month baby carrying compound heterozygous genotype of mutation c.851_854del and c.[1956C>A;1962del].
Mutation c.851_854del was determined from the paternal allele and the other was from the maternal allele. The approximate positions of alterations in citrin caused by SLC25A13 mutations indicated that mutation c.851_854del which alters the coding frame, is a 4-bp (GTAT) deletion from nucleotide 851 to 854 in exon 9, predicting a frameshift and introduction of a stop codon at position 286, leading to truncation of the protein. Further DNA analysis by direct Sanger sequencing revealed that patient WBCT170102 was a carrier of mutation c.[1956C>A;1962del] in exon 18. This novel mutation included a missense mutation c.1956C>A and a 1 bp deletion of c.1962del, which were generated from the baby’s mother. According to the nomenclature rules, this complex mutation was described as c.[1956C>A;1962del], predictively leading to the production of a 24 aa elongated citrin molecule. Then, these mutations were screened for her sibling and the results showed that her older sister was a carrier of mutation c.851_854del. Also, her father was heterozygous of mutation c.851_854del. (Fig. 2).
At the first visit, the biochemical result revealed with status of decreased in total protein and albumin, while the bilirubin/direct bilirubin, gamma-glutamyl transferase (GGT), and alkaline phosphatase (ALP) levels were significantly increased (Supplementary Tables S4). Laboratory data at the first examination showed evidence of liver damage. Accordingly, total bilirubin and conjugated bilirubin were elevated, serum alpha-fetoprotein (AFP) was mildly elevated, while GGT was 10–20 times higher than the normal indices. Plasma glucose level of patient WBCT170102 was slightly increased when she was 3-month, however, it turned out to be normal after two months. Besides, lactate level experienced a decrease pattern and the patient was confirmed without having hyperammonemia (>110 μmol/L) as well as other symptoms. Other biochemical tests were within their normal ranges. At 13 months of age, the patient’s biochemical test results seemed to be recovered for almost all the values excepted lactate level.
At that time, amino acid analysis by electrospray MS/MS for patient WBCT170102 could not be conducted.
Case 2 (WBCT210304)
The second case was a 27-day-old baby, who carried a novel mutation c.70–63_133del and was the first child of his family. He had a compound heterozygous genotype of c.851_854del and c.70-63_133del (Fig. 3). These two mutations were confirmed in his parents. Four high-frequency mutation screenings detected the paternal mutation c.851_854del and sequencing analysis identified maternal mutation c.70-63_133del. Mutation c.851_854del was a four base-pair deletion in exon 9, while c.70-63_133del was a 127 base-pair deletion 64 bp in exon 3 and 63 bp in intron 2. A frameshift mutation was found in exon 3 and intron boundary, but this mutation had not been identified exactly by routine sequencing primer because the frameshift sequence was from the beginning of this sequence, leading to the hypothesis that this mutation overlapped with the 3′-primer sequence. To confirm this mutation, additional primer pairs for the 3′-region of exon 3 were designed, then PCR and direct sequencing for this exon and intron adjacent were performed. The PCR product of these primers showed that the patient and his mother had two bands ~539 bp and 412 bp. Further DNA analysis for this exon by direct sequencing revealed a deletion variant c.70-63_133del (127-bp deletion). Sequencing analysis discovered a 127 bp deletion, involving the 64 nucleotides in exon 3 and 63 nucleotides in the adjacent intronic sequences of exon 3 (intron 2), along with a 63 bp deletion from the first nucleotide of exon 3. According to the nomenclature rules [16], this variant was described as c.70-63_133del, leading to be aberrant RNA and the production of a truncated citrin molecule.
Of this patient, the biochemical results and laboratory data at the first examination showed almost in similar pattern to the 1st patient with total protein, albumin, bilirubin, GGT, ALP, and lactate levels, whose was also with the evidence of liver damage. However, this patient was presented with significant elevations of AST (at baseline) and AFP (at 2.4-month-old), with AST levels usually higher than ALT levels throughout the period. At 15.3 months of age, almost all the indices were recovered compared to these of the baseline, excepted lactate level. (Supplementary Tables S5).
In patient WBCTS210304, the paternal SLC25A13 allele harbored mutations c.851_854del which was detected by PCR-RFLP, while the maternal novel mutation, c.70-63_133del, was detected by direct DNA sequencing, predictively leading to the production of a truncated citrin molecule (Fig. 3). The most biochemical features of the patient were an abnormal aa pattern, with significant elevation of citrulline concentrations (Supplementary Table S6). Arginine, tyrosine, lysine, and ornithine were also two times higher than the normal range. Other amino acids were within or near their normal ranges. C0, C16, C18:1, and C18:2 slightly elevated than the control range.
Discussion
Mutation identification and genotype
The current study has involved 292 unrelated NICCD patients from the Northern area of Vietnam. The CD mutation spectrum was majorly dominated by c.851_854del (mut I) with 247 homozygous and 42 heterozygous carriers, which accounted for 91.78% of identified mutant alleles. Interestingly, among 12 variants, two novel mutations c.70-63_133del (p.Y24_72Ifs*10) and c.[1956C>A;1962del] (p.[N652K;F654Lfs*45]) have been firstly reported. Moreover, two patients carrying novel mutations were described from their genetic diagnoses to clinical manifestation, including biochemical results proving the liver damage.
SLC25A13 mutations related to CD disorder were widely reported to be specifically populated among individuals of East Asian ancestry such as Japan, China, Taiwan, Korea, and Vietnam [1, 7, 10, 17,18,19] [20,21,22], specializing in East Asia [7]. There were a few cases reported sporadically in other regions. In 2009, five novel mutations of SLC25A13 gene were reported in a study, those appearances seemed to be specific for each ethnicity without overlapping among CD patients: two mutations were defined in all five French-Canadians, three others appeared respectively in one Tunisian, one Pakistanian, and one Northern European [23]. Also, a Bulgarian case of NICCD was the first case identified in 2014 carrying a compound heterozygous genotype of one novel and one recurrent allele: c.1081C>T (p.R361*) and c.74C>A (p.A25E) [24]. In Turkey, five cases of NICCD and CTLN2 with novel mutations were reported in 2019 [25]. These mentioned studies have supported the notion that CD is somehow a pan-ethnic disorder. Previously, a Vietnamese NICCD patient was mentioned in a publication in 2002 [26]. Then, two other NICCD cases were the first Vietnamese people found to carry the c.851_854del mutation (mut I), lived in Australia and the United States [7]. Furthermore, Tabata et al.’s study discovered four NICCD Vietnamese patients carrying homozygous mutations of mut I in 2008 [17]. To our knowledge, this study is the very first cohort study, officially investigating the SLC25A13 gene mutation spectrum in a Vietnamese population.
Up to now, these high-frequency mutations have been screened by conventional means of procedures at genomic DNA or mRNA level as the basic molecular targets for NICCD diagnosis: PCR, Nested-PCR, Long and Accurate PCR (LA-PCR), and PCR-RFLP with or without restriction enzyme digestion, or by direct sequencing [17]. If only one or none such mutation was identified in NICCD highly suspicious patients, Sanger sequencing or target Next Generation Sequencing (NGS) of all the SLC25A13 exons and their flanking sequences [1, 7, 17, 21, 27,28,29,30] or entire open reading frames of SLC25A13 cDNA would be performed to identify the possible novel mutation [28]. Multiplex Ligation-dependent Probe Amplification (MLPA) should be considered a test of choice for molecular diagnosis of CD when the sequencing result is inconclusive [29]. In the current study, four mutations I, III, X, and XIX were the target mutations screened by PCR-RFLP, subsequently, undetected patients would be sequenced for whole 18 exons and their adjacent introns of the SLC25A13 gene.
In the Japanese population, four mutations screened by PCR technique (I, III, X, and XIX) were reported as the most common mutations, of which, mutation I (c.851_854del) and mutation III (c.1638_1660dup), together with II (IVS11-1G>A) were first discovered in 1999 among Japanese CTLN2 patients [1]. Another mutation, XIX (IVS16ins3kb), was initially discovered in 2008 [17]. Additionally, these four mutations (I, III, X, and XIX) were majorly identified in several studies in East Asia, including Japanese, Chinese, Korean, and Vietnamese populations [1, 6, 7, 17]. Accordingly, they were the highest mutations reported in a group of NICCD patients in Japan, Korea [17], and Taiwan [30, 31]. In Chinese studies, I, III, X, and XIX mutations accounted for the highest frequencies with 83.19%, 84.47%, and 87.9%, respectively [27, 32]. According to Yed, mutation I, III, and X, but not II, play major roles in Taiwanese infants with NICCD [30], while mutation II was the most common mutation SLC25A13 in Japan [17, 30]. A recent study in Japan as a nationwide scale with 222 CD patients showed that only 2 variants c.1177 + 1G>A (mut II) and c.1638_1660dup (mut III) overlapped with our 12 defined variants, with a much higher frequency of 34% vs. 0.56% and 3% vs. 0.75%, respectively [33]. Overall, the results of this study revealed that almost all participants (99.7%) carrying at least one of these four mutations, with 84.59% patients carrying homozygous genotype of mut I/I and 14.38% patients carrying compound heterozygous of mut I.
Mutation c.851_854del (mut I) appeared as the most alleles detected in our study with 91.78%. This mutation at exon 9 is said to produce a truncated protein, p.R284Rfs*3, which was also reported to be a mutation with the highest prevalence in Asian [1, 7, 17, 18, 21, 27, 32].
The aforementioned studies suggest the varying prevalences of these four common mutations between nations, however, they still also supported the suggestion of prenatal and postnatal screening for these high-frequency mutations initially for the quick molecular diagnosis of CD patients [27, 28, 32, 34]. Furthermore, direct sequencing of the 18 exons of SLC25A13 gene and their adjacent intronic regions could be done to identify the remaining micro-mutations despite their low prevalence reported [27, 32], which accounted for 2.21% in our study.
Identification of two novel mutations
This study had been reported for the first time of two novel mutations: c.[1956C>A;1962del] (p.[N652K;F654Lfs*45]) and c.70-63_133del (p.Y24_72Ifs*10). Mutation c.[1956C>A;1962del] (p.[N652K;F654Lfs*45]) is a 1-bp heterozygote deletion in exon 18 of SLC25A13 that results in a frameshift at codon 654 and the addition of 24 new amino acids, consequently, a novel stop codon would be introduced at position 690. The c.1962del mutation is predicted to affect the process of turning pre-RNA into mature RNA. The mutations were detected by direct sequencing using genomic DNA with 18 primer pairs.
Besides, mutation c.70-63_133del (p.Y24_72Ifs*10) was found with a 127 bp heterozygote deletion in intron 2 and exon 3 of SLC25A13 gene, that resulted in a frameshift at codon 24 and a novel stop codon at position 34. Therefore, the c.70-63_133del mutation might affect the entire functional region of this gene including four putative EF-hand domains (residues 28−39, 66−77, 100−111, and 171−182), which are conserved in calcium-binding proteins, and transmembrane (TM)-spanning regions (TM1-TM6) [1]. To address whether and how mutation c.70-63_133del (p.Y24_72Ifs*10) leads to a premature stop codon that resulted in a truncated protein and how it affected the splicing process of the relevant pre-mRNA molecules, cDNA cloning analysis of the transcripts from the affected SLC25A13 alleles is suggested being performed [32]. Unfortunately, mRNA analysis had not been analyzed, indeed it is also the limitation of our study. Regarding the effect of c.70-63_133del on the splicing process, deletion mutations lead to the removal of the connection bar of exon 2 and exon 3, consequently, the produced mRNA would lack exon 3 (exon 3 skipping) [10, 28, 35]. There is a hypothesis that when the skipping of exon 3 occurs, exon 2 would connect to exon 4 and create a stop codon, which is 10 amino acids away from the mutated amino acid. Thus, the normal SLC25A13 polypeptide chain with 2028 bp in length would be turned to be the chain with 239 bp of length and that leads to the dysfunctional protein derived from abnormal mRNA (aberrant RNA).
The functional bioinformatic tools also significantly contributed to confirm the pathogenicity of these new mutations, laying the foundation for further clinical research [36,37,38,39] [40, 41]. None of these novel mutations are observed among normal populations, as determined when we searched the public databases, 1000 genomes (http://browser.1000genomes.org/index.html) and Exome Aggregation Consortium (ExAC) (Cambridge, MA, USA) (http://exac.broadinstitute.org). Mutation p.[N652K;F654Lfs*45] was found in a female patient and mutation p.Y24_72Ifs*10 (aberrant RNA) was found in a male one. Both were admitted with clinical features of prolonged jaundice in the infantile period, however, only male patient had MS-MS analysis. The result of MS-MS indicated the importance of newborn screening using this technology [42].
Tamamori and colleagues reported that blood citrulline levels in NICCD neonates began to increase immediately after birth and this would be followed by rises in amino acids, galactose level, and the condition of cholestasis due to hepatic dysfunction [43]. Such conditions of jaundice, high value of citrulline, and high levels of other amino acids (Met, Tyr, Ala, His…) were previously proven to occur in CD patients [44, 45]. Of two patients carrying novel mutations in our study, only one patient, WBCTS210304, had elevated citrulline level, the second patient did not reveal the abnormal in citrulline, galactose or cholestasis condition. However, a more efficient method to detect NICCD infants would be to measure citrulline by tandem mass spectrometry during newborn screening [23, 45]. Additionally, the novel mutations should be performed in vivo and in vitro studies to confirm their possible pathogenic effect [28, 35].
Many studies have demonstrated efficacy in the treatment of CD, especially in infants less than 1 year of age [3, 46], and hence the application of expanded neonatal screening, genetic screening, and genetic counseling play an important role [47]. Delayed diagnosis could lead to poor treatment and prognosis [44]. In Vietnam, the treatment for NICCD children is recommended with MCT milk combined with a low-carbohydrate diet. For NICCD patients with severe liver dysfunction, supportive treatment, and liver transplantation for patients with end-stage liver disease were provided.
This is the first study to officially investigate CD patients in Vietnamese population. However, our study has accounted for several limitations. First, the patients in this study were collected from a hospital for children in the North of Vietnam, while CD patients might have been across area from the South to the North, leading to the incomprehensive conclusion for SLC25A13 mutation spectrum for Vietnamese population. In the current study, cases were almost NICCD patients while the older cases might be missed out due to some reasons, such as criteria for screening for CD children in Vietnam, or during the study collection, only infant child was focused because of their significant symptoms at birth. Also, only suspicious patients with CD were examined while other asymptomatic people could have been missed their diagnoses. In Vietnam, the CD diagnosis still challenged physicians, especially those who were not specialists in this disorder, lack of experience due to its low prevalence, or low settings in advanced diagnostic tools. For the genetic analysis, almost all the patients were discovered by PCR/PCR-RFLP for target 4 common mutations without being further sequenced for the whole 18 exons as well as the adjacent introns of the SLC25A13 gene. This limitation leads to the possibility of missing other mutations existing in SLC25A13 gene of these patients, that the mutations could be more diverse compared to the current report.
In conclusion, the study describes for the first time the molecular spectrum of CD in a cohort of 292 Vietnamese patients, laying a foundation for our subsequent clinical investigation. The results of this study indicate that SLC25A13 gene mutations play an important role in Vietnamese children with intrahepatic cholestasis. The novel mutation identified in this study enriched the SLC25A13 mutation spectrum and provided reliable laboratory evidence not only for subsequent clinical investigation but also for laying a foundation for the genetic counseling of their families in the future. Future studies to expand the study subjects such as patients with other age ranges and geographical areas across Vietnam should be done to generalize the mutation spectrum related to CD disorder.
Data availability
The datasets generated and analyzed during the current study are available in the ClinVar repository, accession numbers to datasets: SCV002546525-SCV002546535 and SCV002546358.
References
Kobayashi K, Sinasac DS, Iijima M, et al. The gene mutated in adult-onset type II citrullinaemia encodes a putative mitochondrial carrier protein. Nat Genet. 1999;22:159–63.
Kimelman D, Confino R, Confino E, et al. Do patients who achieve pregnancy using IVF-PGS do the recommended genetic diagnostic testing in pregnancy? J Assist Reprod Genet. 2018;35:1881–5.
Saheki T, Kobayashi K, Iijima M, et al. Adult-onset type II citrullinemia and idiopathic neonatal hepatitis caused by citrin deficiency: involvement of the aspartate glutamate carrier for urea synthesis and maintenance of the urea cycle. Mol Genet Metab. 2004;81:S20–6.
Andres JM, Haafiz AB. Neonatal cholestasis. Gastroenterology and nutrition: neonatology questions and controversies. 2nd ed. Elsevier; 2012. p. 251–91.
Zhang ZH, Lin WX, Zheng QQ, et al. Molecular diagnosis of citrin deficiency in an infant with intrahepatic cholestasis: identification of a 21.7kb gross deletion that completely silences the transcriptional and translational expression of the affected SLC25A13 allele. Oncotarget. 2017;8:87182–93.
Kobayashi K, Bang LuY, Xian LiM, et al. Screening of nine SLC25A13 mutations: their frequency in patients with citrin deficiency and high carrier rates in Asian populations. Mol Genet Metab. 2003;80:356–9.
Lu YB, Kobayashi K, Ushikai M, et al. Frequency and distribution in East Asia of 12 mutations identified in the SLC25A13 gene of Japanese patients with citrin deficiency. J Hum Genet. 2005;50:338–46.
Tran NH, Nguyen Thi TH, Tang HS, et al. Genetic landscape of recessive diseases in the Vietnamese population from large-scale clinical exome sequencing. Hum Mutat. 2021;42:1229–38.
Grünert SC, Schumann A, Freisinger P, et al. Citrin deficiency mimicking mitochondrial depletion syndrome. BMC Pediatr. 2020;20:518.
Saheki T, Kobayashi K, Iijima M, et al. Pathogenesis and pathophysiology of citrin (a mitochondrial aspartate glutamate carrier) deficiency. Metab Brain Dis. 2002;17:335–46.
Saheki T, Song YZ. Citrin deficiency. In: Adam MP, Everman DB, Mirzaa GM, Pagon RA, Wallace SE, Bean LJH, et al., editors. GeneReviews(®). Seattle (WA): University of Washington, Seattle; 1993. Copyright © 1993-2022, University of Washington, Seattle. GeneReviews is a registered trademark of the University of Washington, Seattle. All rights reserved.
Wang L-Y, Chen N-I, Chen P-W, et al. Newborn screening for citrin deficiency and carnitine uptake defect using second-tier molecular tests. BMC Med Genet. 2013;14:24.
Hayasaka K. Metabolic basis and treatment of citrin deficiency. J Inherit Metab Dis. 2021;44:110–7.
Zeng Q, Yang Y, Luo J, Xu J, Deng C, Yang Y. et al. Rapid Genetic Diagnosis of Citrin Deficiency by Multicolor Melting Curve Analysis. Front Pediatr. 2021;9:654527
Fu HY, Zhang SR, Wang XH, et al. The mutation spectrum of the SLC25A13 gene in Chinese infants with intrahepatic cholestasis and aminoacidemia. J Gastroenterol. 2011;46:510–8.
den Dunnen JT, Dalgleish R, Maglott DR, et al. HGVS recommendations for the description of sequence variants: 2016 update. Hum Mutat. 2016;37:564–9.
Tabata A, Sheng JS, Ushikai M, et al. Identification of 13 novel mutations including a retrotransposal insertion in SLC25A13 gene and frequency of 30 mutations found in patients with citrin deficiency. J Hum Genet. 2008;53:534–45.
Chong SC, Lo P, Chow CW, et al. Molecular and clinical characterization of citrin deficiency in a cohort of Chinese patients in Hong Kong. Mol Genet Metab Rep. 2018;17:3–8.
Zhang L, Li Y, Shi W, et al. Identification of a novel splicing mutation in the SLC25A13 gene from a patient with NICCD: a case report. BMC Pediatr. 2019;19:348.
Wongkittichote P, Sukasem C, Kikuchi A, et al. Screening of SLC25A13 mutation in the Thai population. World J Gastroenterol. 2013;19:7735–42.
Oh SH, Lee BH, Kim GH, et al. Biochemical and molecular characteristics of citrin deficiency in Korean children. J Hum Genet. 2017;62:305–7.
Kikuchi A, Arai-Ichinoi N, Sakamoto O, et al. Simple and rapid genetic testing for citrin deficiency by screening 11 prevalent mutations in SLC25A13. Mol Genet Metab. 2012;105:553–8.
Dimmock D, Maranda B, Dionisi-Vici C, et al. Citrin deficiency, a perplexing global disorder. Mol Genet Metab. 2009;96:44–9.
Avdjieva-Tzavella DM, Ivanova MB, Todorov TP, et al. First Bulgarian case of citrin deficiency caused by one novel and one recurrent mutation in the SLC25A13 gene. Genet Couns. 2014;25:271–6.
Kose MD, Kagnici M, Ozdemir TR, et al. Clinical findings in five Turkish patients with citrin deficiency and identification of a novel mutation on SLC25A13. J Pediatr Endocrinol Metab. 2020;33:157–63.
Yamaguchi N, Kobayashi K, Yasuda T, et al. Screening of SLC25A13 mutations in early and late onset patients with citrin deficiency and in the Japanese population: Identification of two novel mutations and establishment of multiple DNA diagnosis methods for nine mutations. Hum Mutat. 2002;19:122–30.
Song YZ, Zhang ZH, Lin WX, et al. SLC25A13 gene analysis in citrin deficiency: sixteen novel mutations in East Asian patients, and the mutation distribution in a large pediatric cohort in China. PLoS One. 2013;8:e74544.
Zhang ZH, Zhao XJ, Song YZ, et al. Cloning and sequence analysis of SLC25A13 transcripts in human amniocytes. Transl Pediatr. 2012;1:85–90.
Lau NKC, Lee HHC, Chen SPL, et al. In-house multiplex ligation-dependent probe amplification assay for citrin deficiency: analytical validation and novel exonic deletions in SLC25A13. Pathology. 2021;53:867–74.
Yeh JN, Jeng YM, Chen HL, et al. Hepatic steatosis and neonatal intrahepatic cholestasis caused by citrin deficiency (NICCD) in Taiwanese infants. J Pediatr. 2006;148:642–6.
Lee NC, Chien YH, Kobayashi K, et al. Time course of acylcarnitine elevation in neonatal intrahepatic cholestasis caused by citrin deficiency. J Inherit Metab Dis. 2006;29:551–5.
Lin WX, Zeng HS, Zhang ZH, et al. Molecular diagnosis of pediatric patients with citrin deficiency in China: SLC25A13 mutation spectrum and the geographic distribution. Sci Rep. 2016;6:29732.
Kido J, Häberle J, Sugawara K, et al. Clinical manifestation and long-term outcome of citrin deficiency: report from a nationwide study in Japan. J Inherit Metab Dis. 2022;45:431–44.
Shi S, Tang X, Shi Z, et al. [Analysis of SLC25A13 gene mutations and prenatal diagnosis for 20 families affected with citrin deficiency]. Zhonghua Yi Xue Yi Chuan Xue Za Zhi. 2018;35:475–9.
Lin W-X, Deng L-J, Liu R, et al. Neonatal intrahepatic cholestasis caused by citrin deficiency: in vivo and in vitro studies of the aberrant transcription arising from two novel splice-site variants in SLC25A13. Eur J Med Genet. 2021;64:104145.
Adzhubei IA, Schmidt S, Peshkin L, et al. A method and server for predicting damaging missense mutations. Nat Methods. 2010;7:248–9.
Choi Y, Sims GE, Murphy S, et al. Predicting the functional effect of amino acid substitutions and indels. PLoS One. 2012;7:e46688.
Lubeck E, Coskun AF, Zhiyentayev T, et al. Single-cell in situ RNA profiling by sequential hybridization. Nat Methods. 2014;11:360–1.
Choi Y, Chan AP. PROVEAN web server: a tool to predict the functional effect of amino acid substitutions and indels. Bioinformatics. 2015;31:2745–7.
Adzhubei I, Jordan DM, Sunyaev SR. Predicting functional effect of human missense mutations using PolyPhen-2. Curr Protoc Hum Genet. 2013;Chapter 7:Unit7.20. https://doi.org/10.1002/0471142905.
Chen JL, Zhang ZH, Li BX, et al. Bioinformatic and functional analysis of promoter region of human SLC25A13 gene. Gene. 2019;693:69–75.
Shigematsu Y, Hirano S, Hata I, et al. Newborn mass screening and selective screening using electrospray tandem mass spectrometry in Japan. J Chromatogr B Anal Technol Biomed Life Sci. 2002;776:39–48.
Tamamori A, Okano Y, Ozaki H, et al. Neonatal intrahepatic cholestasis caused by citrin deficiency: severe hepatic dysfunction in an infant requiring liver transplantation. Eur J Pediatr. 2002;161:609–13.
Hu SW, Lu WL, Chiang IP, Wu SF, Wang CH, Chen AC. Neonatal Intrahepatic Cholestasis Caused by Citrin Deficiency with SLC25A13 Mutation Presenting Hepatic Steatosis and Prolonged Jaundice. A Rare Case Report. Medicina (Kaunas). 2021;57:1032.
Hao H, Li S, Wu S, et al. Application of high-throughput sequencing technologies with target capture/target next-generation sequencing in diagnosis of neonatal intrahepatic cholestasis caused by citrin deficiency (NICCD). Int J Clin Exp. 2017;10:3480–7.
Saheki T, Moriyama M, Funahashi A, Kuroda E. AGC2 (Citrin) Deficiency-From Recognition of the Disease till Construction of Therapeutic Procedures. Biomolecules. 2020;10:1100.
Zhang R, Qiang R, Song C, et al. Spectrum analysis of inborn errors of metabolism for expanded newborn screening in a northwestern Chinese population. Sci Rep. 2021;11:2699.
Acknowledgements
The authors appreciate the participation of the patient’s family in this study. This work was supported by the Vietnam National Children’s Hospital, Ministry of Health.
Author information
Authors and Affiliations
Contributions
M-HNT designed the study, coordinated the study, analyzed data, and wrote the manuscript. A-HNP examined the patients and collected the samples. P-MNT and D-NN performed experiments. H-ST, HG, H-NN, and Y-TL helped with manuscript preparation. M-DT helped to revise the manuscript. All authors reviewed the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Ethics approval
All the methods were obtained in accordance with relevant guidelines and regulations and approved by the Research Institute for Child Health (RICH) - Vietnam National Children’s Hospital (VNCH) and Hanoi University of Public Health (HUPH), Ministry of Health, Hanoi, Vietnam. Informed consent for genetic analysis was obtained from the child’s parent or guardian prior.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Nguyen, MH.T., Nguyen, AH.P., Ngo, DN. et al. The mutation spectrum of SLC25A13 gene in citrin deficiency: identification of novel mutations in Vietnamese pediatric cohort with neonatal intrahepatic cholestasis. J Hum Genet 68, 305–312 (2023). https://doi.org/10.1038/s10038-022-01112-2
Received:
Revised:
Accepted:
Published:
Version of record:
Issue date:
DOI: https://doi.org/10.1038/s10038-022-01112-2
