
Abstract
The vast majority of cellular processes are interconnected in a manner that facilitates the overall function of the cell within its tissue environment. It has become evident that the dynamic interplay between metabolic processes and the regulation of genomic activities, including gene expression, is contingent on metabolite levels and factors that govern cellular distribution and compartmentalization. The advent of rapid technological and biophysical advances over the past two decades has yielded a compendium of factors, including metabolites and genes, that have provided extensive insight into their interrelationship. Here we discuss and summarize the many metabolites that have been experimentally shown to directly influence chromatin factors and epigenetic patterns. We aim to provide a comprehensive overview of the temporal and spatial dynamics of these processes within the cell, emphasizing the significance of metabolite abundance and the intricate orchestration of these processes during ontogeny and disease progression. The influence of lifestyle factors, such as diet and environmental exposures, on metabolite levels and their potential implications for therapeutic interventions is a subject of particular interest. The intricate interplay between metabolism and the epigenome in cancer offers a fertile ground for further research. By elucidating the manner in which metabolic fluctuations influence the epigenetic landscape, novel therapeutic approaches that target both metabolic and epigenetic pathways may emerge as promising avenues for cancer treatment.
Similar content being viewed by others
Introduction
Metabolism and epigenetics, once seen as distinct processes, are now recognized as deeply intertwined, especially in cancer biology. Key metabolites such as S-adenosylmethionine (SAM), acetyl-CoA and nicotinamide adenine dinucleotide (NAD) serve as direct coenzymes or substrates for epigenetic enzymes, tightly linking cellular metabolic states to epigenetic regulation. Metabolic reprogramming in cancer often disrupts this balance, producing oncometabolites such as 2-hydroxyglutarate (2-HG), succinate and fumarate, which competitively inhibit epigenetic regulators and cause widespread epigenetic deregulation1. Beyond well-characterized oncometabolites, unconventional metabolites such as sarcosine, glycine, hypotaurine, kynurenine and methylglyoxal (MGO) are now being explored for their roles in epigenetic regulation and cancer progression, opening novel avenues for investigation2,3,4,5,6,7. Cellular metabolism transitions from coordinated processes during development to instability with aging, external exposures and genetic mutations. This metabolic disruption leads to epigenetic changes, clonal selection and disease. In cancer, metabolic adaptation is exploited for uncontrolled growth, with tumor cells hijacking developmental pathways and evading constraints through epigenetic plasticity8. Lifestyle factors such as diet, stress and environmental exposures influence cellular pools of key metabolites such as SAM, acetyl-CoA and NAD, further modulating the epigenome and providing additional opportunities for therapeutic intervention9.
Despite significant progress, studying transient metabolite–enzyme interactions remains challenging, but advanced techniques such as high-resolution structural studies and computational modeling are crucial for understanding these dynamics. This Review explores the interplay between metabolism and the epigenome in cancer, focusing on key metabolites, oncometabolites and dietary interventions. It addresses the challenges in studying metabolite–epigenome interactions and highlights emerging therapeutic strategies, aiming to provide insights into how metabolic shifts shape the epigenetic landscape and open new therapeutic avenues.
Metabolites: the unsung authors of the epigenome
Epigenetic modifications, such as DNA methylation and histone modifications, serve as a dynamic ‘code’ that governs gene expression without altering the underlying DNA sequence9. Whereas enzymes such as DNA methyltransferases (DNMTs), histone acetyltransferases (HATs) and histone demethylases are the molecular ‘scribes’ of this code, metabolites act as both the ink and erasers, providing the necessary chemical groups for these modifications or removing them10. This interplay forms the foundation of how metabolites influences the epigenome.
What are metabolites doing in the nucleus?
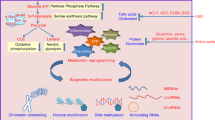
Metabolites, typically seen as intermediates in metabolic pathways, have a profound influence on epigenetic regulation (Fig. 1). They act as substrates, cofactors or inhibitors for key epigenetic enzymes, thereby directly shaping the epigenome11. Intriguingly, several metabolites bind directly to epigenetic modulators to regulate their activity, whereas others work indirectly by modulating enzyme availability or functionality. Beyond these roles, metabolites may also orchestrate the epigenome by coordinating the establishment and maintenance of epigenetic marks in response to changes in cellular metabolism, nutrient availability and environmental cues12.

All metabolic pathways are compartmentalized but share metabolites to neighboring pathways. The pathways that contribute to epigenetic activities are depicted with an arrows, where the thick-stroke lines contribute the most (Pathway of Human Metabolism, v10.23).
SAM: the master methyl donor in cancer epigenetics
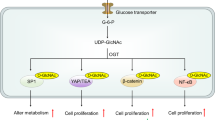
SAM is a central metabolite synthesized through the methionine cycle, serving as the universal methyl donor for DNA, RNA and histone methylation. SAM directly fuels the activity of DNMTs and histone methyltransferases (HMTs) and facilitates posttranslational modifications (PTMs) such as the methylation of lysine and arginine residues in nonhistone proteins (Fig. 2). SAM production is intricately linked to one-carbon metabolism, where nutrients such as methionine, serine and folate regulate its synthesis via methionine adenosyltransferase (MAT) enzymes. Once SAM donates a methyl group, it is converted into S-adenosylhomocysteine (SAH), a potent inhibitor of DNMTs and HMTs, highlighting the importance of maintaining the SAM/SAH ratio for proper epigenetic regulation13,14. High SAM levels enhance DNMT and HMT activity, promoting the methylation of DNA and histones, whereas the accumulation of SAH inhibits these enzymes. SAM availability also influences the activity of demethylases such as TETs and histone demethylases, which are sensitive to the metabolic environment15. Thus, SAM levels act as a key metabolic checkpoint controlling both methylation and demethylation processes. In cancer, the one-carbon metabolic pathway is frequently upregulated to support rapid cell division, with enzymes such as phosphoglycerate dehydrogenase (PHGDH) enhancing serine biosynthesis, which elevates SAM levels. This metabolic shift leads to the hypermethylation of tumor-suppressor genes and altered histone modifications, promoting tumor progression16. Key amino acid transporters, such as LAT1 and LAT4, are overexpressed in tumors to facilitate methionine uptake, essential for SAM production and subsequent cellular proliferation and differentiation17. In lung cancer, for example, LAT1 overexpression increases SAM abundance, which enhances the activity of HMT EZH2 and boosts H3K27me3 levels, further promoting tumor growth18.

Several metabolites are cofactors or transient coenzymes (cosubstrates) to epigenetic enzymes, and several are inhibitors of their activities. The stars depicts major metabolic contributors in epigenetic regulation. Molecular abbreviations are NAMN Nicotinic acid mononucleotide, NaAD Nicotinic acid adenine dinucleotide, NMN Nicotinamide mononucleotide, NAD+ Nicotinamide adenine dinucleotide, NAM Nicotinamide, NR Nicotinamide riboside, MTHF Methylenetetrahydrofolate, THF Tetrahydrofolate, mC Methylated cytosine, H3me Methylated histone H3, H3ac Acetylated histone H3, HDAC Histone deacetylase, HAT Histone acetyltransferase, KDM Lysine demethylase, TET Methylcytosine dioxygenase, α-KG Alpha-ketoglutarate.
DNA hypomethylation can activate oncogene transcription, thereby promoting carcinogenesis and tumor development. In cancers with low SAM, replenishment helps restore methylation balance rather than merely increasing methylation levels. SAM acts as a methyl donor in numerous methylation reactions and inhibits intracellular demethylase activity, which results in the hypermethylation of DNA. In a study on gastric cancer, SAM treatment was shown to induce hypermethylation of the vascular endothelial growth factor (VEGF)-C promoter. VEGF-C expression, typically elevated in gastric cancer, was downregulated following SAM treatment19. The VEGF-C promoters in MGC-803, BGC-823 and SGC-7901 gastric cancer cells, which normally express VEGF-C, were nearly unmethylated. However, after SAM treatment, these promoters were highly methylated, leading to the downregulation of VEGF-C expression. This resulted in a significant inhibition of cancer cell growth both in vitro and in vivo. SAM’s ability to regulate VEGF-C methylation underscores its potential as a therapeutic agent for silencing oncogenes and blocking cancer progression19. Dietary inputs and environmental factors influence SAM availability. For instance, restricting dietary methionine decreases SAM levels, suppresses EZH2 activity (an H3K27 methyltransferase) and inhibits tumor growth20. Similarly, reducing threonine metabolism lowers SAM synthesis, affecting H3K4me3 levels and cell proliferation21. The methionine salvage pathway, where methylthioadenosine is recycled into methionine and SAM, further supports SAM regeneration. However, the loss of methylthioadenosine phosphorylase in cancers impairs this process and sensitizes cells to PRMT5 inhibition, underscoring a therapeutic vulnerability22. SAM has demonstrated anticancer potential across various malignancies. In hepatocellular carcinoma (HCC), SAM selectively inhibits the growth, transformation and invasiveness of cancer cells, sparing normal liver cells. Methylomic and transcriptomic analyses reveal that SAM downregulates oncogenic pathways and hypermethylates metastasis-promoting genes such as STMN1 and TAF1510. Similarly, in breast cancer and osteosarcoma, SAM reduces tumor proliferation, migration, invasion and metastasis in vitro and decreases tumor volume in vivo. Epigenome-wide association studies further reveal that SAM hypermethylates genes involved in prometastatic signaling and tumor progression, providing a mechanism for SAM’s therapeutic effects23. SAM’s potential in cancer therapy highlights its central role in linking metabolism to epigenetics, with implications for regulating key epigenetic modifications. Its synthesis and activity are influenced by intracellular metabolic pathways and external factors, such as nutrient availability, which directly modulate SAM levels. By targeting the one-carbon cycle or optimizing SAM production through dietary or pharmacological interventions, SAM can be harnessed for therapeutic purposes.
Acetyl-CoA metabolism and epigenetic regulation in cancer
Acetyl-CoA is a central metabolite essential for processes such as histone acetylation, a key epigenetic regulator in cancer (Fig. 2). In cancer cells, fatty acid oxidation produces acetyl-CoA within mitochondria, which cannot directly cross the mitochondrial membrane. Instead, acetyl-CoA is converted to citrate in the tricarboxylic acid (TCA) cycle, exported to the cytosol and then converted back to acetyl-CoA by ATP citrate lyase (ACLY), linking fatty acid oxidation indirectly to ACLY activity and histone acetylation, independent of glucose metabolism24. In addition, the carnitine/acyl-carnitine shuttle system facilitates the transport of acetyl-CoA from mitochondria to the nucleus, where it supports epigenetic modifications and enhances metabolic flexibility25. In hypoxic tumor environments, acetate metabolism further boosts acetyl-CoA production through acetyl-CoA synthetase 2 (ACSS2), ensuring continuous histone acetylation even during metabolic stress26. Moreover, glutamine metabolism contributes to acetyl-CoA production through reductive carboxylation, particularly in cells with impaired oxidative phosphorylation27. In various cancers, elevated ACLY expression is linked to increased histone acetylation, driving oncogenes such as MYC and HIF-1α, which play pivotal roles in tumor progression24. Furthermore, acetyl-CoA metabolism also regulates immune evasion by promoting the expression of programmed cell death-ligand 1 (PD-L1), a key immune checkpoint protein. Studies have shown that high acetyl-CoA metabolism correlates with reduced antitumor immunity, whereas inhibiting ACLY reactivates cytotoxic T cells, enhancing the effectiveness of immunotherapy28. In pancreatic ductal adenocarcinoma, the acetyl-CoA metabolism is critical in tumor progression. A recent study observed that histone H4 acetylation is elevated in pancreatic acinar cells harboring Kras mutations, even before the appearance of premalignant lesions. This upregulation of acetyl-CoA in KRAS-mutant acinar cells supports acinar-to-ductal metaplasia, a crucial step in tumorigenesis. Interestingly, the pancreas-specific loss of ACLY inhibits acinar-to-ductal metaplasia and suppresses tumor formation29. In pancreatic ductal adenocarcinoma cells, the activation of AKT–ACLY signaling promotes histone acetylation, stimulating cell proliferation and tumor growth. Both BET inhibition and statin treatment, which target acetyl-CoA-dependent processes, suppress these effects, highlighting the therapeutic potential of targeting the acetyl-CoA metabolism in cancer29.
Collectively, these findings underscore the vital role of acetyl-CoA metabolism in promoting cancer cell plasticity, tumorigenesis and immune evasion. Given its central role in both metabolic and epigenetic regulation, targeting acetyl-CoA metabolism presents a promising strategy for treating KRAS-driven cancers and overcoming therapeutic resistance.
Epigenetic enzyme regulation via cosubstrates
The function of many enzymes, including chromatin remodelers, depends on coenzymes such as NAD+. Specifically, the term ‘cosubstrate’ is used when a coenzyme temporarily binds to an enzyme and leaves it in an altered state. NAD+ is an oxidizing agent that accepts a hydride ion and a proton (H+) to become NADH or a phosphate group to become NADP. A lack of critical cosubstrates leads to altered epigenetic states that are correlated with disease states, such as cancer.
NAD+ as a cosubstrate in histone deacetylation
Histone deacetylation, a crucial mechanism in regulating chromatin structure and gene expression, is mediated by four classes of histone deacetylases (HDACs): classes I, II, III and IV11. While classes I, II and IV HDACs are zinc dependent, class III HDACs, known as sirtuins, utilize NAD+ as a cosubstrate. During the deacetylation process, NAD+ is cleaved into 2′-O-acetyl-ADP-ribose and nicotinamide (NAM), with NAM being recycled into NAD+ through the NAD+ salvage pathway, a crucial source of NAD+ in the cell30.
NAD+ and cancer metabolism
In rapidly proliferating cancer cells, the upregulation of NAM phosphoribosyltransferase (NAMPT) ensures sufficient NAD+ to sustain metabolic demands and support sirtuin function. In the salvage pathway, NAMPT converts NAM into NAM mononucleotide (NMN), which is then converted into NAD+ by NMN adenylyltransferases (NMNATs) (Fig. 2). NAMPT, the rate-limiting enzyme in this pathway, is frequently upregulated in cancers, including colorectal tumors, highlighting its potential as a therapeutic target30,31. Elevated NAMPT activity is crucial for maintaining NAD+ levels in rapidly proliferating cancer cells, supporting metabolic reprogramming, such as enhanced glycolysis, a hallmark of cancer cells32. This metabolic shift involves the conversion of NAD+ to NADH, which reduces the NAD+/NADH ratio, thereby diminishing sirtuin activity. The resulting decrease in sirtuin function leads to histone hyperacetylation and altered gene expression patterns that promote tumorigenesis30. Nutrient availability significantly influences NAD+ metabolism. For example, calorie restriction increases the NAD+/NADH ratio and enhances sirtuin-mediated deacetylation, which has been associated with tumor suppression32. Conversely, high-fat diets reduce NAD+ levels and sirtuin activity, contributing to a metabolic environment that supports cancer progression. Furthermore, the three NAD+-producing NMNATs—NMNAT1, NMNAT2 and NMNAT3—are localized in distinct cellular compartments: NMNAT1 in the nucleus, NMNAT2 in the Golgi complex and NMNAT3 in the mitochondria33. This compartmentalization ensures the precise regulation of NAD+ levels within specific cellular regions, which may influence sirtuin activity and chromatin dynamics. Given NAD+’s central role in regulating histone deacetylation and its impact on metabolic reprogramming, targeting NAD+ metabolism and its regulatory enzymes, particularly NAMPT, offers promising therapeutic avenues for cancer treatment. Modulating NAD+ levels could shift the epigenetic landscape, either promoting or inhibiting tumorigenesis, depending on the cancer cell’s metabolic context33.
Oncometabolites: when good molecules go bad
Oncometabolites are metabolites that, when accumulated due to mutations in metabolic enzymes, contribute to the initiation and progression of cancer. These metabolites, typically involved in normal cellular processes, can become ‘bad’ molecules in the context of cancer, as they disrupt cellular metabolism, epigenetic regulation and overall cell function34. Oncometabolites can reprogram the epigenome by directly or indirectly interfering with key epigenetic modulators, thereby altering gene expression and promoting oncogenesis.
2-HG
The metabolite 2-HG, a metabolite structurally resembling α-ketoglutarate (α-KG), plays a key role in cancer, particularly in tumors with isocitrate dehydrogenase (IDH1/2) mutations. 2-HG exists as two enantiomers: ᴅ-2-HG (R-enantiomer), produced by mutant IDH1/2 enzymes, and ʟ-2-HG (S-enantiomer), which accumulates under conditions such as hypoxia and acidity or through the activity of enzymes such as PHGDH, MDH1/2 and LDHA34. Mutations in IDH1/2 lead to the neomorphic conversion of α-KG into ᴅ-2-HG, resulting in its accumulation and inhibition of α-KG-dependent dioxygenases such as TET2 and Jumonji-C histone demethylases. This leads to widespread DNA hypermethylation and histone modifications, contributing to tumorigenesis. In clear cell renal cell carcinoma, the loss of ʟ-2-HG dehydrogenase (L2HGDH), the enzyme responsible for degrading ʟ-2-HG, results in its accumulation as an oncometabolite and promotes oncogenic phenotypes34. In IDH1/2-mutant cancers, including gliomas and acute myeloid leukemia (AML), the mutant enzymes specifically produce ᴅ-2-HG, not ʟ-2-HG35. By contrast, wild-type IDH1/2 enzymes catalyze the oxidative decarboxylation of isocitrate to α-KG and do not produce 2-HG under normal physiological conditions. Accumulated ᴅ-2-HG competitively inhibits α-KG-dependent enzymes, leading to epigenetic alterations such as increased H3K9me3 and H3K27me3, gene silencing and impaired differentiation36. Inhibiting mutant IDH enzymes with drugs such as ivosidenib (AG-120) and enasidenib (AG-221) in IDH1/2-mutant cancers restores α-KG levels, reverses 2-HG-induced epigenetic changes and promotes differentiation, highlighting 2-HG as both a diagnostic marker and therapeutic target37.
Lactate
Lactate, a key metabolic byproduct of glycolysis, is critical in cancer metabolism. Glycolysis, the process by which cells generate energy in the absence of sufficient oxygen, a hallmark of cancer cells known as the Warburg effect, leads to lactate accumulation38. Lactate acidifies the tumor microenvironment, promoting invasion and metastasis, while also acting as a signaling molecule. It also induces histone lactylation, an epigenetic modification that alters chromatin structure and gene expression38. Lactate-driven histone lactylation regulates cancer progression by promoting oncogene activation. In breast cancer, it enhances c-Myc expression and drives oncogene splicing via SRSF10, fueling tumor cell proliferation39. Similarly, in AML, lactate accumulation promotes PD-L1 expression via the STAT5–lactate–PD-L1 axis, facilitating immune evasion and resistance to immunotherapy40. In HCC, lactylation affects TCA cycle enzymes, linking metabolism and epigenetic regulation41. In ocular melanoma, increased histone lactylation correlates with poor prognosis, enhancing YTHDF2 expression, destabilizing tumor-suppressor mRNAs and accelerating tumor growth42. These findings highlight lactate as a key oncometabolite in cancer, driving metabolic reprogramming and epigenetic changes that promote tumor progression. Targeting lactate-driven pathways offers promising therapeutic strategies to inhibit cancer growth.
Succinate
Succinate dehydrogenase (SDH) is a key in mitochondrial metabolism and tumor suppression. Mutations in SDH subunits impair succinate oxidation, leading to succinate accumulation, which activates oncogenic pathways such as STAT3 and EMT in colorectal cancer and drives angiogenesis in gastric cancer through VEGF signaling via GPR9143. Succinate reprograms T cell metabolism and epigenetics to promote proinflammatory states. It also alters histone and DNA methylation by inhibiting α-KG-dependent dioxygenases, such as TET enzymes and histone demethylases44. Despite these findings, succinate’s epigenetic effects appear context dependent, as several studies observed limited DNA methylation changes in succinate-treated cells45,46. Moreover, high succinate levels in HepG2 cells increase caspase activity with minimal genotoxicity, whereas fumarate induces DNA fragmentation and chromosomal instability, highlighting distinct oncogenic mechanisms45. These findings underscore the role of succinate as a retrograde signaling molecule, that is, a mitochondria-derived metabolite that communicates mitochondrial dysfunction or metabolic status to the nucleus and cytoplasm. Succinate has been demonstrated to promote cancer progression and immune modulation via metabolic, epigenetic and oncogenic pathways through such signaling. This provides new therapeutic insights.
Fumarate
Fumarate, a key TCA cycle intermediate, is crucial for cellular metabolism. Its accumulation due to fumarate hydratase (FH) deficiency or mutation drives oncogenesis, making fumarate a significant oncometabolite47. Fumarate is normally metabolized by FH. In the event that FH loss has been rendered inactive, there is an accumulation of fumarate, which disrupts metabolic balance and contributes to the progression of cancer. As an oncometabolite, fumarate stabilizes HIF-1α by inhibiting prolyl hydroxylases, promoting angiogenesis and tumor progression through genes such as VEGF and impacting epigenetic regulation, DNA repair and immune evasion3,47,48. Fumarate inhibits TET enzymes, impairing DNA demethylation and causing DNA hypermethylation, which silences tumor-suppressor genes and activates oncogenes, further promoting cancer progression49. Fumarate-driven epigenetic alterations promote a malignant phenotype, enhancing cell proliferation, apoptosis resistance and metastasis. It also impacts DNA repair by interacting with histone variant H2A.Z at DNA double-strand breaks, inhibiting KDM2B and enhancing DNA-PK accumulation, which promotes nonhomologous end-joining for DNA repair and cell survival47,49. Fumarate supports DNA repair, maintaining genomic stability and cancer cell survival, especially after radiation. It also influences the tumor microenvironment, modulating immune cell function and promoting immune evasion. Accumulation due to FH mutations or loss is linked to hereditary cancers such as hereditary leiomyomatosis and renal cell carcinoma, driving aggressive tumors with early metastasis and poor prognosis47,50. Fumarate is also implicated in sporadic cancers, such as clear cell carcinomas and colorectal cancer, where its accumulation drives DNA hypermethylation and epigenetic changes, contributing to the malignant phenotype47,51. In clear cell carcinoma, FH inactivation causes fumarate accumulation, altering metabolism, promoting angiogenesis and enhancing cancer cell survival, accelerating tumor progression. Fumarate’s dual role in epigenetics and DNA repair highlights its potential as a therapeutic target for tumor progression and therapy resistance47,51.
Modifications induced by oncometabolites
Oncometabolites such as ʟ-lactate drive cancer progression by inducing various PTMs, including lactylation. One key example is the ʟ-lactylation of adenylate kinase 2, which reduces its activity, disrupting energy homeostasis and HCC cell proliferation41. In addition, ᴅ-lactylation, a ᴅ-lactate-related modification, involves the nonenzymatic transfer of ᴅ-lactate to lysine residues. It arises from MGO conversion to lactoylglutathione by glyoxalase 1 (GLO1), followed by hydrolysis by glyoxalase 2 (GLO2), producing ᴅ-lactate. ᴅ-lactylation predominantly affects glycolytic enzymes, leading to a global reduction in glycolytic activity, with nearly 350 proteins identified as modified52.
Beyond lactate, fumarate and succinate, key TCA cycle metabolites, contribute to additional PTMs such as succination and succinylation. In succination, fumarate forms adducts with cysteine thiol groups, such as in KEAP1, which stabilizes NRF2 and activates an antioxidant program, aiding tumor survival under oxidative stress53. Other important targets of succination include the tumor suppressor PTEN and gasdermin D, with implications for apoptosis and inflammatory cell death, respectively54. Furthermore, fumarate-induced succination depletes glutathione, disrupting the cellular redox balance and contributing to cancer progression55. Similarly, succinylation, the transfer of succinyl groups from succinyl-CoA to lysine residues, regulates chromatin dynamics and mitochondrial protein function, influencing key processes in cancer development56. Elevated succinyl-CoA, driven by SDH mutations, leads to aberrant succinylation, contributing to apoptosis resistance and metabolic reprogramming, especially in IDH1/IDH2-mutant cancer cells56. Collectively, oncometabolites such as lactate, fumarate and succinate contribute to PTMs (specifically lactylation, succination and succinylation, respectively) that link metabolic reprogramming with epigenetic changes. These modifications influence cellular behavior, promoting tumorigenesis by rewiring metabolic and regulatory networks. Their interplay underscores their role in cancer progression and as potential therapeutic targets. A summary of oncometabolites and their effects on cancer epigenetics is provided in Table 1.
Uncovering metabolic drivers beyond tradition
Sarcosine
Sarcosine (N-methylglycine), a metabolite linked to one-carbon metabolism, has been explored as a potential biomarker for prostate cancer progression. Initial studies showed that urinary sarcosine levels increased with cancer progression and metastasis, raising its diagnostic potential. However, later research revealed inconsistencies, as sarcosine failed to reliably differentiate prostate cancer from benign conditions or healthy individuals, offering no advantage over existing markers such as PCA357,58,59. Despite its limited diagnostic value, a large study found a modest link between high serum sarcosine levels and reduced prostate cancer risk, highlighting the complexity of its role in cancer biology59. Beyond being a potential biomarker, sarcosine has been shown to affect epigenetic regulation. It increases levels of SAM, a key methyl donor and promotes CpG island methylation in prostate cells60. These findings suggest that sarcosine may act as an epigenetic modifier, influencing gene silencing and cellular behavior in prostate cancer. Although its diagnostic value is limited, its potential role in cancer epigenetics warrants further research into its biological functions and therapeutic implications.
Glycine
Untargeted liquid chromatography–tandem mass spectrometry metabolomics of NCI-60 cancer cell lines highlights glycine’s crucial role in cancer cell proliferation6. Glycine supports purine biosynthesis and provides one-carbon units for DNA and histone methylation, essential for epigenetic regulation, by contributing to SAM synthesis6. Cancer cells amplify glycine dependence by upregulating transporters for glycine, serine, methionine and glutamine, driving metabolic and epigenetic reprogramming6. In non-small-cell lung cancer, glycine decarboxylase (GLDC) overexpression rewires glycolysis and glycine metabolism, driving tumorigenesis through metabolic and epigenetic shifts. Sarcosine supplementation restores proliferation in GLDC-deficient cells, highlighting cancer’s metabolic adaptability61. These finding positions glycine as a potential oncometabolite, bridging metabolism and epigenetic regulation in cancer. However, its role in tumorigenesis remains unclear, requiring further research to uncover its mechanisms and therapeutic potential.
Hypotaurine
Hypotaurine emerges as a key oncometabolite in glioblastoma (GBM), with levels increasing alongside tumor grade, as revealed by capillary electrophoresis–mass spectrometry. Elevated homocysteic acid, which inhibits cysteine sulfinic acid decarboxylase, disrupts the conversion of cysteinesulfinate to hypotaurine and results in U-251 GBM cell arrest, highlighting the metabolic dysregulation in GBM progression4.
Hypotaurine drives GBM progression by inhibiting HIF-1α hydroxylation, activating hypoxia signaling and promoting a highly aggressive metabolic state. Its antagonist, taurine, counteracts this effect, suppressing hypotaurine synthesis and slowing tumor growth in xenograft models. In addition, 2-aminoethanethiol dioxygenase (ADO) catalyzes hypotaurine production from cysteamine, with its expression tightly linked to GBM progression. CRISPR–Cas9 knockout of ADO significantly impairs GBM cell proliferation and tumor growth, highlighting hypotaurine as a critical oncometabolite and potential therapeutic target13,62. These findings establish hypotaurine as a potential diagnostic and prognostic biomarker while positioning ADO and hypotaurine metabolism as promising therapeutic targets for GBM. Though its role in GBM progression is evident, its impact on epigenetic regulation remains unexplored. Given its influence on hypoxia signaling and metabolism, hypotaurine may also drive epigenetic changes affecting gene expression. Investigating its role in DNA methylation, histone modifications and chromatin remodeling could unveil new therapeutic avenues in epigenetic-based cancer treatments.
Kynurenine
Kynurenine, a tryptophan-derived metabolite, is pivotal in cancer metabolism. It serves as a precursor for nicotinic acid and NAD synthesis, essential for energy metabolism and DNA repair2. Recent studies show that the oncogenic transcription factor MYC boosts the expression of tryptophan transporters (SLC1A5 and SLC7A5) and AFMID, promoting kynurenine synthesis and enhancing its metabolic flux in cancer cells63. Moreover, kynurenine activates the AHR transcription factor, which regulates growth-promoting genes, further linking it to cancer progression2. Elevated levels of kynurenine increase the synthesis of nicotinic acid, leading to higher concentrations of NADH and NADPH, which have been shown to be overexpressed in tumors harboring mutant p5364. Given its critical role in energy metabolism and DNA repair, kynurenine emerges as a potential oncometabolite. The enhanced cellular uptake of tryptophan and its subsequent metabolism through the kynurenine pathway has been confirmed using high-performance liquid chromatography–tandem mass spectrometry metabolomics, highlighting its relevance in cancer cell metabolism63. Kynurenine’s impact on NAD+ biosynthesis suggests it may influence epigenetic processes by affecting sirtuin enzymes, which regulate histone deacetylation and chromatin remodeling. This information suggests that kynurenine may be a promising target for therapeutic strategies that modulate both metabolic and epigenetic pathways in cancer.
MGO
MGO, a byproduct of glycolysis, is elevated in cancer owing to increased glycolytic flux. As a precursor to advanced glycation end products (AGEs), MGO contributes to protein glycation, which is linked to cancer and other pathologies65. MGO modifies proteins at arginine residues, forming AGEs such as argpyrimidine and hydroimidazolone66. These modifications have been linked to cancer progression, particularly through the disruption of protein functions and cellular signaling pathways65. Recent studies reveal that MGO suppresses the detoxification enzyme glyoxalase 1, leading to nuclear localization of the transcriptional coactivator Yes-associated protein (YAP) and inhibition of the Hippo tumor-suppressor pathway in breast cancer cells, promoting uncontrolled cell proliferation67,68. Furthermore, MGO levels oscillate during the cell cycle, with higher concentrations observed in the G1 phase, suggesting its involvement in cell cycle regulation3. In addition, MGO has been shown to activate checkpoint kinases Chk1 and Chk2 in an AGE-dependent manner, highlighting its role in DNA damage response pathways3,69.
MGO modifies histones through glycation, potentially altering chromatin structure and gene expression in breast cancer. AGEs from MGO may disrupt DNA repair and transcription, influencing the epigenetic landscape. This positions MGO as a potential oncometabolite, warranting further investigation into its role in cancer metabolism and epigenetics. A summary of putative oncometabolites is provided in Table 2.
Unraveling transient metabolite–protein interactions: challenges, emerging technologies and unanswered questions
Metabolite–enzyme interactions play a key role in metabolism, epigenetic regulation and cancer progression. However, their transient and dynamic nature, driven by weak noncovalent forces, makes them difficult to study. Conventional methods such as mass spectrometry and metabolomics provide valuable snapshots but struggle to link metabolites with their protein interactors owing to dissociation or low sensitivity for low-abundance complexes9. Similarly, chromatin immunoprecipitation, optimized for studying DNA–protein or histone–protein complexes, cannot stabilize or detect interactions involving small metabolites such as acetyl-CoA or SAM70. The discovery of oncometabolites, such as 2-HG in IDH-mutant cancers and fumarate/succinate in SDH- and FH-deficient cancers, underscores the critical role of dysregulated metabolite–enzyme interactions in epigenetic reprogramming and tumorigenesis34. Despite this, many oncometabolites remain undiscovered, and their contributions to epigenetic regulation and cancer progression are probably underestimated owing to methodological limitations. Emerging technologies, such as proximity labeling techniques (for example, BioID and APEX) and cross-linking mass spectrometry (XL-MS), offer new ways to capture these transient interactions71. XL-MS, for example, has successfully mapped acetyl-CoA interactions with chromatin-modifying enzymes, illustrating how metabolite–protein interactions are integral to both metabolism and epigenetic regulation72. However, XL-MS faces challenges, such as nonspecific interactions and the need for high concentrations of cross-linkers, which can perturb native cellular conditions72. Despite these advances, significant questions remain. What novel metabolite–protein interactions are critical to chromatin regulation and cancer progression but remain undetectable with current tools? Can selective cross-linkers or engineered probes be developed to stabilize metabolite–enzyme interactions under physiological conditions? How can spatially compartmentalized metabolites in distinct cellular regions be studied in their native environments? By integrating chemical biology, structural proteomics and computational modeling, we can uncover new oncometabolites as potential cancer therapeutic targets. Addressing these challenges is crucial for advancing our understanding of cancer metabolism and epigenetics.
New frontiers in research
Recent advances in research tools are opening new frontiers in understanding the dynamic interplay between metabolites and epigenetic regulation. Single-cell multiomics technologies, which combine genomic, transcriptomic, proteomic and metabolomic data at the single-cell level, enable researchers to capture cellular heterogeneity and map complex molecular networks in unprecedented detail73. These platforms offer powerful insights into the spatial and temporal variations of metabolite levels, facilitating the identification of key metabolic regulators that drive epigenetic modifications73. Alongside this, artificial-intelligence-driven molecular simulations are proving essential in predicting metabolite–protein interactions and modeling complex biochemical pathways74. These tools may provide a computational framework to understand the mechanisms underpinning cellular metabolism and epigenetic changes with greater precision. Another major innovation is isotope-labeled metabolomics, which enables the tracking of specific metabolites in real time, providing insights into metabolic flux and its impact on gene expression and chromatin remodeling75. However, despite these advances, one of the most pressing challenges remains the integration of real-time metabolite tracking with epigenomic profiling. To truly capture the dynamic relationship between metabolism and epigenetics, future research must develop approaches that can synchronize metabolic changes with chromatin modifications as they occur in living cells. Integrating these data in a seamless and real-time manner would provide deeper insights into how metabolic reprogramming shapes gene expression, cellular fate and disease progression, particularly in cancer and other metabolic disorders.
Order to chaos as revealed by metabolic shifts in aging and cancer
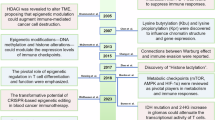
Cellular metabolism undergoes dynamic transitions throughout development, from embryonic stem cells to differentiated tissues8. During early development, metabolic and epigenetic programs are highly coordinated, ensuring controlled differentiation and tissue homeostasis8. This structured process is maintained by precise genetic programs, metabolic adaptation and paracrine signaling, allowing cells to respond to physiological stimuli in an orderly manner. However, as tissues age, external exposures (such as ultraviolet (UV) radiation), hormonal fluctuations and accumulated genetic mutations introduce metabolic instability. These disruptions contribute to epigenetic deterioration, clonal selection and eventually disease etiology76 (Fig. 3). Cancer represents an extreme example of this shift, where metabolic adaptation is no longer regulated but exploited for uncontrolled growth. Tumor cells hijack developmental metabolic pathways; however, instead of following an orchestrated differentiation program, they remain in a state of epigenetic plasticity, evading cellular constraints. Understanding how these metabolic networks transition from order to chaos, particularly in aging and neoplasia, could provide critical insights into therapeutic strategies aimed at restoring homeostasis and targeting metabolic vulnerabilities in cancer progression (Fig. 3).

Broad categories of events and their quantitative contribution over time from embryonic stem cells to adult stem cells to functional tissue. The homeostasis and integrity of tissues in the body, in large part driven by age-related processes, contributes to disease etiology. Cancer develops when homeostasis is compromised and chaos ensues, leading to processes that resemble the events that allow embryonic stem cells to proliferate, awaiting signaling leading to cellular orchestration and differentiation.
Insights into DNMT–metabolite interactions and lipid metabolism
As discussed, distinct metabolites directly influence epigenetic modifications, mostly as cofactors. However, metabolites could also influence the epigenome as competitive and/or noncompetitive inhibitors of chromatin remodelers, possibly through altered metabolic output and shift in concentration leading to unwanted protein–metabolite interactions. We performed an untargeted metabolite analysis of cancer cell lines overexpressing FLAG-tagged DNMT3A and DNMT1, followed by co-immunoprecipitation, which provided us with chemical categories of molecules that would follow this pattern. Preliminary analysis based on UV and dimethyl pimelimidate cross-linking revealed associations with lipid-like molecules and organic metabolites using mass spectrometry. These interactions suggest that abundant metabolites could act as reversible inhibitors that negatively influence DNMT activity. Lipid-like molecules may influence enzymatic function as competitive inhibitors through hydrophobic or electrostatic interactions, potentially altering DNA methylation dynamics. Organic molecules, including aromatic or amino acid derivatives, could act as both competitive inhibitors or allosteric regulators, interfering with substrate binding or catalysis. Supporting this, a recent epigenome-wide association study of leukocyte DNA methylation identified significant associations with metabolic measures, particularly those related to lipoproteins. The study highlighted CpG sites in genes such as ABCG1 and PHGDH, which were linked to lipid metabolism and metabolic diseases, including obesity and myocardial infarction. These genes also play a direct role in lipid transport (ABCG1) and amino acid metabolism (PHGDH), thereby highlighting a regulatory feedback loop. This finding lends further support to the notion that epigenetic regulation, facilitated by metabolites, is crucial for lipid metabolism and its associated disease pathways77. Furthermore, research examining the effects of high-glucose culture conditions on hepatocytes indicated that 25-hydroxycholesterol activates DNMT1, modulating genes involved in lipid metabolism. This suggests a direct role of metabolites, such as 25-hydroxycholesterol, in epigenetically regulating lipid metabolism. Similarly, another study found that maternal circulating lipids correlate with infant DNA methylation patterns, further supporting the hypothesis that metabolites have long-lasting epigenetic effects influencing metabolic health78. In addition, a study tracking 40 mother–infant dyads found significant correlations between maternal plasma metabolites, such as very-long-chain fatty acids and medium-chain acylcarnitines, and infant DNA methylation patterns79. These findings suggest maternal metabolites shape fetal epigenetics and long-term health. Exploring DNMT–lipid interactions through lipidomics and structural proteomics could reveal novel metabolic–epigenetic mechanisms, paving the way for targeted therapies in metabolic disorders and cancer.
Dietary influence on epigenetics and cancer prevention
Dietary patterns influence cancer development and prevention by modulating epigenetic and metabolic pathways. Bioactive compounds from food interact with stress and environmental factors, shaping the epigenetic landscape. Cancer is marked by aberrant DNA methylation, characterized by global hypomethylation, which contributes to chromosomal instability and can lead to the activation of oncogenes. Simultaneously, gene-specific hypermethylation, particularly in the regulatory regions of tumor-suppressor genes, results in their silencing and promotes tumorigenesis80. This dual nature of DNA methylation underscores the potential of diet to counteract these changes, offering avenues for chemoprevention strategies80 Bioactive compounds such as epigallocatechin gallate (green tea), curcumin (turmeric) and quercetin (apples, onions) modulate epigenetics. Epigallocatechin gallate inhibits DNMTs, restoring tumor-suppressor genes such as CDKN2A and MGMT81. Similarly, quercetin and curcumin synergistically trigger global hypomethylation and apoptosis in cancer cells by disrupting mitochondrial function and cell cycle regulation80,82,83. Among dietary bioactives, phytoestrogens such as stilbenes, including resveratrol and pterostilbene and isoflavones such as genistein, act as natural endocrine modulators with significant epigenetic effects. The effect of stilbenes on the remodeling of DNA methylation and histone modifications have been demonstrated in the context of breast cancer and other models. These effects include the silencing of oncogenes and the restoration of tumor-suppressor activity84,85,86. Notably, despite growing evidence of their epigenetic effects, the influence of such compounds on epigenetic metabolites and their direct connection to chromatin remodeling remains insufficiently studied. However, challenges such as poor bioavailability and the need for effective systemic concentrations of these compounds limit their therapeutic application80. Advancing drug delivery and dietary formulations is key to overcoming these limitations. Environmental pollutants, tobacco smoke and endocrine disruptors further amplify epigenetic disruptions, worsening dietary imbalances12. Chronic stress disrupts epigenetic regulation by activating the hypothalamic–pituitary–adrenal axis, elevating cortisol and altering DNA methylation and histone modifications87. These stress-induced epigenetic changes can synergize with environmental carcinogens to drive tumorigenesis, underscoring the need to address psychosocial and environmental factors alongside diet. For example, one-carbon metabolism, crucial for DNA, RNA and histone methylation, relies on B vitamins (that is, B6, B9 and B12), methionine and choline80,83. Nutrient deficiencies and genetic polymorphisms, such as the MTHFR C677T variant, represent a significant cause for concern. This prevalent mutation has been demonstrated to reduce the activity of the methylenetetrahydrofolate reductase enzyme, thereby impairing folate metabolism83. Elevated homocysteine levels, impaired DNA repair and weakened immune responses are among the downstream effects of such disruptions10,44. Environmental toxins, such as heavy metals and persistent organic pollutants, can also interfere with one-carbon metabolism, compounding these12. Optimizing methyl-donor intake through diet may restore epigenetic balance and reduce cancer risk. Nutritional epigenomics offers a promising, accessible prevention strategy, with early life interventions supporting lifelong epigenetic stability, especially in high-risk populations80. Integrating bioactive dietary compounds with lifestyle changes—such as stress management, regular exercise and reduced exposure to environmental carcinogens—could synergistically reverse malignant epigenetic patterns12. Advancing precision nutrition for cancer prevention and therapy requires understanding the molecular mechanisms of dietary–epigenetic interactions, overcoming bioavailability challenges and exploring the interplay between genetic, epigenetic and environmental factors. This will enable targeted, personalized dietary strategies to reduce cancer incidence and improve outcomes.
Dietary interventions as modulators of cancer metabolism and epigenetics
Recent studies suggest that dietary interventions, such as the ketogenic diet, may help combat cancer by targeting metabolic pathways and epigenetic modifications. This high-fat, low-carbohydrate diet lowers glucose and insulin levels while increasing ketone bodies such as β-hydroxybutyrate, which can inhibit tumor growth in certain models88,89. The ketogenic diet’s effects vary by context, with several studies linking it to increased tumor growth. Recent research shows it promotes metastasis in mice via BACH1, which activates prometastatic genes such as hyaluronidase 1. Inhibiting BACH1 mitigates this effect, while ATF4 enhances BACH1’s activity, highlighting potential risks for patients with cancer90.
Of note, methionine restriction alters DNA and histone methylation, influencing tumor suppression and chemotherapy sensitivity (for example, 5-fluorouracil). While promising, it may have adverse effects in some rodent models, highlighting the need for cautious application91. Similarly, glutamine, an essential amino acid for cancer cell metabolism, has been found to influence epigenetic marks, such as histone methylation, suggesting that glutamine modulation could be a potential strategy to disrupt cancer progression. Choline, a vital methyl donor in one-carbon metabolism, plays a critical role in cancer epigenetics. Its depletion disrupts DNA methylation and histone modifications, which can drive oncogenic reprogramming. Similarly, dietary fructose exacerbates tumorigenesis by promoting lipogenesis and histone acetylation, activating cancer-associated pathways92. In parallel, the gut-microbial metabolism of choline alters host methylation, metabolism and even behavior, as demonstrated in engineered microbial systems. Together, these findings emphasize the interplay between diet, microbial metabolism and epigenetic regulation in cancer and metabolic health. Recent advances in machine learning enable personalized health interventions by classifying individuals on the basis of metabotypes and lifestyle traits. These data-driven approaches might support precision medicine and chronic disease prevention93. These findings collectively suggest that dietary interventions targeting metabolic and epigenetic changes show promise as cancer therapies. However, their complex, context-dependent effects require further research to optimize clinical application and develop personalized treatments.
Conclusions and future directions
The intricate relationship between metabolism and epigenetics represents a transformative area in cancer research, providing new insights into how cellular metabolic states shape gene regulation and tumor progression. This Review underscores the diverse roles of key metabolites, oncometabolites and dietary factors in modulating the epigenome, highlighting their potential as biomarkers and therapeutic targets. Despite significant advancements, several unanswered questions and technical challenges remain. One of the most pressing issues is the transient nature of metabolite–enzyme interactions, which complicates their study under physiological conditions. Current technologies, though innovative, often lack the sensitivity or precision required to capture these fleeting associations. Advancing methods such as isotope-labeled metabolomics, single-cell multiomics and spatially resolved epigenomics will be crucial to overcoming these barriers. Moreover, identifying unconventional metabolites with potential epigenetic roles, such as sarcosine and hypotaurine, opens new research avenues. Expanding the scope of the investigation beyond well-characterized molecules such as SAM and acetyl-CoA to explore the broader metabolome will probably uncover novel regulatory mechanisms and therapeutic opportunities. Cellular metabolism undergoes coordinated transitions from embryonic stem cells to differentiated tissues to maintain homeostasis. As tissues age, external exposures, hormonal changes and genetic mutations disrupt metabolic stability, contributing to epigenetic deterioration and cancer. In cancer, tumor cells exploit developmental metabolic pathways, maintaining epigenetic plasticity. Understanding how these metabolic networks shift from order to chaos, especially in aging and cancer, can inform therapeutic strategies targeting metabolic vulnerabilities. Regarding translational research, dietary interventions represent an exciting frontier for modulating cancer metabolism and epigenetics. Precision nutrition strategies tailored to individual metabolic profiles could optimize therapeutic outcomes, particularly when combined with pharmacological agents targeting epigenetic regulators. However, the context-dependent effects of dietary components underscore the need for personalized approaches based on tumor type, genetic background and metabolic status. Looking forward, integrating artificial-intelligence-driven computational modeling or machine learning with experimental research will be a game-changer, enabling the prediction and validation of metabolite–protein interactions at unprecedented scales. These tools could also facilitate the development of small-molecule inhibitors or activators specifically targeting metabolite-driven epigenetic dysregulation. Understanding the metabolism–epigenetics interplay can drive breakthroughs in cancer biology and personalized therapies. Bridging knowledge gaps and leveraging emerging technologies will unlock the full potential of this axis for cancer diagnostics, prevention and treatment.
References
Lanzetti, L. Oncometabolites at the crossroads of genetic, epigenetic and ecological alterations in cancer. Cell Death Differ. 31, 1582–1594 (2024).
Beadle, G. W., Mitchell, H. K. & Nyc, J. F. Kynurenine as an intermediate in the formation of nicotinic acid from tryptophane by neurospora. Proc. Natl Acad. Sci. USA 33, 155–158, https://doi.org/10.1073/pnas.33.6.155 (1947).
Beyoglu, D. & Idle, J. R. Metabolic rewiring and the characterization of oncometabolites. Cancers https://doi.org/10.3390/cancers13122900 (2021).
Gao, P. et al. Hypotaurine evokes a malignant phenotype in glioma through aberrant hypoxic signaling. Oncotarget 7, 15200–15214 (2016).
Ippolito, L., Morandi, A., Giannoni, E. & Chiarugi, P. Lactate: a metabolic driver in the tumour landscape. Trends Biochem. Sci. 44, 153–166 (2019).
Jain, M. et al. Metabolite profiling identifies a key role for glycine in rapid cancer cell proliferation. Science 336, 1040–1044 (2012).
Sreekumar, A. et al. Metabolomic profiles delineate potential role for sarcosine in prostate cancer progression. Nature 457, 910–914 (2009).
Mahmoud, A. I. Metabolic switches during development and regeneration. Development https://doi.org/10.1242/dev.202008 (2023).
Gao, Y. et al. Crosstalk between metabolic and epigenetic modifications during cell carcinogenesis. iScience 27, 111359 (2024).
Wang, Y., Sun, Z. & Szyf, M. S-adenosyl-methionine (SAM) alters the transcriptome and methylome and specifically blocks growth and invasiveness of liver cancer cells. Oncotarget 8, 111866–111881 (2017).
Schvartzman, J. M., Thompson, C. B. & Finley, L. W. S. Metabolic regulation of chromatin modifications and gene expression. J. Cell Biol. 217, 2247–2259 (2018).
Ho, S. M. et al. Environmental epigenetics and its implication on disease risk and health outcomes. ILAR J. 53, 289–305 (2012).
Dai, Z., Ramesh, V. & Locasale, J. W. The evolving metabolic landscape of chromatin biology and epigenetics. Nat. Rev. Genet. 21, 737–753 (2020).
Newman, A. C. & Maddocks, O. D. K. One-carbon metabolism in cancer. Br. J. Cancer 116, 1499–1504 (2017).
Chen, C., Wang, Z. & Qin, Y. Connections between metabolism and epigenetics: mechanisms and novel anti-cancer strategy. Front. Pharm. 13, 935536 (2022).
Locasale, J. W. et al. Phosphoglycerate dehydrogenase diverts glycolytic flux and contributes to oncogenesis. Nat. Genet. 43, 869–874 (2011).
Haase, C., Bergmann, R., Fuechtner, F., Hoepping, A. & Pietzsch, J. ʟ-type amino acid transporters LAT1 and LAT4 in cancer: uptake of 3-O-methyl-6-18F-fluoro-L-dopa in human adenocarcinoma and squamous cell carcinoma in vitro and in vivo. J. Nucl. Med. 48, 2063–2071 (2007).
Dann, S. G. et al. Reciprocal regulation of amino acid import and epigenetic state through Lat1 and EZH2. EMBO J. 34, 1773–1785 (2015).
Da, M. X., Zhang, Y. B., Yao, J. B. & Duan, Y. X. DNA methylation regulates expression of VEGF-C, and S-adenosylmethionine is effective for VEGF-C methylation and for inhibiting cancer growth. Braz. J. Med Biol. Res 47, 1021–1028 (2014).
Huo, M., Zhang, J., Huang, W. & Wang, Y. Interplay among metabolism, epigenetic modifications, and gene expression in cancer. Front. Cell Dev. Biol. 9, 793428 (2021).
Shyh-Chang, N. et al. Influence of threonine metabolism on S-adenosylmethionine and histone methylation. Science 339, 222–226 (2013).
Kryukov, G. V. et al. MTAP deletion confers enhanced dependency on the PRMT5 arginine methyltransferase in cancer cells. Science 351, 1214–1218 (2016).
Parashar, S. et al. S-adenosylmethionine blocks osteosarcoma cells proliferation and invasion in vitro and tumor metastasis in vivo: therapeutic and diagnostic clinical applications. Cancer Med. 4, 732–744 (2015).
Guertin, D. A. & Wellen, K. E. Acetyl-CoA metabolism in cancer. Nat. Rev. Cancer 23, 156–172 (2023).
Izzo, L. T. et al. Acetylcarnitine shuttling links mitochondrial metabolism to histone acetylation and lipogenesis. Sci. Adv. 9, eadf0115 (2023).
Schug, Z. T. et al. Acetyl-CoA synthetase 2 promotes acetate utilization and maintains cancer cell growth under metabolic stress. Cancer Cell 27, 57–71 (2015).
Mullen, A. R. et al. Reductive carboxylation supports growth in tumour cells with defective mitochondria. Nature 481, 385–388 (2011).
Wang, H. et al. Nucleo-cytosolic acetyl-CoA drives tumor immune evasion by regulating PD-L1 in melanoma. Cell Rep. 43, 115015 (2024).
Carrer, A. et al. Acetyl-CoA metabolism supports multistep pancreatic tumorigenesis. Cancer Discov. 9, 416–435 (2019).
Covarrubias, A. J., Perrone, R., Grozio, A. & Verdin, E. NAD+ metabolism and its roles in cellular processes during ageing. Nat. Rev. Mol. Cell Biol. 22, 119–141 (2021).
Lucena-Cacace, A., Otero-Albiol, D., Jimenez-Garcia, M. P., Munoz-Galvan, S. & Carnero, A. NAMPT is a potent oncogene in colon cancer progression that modulates cancer stem cell properties and resistance to therapy through Sirt1 and PARP. Clin. Cancer Res. 24, 1202–1215 (2018).
Canto, C. & Auwerx, J. Targeting sirtuin 1 to improve metabolism: all you need is NAD. Pharm. Rev. 64, 166–187 (2012).
Zhu, Y., Liu, J., Park, J., Rai, P. & Zhai, R. G. Subcellular compartmentalization of NAD+ and its role in cancer: a sereNADe of metabolic melodies. Pharm. Ther. 200, 27–41 (2019).
Yong, C., Stewart, G. D. & Frezza, C. Oncometabolites in renal cancer. Nat. Rev. Nephrol. 16, 156–172 (2020).
Dang, L. et al. Cancer-associated IDH1 mutations produce 2-hydroxyglutarate. Nature 462, 739–744 (2009).
Lu, C. et al. IDH mutation impairs histone demethylation and results in a block to cell differentiation. Nature 483, 474–478 (2012).
Rohle, D. et al. An inhibitor of mutant IDH1 delays growth and promotes differentiation of glioma cells. Science 340, 626–630 (2013).
Li, Z. et al. Lactate in the tumor microenvironment: a rising star for targeted tumor therapy. Front. Nutr. 10, 1113739 (2023).
Pandkar, M. R., Sinha, S., Samaiya, A. & Shukla, S. Oncometabolite lactate enhances breast cancer progression by orchestrating histone lactylation-dependent c-Myc expression. Transl. Oncol. 37, 101758 (2023).
Huang, Z. W. et al. STAT5 promotes PD-L1 expression by facilitating histone lactylation to drive immunosuppression in acute myeloid leukemia. Signal Transduct. Target Ther. https://doi.org/10.1038/s41392-023-01605-2 (2023).
Yang, Z. et al. Lactylome analysis suggests lactylation-dependent mechanisms of metabolic adaptation in hepatocellular carcinoma. Nat. Metab. 5, 61–79 (2023).
Xiong, J. et al. Lactylation-driven METTL3-mediated RNA m6A modification promotes immunosuppression of tumor-infiltrating myeloid cells. Mol. Cell 82, 1660–1677.e1610 (2022).
Mu, X. et al. Oncometabolite succinate promotes angiogenesis by upregulating VEGF expression through GPR91-mediated STAT3 and ERK activation. Oncotarget 8, 13174–13185 (2017).
Wang, P., Chen, L. L., Xiong, Y. & Ye, D. Metabolite regulation of epigenetics in cancer. Cell Rep. 43, 114815 (2024).
Wentzel, J. F. et al. Exposure to high levels of fumarate and succinate leads to apoptotic cytotoxicity and altered global DNA methylation profiles in vitro. Biochimie 135, 28–34 (2017).
Cramer-Morales, K. L., Heer, C. D., Mapuskar, K. A. & Domann, F. E. Succinate accumulation links mitochondrial MnSOD depletion to aberrant nuclear DNA methylation and altered cell fate. J. Exp. Pathol. 1, 60–70 (2020).
Valcarcel-Jimenez, L. & Frezza, C. Fumarate hydratase (FH) and cancer: a paradigm of oncometabolism. Br. J. Cancer 129, 1546–1557 (2023).
Sciacovelli, M. et al. Fumarate is an epigenetic modifier that elicits epithelial-to-mesenchymal transition. Nature 537, 544–547 (2016).
Jiang, Y. et al. Local generation of fumarate promotes DNA repair through inhibition of histone H3 demethylation. Nat. Cell Biol. 17, 1158–1168 (2015).
Menko, F. H. et al. Hereditary leiomyomatosis and renal cell cancer (HLRCC): renal cancer risk, surveillance and treatment. Fam. Cancer 13, 637–644 (2014).
Giallongo, S. et al. The pleiotropic effects of fumarate: from mitochondrial respiration to epigenetic rewiring and DNA repair mechanisms. Metabolites https://doi.org/10.3390/metabo13070880 (2023).
Gaffney, D. O. et al. Non-enzymatic lysine lactoylation of glycolytic enzymes. Cell Chem. Biol. 27, 206–213.e206 (2020).
Adam, J. et al. Renal cyst formation in Fh1-deficient mice is independent of the Hif/Phd pathway: roles for fumarate in KEAP1 succination and Nrf2 signaling. Cancer Cell 20, 524–537, https://doi.org/10.1016/j.ccr.2011.09.006 (2011).
Ge, X. et al. Fumarate inhibits PTEN to promote tumorigenesis and therapeutic resistance of type2 papillary renal cell carcinoma. Mol. Cell 82, 1249–1260.e1247 (2022).
Zheng, L. et al. Fumarate induces redox-dependent senescence by modifying glutathione metabolism. Nat. Commun. 6, 6001 (2015).
Li, F. et al. NADP+-IDH mutations promote hypersuccinylation that impairs mitochondria respiration and induces apoptosis resistance. Mol. Cell 60, 661–675 (2015).
Jentzmik, F. et al. Sarcosine in urine after digital rectal examination fails as a marker in prostate cancer detection and identification of aggressive tumours. Eur. Urol. 58, 12–18 (2010).
Wu, H. et al. GC/MS-based metabolomic approach to validate the role of urinary sarcosine and target biomarkers for human prostate cancer by microwave-assisted derivatization. Anal. Bioanal. Chem. 401, 635–646 (2011).
Khan, A. P. et al. The role of sarcosine metabolism in prostate cancer progression. Neoplasia 15, 491–501 (2013).
Strmiska, V. et al. Sarcosine is a prostate epigenetic modifier that elicits aberrant methylation patterns through the SAMe–Dnmts axis. Mol. Oncol. 13, 1002–1017 (2019).
Zhang, W. C. et al. Glycine decarboxylase activity drives non-small cell lung cancer tumor-initiating cells and tumorigenesis. Cell 148, 259–272 (2012).
Shen, D. et al. ADO/hypotaurine: a novel metabolic pathway contributing to glioblastoma development. Cell Death Discov. 7, 21 (2021).
Venkateswaran, N. et al. MYC promotes tryptophan uptake and metabolism by the kynurenine pathway in colon cancer. Genes Dev. 33, 1236–1251 (2019).
Rather, G. M., Pramono, A. A., Szekely, Z., Bertino, J. R. & Tedeschi, P. M. In cancer, all roads lead to NADPH. Pharm. Ther. https://doi.org/10.1016/j.pharmthera.2021.107864 (2021).
Leone, A. et al. The dual-role of methylglyoxal in tumor progression – novel therapeutic approaches. Front Oncol. https://doi.org/10.3389/fonc.2021.645686 (2021).
Lin, J. A., Wu, C. H., Lu, C. C., Hsia, S. M. & Yen, G. C. Glycative stress from advanced glycation end products (AGEs) and dicarbonyls: an emerging biological factor in cancer onset and progression. Mol. Nutr. Food Res. 60, 1850–1864 (2016).
Koo, J. H. & Guan, K. L. Interplay between YAP/TAZ and metabolism. Cell Metab. 28, 196–206 (2018).
Wang, Z. et al. The Hippo–TAZ axis mediates vascular endothelial growth factor C in glioblastoma-derived exosomes to promote angiogenesis. Cancer Lett. 513, 1–13 (2021).
Kani, S. et al. Chk2 kinase is required for methylglyoxal-induced G2/M cell-cycle checkpoint arrest: implication of cell-cycle checkpoint regulation in diabetic oxidative stress signaling. Genes Cells 12, 919–928 (2007).
Small, E. C. et al. Chromatin immunoprecipitation (ChIP) to study DNA–protein interactions. Methods Mol. Biol. 2261, 323–343 (2021).
Wu, S., Zhang, S., Liu, C. M., Fernie, A. R. & Yan, S. Recent advances in mass spectrometry-based protein interactome studies. Mol. Cell Proteom. 24, 100887 (2025).
Piersimoni, L., Kastritis, P. L., Arlt, C. & Sinz, A. Cross-linking mass spectrometry for investigating protein conformations and protein–protein interactions horizontal line a method for all seasons. Chem. Rev. 122, 7500–7531 (2022).
Mansoor, S., Hamid, S., Tuan, T. T., Park, J. E. & Chung, Y. S. Advance computational tools for multiomics data learning. Biotechnol. Adv. 77, 108447 (2024).
Tambi, R. et al. Artificial intelligence and omics in malignant gliomas. Physiol. Genomics 56, 876–895 (2024).
Chokkathukalam, A., Kim, D. H., Barrett, M. P., Breitling, R. & Creek, D. J. Stable isotope-labeling studies in metabolomics: new insights into structure and dynamics of metabolic networks. Bioanalysis 6, 511–524 (2014).
Lopez-Otin, C., Blasco, M. A., Partridge, L., Serrano, M. & Kroemer, G. The hallmarks of aging. Cell 153, 1194–1217 (2013).
Gomez-Alonso, M. D. C. et al. DNA methylation and lipid metabolism: an EWAS of 226 metabolic measures. Clin. Epigenetics https://doi.org/10.1186/s13148-020-00957-8 (2021).
Wang, Y. et al. High glucose induces lipid accumulation via 25-hydroxycholesterol DNA-CpG methylation. iScience 23, 101102 (2020).
Marchlewicz, E. H. et al. Lipid metabolism is associated with developmental epigenetic programming. Sci. Rep. 6, 34857 (2016).
Tollefsbol, T. O. Dietary epigenetics in cancer and aging. Cancer Treat. Res 159, 257–267 (2014).
Agarwal, A., Kansal, V., Farooqi, H., Prasad, R. & Singh, V. K. Epigallocatechin gallate (EGCG), an active phenolic compound of green tea, inhibits tumor growth of head and neck cancer cells by targeting DNA hypermethylation. Biomedicines https://doi.org/10.3390/biomedicines11030789 (2023).
Mason, J. B. Biomarkers of nutrient exposure and status in one-carbon (methyl) metabolism. J. Nutr. 133, 941S–947S (2003).
Lyon, P., Strippoli, V., Fang, B. & Cimmino, L. B vitamins and one-carbon metabolism: implications in human health and disease. Nutrients https://doi.org/10.3390/nu12092867 (2020).
Beetch, M. et al. Pterostilbene leads to DNMT3B-mediated DNA methylation and silencing of OCT1-targeted oncogenes in breast cancer cells. J. Nutr. Biochem. 98, 108815 (2021).
Kurzava Kendall, L., Ma, Y., Yang, T., Lubecka, K. & Stefanska, B. Epigenetic effects of resveratrol on oncogenic signaling in breast cancer. Nutrients https://doi.org/10.3390/nu16050699 (2024).
Chen, M. et al. Maternal soybean genistein on prevention of later-life breast cancer through inherited epigenetic regulations. Carcinogenesis 43, 190–202 (2022).
Palma-Gudiel, H. et al. HPA axis regulation and epigenetic programming of immune-related genes in chronically stressed and non-stressed mid-life women. Brain Behav. Immun. 92, 49–56 (2021).
Talib, W. H. et al. Ketogenic diet in cancer prevention and therapy: molecular targets and therapeutic opportunities. Curr. Issues Mol. Biol. 43, 558–589 (2021).
Xiao, Y. L., Gong, Y., Qi, Y. J., Shao, Z. M. & Jiang, Y. Z. Effects of dietary intervention on human diseases: molecular mechanisms and therapeutic potential. Signal Transduct. Target Ther. https://doi.org/10.1038/s41392-024-01771-x (2024).
Su, Z., Liu, Y., Xia, Z., Rustgi, A. K. & Gu, W. An unexpected role for the ketogenic diet in triggering tumor metastasis by modulating BACH1-mediated transcription. Sci. Adv. 10, eadm9481 (2024).
Kubota, Y. et al. Synergy of combining methionine restriction and chemotherapy: the disruptive next generation of cancer treatment. Cancer Diagn. Progn. 3, 272–281 (2023).
Zhou, P. et al. High dietary fructose promotes hepatocellular carcinoma progression by enhancing O-GlcNAcylation via microbiota-derived acetate. Cell Metab. 35, 1961–1975.e1966 (2023).
Higuera-Gomez, A. et al. Computational algorithm based on health and lifestyle traits to categorize lifemetabotypes in the NUTRiMDEA cohort. Sci. Rep. 14, 24835 (2024).
Noushmehr, H. et al. Identification of a CpG island methylator phenotype that defines a distinct subgroup of glioma. Cancer Cell 17, 510–522 (2010).
Turcan, S. et al. IDH1 mutation is sufficient to establish the glioma hypermethylator phenotype. Nature 483, 479–483 (2012).
Cai, M. et al. Understanding the contribution of lactate metabolism in cancer progress: a perspective from isomers. Cancers https://doi.org/10.3390/cancers15010087 (2022).
Acknowledgements
This work was supported by a postdoctoral grant (to A.V.) and research grant nos. 2210530 and 2510450 (to A.M.L.) from the National Cancer Center of Korea Graduate School of Cancer Science and Policy (NCC-GCSP).
Author information
Authors and Affiliations
Contributions
A.V. and A.M.L. conceptualized the manuscript. A.V. conducted literature reviews and drafted the manuscript; both authors participated in revision and editing. A.M.L. prepared the figures.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Verma, A., Lindroth, A.M. The emerging intertwined activities of metabolism and epigenetics unveils culprits and prospects in cancer. Exp Mol Med 57, 1928–1939 (2025). https://doi.org/10.1038/s12276-025-01537-7
Received:
Revised:
Accepted:
Published:
Version of record:
Issue date:
DOI: https://doi.org/10.1038/s12276-025-01537-7
