
Abstract
Astroglia, an extended class of homeostatic and defensive cells of the central nervous system (CNS), contribute to the pathogenesis of all known neurological and neuropsychiatric disorders. The pathophysiology of astrocytes is complex, mutable, disease and disease-stage specific. In neuroinflammatory lesions and in various chronic conditions, astrocytes undergo an evolutionary conserved defensive remodeling known as reactive astrogliosis, which produces highly heterogeneous reactive astrocytic phenotypes. Broadly, reactive astrogliosis can be classified into proliferative anysomorphic barrier-forming astrogliosis characteristic of traumatic CNS lesions and nonproliferative isomorphic gliosis widely manifested in chronic neuropathologies. In addition, in many pathologies, astrocytes undergo atrophy and asthenia with resulting loss of homeostatic support and neuroprotection precipitating neuronal damage. Reactive and atrophic astrocytes may coexist or emerge in sequence in a disease-stage-dependent manner. Several classes of astrocyte-specific molecules and processes implicated in various diseases of the CNS represent therapeutic targets. Astrocyte-specific therapeutic strategies may improve both disease-preventing and disease-modifying therapeutic outcomes.
Similar content being viewed by others
From neuronocentrism to the inclusive brain—the key for therapeutic success
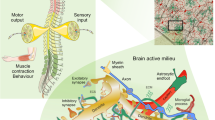
Cognitive impairments are caused by many pathologies affecting the ability to think, concentrate, remember or make decisions. Diseases of the brain, which lead to cognitive and neurological deficiencies and limit the quality of life in the aging global population, represent the main therapeutic challenge of the twenty-first century. There are no effective therapeutic strategies for most of the major disorders of the brain, including ischemic, toxic, autoimmune, neurodevelopmental, neuropsychiatric, malignant and neurodegenerative pathologies; for many of these, neither disease-preventing nor disease-modifying medicines exist. This reflects the complexity of the human nervous system forged by ~500 million years of evolution, which assembled 200 billion of highly heterogeneous cells into intricate networks capable of lifelong structural and functional plasticity. The brain cells include executive neurons capable of long-range fast signaling connecting the brain to the body, the homeostatic and defensive neuroglia, and cells of the brain vasculature. The segregation of functions between neurons and neuroglia emerged early in evolution1. Invertebrates possess many types of neuroglial cells that create brain–body barriers, protect and support axons, and produce complex defensive responses to external insults. In vertebrates and mammals, neuroglia underwent further advancement, becoming the main element responsible for nervous tissue homeostasis and protection.
The human central nervous system (CNS) contains three main types of neuroglia: the homeostatic and neuroprotective astroglia, the myelinating oligodendroglia and the defensive microglia2,3,4,5,6,7. All cells in the brain are linked by multiple feedforward and feedback connections, which maintain the function, versatility and plasticity of the neural tissue8. In response to pathological insults, neuroglial cells actively change to protect the nervous tissue; at the same time, loss of function of neuroglia compromises the resilience of the brain and causes neuronal damage9. The last century witnessed seminal discoveries of ion channels that explained neuronal excitability, information processing, and plasticity, leading to the predominance of neuron-centric views in neurology, psychiatry and neuropharmacology. Consequently, drugs solely targeting neuronal pathways were conceived and developed, whereas neuroglia remained overlooked. This represents a therapeutic gap, which needs to be addressed. In this Review, we focus on astroglia; we briefly introduce these cells from an evolutionary and functional perspective, we present the general pathophysiology of astroglia, and finally we discuss how to target astroglial cells in cognitive brain disorders.
Astrocytes—guardians and housekeepers of the brain
The concept of neuroglia as a connective tissue of the CNS was introduced by Rudolf Virchow10,11. The introduction of the Golgi black staining technique12 revealed the diversity and heterogeneity of neuroglia populating the brain and spinal cord. Many of these cells, when stained with the Golgi technique, have a star-like appearance, which prompted Michal von Lenhossék to coin the name astrocyte13. Astrocytes belong to a larger class of astroglia (Fig. 1), which includes various types of parenchymal astrocytes (protoplasmic, fibrous, velate, marginal and so on), radial astrocytes (Bergmann glia of the cerebellum, Műller glia of the retina, and radial stem astrocytes of the neurogenic niches) and ependymoglia (ependymocytes, tanycytes and choroid plexus cells). The shapes of astroglia are diverse and vary across brain regions14. Protoplasmic astrocytes have a highly complex spongioform (not star-like, which is an artifact of staining with the Golgi technique or immunolabeling with cytoskeletal antibodies) shape defined by a mass of peripheral processes known as leaflets15,16; long processes of fibrous astrocytes align with axons in the white matter and contact nodes of Ranvier17, whereas Müller glia resemble pillars that span the whole thickness of the retina and integrate retinal neurons into independent functional units and acting as light guides18,19. Although morphologically heterogeneous (Fig. 2), the common function of astroglia is the preservation of the brain homeostasis, neuroprotection and brain defense. The core functions of astrocytes include controlling the homeostasis of neurotransmitters (through uptake, catabolism and the supply of precursors), K+ buffering, metabolic support, scavenging of reactive oxygen species (ROS), regulation of water transport and interstitial fluid, formation—through perivascular endfeet—of the parenchymal component of the blood–brain barrier (glia limitans perivascularis), contribution to chemosensing, regulation of energy homeostasis, participation in transmitophagy, and many other processes14. For that, astrocytes are equipped with many receptors (to sense the environment) and numerous transporters that are the backbone for astrocytic homeostatic function; many of these transporters are Na+-dependent, and hence, the astrocytic α2-containing Na+–K+ pump is central for astrocytes homeostatic capacity20. The complement of receptors and transporters varies substantially between brain regions and is regulated by the immediate neurochemical environment.

The plates show diff feremt types of astroglial cells with typical morphology.

A Diversity of astrocyte shapes in the human fetal cortex revealed by Golgi staining. Reproduced from ref. 673. B Morphology of thalamic astrocytes labeled with membrane-targeted GFP. Astrocytes were transduced with AAV-GfaABC1D-Lck-GFP, which drives astrocyte-specific expression under the minimal GfaABC1D promoter, enabling clear visualization of fine membrane processes and leaflet structures. Image is courtesy of Prof. Eunji Cheong, Yonsei University, Republic of Korea. C Rodent astrocytes from different brain regions (i, entorhinal cortex; ii, prefrontal cortex; iii, CA1 area of hippocampus) immunolabeled with antibodies against GFAP, which visualizes soma and major branches. Reproduced, with permission, from ref. 14. D Morphology of the cerebellar Bergmann glial cell. (i) Fluorescence light micrograph of a dye-injected Bergmann glial cell is shown; the red square (20 × 20 mm) corresponds to the portion that was reconstructed from consecutive ultrathin sections. (ii) One of the lateral appendages, arising directly from fiber with all the other side branches omitted for clarity. (iii) The same appendage as shown in (ii), but with one of the appendages marked by blue. Reproduced, with permission from ref. 674. E Astrocytes from human and mouse brains. (i) Astrocyte in the human anteroventral thalamic nucleus with long processes contacting blood vessels. (ii) Astrocyte in the human presubiculum. (iii) Astrocytes in the mouse subicular complex. All images show vimentin immunoreactivity (maximum intensity z-projections: (i) 20 μm thick; (ii) and (iii) 17.4 μm thick). Note the difference in size between human and mouse astrocytes. Images courtesy of Prof. Tim Viney, University of Oxford, UK.
Parenchymal astrocytes (protoplasmic, fibrous, Bergmann glia, Müller glia and so on) establish intricate contacts with synapses, creating a multipartite (and multicellular) synaptic complex—also known as the synaptic cradle—that oversees synaptogenesis, synaptic maturation, maintenance and extinction21,22,23,24. Astrocytes support and regulate synaptic transmission through many astrocyte-specific mechanisms25. Astrocytic glutamate transporters, for example, define the kinetics of glutamate in the synaptic cleft, while astrocytic glutamine is obligatory for neuronal production of glutamate and GABA26,27. Astrocytes are intimately interconnected with all cells of the nervous tissue (the brain active milieu15), with numerous feedback and feedforward signals contributing to the integration of a broad variety of cellular functions in the CNS.
The evolutionary advancement of astroglia is a defining feature of the human brain
The evolutionary history of neuroglia begins with the advent of bilateralia; the existence of neural supportive cells in cnidarians and ctenophores remains debatable28,29,30. The first neuroglial cells were associated with sensory organs known as sensilla in roundworms and flatworms; notably, these glia–neuron sensory units are evolutionarily conserved from Caenorhabditis elegans to humans31. Parenchymal glia emerged in some Platyhelminthes; in particular, glial cells associated with the neuropil and synapses populate the brains of planarians. Neuroglia are present in all Ecdysozoa and Lophotrochozoa, are well developed in Annelida, and are even more developed and highly diverse in Arthropoda, particularly in insects and crustaceans. In Drosophila, for example, up to 36 distinct glial cell types were distinguished; some of these cells create brain–body barriers (perineurial and subperineurial surface glial cells), others cover neuronal somata (cortex glia) and some ensheath synapses in neuropil (astroglia-like cells)32,33. In Echinodermata (which share the common ancestor with Chordata), the radial glia emerged, which signals the appearance of the layered cytoarchitecture. Increase in the brain thickness took place in parallel with the emergence and diversification of parenchymal astrocytes, which become larger and more complex in the brain of Homo sapiens.
Cell morphology is deeply entwined with function, and interspecies comparisons of cell shape may instruct on broad trends in evolution. Likewise, while many evolutionary changes may be related to the generation of new genes, it is broadly recognized that the bulk of interspecies differences arise corollary to changes in gene activity. Moreover, brain evolution is intimately linked to neurological disorders, many of which are specific to humans34,35. Comparative studies of morphology demonstrated an explosion of astrocytes of various sizes, specific shapes (interlaminar astrocytes) and complexity in the human brain36,37,38,39,40 (Fig. 3). Single-cell RNA sequencing (RNA-seq) further shows that, across the large evolutionary distances separating humans from monkeys and rodents, astrocytes, like other neuroglial cells, exhibit greater transcriptomic changes than neuronal cells41,42,43,44. Hence, changes in the astrocyte lineage are instrumental for brain evolution.

a 3D reconstructions of protoplasmic astrocytes filled with fluorescent dye Alexa 594 from adult mouse and human. Modified and reproduced from refs. 134,135. b Primate iAstrocytes recapitulate evolutionary features of fetal brain astrocytes. Top: experimental strategy to obtain primate (human, chimpanzee and macaque) iAstrocytes. Middle: human iAstrocytes feature more complex morphology than higher primates. Representative examples of CD44-immunostained iAstrocytes from Human (left two images) and chimpanzee (right two images). Scale bar, 50 μm. Bottom right: total cell area of iAstrocytes derived from distinct donors (ELE10, n = 51; ELE30, n = 50; AG93, n = 62; AG94, n = 50; SandraA, n = 49; Mandy04, n = 49; Mandy06, n = 51; Becky, n = 47. P: two-sided t-test; ***P < 0.001). Bottom middle: number of primary projections in iAstrocytes per line. Only cells featuring clear-cut projections were considered (ELE10, n = 35; ELE30, n = 26; AG93, n = 17; AG94, n = 19; SandraA, n = 13; Mandy04, n = 18; Mandy06, n = 22; Becky, n = 13. P: two-sided t-test; ***P < 0.001. Bottom: Sholl analysis of iAstrocytes. Intersections were measured every 5 μm in 200-μm radius from the cell soma (concentric circles; ncells 20 (human), 14 (chimpanzee); 8 (macaque); P: two sample Kolmogorov-Smirnov test: Hs versus Pt P = 0.008; Hs versus Mm P = 0.00004; Individual intersections pairs: t-test (***P < 0.0001, **P < 0.001, * **P < 0.01). Modified and reproduced from ref. 45.
Hundreds of differentially expressed genes (DEGs, both protein-coding and noncoding) between human and nonhuman primate samples were identified by RNA-seq of stem cell-derived astrocytes (iAstrocytes)45. In particular, the upregulation of the Hippo-pathway-regulated transcription factor TEAD3 in the human lineage contributes to the increased size and complexity of human astrocytes. Moreover, human astrocytes showed upregulation of genes implicated in the formation of extracellular vesicles (EVs), and indeed human iAstrocytes produce more EVs than chimpanzee and macaque iAstrocytes. Remarkably, treating macaque iAstrocytes with human iAstrocyte-derived EVs leads to increased size and complexity of the macaque cells, highlighting a previously overlooked role of EVs—and, evidently, the cargo they carry—in brain evolution45. Altogether, numerous genes are activated in astrocyte evolution translating into the gain of human-specific features of these cells. Some of these genes can be linked to human-specific neurological disorders, and understanding how they change in evolution can help to explain the natural history of these diseases.
Diversity of astrocytes—cellular, genetic and molecular features
The remarkable morphological diversity of astroglia has been noted from the very early studies; the invention of Golgi ‘reazione nera’12,46 allowed visualization of many morphotypes of radial (such as Bergmann glia or interlaminar astrocytes) and parenchymal astrocytes (protoplasmic, fibrous, velate and others; Figs. 1 and 2). Whether this morphological diversity translates into molecular heterogeneity and, consequently, functional specialization remained less clear. For a long time, astrocytes were considered a rather homogeneous cell population across the entire brain.
As discussed earlier, the archetypal role of astrocytes is to support and protect nervous tissue. In doing so, astrocytes dynamically modulate their interactions with neurons and synapses, with other glial cells, and with the vascular network that supplies nutrients to neurons, synapses and glia, while also regulating local blood flow15,47,48. To serve these manifold functions, astrocytes retain remarkable, life-long cellular plasticity. Moreover, subsets of astrocytes recruited to neuronal circuits under focal demand can episodically undergo cell-state changes, reflected in bursts of transcriptional activity that rapidly re-express gene sets—whether encoding cytoskeletal proteins, transmembrane transporters or antioxidant enzymes—tailored to their microenvironmental stimuli49,50,51. For this very reason, the molecular classification of astrocytes, and the definition of the spatial segregation of their subtypes, if any, are not only more complex than previously thought, but also paramount to dissect the diversity and specificity of mechanisms underlying their cellular plasticity.
Even though the morphological classification of astrocytes is well developed (Fig. 1), these cells have long been—and still often are—functionally classified by default as either ‘resting-state’ or ‘activated’ in response to a stimulus, whether toxic or benign. These definitions are confusing, because astrocytes never ‘rest’ in the healthy brain, while their shape and function are tightly linked and interdependent. Instead, they dynamically respond to physiological stimuli and mount multiple signaling, homeostatic, morphological and secretory responses14,52,53,54. The classification of healthy astrocytes based on their molecular signatures, together with their specific locations, has only begun to evolve—particularly through lineage-tracing studies of radial glia domains, whose neuronal and astrocyte progeny express remarkably similar homeobox gene sets, migrate along identical routes and form microcircuits55,56,57. Despite these remarkable observations, an ensuing avalanche of single-cell RNA-seq studies first molecularly separated astrocytes as a quasi-homogeneous cell group from neurons, oligodendroglia, microglia and vascular cell lineages. More recently, the subgrouping of astrocytes through single-most important molecular features in physiological states or upon genetic manipulations was accomplished58,59,60. Nevertheless, ambiguities remain regarding the bona fide subclasses of astrocytes and their spatial distribution in the nervous system under physiological conditions. This is largely due to the difficulty of distinguishing ‘cell identity feature sets’ from ‘cell-state markers’ in transcriptomic data, with classical correlation analyses relying heavily on statistical differences to segregate cell identities. For astrocytes, however, the most pronounced changes in gene expression are typically associated with their engagement in specific tasks, rather than with their developmental trajectories or spatially defined identities.
The above gaps in appreciating astrocyte diversity arguably reflect the neuron-centric strategies for the molecular interrogation of cellular features used today, ranging from genetic access to astrocyte subgroups and their interrogation by chemical probes or optical stimulation. A restriction that long existed in most algorithms for the analysis of single-cell RNA-seq data is dependence on the gradual enrichment (aggregation) of features for cell annotation when performing unsupervised feature selection based on variability. For neuronal features, a priori assumptions include (1) regionally specific transcriptional signatures, (2) stable developmental endpoints (that is, no dedifferentiation and activity-dependent neurogenesis), (3) stable subsets of function-defining markers, be these neurotransmitter transporters, metabolizing enzymes or neuropeptides61,62, and (4) receptor repertoires for prospective circuit embedding (Fig. 4a). These characteristics do not apply to most astrocytes, except for a few specialized subtypes (for example, tanycytes, ependymocytes that line the walls of the ventricles and central canal, and Bergmann and Müller glia63).

a Single-cell biology for neurons stipulates an enrichment approach built on the cohesive alignment of gene regulatory networks driving cell identification developmentally, to the molecular underpinnings of neurotransmitter and neuropeptide systems that set neurons apart from their peers. b Such gene enrichment is unlikely to suffice for astrocytes because their most distinct gene regulatory networks are related to cellular metabolism and homeostatic adaptation. Therefore, a reductionist approach, in which layers of complexity are sequentially removed, could reveal genuine transcriptionally distinct and positionally segregated subtypes through bona fide subtype-defining genetic features. c Feature-based reconstruction of astrocyte identity. On average, five or more unique features specify spatially segregated astrocytes at a confidence level of >98%. Thus, by iteratively overlaying genes through either single-molecule fluorescence in situ hybridization or spatial transcriptomics, can reveal spatially segregated astrocyte subsets. Note that the addition of each gene increases fidelity while proportionately reducing noise.
It is therefore appealing to harness a reverse approach that relies on the stepwise restriction of features64 (Fig. 4b). First, the removal of genes indiscriminately expressed by astrocytes to retain their cytoarchitectural and fundamental signaling layouts (for example, S100b) or to encode their generic functions (for example, neurotransmitter metabolism and uptake (EAAT1,2, Adcyap1, Glul, Aqp4 and Slc38a3), syncytial connectivity (Gja1), energy metabolism (Apoe, Aldh1l1 and Aldoc) and trophic factors (Ntrk2, Egfr and Fgf3) eliminates ‘stability bias’. Second, context-dependent genes that are historically called ‘inducible’ can be filtered out by comparing RNA-seq datasets on astrocytes under experimental versus control conditions, regressing out inducible genes that either change stochastically from one experiment to another (for example, Ntrk2 and Apoe) or are commonly associated with specific activity states (for example, Gfap and Fos). At this point, the remaining gene set integrates both developmental information—such as transcriptional signatures that characterize distinct proliferative domains—and spatial information, including gene regulatory networks restricted to anatomical loci, along with a flexible repertoire of metabolic and allostatic genes. In other words, such regressive analysis reveals stable molecular marks unifying relatively small numbers of astrocytes that otherwise would remain deeply buried in the conventional RNA-seq data. Henceforth, the expression of stable yet divergent gene regulatory networks can be assigned to specify locations without being negated due to the ‘dilution effect’ of direct unsupervised analysis. By analogy, astrocyte identities might be best resolved by consecutively peeling away layers—like an onion—until the core structure is revealed (Fig. 4b). Once reaching the innermost make-up of astrocytes, not only homeobox genes but also those that are harmonious with the roles of local circuits (hormone receptors, neuropeptide receptors and synapse-specific neuromodulatory units) may define the genuine spatial heterogeneity of astrocytes. Nevertheless, single genes will not suffice to segregate and spatially confine astrocytes. Instead, an iterative process of overlaying core genes should be used to achieve sufficient confidence in identifying molecularly and spatially segregated stable astrocyte subclasses (using five or more genes can provide near-absolute confidence in regions where a high-fidelity reference is available; Fig. 4c).
Thus, efforts of the neuroscience community through molecular and experimental profiling of astrocytes open opportunities to redefine ‘astrocyte identity’ by using an iterative classifier. This approach recognizes astrocytes as progeny of radial glia—born after neurons—and retaining unique positional genes that enable their regionalization, allowing them not only to co-evolve with neurons derived from the same progenitor niche but also to optimally support specialized circuit functions. It is no longer surprising, therefore, that focal silencing (or ablation) of astrocytes renders neurons inapt to control specific behaviors, causally implicating astrocytes in driving higher-order brain functions65,66,67,68.
Astrocytes dynamically control excitation and inhibition of neuronal networks
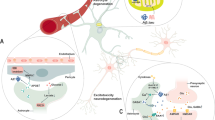
Coordination of glutamatergic (mainly excitatory) and GABAergic (mainly inhibitory) inputs is of fundamental importance for the activity of neuronal networks and therefore required for normal cognitive function. Both processes localize to the plasmalemma and arise from the activity of ion channels and ionotropic receptors, both being further tuned by a panoply of metabotropic receptors. Maintaining stable electrochemical gradients and preserving ion homeostasis in the brain are of fundamental importance for proper neuronal function and cognitive processes. The activity of ion channels is tightly regulated by transmembrane ion gradients, and even modest fluctuations in extracellular ion concentrations can markedly affect neuronal excitability and synaptic transmission and integration. Astrocytes are central for regulating the ion composition of interstitium, the ionostasis, which is intimately associated with neuronal excitability. Recent in vivo mouse studies demonstrate that the composition of interstitial ions shifts according to the prevailing brain state. During wakefulness, extracellular K⁺ concentration is increased, whereas Ca²⁺ and Mg²⁺ concentrations are decreased; the reverse is observed during sleep69. Astrocytic cytosolic Cl⁻ concentration ([Cl⁻]i) also undergoes marked state-dependent variations: [Cl⁻]i remains consistently high during sleep but experiences pronounced fluctuations in wakefulness, culminating in lower mean levels of [Cl⁻]i (ref. 70).
Voltage-gated ion channels that mediate membrane excitability and action potential generation and propagation are highly sensitive to ion gradients and membrane polarization; even relatively small fluctuations in extracellular K+ concentration affect membrane potential and, hence, neuronal excitability. Astrocytes control extracellular K+ primarily through the astrocyte-specific α2-subunit containing Na+–K+ pump and astrocytic inward rectifying Kir4.1 K+ channels71. Inhibition (that is, membrane hyperpolarization) in the adult CNS is mediated by Cl− ions fluxes through several types of Cl− channels and ionotropic GABA and glycine receptors. Cytosolic Cl- in neurons is low (~5 mM), and hence, Cl− channels mediate Cl− influx and hyperpolarization, while the long-lasting activity of these channels depletes the extracellular Cl−. Astrocytes to the contrary have high cytosolic Cl− (30–50 mM)70,72, and when astrocytic Cl− channels are opened (incidentally, astrocytic perisynaptic membranes contain GABAA receptors) Cl− efflux is generated to maintain high extracellular Cl− concentration and sustain inhibitory transmission70.
Synaptic transmission, a fundamental aspect of neuronal coordination and cellular excitability, is dependent on both glutamatergic (excitatory) and GABAergic (inhibitory) systems. These systems are regulated by astrocytes, which, through the glutamate(GABA)–glutamine shuttle, remove extracellular glutamate and supply neurons with glutamine, from which both glutamate and GABA are synthesized73,74,75. At glutamatergic synapses, astrocytes remove the majority of the released glutamate by excitatory amino acid transporters 1 and 2 (EAAT1 and EAAT2/SLC1A2 and SLC1A3)75, which is crucial for maintaining synaptic transmission76,77.
At the same time, glutamate can be released from astrocytes78 through several mechanisms including exocytosis53,79, Sxc− cystine/glutamate antiporter80 or diffusion through plasmalemmal channels, such as Bestrophin-1 (Best-1) anion channels or two-pore-domain potassium channel TREK-181. Astrocytes also regulate the neuronal excitability by modulating tonic N-methyl-D-aspartate (NMDA) receptor currents, by releasing glycine and contributing to extracellular level of D-serine, both being allosteric co-agonists of NMDA receptors through the release of glutamate and contributing to extracellular levels of NMDA receptors82,83. Tonic NMDA receptors currents increase intrinsic neuronal excitability by facilitating action potential generation and integration of dendritic excitatory inputs84; similarly, astrocytes can release homocysteic acid, which acts as an agonist of NMDA receptors85,86.
Astrocytes mediate tonic GABA inhibition in several brain regions, including the thalamus and cerebellum87,88, and supply extracellular Cl− to maintain inhibitory Cl− currents70. It is also noteworthy that the astrocytic cytosolic Cl⁻ concentration [Cl−]i varies not only with wake–sleep cycles but also in response to movement onset and sensory stimulation70. Such dynamic changes highlight the importance of tightly maintained astrocytic Cl− homeostasis for supporting normal neuronal function, especially under prolonged or repetitive bouts of synaptic activation and inhibition. Although the molecular machinery responsible for regulating astrocytic [Cl−]i and orchestrating its brain-state dependence has yet to be fully deciphered, a deeper understanding of these processes may offer valuable therapeutic avenues for modulating brain states and addressing a variety of neurological disorders.
In the healthy brain, astrocytes contribute to tonic inhibition by synthesizing and releasing GABA through nonsynaptic mechanisms. Astrocytic monoamine oxidase B (MAO-B) and diamine oxidase (DAO) catalyze the conversion of putrescine to GABA (Fig. 5). Release of GABA from astrocytes is mediated by nonvesicular pathways, including diffusion through Best-1 channels or through reversed GABA transporters; astrocyte-derived GABA acts through extrasynaptic GABA receptors. The Best-1 channels are localized in astrocytic leaflets and are opened by relatively moderate increases in [Ca2+]i at the resting membrane potential89; notably, Best-1 channel also localize to the plasma membranes of neurons90,91. Astroglial GABA transporters (mainly GAT3/SLC6A1192) contribute to tonic inhibition by regulating extracellular GABA levels. While GAT3 is primarily responsible for GABA uptake, it can also function in reverse mode to release GABA93,94,95. The reversal of GAT3 is controlled by [GABA]i, membrane potential and [Na+]i: both depolarization and an increase in [Na+]i favor the reverse mode20. Neuronal tonic GABA currents tend to reduce neuronal excitability, decrease neuronal spiking and refine information processing by dampening excitatory potentials88,96,97,98; transporter-mediated GABA release has been implicated in this tonic inhibition99,100,101,102, although some studies suggest that such a mechanism may not be physiologically relevant103,104. Moreover, GABA transporters are expressed in oligodendrocytes and microglia, which may also contribute to extracellular GABA dynamics105,106.

See text for further details.
Because astrocytic [Cl−]i is critical for regulating neuronal inhibition, a key question arises: are [Cl−]i shifts merely a byproduct of distinct neuronal activity patterns in different brain states, or do they actively shape neuronal excitability on a state-dependent basis? The equilibrium potential of GABAA receptors (EGABA, which is essentially ECl) in pyramidal neurons varies with the time of day, shifting toward hyperpolarization during sleep and adopting more depolarized values in wakefulness107. This shift in EGABA hinges on neuronal Cl⁻ gradients, governed by the levels of [Cl⁻]ᵢ and [Cl⁻]ₒ. Sleep deprivation induces upregulation of the main Cl⁻-accumulating transporter, NKCC1/SLC12A2, resulting in increased neuronal Cl⁻ accumulation—a reversible process modified by the NKCC1 inhibitor bumetanide107. At the same time, EGABA also depends on [Cl⁻]ₒ in the synaptic cleft, which is regulated by astrocytic Cl⁻ transport mechanisms and astrocytic Cl⁻ release. Elevated astrocytic [Cl−]i during sleep sustains a more hyperpolarized EGABA, whereas decreased [Cl−]i in wakefulness restricts Cl⁻ availability, thereby shifting EGABA to more positive values. These findings highlight the fundamental role of astrocytes in coordinating glutamate, GABA and Cl− to adjust neuronal excitability across brain states70.
To summarize, astrocytes possess multiple pathways contributing to the regulation, both rapid and long-term, of neuronal excitability.
Astroglia, neuroprotection and cognitive reserve
Astroglia-driven homeostatic pathways are central for neuroprotection, through both core homeostatic mechanisms and inducible responses to environmental challenges, stress and pathological insults. Moreover, astrocytes contribute to—and to a large extent define—cognitive reserve, which in turn influences the neurological and cognitive outcomes of all CNS disorders as well as physiological aging. The concept of cognitive reserve was introduced by Yaakov Stern108,109 to account for a well-known absence of direct correlation between the degree of damage to the CNS and cognitive as well as neurological outcome. In particular, cognitive reserve defines cognitive longevity in aging and neurodegenerative disorders.
Cognitive reserve is made from several components: (1) the brain reserve (which reflects the anatomical individual differences quantified by the number of neurons and synapses), (2) the brain maintenance supported by all homeostatic pathways from cellular to organ level), (3) the brain resilience (the ability to withstand stress without mounting the pathology) and (4) the brain compensation directly linked to the regenerative capacity of the nervous tissue. All these components are shaped by the interplay between genetic factors and lifelong experience: in particular, physical exercise, healthy diet and intellectual engagement increase cognitive reserve, whereas diseases and stressors that lead to the accumulation of pathological burden decrease it110,111,112.
Homeostatic, protective and regenerative functions of neuroglia are central for defining the cognitive reserve113. Astrocytes, in particular, contribute to the brain reserve through life-long neurogenesis114,115 as well as through initiating and regulating synaptogenesis, synaptic maturation and synaptic extinction52,116,117. Astrocytes are central to the brain maintenance through (1) controlling ionostasis of major ions (Na+, K+, Ca2+ and Cl−) that define neuronal excitability; (2) providing clearance of major neurotransmitters (glutamate, GABA, and monoamines) and supplying neurons with obligatory precursors of neurotransmitters and neuromodulators (glutamine and L-serine); (3) providing neural cells with energy substrates; and (4) limiting oxidative stress through contributing to biosynthesis of glutathione and recycling of ascorbic acid113. Through all these mechanisms, astrocytes also support synaptic transmission and synaptic plasticity. Finally, astrocytes are fundamental for brain resilience and brain compensation through numerous neuroprotective systems (again glutamate clearance, K+ buffering and ROS scavenging), through reactive astrogliosis, which protects the nervous tissue against acute lesions and limits chronic disorders118, and through supporting postlesional regeneration119,120.
General pathophysiology of astroglia
Classifying astroglial pathophysiology
The role of astrocytes in neuropathology is complex and mutable; astrocytes may undergo multiple progressive and/or regressive changes, exhibiting numerous pathological phenotypes that can coexist within the same disorder, be context- or region-specific, or influence one another during disease progression. Astroglial pathophysiology can be broadly classified into: (1) astroglial reactivity or reactive astrogliosis; (2) astroglial atrophy and functional asthenia; (3) astroglial degeneration and death; and (4) astrocytopathies with aberrant pathological astrocytes9.
Reactive astrogliosis (from Greek glia and -osis, meaning ‘glial process’), which forms a barrier between the brain parenchyma and acutely injured tissue, was described in detail by Pío del Río-Hortega and Wilder Penfield in the 1920s121,122,123. Reactive astrogliosis is an evolutionarily conserved, graded, context-specific and multistage defensive response of astrocytes to neuropathology governed by complex molecular programs that translate into biochemical, morphological, metabolic and physiological features of astroglia, leading to an upregulation or loss of homeostatic cascades, or to the gain of new protective or regenerative functions9,118. There are many distinct or converging reactive phenotypes with idiosyncratic transcriptomic and molecular signatures124,125,126,127,128; the popular dichotomic division into A1–A2 astrocytes is misleading and has been refuted by the glial community118. Conceptually, reactive astrogliosis is subclassified into (1) proliferative, anisomorphic (that is, with loss of territorial domains and profound morphological remodeling), and (2) nonproliferative isomorphic (that is, with preservation of territorial domain organization). The former is the feature of inflammatory process in response to brain trauma of various etiologies (mechanical, ischemic, bacterial and autoimmune) and results in the formation of perilesional borders, and ultimately of the glia limitans perilaesiones. The latter contributes to pathogenesis of chronic neurological disorders9,129,130,131,132. This classification is very broad, and it does not account for the diversity of reactive phenotypes; in-depth characterization of the latter is needed for more precise stratification of astrocytic reactivity.
Astroglial structural atrophy and functional asthenia (from Greek ἀσθένεια, meaning lack of strength, weakness or feebleness, that is, loss of function) is widely present across neurological disorders. Asthenic and atrophic changes are accumulating with aging133, thus reducing neuroprotection and lowering brain resilience and therefore increasing the susceptibility to age-dependent neurodegenerative disorders. Structural atrophy of astrocytes leads to a decrease in synaptic coverage and synaptic maintenance thus affecting both excitatory–inhibitory balance and synaptic plasticity134,135. Atrophy of astrocytic endfeet leads to the loss of perivascular coverage and compromises the blood–brain barrier136. Deficient glutamate clearance drives neuronal damage in neurotoxic disorders such as Wernicke–Korsakoff encephalopathy or hepatic encephalopathy137,138,139, whereas diminished K+ buffering together with insufficient glutamate clearance leads to spreading depression, migraine and epilepsy140,141,142. Structural atrophy of astrocytes is a common sign of stress-induced depression, while manipulation with plasmalemmal linker ezrin, which controls the extension of peripheral astrocytic leaflets, alleviates depressive-like behaviors143,144,145,146,147. Functional deficiency of astrocytic glutamate clearance and K+ buffering is the primary cause of neuronal damage in neurodegenerative diseases, most notably in amyotrophic lateral sclerosis (ALS) and Huntington’s disease (HD)148,149,150,151,152,153, whereas functional asthenia of astrocytes may exacerbate β-amyloid pathology in the context of Alzheimer’s disease (AD)154. Fundamentally, the loss of astrocytic support and neuroprotection rather than the emergence of ‘toxic’ phenotypes takes a leading role in mediating neuronal damage and death across all types of neuropathology.
Astrocytopathies cover the yet poorly characterized pathological changes leading to the emergence of aberrant astrocytes that act as primary pathophysiological entities driving the disease. These are represented by genetic astroglial leukodystrophies, in which aberrant astrocytes fail to support myelination, leading to profound lesions of white matter155. Aberrant astrocytes expressing markers of both astrocyte and microglia have been detected in ALS, in stroke and in dementia with Lewy bodies156,157. Finally, degeneration of astrocytes leads to profound morphological changes, including process fragmentation or clasmatodendrosis, signaling irreparable damage and cell death; clasmatodendrosis is observed in several neuropathologies, including infections, trauma and neurodegeneration158.
Rethinking CNS scarring
In contrast to the healthy brain, where astrocytes have historically struggled for recognition against the neuronocentric perspective, their role in the CNS scarring following brain tissue damage has been often overstated. For much of the last century, the term ‘glial scar’ has dominated the discourse on this topic. While this term includes microglia and oligodendrocyte precursor cells, it has primarily been used to describe reactive astrocytes flanking the lesion border. Regrettably, the glial scar became an oversimplified descriptor for the entire CNS scar, overlooking the critical contribution of the fibrotic component.
Another long-standing misconception was that the astrocytic ‘scar’ impedes tissue repair and axonal regrowth. However, recent findings indicate that reactive astrocytes play a protective role by containing damage and preventing its spread beyond the lesion site159,160. Injuries to the CNS often compromise the integrity of the blood–brain or blood–spinal cord barriers, leading to increased permeability, immune cell infiltration, and the release of inflammatory mediators that exacerbate neurodegeneration. Astrocytes are essential in re-establishing brain barriers after injury159. Furthermore, border-forming astrocytes can extend their processes across the lesion, creating a scaffold that bridges the fibrotic scar and facilitates axonal regeneration161,162,163.
This evolving perspective has shifted attention beyond the astrocytic ‘scar’, recognizing the fibrotic core as a distinct component of CNS scarring119. In larger lesions, such as ischemic brain injuries or traumatic spinal cord damage, fibroblasts and macrophages heavily populate the lesion site164,165. Unless the injury breaches the meninges, scar-forming fibroblasts originate from perivascular sources within CNS tissue. These perivascular fibroblasts, along with pericytes, are recruited locally in a lesion-dependent manner166. Fibroblast activation and extracellular matrix deposition are crucial for wound closure and tissue integrity restoration, as well as to the formation of fibrotic scar formation in the adult mammalian CNS164,167. Notably, the African spiny mouse remains the only known exception, displaying an alternative regenerative response without persistent fibrotic scarring168,169.
Following wound contraction, scar formation is orchestrated through complex astrocyte–fibroblast interactions. Reactive astrocytes express ephrin-B2, while stromal fibroblasts express the congruent receptor EphB2, guiding the formation of a distinct lesion border that segregates glial and fibrotic compartments170. Border-forming astrocytes are recruited locally through the remodeling of astrocytes in the vicinity of the lesion, which proliferate, become reactive and upregulate glial fibrillary acidic protein (GFAP)171. After spinal cord injuries, border-forming astrocytes may also arise from proliferating ependymal cells, contributing to the formation of a protective glial border172,173. These astrocytes secrete growth factors that mitigate secondary damage and promote tissue stabilization174. Experimental reduction of border-forming astrocytes leads to an expansion of the fibrotic scar core and further limits axonal regeneration161,174,175. Conversely, attenuation of fibrotic scarring enhances axonal regeneration and improves sensorimotor function recovery176. A complete blockade of the fibrotic response, however, impairs wound healing and results in structural tissue defects167.
This refined understanding of CNS scarring underscores the dynamic roles of both astrocytes and fibroblasts, shifting the paradigm from a simplistic view of the glial scar to a more nuanced appreciation of the interplay between glial and fibrotic components. Future research will be critical in elucidating how these cellular interactions can be modulated to optimize recovery and regeneration after CNS injury.
Astrocytes in CNS disorders
Cognitive impairment is a frequent outcome of a wide range of disorders, which affect individual’s ability to think, concentrate, remember or make decisions. In this Review, we focus specifically on diseases of the CNS directly linked to cognitive disturbances. Astrocytes are the main actors in all neurological, neurodegenerative and psychiatric diseases (Table 1). Here, we present a brief account of the major pathological roles of astrocytes in several diseases affecting cognition. We included aging as the main risk factor for cognitive disorders; we also included neuropathic pain, which, of course, has peripheral roots, but the main pathology is localized to the spinal cord. Neuropathic pain impairs cognitive abilities through persistent suffering and is largely driven by astrocytic pathology.
Aging
Physiological aging is not a pathology, and yet it is the major risk factor for many diseases of cognition, most notably for neurodegeneration. Aging could be defined as a progressive decline of structural integrity and functional capacity of all organs and systems that weakens organism defenses and increases the vulnerability to diseases. Physiological aging is characterized by a general decline in neuroglial function177, and astrocytes follow this trend. Cell-specific transcriptomics reveal pronounced age-dependent changes in glial gene expression, whereas the neuronal transcriptome shows only minor changes178,179,180. The number of astrocytes does not change in old brains of humans, marmosets and rodents181,182,183; however, the size and complexity of cortical and hippocampal astrocytes decrease substantially with age134,135,183. A decrease in astrocytic size, territorial domain, complexity and synaptic coverage impairs homeostatic support of synaptic transmission thus affecting synaptic plasticity133,134. All major homeostatic functions of astrocytes—including metabolic support, neurotransmitter and precursor homeostasis and metabolism, cholesterol synthesis, water transport, support of the glymphatic system, neurogenesis, neuroprotection and defense—decline with aging133,182.
Acute neurodegeneration—neurotrauma, stroke, autoimmune diseases and infection
Acute traumatic lesions of the CNS trigger neurological and cognitive symptoms directly linked to the death of neural cells—particularly neurons and oligodendrocytes—destroying both information-processing units and the connectome; thus, such lesions can be considered a form of acute neurodegeneration. These lesions are caused by mechanical, ischemic, infectious or autoimmune attack and are fundamentally characterized by prominent neuroinflammation. Astrocytes are key cellular elements of acute brain trauma, which triggers proliferative, isomorphic astrogliosis9,118,130,131. The peak of astrocyte proliferation at the trauma perimeter occurs 2–7 days after the traumatic event; over time, this proliferation gradually subsides184. Reactive astrocytes form the perilesional border essential for wound closure, proper scar formation and postlesional regeneration; after the resolution of neuroinflammation, astrocytes form glia limitans perilaesiones185.
Astrocytes begin responding to an injury as early as 3 h after ischemic insult. By 3 days post-ischemia, astrocytes exhibit strong reactive phenotypes, including high inducible nitric oxide synthase (iNOS) expression and pronounced morphological changes. Around day 5, astrocytes elongate and maintain iNOS expression, and by day 7, they start to organize a barrier structure, which becomes fully matured within approximately 1 month186. During this process, astrocyte-derived type I collagen is upregulated, coinciding with the onset of neuronal death. SPARC (secreted protein acidic and rich in cysteine), a critical regulator of collagen synthesis, cell–matrix interaction and tissue remodeling, is co-expressed with type I collagen during this phase. In vitro experiments using separate primary cultures of astrocytes and cortical neurons demonstrated that type I collagen induces neuronal death through integrin signaling pathways.
In multiple sclerosis (MS), reactive astrocytes similarly surround the lesions, form borders and support the formation of fibrotic scar, although astrocytes present asthenia and loss of homeostatic functions187,188. In neuromyelitis optica, astrocytes are primary targets for anti-AQP4 antibodies, and thus undergo death and loss of function; the secondary astrogliosis may be generated around lesions189,190. A wall of reactive astrocytes also surrounds brain abscesses, with reactive astrogliosis being a prominent feature of brain infectious damage191. Diffuse astrocytic reactivity is also observed in sepsis-induced encephalopathies132.
Chronic neurodegeneration
Alzheimer’s disease
Astrocytes undergo complex and spatially segregated changes in the course of AD; these changes include reactive remodeling, atrophy with functional asthenia, astrocyte degeneration and clasmatodendrosis192,193,194. Extracellular deposition of β-amyloid and formation of plaques trigger reactive nonproliferative isomorphic astrogliosis. Reactive astrocytes surround senile plaques (both in post-mortem tissues and in animal models) forming (together with microglia) a loose perimeter distinct from astrocytic palisades in traumatic brain injury. These reactive glial barriers are, arguably, protective against β-amyloid toxicity195,196, whereas suppression of reactive astrogliosis exacerbates pathology in AD animal models197. Atrophic astrocytes have been detected in human post-mortem brains in all Braak tau disease stages of AD, while the loss of astrocytic homeostatic functions contributes to AD pathogenesis133,198,199.
Astrocytes are sensitive to oxidative stress and the astrocyte enzymes glutamine synthetase (GS) and brain creatine kinase (CKB) are among the oxidatively modified proteins in AD200,201. CKB is involved in maintaining local ATP reserves, while GS contributes to extracellular glutamate homeostasis and glutathione production. Disturbance of this key function of astrocytes may affect neurotransmission and drive excitotoxicity. Modification of CKB is associated with β-amyloid depositions and a loss of function of APOE as a scavenger for lipid peroxidation-derived aldehydes202 as both APOE deletion203 and high levels of lipid peroxidation products are associated with increased lipoxidative modifications of CKB, GS, vimentin and GFAP204.
Parkinson’s disease
Astrocyte pathology in Parkinson’s disease (PD) is mainly manifested by functional deficiency and loss of neuroprotection; all in all, reactive astrogliosis is limited and most likely secondary, induced primarily by neuronal death205,206. In some familial forms of PD, astrocytic atrophy was detected. In particular, in PD associated with PRKN mutation, a significant decrease in GFAP expression was found in both human post-mortem samples and organoids207. Similarly, astrocytes derived from induced pluripotent stem cells reprogrammed from PD patients with LRRK2 mutation showed morphological atrophy208. A decrease in astrocytic complexity was also observed in the late-stage post-mortem PD brains209. Pathological astrocytes in the context of PD have deficient mitochondrial function208 and decreased transmitophagy, the latter being instrumental for supporting energy production in dopaminergic neurons210. Protoplasmic astrocytes may accumulate and remove α-synuclein, thus exercising neuroprotection211, although overload with α-synuclein may cause mitochondrial damage while aggregated α-synuclein may spread through astrocytic syncytia212,213.
Huntington’s disease
HD is a monogenetic neurodegenerative disorder caused by a single dominant allele of the huntingtin gene containing an expanded number of CAG (cytosine, adenine, guanine) repeats; the disease develops when the number of repeats exceeds 40 (refs. 214,215). Astrocytes derived from human embryonic stem cells obtained from mutant Huntingtin (mHTT) embryos and grafted into mouse corpus callosum demonstrated aberrant differentiation and atrophy216. Morphological atrophy and retraction of astrocytic processes from cortico-striatal projection synapses (the first to be affected in HD) have also been reported in a HD mouse model217. In HD, astrocytes undergo functional asthenia manifested in the deficient K+ buffering and glutamate clearance. The transcriptome of HD mouse model striatal astrocytes reveal significant downregulation of Kir4.1, glutamate transporters and molecules of Ca2+ signaling. These astrocytes have a depolarized resting membrane potential and higher input resistance, reflecting a decrease in their size150. A decrease in the expression of Kir4.1 in striatal astrocytes was also verified in human HD samples151,218. Likewise, the expression of EAAT2 is significantly downregulated in human HD tissue and in mouse HD models152,153; HD astrocytes also produce less glutamine, thus affecting glutamate and GABA neuronal pools219. Specific expression of mutated Huntingtin with 160 CAG repeats in astrocytes resulted in a decrease in the expression of EAAT2 and in the emergence of a HD phenotype220. At the same time, astrocyte-specific ablation of mutant Huntingtin in mice that constitutively express this protein in all cells mitigated disease symptoms and slowed disease progression221, thus highlighting astrocytic contribution to the pathogenesis of HD.
Frontotemporal dementia—clustering of pathological astrocytes
Frontotemporal dementia (FTD) is the second most prevalent cause of early-onset dementia and is characterized by profound region-specific neurodegeneration. Anterior brain regions (for example, frontal and temporal lobes) are severely damaged, whereas posterior brain regions (for example, occipital lobe) are seemingly unaffected. FTD can occur sporadically (late-onset) or due to genetic mutations. About one-third of the genetic FTD cases are caused by an autosomal-dominant genetic mutation in either progranulin (GRN), microtubule-associated protein tau (MAPT) or chromosome 9 open reading frame 72 (C9orf72)222,223,224.
Astrocytes, arguably, are among the most affected cells in the brains of patients with FTD-GRN225,226. Pathological astrocytes are distinguished by the expression of a specific marker WD Repeat Domain 49 (WDR49), while immunohistochemistry demonstrated that WDR49-positive astrocytes form clusters that are scattered randomly throughout the cortex of patients with FTD225,226. These WDR49-positive astrocytes were exclusively found in brain regions with neurodegeneration (frontal and temporal cortex), and not in the occipital cortex of patients with FTD, indicating that these cells are topically related to neuronal loss. A follow-up study investigated the distribution of WDR49-positive astrocytes in other subtypes of FTD and in AD, and showed that WDR49-positive astrocytes are most abundant in FTD-GRN, have a different morphology in FTD with TDP43 versus TAU pathology, and colocalize with senile plaques in AD226.
At the same time, evidence for astrogliosis in mouse models for FTD is variable and probably reflects underlying genetic mutations227. In Grn−/− mice, but not Grn+/− mice, increased GFAP immunoreactivity was observed in the thalamus, hippocampus, cortex and amygdala228. Conversely, no increase in GFAP immunoreactivity was identified in C9orf72−/− mice229. The brains from (G4C2)66 mice, another model for C9-FTD/ALS, showed signs of astrogliosis in the cortex230. In vitro, astrocytes derived from induced pluripotent stem cells obtained from patients with FTD-MAPT showed hypertrophy, elevated GFAP protein levels, disease-associated changes in TAU expression, increased vulnerability to oxidative stress, and protein ubiquitination231. The presence of GFAP was detected in the cerebrospinal fluid (CSF) and plasma from symptomatic FTD-GRN, and this correlated with age227,232. In the presymptomatic period, higher GFAP plasma concentrations correlated with a lower cognitive score and lower brain volumes, suggesting that GFAP expression is increased in the late presymptomatic period227.
Amyotrophic lateral sclerosis
Functional insufficiency and pathological remodeling of astrocytes play a central role in driving motor neuron death in ALS. Several pathological phenotypes including reactive, degenerating, aberrant and atrophic astrocytes are present in ALS156,233,234,235,236. Astrocytes derived from ALS post-mortem tissues trigger the death of neurons in co-cultures237,238, whereas transplantation of astrocytes derived from pluripotent stem cells obtained from patients with ALS into the spinal cord of mice resulted in the degeneration of motor neurons and motor deficits239. Using the human ALS-linked superoxide dismutase 1 (SOD1G93A) mutant gene to replicate ALS in mice further highlighted the primary role of astrocyte: astrocyte-selective silencing of SOD1 alleviated ALS symptoms and increased life span240,241, whereas restricted expression of SOD1 in neurons did not cause pathology242,243. Moreover, grafting fetal astrocytes bearing SOD1G93A to the spinal cord of healthy mice precipitated ALS-like pathology244, while transplanting healthy astrocytes slowed ALS progression in rats carrying mutant SOD1G93A (ref. 245). The primary damaging effect is associated with a profound loss of astrocytic glutamate clearance, leading to excitotoxicity246,247. In patients with ALS, the expression of EAAT2 is decreased by up to 90% (refs. 149,248), and a similar decrease was observed in SOD1G93A mice148,249,250. Aberrantly proliferating astrocytes that express both astrocytic and microglial markers exhibit toxicity in vitro. They display low levels of intermediate filaments, numerous microtubules, abundant secretory vesicles and lipid droplets, and lack homeostatic functions. Neurotoxicity of these cells can be mediated by the overproduction of ROS236,251,252.
Astrocytotauopathies
Astrotauopathies, characterized by tau accumulation predominantly in astrocytes, are classified, according to histopathology, into (1) astrocytic plaques, (2) tufted astrocytes, (3) ramified astrocytes, (4) globular astroglial inclusions, (5) thorn-shaped astrocytes and (6) granular/fuzzy astrocytes253. Astrocytic plaques, which appear in the form of fuzzy short argyrophilic processes arranged annularly with fine collaterals at vertical or sharp angles, are the hallmark of corticobasal degeneration, a primary tauopathy253,254. Tufted astrocytes identified by phosphorylated-tau immunoreactivity in the proximal segments of astrocytic processes are a histopathological hallmark of progressive supranuclear palsy, another rare primary tauopathy. Ramified atrophic astrocytes distinguish Pick’s disease, also known as frontotemporal lobular degeneration255, while globular astroglial tau inclusions are specific for FTD256. Finally, thorn-shaped and granular-fuzzy astrocytes are found in aging-related tau astrogliopathy, ARTAG257.
Autoimmune astrocytopathies
Encephalopathies associated with the generation of antiself antibodies against two astrocyte-specific proteins GFAP and vimentin were discovered quite recently. In 1996, encephalopathy triggered by GFAP autoantibodies (GFAP-A) was described258. The presence of anti-GFAP antibodies in serum and cerebrospinal fluid is a critical diagnostic criterion, as clinical manifestations are nonspecific and can include movement disturbances, paresthesia, visual impairment, brainstem syndrome and autonomic dysfunction259,260,261,262. The clinical manifestations of GFAP-A are highly diverse and include encephalitis, myelitis and meningitis262. The second autoimmune astrocytopathy caused by anti-vimentin autoantibodies was described in 2025263. It has a clinical picture of unidentified meningoencephalitis with the involvement of several brain regions and lesions in bilateral corticospinal tracts. The CSF of patients stained astrocytes and ependymoglia in the rodent brains, again highlighting pathophysiological contribution of astroglial cells. The cellular pathophysiology of these diseases is unknown.
Neuropathic pain
While neuronal hyperexcitability has traditionally been the focus of neuropathic pain research, the past two decades have seen growing interest in the role of astrocytes. In rodent models of neuropathic pain, reactive astrogliosis was well characterized in the spinal dorsal horn. Expression of GFAP is markedly upregulated after nerve injury and remains elevated for several weeks to months264,265. Reactive astrocytes contribute to pain hypersensitivity through activation of the JAK–STAT3 pathway266 with consequent release of cytokines, including tumor necrosis factor (TNF), interleukin (IL)-1β (IL-1β) and C–C motif chemokine 2 (CCL2)267,268,269,270. Reactive astrocytes also contribute to neuropathic pain through MAO-B-mediated GABA synthesis, as described in the following sections.
Psychiatric disorders
Mood disorders and stress-induced depression
Astroglial pathology in major depressive disorders in human patients and stress-induced depressive behaviors in animals is represented by a significant decrease in the numbers and complexity of astrocytes in several brain regions, including, in particular, the prefrontal cortex, which is responsible for stress processing144,146,271,272. Other brain regions are affected, too; in the hippocampus, for example, reduced levels of glutamate transporters were found in animal models with depressive-like behavior273,274,275,276. In the hypothalamus, early-life stress resulted in a marked atrophy of astrocytes, aberrant purinergic signaling and limited supply of L-lactate, leading to a pathological activity of orexin neurons driving aberrant behaviors277. A decrease in astrocytic presence in the brain reduces homeostatic support of nervous tissue, while decreased astrocytic synaptic coverage affects synaptic transmission and plasticity, which are translated into depressive symptoms and behaviors143. Reduced glutamate uptake278 and increased release of GABA279 also affect synaptic transmission and plasticity, arguably contributing to the pathophysiology of depression. Astrocytic atrophy and asthenia are causal for depression: selective ablation of astrocytes or downregulation of astrocytic homeostatic molecules such as glutamate transporters or connexins is sufficient to trigger depressive-like behaviors in experimental animals280,281,282,283,284. Glutamate homeostasis, supported by astrocytes, is of particular relevance; aberrant glutamate clearance retunes synaptic transmission77, promotes dendritic shrinkage and is associated with depression in animals and humans285,286,287. Anti-depressant treatment or pharmacological stimulation of glial glutamate transporters alleviates aberrant behaviors and restores astrocytic morphology146,284,288. Thus, depression can be considered a primary astrocytopathy. The emergence of depressive symptoms and behaviors is, however, highly individual, and the same amount of stress produces different outcomes in both patients and animal models289,290,291,292. This resilience to stress is also linked to astrocytes. Manipulating the expression level of the astrocyte-specific plasma membrane–microfilament linker ezrin alters resilience to stress147. In bipolar disorder, the number of GFAP-positive astrocytic profiles decreases, accompanied by astrocytic atrophy271,293,294. Asthenic astrocytes may be linked to a hyperactive glutamatergic transmission (due to the reduced glutamate clearance observed in bipolar disorder295,296, reduced neuroprotection and metabolic abnormalities297.
Schizophrenia
Astrocytic pathology in schizophrenia manifests as altered astrocyte-specific protein expression, impaired differentiation and disrupted neuron–astrocyte interactions. Post-mortem analyses of the dorsolateral prefrontal cortex reveal elevated GFAP levels, particularly in individuals with psychotic symptoms, while other astrocytic markers remain unchanged, suggesting astrocyte-specific malfunction unrelated to antipsychotic treatment298. Developmental astrocyte abnormalities in humanized glial chimeric mice, transplanted with astrocytes from patients with schizophrenia, showed delayed differentiation and atrophic morphologies299,300. Gene expression studies highlight increased cortical astrocytic activity, coupled with parvalbumin interneuron loss, suggesting a role for astrocytes in neuronal network destabilization301. Malfunction of astrocyte-mediated GABAergic regulation is supported by findings in GABRB2-knockout mice, which exhibit astrocytic atrophy, degeneration of parvalbumin-expressing cortical interneurons, neuroinflammation and oxidative stress, aligning with key pathological features of schizophrenia302. Astrocytic differentiation deficits in schizophrenia are driven by upregulated inhibitors of the bone morphogenetic protein (BMP) pathway, which prevent proper maturation. Targeting BMP/SMAD4 and repressor element-1 silencing transcription factor (REST) pathways restored astrocyte function, presenting a potential therapeutic approach303. Astrocytes are targets of lithium antipsychotic treatment, with pharmacogenomic analyses identifying lipoxygenases LOX and peroxisome proliferator-activated receptor γ serving as regulators of astrocytic morphology and function, offering novel therapeutic directions304. Astrocyte–neuron interactions are crucial for synaptic regulation, with astrocytes enhancing synaptic gene expression and synaptic plasticity. These interactions are disrupted in schizophrenia, as reflected by dysregulation of synaptic adhesion genes and cholesterol metabolism, both being fundamental for synaptic integrity305,306. Thus, astrocytes are potential targets for schizophrenia therapy.
Epilepsy and migraine
Epilepsy
Epilepsy and seizures reflect aberrant electrical activity in the brain. In epilepsy, astrocytes undergo a peculiar form of reactivity associated with an increased GFAP expression and atrophy of peripheral processes307,308. This atrophy is paralleled with asthenia: astrocytes in epileptic brains show significant downregulation of Kir4.1 channels, which affects K+ buffering and facilitates seizures309. Conditional knockout of Kir4.1 channels compromised K+ buffering, triggering seizures and an epileptic phenotype310,311. In humans, epilepsy is linked to several missense mutations, loss-of-function mutations or single-nucleotide polymorphisms in the KCNJ10 gene encoding Kir4.1309.
Pathogenesis of epilepsy also included deficient glutamate clearance and insufficient inhibition; epileptic tissue has high glutamate content312 reflecting a 20–40% reduction in the expression of astrocytic glutamate transporters in the hippocampi of patients with temporal lobe epilepsy313. Likewise, downregulation of astrocytic glutamate transporters is confirmed in animal models of the disease142,314,315, whereas genetic deletion of transporters triggers seizures313,316. Epileptic astrocytes are also characterized by partial loss of glutamate synthetase and reduced GABA availability317, increased expression of astrocyte-specific adenosine kinase—which limits adenosine levels and further decreases adenosine A1 receptor-mediated inhibition318—loss of AQP4 polarization at astrocytic endfeet319 and downregulation of astrocytic Cx43; these changes collectively contribute to increased epileptiform activity320,321. A gain-of-function mutation in the anion conductance of EAAT1 in astrocytes is associated with spontaneous seizures322. This mutation reduces astrocytic Cl⁻ levels and trigger apoptosis323. Deletion of NKCC1 in astrocytes, which reduces astrocytic Cl⁻, leads to a lower seizure threshold324. Finally, the loss of gap junctional coupling among astrocytes lowers the seizure threshold by disturbing normal ionostasis325. Notably, astrocytic uncoupling has also been shown to directly affect inhibitory transmission326.
Migraine
Monogenic familial hemiplegic migraine (FHM) type 2 is a primary genetic astrocytopathy caused by loss-of-function mutations in the ATP1A2 gene encoding the α2 subunit of Na+/K+ ATPase (the NKA) expressed solely in astrocytes327. FHM type 2 belongs to migraine with aura, which is associated with spreading depression328. The α2-containing NKA is the principal molecule of K+ buffering and is linked to regulation of expression of astrocytic glutamate transporters329. Astrocytes in FHM type 2-mutant expressing mouse demonstrated deficient K+ buffering and glutamate clearance, which also contribute to the initiation of spreading depression329.
Primary astrocytic leukodystrophies
Alexander disease (AxD), the first known monogenic disease originating in astrocytes, is caused by toxic gain-of-function mutations in GFAP manifested by severe white matter damage330,331. While the molecular pathogenesis of AxD remains incompletely understood, massive reactive astrogliosis with Rosenthal fibers composed of aggregated GFAP, vimentin, nestin, αB-crystallin and small heatshock proteins together with prominent leukodystrophy are the pathological hallmarks, with the pathophysiological picture including mitochondrial malfunction, redox imbalance and susceptibility to oxidative stress331,332,333. Increased stress susceptibility and impaired differentiation of astrocytes and neurons was found in co-cultures and in neural organoids generated from AxD patient-derived induced pluripotent stem cells with AxD-causing GFAPR239C mutation334.
Vanishing white matter disease is an autosomal recessive, polygenic disorder caused by mutations in five genes (EIF2B1, EIF2B2, EIF2B3, EIF2B4 and EIF2B5) encoding the eukaryotic translation initiation factor eIF2B, a key control point for protein synthesis in all eukaryotes335. The pathophysiology of vanishing white matter disease is cell specific and primarily targeting astrocytes. Atrophic astrocytes with reduced complexity and blunt and coarse processes are present in the white matter and are characterized by impaired GFAP-related structures, suppressed expression of S100B, aberrant maturation and loss of astrocytic functions ultimately translated into white matter disintegration336,337.
Megalencephalic leukoencephalopathy with subcortical cysts (MLC), is another primary leukodystrophy caused by recessive pathogenesis variants in the genes MLC1 (Modulator of VRAC Current 1) or HEPACAM (Hepatic and Glial Cell Adhesion Molecule, previously known as GlialCAM)338. In astrocytes, GlialCAM and MLC1 assemble into a functional unit, interacting with Na+–K+ ATPase, SLC ionic transporters, Kir4.1 channels, connexins, AQP-4 water channels, volume-regulated anion channels (VRAC) and transient receptor potential vanilloid 4 channel (TRPV4), and are also associated with the dystrophin–glycoprotein complex regulating endfeet channels and transporters339,340,341. In MLC, astrocytes show impaired K+ buffering, glutamate uptake and ionic homeostasis, which translate into white matter damage in a yet uncharacterized way339,342,343.
Imaging human reactive astrocytes
The rapid development of in vivo molecular imaging techniques such as positron emission tomography (PET) has made it possible to visualize astrocytes in the brain of living humans and to study changes in astrogliosis in brain disorders. Several astrocyte PET tracers currently in clinical and research use include [11C]deprenyl, [11C] BU99008, [18F] SMBT-1, [11C] 25.1188 and [11C] acetate (Table 2 and Fig. 6) and are described in more detail below. However, developing methods for in vivo visualization of astroglia is challenging, as reactive astrocytes undergo remodeling into diverse states with varying functions and properties118. We may therefore expect multiple subtypes of astrocytes in different diseases and stages of different disorders.

a Chemical structures of different PET tracers for visualizing astrogliosis in the human brain. b Higher [11C]deprenyl PET binding (astrogliosis) in a patient with MCI (amyloid positive; prodromal AD) compared with a patient with AD. Reproduced, with permission, from ref. 353. c GFAP antibody staining of astrocytes in hemispheric section of AD brain showing high astrogliosis in hippocampus/entorhinal cortex. d High [11C]deprenyl PET binding (astrogliosis) in an APPswe mutation carrier 10 years before expected clinical symptoms compared with a nonmutation carrier (photo courtesy of Nordberg Translational Imaging Lab). e Reactive astrogliosis in the AD disease continuum. The light-blue curve illustrates increased [11C] deprenyl PET binding in preclinical AD as the first wave of reactive astrogliosis. When plaque deposition (orange curve) increases, the astrocyte PET curve declines. GFAP levels in plasma increase (teal curve) with β-amyloid deposition in the brain. This is followed by tau pathology (green curve) and a second wave of reactive astrogliosis, indicative of the later stages of AD. Reproduced, with permission, from ref. 360.
[11C] deprenyl PET labeling astrocytic MAO-B to visualize astrogliosis
MAO-B is a mitochondrial enzyme responsible for the oxidative deamination of monoamines, including dopamine, norepinephrine and phenylethylamine344. Although traditionally associated with neuronal neurotransmitter metabolism, MAO-B is expressed predominantly (if not exclusively) in astrocytes, where it plays a central role in reactive astrogliosis and neuroinflammation345,346. In the astrocytes, MAO-B is localized to the outer mitochondrial membrane and contributes to metabolic regulation and oxidative stress responses347. MAO-B is highly expressed in brain regions susceptible to neurodegenerative pathology, including the striatum, hippocampus and cortex348. Increasing evidence suggests an upregulation of MAO-B in reactive astrocytes in neuroinflammatory pathologies as well as in many chronic neurological disorders345; this has motivated efforts to measure MAO-B as a marker of astrogliosis and prompted the development of the first PET tracer, [11C] d-l-deprenyl -l-dL deprenyl, as a selective target for imaging reactive astrocytes.
[11C] deprenyl PET
Deprenyl is an irreversible MAO-B inhibitor with a well-established use in the treatment of patients with PD. The concentration of the ligand used in PET studies is, however, 100–1,000 times lower than its pharmacological dose. In the initial studies in patients with epilepsy, [11C] d-l-deprenyl PET demonstrated a lower uptake in the temporal lobe compared with healthy controls and turned out to be a useful method to diagnose temporal lobe epilepsy349. An increased astrogliosis measured as an enhanced uptake of [11C] d-l-deprenyl was observed in the pons and white matter of patients with ALS350. When patients with Jakob–Creutzfeldt disease underwent PET imaging with [11C] d-l-deprenyl and [18F]fluorodeoxyglucose ([18F]FDG), increased astrogliosis and decreased cerebral glucose metabolism were observed in the frontal, occipital and parietal cortices and cerebellum351,352. Autopsy studies confirmed a good correlation between the in vivo measured high [11C] d-l-deprenyl binding with high reactive astrogliosis measured by GFAP immunostaining at autopsy.
Hypertrophic reactive astrocytes surrounding senile plaques can be seen in autopsy tissue of patients with AD; hence, it was logical to perform [11C] d-l-deprenyl as well as amyloid [11C]-labeled Pittsburgh Compound-B ([11C] PIB) PET in patients with AD and mild cognitive impairment (MCI)353. It was quite unexpected that patients with MCI with pathological β-amyloid load revealed by PIB positive binding demonstrated higher [11C] d-l-deprenyl uptake, indicating more prominent astrogliosis compared with patients with AD as well as with MCI amyloid-negative patients and age-matched healthy controls353. All individuals also underwent [18F] FDG PET, which showed pronounced hypometabolism in patients with AD compared with the patients with β-amyloid positive MCI. These observations created the initial hypothesis of prominent astrogliosis as an early event in the time course of pathology in the AD continuum354. This initial observation was followed by multitracer PET imaging with [11C] d-l-deprenyl, [11C] PIB and [18F] FDG studies in members of families with known autosomal-dominant familial AD in both presymptomatic and symptomatic carriers as well as in patients with sporadic AD and MCI and healthy individuals355. An inverse relationship was observed in the brain of autosomal-dominant familial AD mutation carriers between [11C] d-l-deprenyl and [11C] PIB PET demonstrating a high uptake of [11C] d-l-deprenyl PET (that is, prominent reactive astrogliosis) already 16–20 years before the onset of clinical symptoms; the [11C] d-l-deprenyl signal declined with increasing β-amyloid PIB binding in the brain. The [11C] d-l-deprenyl uptake nevertheless was high compared with non-mutation carriers at the onset of first cognitive symptoms and showed a negative correlation with [18F] FDG uptake356,357,358. Based upon these observations, a ‘two-wave model of reactive astrogliosis’ in the AD continuum was suggested359,360.
[11C] BU99008 PET
[11C] BU99008 was developed as a PET tracer with high affinity and reversible binding to imidazol I2BS localized in the outer mitochondrial membranes and expressed mainly in astrocytes, with lower neuronal presence361. [11C] BU99008 is considered as a PET ligand with good penetration to the brain and reversible and highly specific binding to I2BS362. A high uptake of [11C] BU99008 was observed in cortical brain regions of patients with AD and those with β-amyloid positive MCI compared with healthy controls, showing a positive correlation with β-amyloid load363. Uptake of [11C] BU99008 in the temporal and parietal cortex in β-amyloid-positive patients positively correlated with [18F] FDG uptake and gray matter volume, while negatively correlating with β-amyloid [18F] florbetaben uptake364. These findings were interpreted as showing reactive astrogliosis at the early stages of AD, whith subsequent astrocytes atrophy and loss of reactivity with higher β-amyloid deposition364. During the early stages of PD, [11C]BU99008 PET showed an increased uptake in frontal, temporal, parietal, occipital and insula cortices and subcortical regions such as caudate, putamen, thalamus and brainstem as compared with healthy controls, whereas in advanced stages these differences disappeared365. Similarly, no differences were observed in [11C] BU99008 uptake in patients with PD and healthy controls365, again indicating that astrogliosis occurs mainly in early stages of the disease.
[18F] SMBT-1 PET
[18F] SMBT-1 is a reversible MAO-B inhibitor developed through lead optimization from the THK-5351 Tau PET tracer366. In patients with AD, a significant increase in the uptake of [18F] SMBT-1 was found compared with β-amyloid-negative controls367. In addition, a positive correlation was observed in patients with AD between astrogliosis measured by [18F] SMBT-1 and amyloid PET uptake in the brain367.
[11C] SL25.1188
[11C] SL25.1188 is a PET tracer based on a reversible MAO-B inhibitor used in psychiatric disorders such as depression368, psychosis369 and post-traumatic stress disorder370, in traumatic brain injury371 and in COVID-19 with chronic depressive and cognitive symptoms372. In PET imaging of patients with traumatic brain injury, an increased [11C] SL25.1188 uptake was observed in the prefrontal and cortical brain regions compared with controls, and this increase showed an inverse correlation with performance on the Trail Making Test, reflecting reduced psychomotor and processing speed371. In patients with COVID-19 uptake of the [11C] SL25.1188 was higher in several brain regions, including the prefrontal cortex, anterior cingulate cortex, hippocampus, dorsal putamen and ventral striatum, compared with healthy control participants, thus revealing astrogliosis as pathogenetic factor372. In patients with psychosis or post-traumatic stress disorder, a slight tendency toward a lower [11C] SL25.1188 uptake compared with controls was noted369,370.
[11C] acetate
Acetate is a substrate for energy generation in astrocytes associated with their metabolism and activity. A significantly higher [11C] acetate uptake was observed in several brain regions, including the entorhinal cortex, hippocampus and temporal cortex in patients with AD compared with controls373. Increased [11C] acetate uptake was paralleled by a significant decrease in [18F] FDG uptake in the same brain regions373. A negative correlation was also observed between cognitive mini mental test score (MMSE) score and [11C] acetate uptake373. In patients with MS, high uptake of [11C] acetate was observed in both gray and white matter, which correlated with the number of magnetic resonance imaging (MRI) lesions374. Increased uptake of acetate observed in the patients is probably driven primarily by reactive astrocytes, which upregulate monocarboxylate transporter 1 (MCT1) both in vivo in the brains of AD model mice and patients, as well as in vitro373. Astrocyte-specific suppression of MCT1 expression significantly reduced the [11C] acetate uptake.
To conclude, multiple astrocyte PET tracers indicate reactive astrogliosis in several brain diseases. Most of the PET studies (Table 1) focused on patients with AD at different stages of the disease continuum. When comparing three astrocytic PET tracers ([11C] d-l-deprenyl, [18F] SMBT-1 and [11C] BU99008), some differences in the sensitivity to pick up changes in astrogliosis during the development of AD were noted. In vitro binding studies using the three PET ligands in post-mortem tissues also revealed differences in binding properties, including multiple binding sites with varying affinities375,376. Hitherto, [11C] d-l-deprenyl is the only astrocytic tracer that demonstrated a high uptake in the presymptomatic carrier of AD 15–20 years before the onset of cognitive symptoms, that is, at the stage when the β-amyloid is rather low. It is therefore possible that soluble oligomeric amyloid triggers the reactive astrogliosis detected by [11C] d-l-deprenyl. An inverse correlation was observed between reactive astrogliosis measured by [11C] d-l-deprenyl and β-amyloid load measured by [11C] PIB PET imaging during the progression of the disease until the onset of clinical symptoms. This highlights the first wave of reactive astrogliosis in AD preceding other pathological markers such as tau deposits, neurodegeneration and cognition. A second wave can also be observed in the later stage of AD with a positive correlation with amyloid load375,376. The second wave was also demonstrated with [18F] SMBT-1 PET showing a positive correlation with β-amyloid PET in patients with AD367. The use of several astrocyte PET tracers might increase the possibility of detecting multiple astrocyte states and lead to the development of subtype-specific astrocytic PET tracers.
Targeting astrocytic molecules and cascades
In this section, we provide an overview of major molecules and molecular cascades expressed predominantly or exclusively in astrocytes. We also narrate the current state of knowledge on the potential drugs that target these molecular entities, representing astrocyte-specific therapeutic strategies (Table 3).
GFAP—targeting reactive astrogliosis
GFAP is the main component of astrocyte cytoplasmic intermediate filaments (also referred to as nanofilaments) (Fig. 2C). These nanofilaments are dynamic structures acting as a signaling system regulating cellular stress responses in health and disease332,377. The upregulation of GFAP and vimentin are hallmarks of astrocyte reactivity and can be seen in neurotrauma, stroke or neurodegenerative diseases, such as AD or ALS332,378, while GFAP mutations result in AxD. Treatment with GFAP-targeted antisense nucleotides mitigates white matter loss and motor impairment, in both mouse379 and rat380 models of AxD. The same approach is being applied to AxD patients in an ongoing phase 1–3 clinical study (Zilganersen, NCT04849741; the drug received FDA fast-track designation; https://ionistrials.com/study/a-study-to-evaluate-the-safety-and-efficacy-of-ion373-in-patients-with-alexander-disease-axd/).
Genetic ablation of GFAP affects reactive astrogliosis. The reactivity of astrocytes in GFAP−/− mice is attenuated after neurotrauma381, whereas GFAP−/− astrocytes show decreased motility382. Using cellular deconvolution of bulk transcriptomics data, GFAP was identified as the main driver of reactive astrogliosis in the peri-infarct region after ischemic stroke383. In GFAP−/− mice, vimentin can compensate for the absence of GFAP384,385, thus masking some phenotypes, which become apparent only when GFAP−/− mice were crossed with a Vim−/− background. Astrocytes of GFAP−/−Vim−/− mice completely lack cytoplasmic nanofilaments, and these mice have provided insights into the role reactive gliosis in the brain, spinal cord and retina in a wide range of neurological diseases332,377,386. The abundance and domain tiling of astrocytes in the CNS of GFAP−/−Vim−/− mice is normal. However, following injury, GFAP−/−Vim−/− astrocytes do not undergo the characteristic hypertrophy of their main cellular processes381 and show a number of molecular features of attenuated reactive gliosis, such as decreased activation of c-FOS and ERK387 as well as less prominent upregulation of the 14-3-3 adapter proteins388. GFAP−/−Vim−/− Müller cells fail to respond to retinal ischemia by increasing the stiffness of their endfeet and inner processes389. GFAP−/−Vim−/− mice have reduced Notch signaling from astrocytes to neural stem/progenitor cells that control neurogenesis in adult mammalian CNS390,391, and GFAP−/−Vim−/− mice were instrumental for the finding that vesicle trafficking in astrocytes depends on vimentin, GFAP and also nestin, another nanofilament protein, which is present in immature as well as reactive astrocytes392,393,394,395,396, but also in some astrocytes in the dentate gyrus of the hippocampus396.
GFAP−/−Vim−/− mice have been essential for defining the concept of context-specific roles of reactive astrocytes and characterizing the disease-specific functions of these cells. Depending on the specific context, reactive gliosis can be either beneficial—promoting improved functional outcomes—or detrimental, leading to maladaptive impairments or neurodegeneration377.
Genetic attenuation of reactive gliosis in GFAP−/−Vim−/− mice has a number of negative consequences. It decreases the resistance of the CNS to severe mechanical stress397,398 and to ischemic injury399,400,401,402. Ischemic stroke induced in GFAP−/−Vim−/− mice by middle cerebral artery transection leads to a more prominent loss of the ischemic penumbra, with less efficient endothelin-3-induced blockage of gap junction communication between astrocytes and decreased glutamate clearance401. Astrocytes in GFAP−/−Vim−/− mice had lower glutamine levels403 and were less resilient when challenged with oxidative stress399. Thus, GFAP and vimentin nanofilaments support neuroprotective functions of astrocytes in acute ischemic stroke and exemplify the positive role of reactive astrocytes. This contention is further supported by the finding that, after retinal ischemia–reperfusion, fewer cells of the inner retina of GFAP−/−Vim−/− mice survived402. The effect of reactive astrogliosis after acute injury depends on the presence of the ischemic penumbra and differs between the immature and adult CNS. The absence of vimentin and GFAP does not affect tissue loss in photothrombotic brain injury—which involves a very limited ischemic penumbra404—nor in neonatal hypoxic-ischemic brain injury, a model of birth asphyxia405. The formation of post-traumatic perilesional astrocytic border is impaired in GFAP−/−Vim−/− mice and is accompanied by slower wound healing after both brain and spinal cord trauma381,403. In addition, GFAP−/−Vim−/− mice show an altered response of astrocytes in neurodegenerative diseases, including limited presence of astrocyte processes in the direct vicinity of β-amyloid plaques, impaired cytokine production, aberrant astrocyte–microglia interactions and faster progression of both AD and Batten disease in the respective mouse models127,197,406. Regeneration of crushed sciatic nerve takes longer in GFAP−/−Vim−/− mice; however, the final outcome is comparable to that of wild-type mice407. The slower wound healing after brain or spinal cord trauma in GFAP−/−Vim−/− mice381,403, may ultimately lead to a better functional outcome, or contribute to neurodegeneration, malfunction and disability377. Perilesional astrocytic border contributes to post-traumatic functional recovery9,161,408. Attenuation of reactive astrogliosis also affects post-ischemic neural plasticity responses: corticospinal tract remodeling and axonal regeneration following photothrombotic stroke are delayed in GFAP−/−Vim−/− mice404. The GFAP−/−Vim−/− mice exhibit maladaptive post-stroke neuronal connectivity, characterized by increased synaptic plasticity in the perilesional region and an altered balance between lost and newly generated neuronal connections in sensorimotor networks, resulting in impaired recovery of sensorimotor functions409.
Genetic attenuation of reactive gliosis in GFAP−/−Vim−/− mice also has a number of positive consequences. Pathological and detrimental neovascularization in oxygen-induced retinopathy is mitigated in GFAP−/−Vim−/− mice397, whereas photoreceptor cell death and monocyte infiltration are both reduced in GFAP−/−Vim−/− mice with sodium hyaluronate-induced degeneration of retina387. In addition, GFAP−/−Vim−/− mice exhibit increased neurogenesis in the dentate gyrus of the hippocampus in the absence of any challenge391,410 and increased memory extinction, conceivably due to the increased rate of the reorganization of the hippocampal circuitry411. The GFAP−/−Vim−/− mice also show increased neurogenesis following hypoxic-ischemic injury to the immature brain405 and after hippocampal deafferentation in adults391, as well as improved regeneration of neuronal axons and synapses after trauma381,412,413. The CNS environment in GFAP−/−Vim−/− mice is more supportive for integration and survival of neural grafts in the retina414 while supporting increased differentiation of transplanted neural stem cells into neurons and astrocytes in hippocampus415. These results suggest that limiting graft-induced reactive gliosis might be the way to increase the success of transplantation of neural grafts or neural progenitor cells in the brain, spinal cord or retina. It remains unclear to what extent the improved graft integration is a consequence of attenuated reactive gliosis or of an altered Notch signaling between resident astrocytes and neural grafts or grafted neural stem/progenitor cells. The appeal of treatment strategies that target and modulate reactive astrogliosis in a context-dependent manner is further supported by recent findings showing that pharmacological modulation of astrocyte reactivity improves neuronal connectivity in the peri-infarct cortex and accelerates motor function recovery after stroke383. Thus, understanding disease-specific astrocyte responses and targeting and modulating them presents an innovative treatment strategy for stroke and other neurological disorders.
Ezrin—controlling astrocytic morphological plasticity
Astrocytic leaflets are highly dynamic; their association with synapses can change rapidly, thus affecting the volume of the synaptic cleft, as well as the efficacy of ion and neurotransmitter buffering and removal. These changes may facilitate or limit neurotransmitter spillover, thus contributing to synaptic plasticity416; retraction of astrocytic processes from synapses may cause aberrant transmission and plasticity134. Swift morphological remodeling of astrocytic leaflets requires plasmalemma–cytoskeleton interactions, mediated in particular by plasmalemmal linker ezrin. Ezrin belongs to the ezrin–radixin–moesin (ERM) family of proteins, which are involved in the assembly and stabilization of membrane–cytoskeletal complexes. This role is critical for controlling membrane dynamics that define cell shape and polarity, the formation of membranous protrusions, and the motility of cellular processes417,418. Ezrin in particular is enriched in microvilli of various types of epithelial cells from the placenta to renal ducts419; ezrin connects plasmalemma with F-actin, thus mediating morphological plasticity420. In the CNS, ezrin is highly expressed in microvilli of ependymal glia and in astrocytic leaflets421; in general, most of ezrin in the CNS is localized to astrocytes422. Experiments in vitro showed that ezrin regulates extension of astrocytic philopodia (analogs of leaflets), while in situ astrocytic ezrin expression follows circadian changes in glutamatergic transmission, thus changing synaptic coverage423. Various learning paradigms increase the expression of ezrin in hippocampal astrocytes, potentially enhancing synaptic coverage424. Depletion of astrocytic ezrin translated in shorter leaflets, decreased astrocytic synaptic coverage and glutamate spillover425. Ezrin also interacts with glutamate transporters and may regulate the efficacy of glutamate uptake426,427.
In pathology, downregulation of ezrin expression is typically associated with—and probably contributes to—astrocytic atrophy, reduced number and volume of astrocytic leaflets, leaflet retraction and decreased synaptic coverage. Astrocytic atrophy, in turn, is linked to cognitive impairments. Astrocyte-specific ezrin knockout triggered cognitive impairments and anxiety-like behaviors424. Decreased ezrin expression coincides with astrocytic atrophy in aging in humans and mice135,424, with stress-induced depression146 (Fig. 7), with systemic inflammation and postoperative cognitive impairments424, and with general anesthesia-induced cognitive abnormalities428. Overexpression of ezrin in astrocytes results in an increased astrocytic morphological presence and improved cognition424. Manipulation with ezrin expression in astrocytes also indicated the resilience of mice to chronic stress: ezrin downregulation increased susceptibility to stress and facilitated emergence of depression-like behaviors, whereas overexpression of ezrin made mice resistant to chronic stress147. Treatment with antidepressants as well as acupuncture at a specific acupoint alleviated depression-induced behaviors in response to chronic stress, increased expression of ezrin and rescued astrocytic atrophy146. In summary, ezrin is an astrocyte-specific molecular target to manage cognitive disorders.

a Representative 3D reconstruction of astrocyte in control mice and mice subjected to chronic unpredictable stress (CUMS), which triggered depressive-like behaviors. b Sholl analysis of astrocytic morphology for control and CUMS groups shows the number of intersections of astrocytic branches with concentric spheres centered in the middle of cell soma. c Left: average length of astrocytic processes (branches). Right: average size of astrocytic domain areas. d Representative 3D reconstruction of astrocytic profiles (red; astrocytes were labeled with virally transfected with mCherry) with Ezrin puncta (green; immunostaining) for control and CUMS groups. e Average fluorescence intensity of ezrin associated with astrocytic profiles. All data are presented as mean ± s.e.m. *P < 0.05, **P < 0.01, ***P < 0.001. The number of experiments is indicated in each column. Reproduced from ref. 146.
Glutamate–glutamine cycle—preventing excitotoxicity
Astrocytes are indispensable for CNS synaptic transmission supplying neurons with glutamine, an obligatory precursor for glutamate and (by proxy) GABA as well as clearing glutamate and, to a lesser extent GABA, from the synaptic cleft and interstitium. The exchange of glutamate, glutamine, and GABA between neurons and astrocytes is mediated by the glutamate(GABA)–glutamine cycle, which is critical for maintaining excitatory and inhibitory neurotransmission and preventing excitotoxicity27,73,429,430. The cycle is composed of plasmalemmal glutamate transporters, GS, glutamate-synthesizing enzymes and glutamine transporters mediating the export (astrocytes) and import (neurons) of the latter (Fig. 8). Astrocytes remove ~80% of glutamate released during synaptic transmission by Na+-dependent EAAT1 and EAAT2 (SLC1A2 and SLC1A3); EAAT1 is mainly expressed in the cerebellum, retina and circumventricular organs, whereas EAAT2 predominates in all other brain regions. Glutamate transporters are mainly localized to perisynaptic astrocytic leaflets at a high density reaching 8,500–12,000/μm2 in the hippocampus431,432,433. GS, expressed specifically in astrocytes, converts glutamate (which is synthesized in astrocytes from glucose) to biologically inactive glutamine, which is exported to extracellular space by Na+-dependent neutral amino acid transporters SNAT3 and SNAT5 (SLC38A3 and SLC38A5) and imported into neurons by SNAT1 and SNAT2 (SLC38A1 and SLC38A2)434,435. All components of the glutamate(GABA)–glutamine cycle are regulated by transmembrane Na+ gradients, which drive the transporters, and by cytosolic Na+ concentration, which influences GS activity436,437.

AAT, aspartate aminotransferase; EAAT1/2, excitatory amino acid transporters 1 and 2; GABA-T, GABA-α-ketoglutaric acid aminotransferase; GAD, glutamic acid decarboxylase; GAT1/3, GABA transporters 1 and 3; GDH, glutamate dehydrogenase; PAG, phosphate-activated glutaminase; SNAT3/5, sodium-dependent neutral amino acid transporters 3 and 5; SSADH, succinic semialdehyde dehydrogenase; TCA, tricarboxylic acid cycle. Reproduced with permission from ref. 4.
Deficient glutamate clearance, usually associated with downregulation of EAAT transporters is one of the primary mechanisms of glutamate excitotoxicity leading to neuronal death (see Table 2 and the previous section). Failure of glutamate uptake is particularly prominent in ALS, in Wernicke encephalopathy and in neurotoxic poisoning with trace metals138,148,248,438. Downregulation of EAAT2 also contributes to the pathogenesis of neurodegenerative diseases, epilepsy, migraine and depression, as described in the previous sections. Incidentally, cognitively preserved patients with AD pathology exhibited higher astrocytic expression of EAAT2439.
Targeting glutamate clearance by boosting the expression and function of EAAT2 is considered to be a promising therapeutic strategy. Expression of EAAT2 protein can be induced by β-lactam antibiotic ceftriaxone, which attenuated symptoms and increased survival of SODG93A ALS model mice440. Ceftriaxone also showed beneficial effects in β-amyloidosis mouse models441,442. A pyridazine derivative LDN-212320, which appears to potentiate EAAT2 translation, demonstrated beneficial effects in SODG93A mice443 and improved cognitive performance in the APPSw,Ind AD mouse model444. Trolox, a derivative of vitamin E, was shown to normalize β-amyloid-induced mislocalization of EAAT2 in astrocytes under the action of445. Treatment with minocycline, dexamethasone and histamine may also increase the expression of EAAT2446. Finally, the benzothiazole Riluzole, which enhances EAAT2 expression and glutamate clearance, has been approved and shown modest beneficial effects on ALS progression447,448.
Complement system—alleviating post-ischemic damage
Whereas the importance of the complement system in defense against pathogens has been recognized for over a century, its homeostatic and non-immune functions—particularly in the CNS—have only begun to be elucidated recently. Astrocytes are the major source of complement system proteins, including the third complement component (C3), and through complement receptors expressed on their cell membrane, astrocytes receive and respond to complement-derived signals sent by other cells449.
Astrocytes express a G-protein-coupled receptor for a complement peptide C3a, C3aR450,451,452 and TLQP-21, a neuropeptide derived from the neurotrophin-inducible protein VGF453,454. The expression of C3aR by astrocytes is increased by ischemia450,451. Activation of C3aR inhibits the adenylyl cyclase pathway, and stimulates the phospholipase C pathway, leading to Ca2+ signaling originating from the endoplasmic reticulum Ca2+ store455. C3a–C3aR signaling modulates the activity of the extracellular signal regulated kinase 1/2 (ERK1/2) cascade including Ras and c-Raf456,457. By inhibiting ERK signaling-mediated apoptotic pathway and caspase-3 cleavage, C3a promotes survival of astrocytes exposed to ischemic stress458. In vitro, C3a induces astrocyte expression of cytokines such as IL-6, IL-8 and nerve growth factor (NGF)459,460,461. Astrocytes are involved in or directly mediate the neuroprotective effects of C3a in vitro462. A study assessing the effects of C3a on expression of Gfap, Nes, C3ar1, C3, Ngf, Tnf and Il1b in naive astrocytes, astrocytes challenged with ischemia and astrocytes exposed to lipopolysaccharide showed that C3a downregulated the expression of Gfap, C3 and Nes in astrocytes after ischemia, and increased the expression of Tnf and Il1b in naive astrocytes and the expression of Nes in astrocytes exposed to lipopolysaccharide, but did not affect the expression of C3ar1 or Ngf463. Evidently, the responses of astrocytes to C3a are context dependent.
Combining genetic loss-of-function (constitutive C3aR ablation) and gain-of-function (transgenic overexpression of C3a in the brain) approaches showed that C3aR downregulates peri-infarct astrocyte reactivity, as assessed by reduced immunoreactivity of GFAP383. Pharmacological modulation of C3aR signaling by daily intranasal treatment of wild-type mice with the C3a peptide for 2 weeks starting 1 week after stroke reduced peri-infarct expression of GFAP (Fig. 9), with this effect being similar to that of transgenic C3a overexpression. Peri-infarct GFAP expression was also reduced in mice that received daily intranasal C3a treatment for 3 weeks and were followed up for 4 weeks after treatment cessation383. Expression of GFAP in peri-infarct cortex at this time point was inversely correlated with improved motor function383. Cellular deconvolution of bulk transcriptomic data from the peri-infarct cortex showed that cells with molecular signature similar to the so-called disease-associated astrocytes, originally described in the vicinity of β-amyloid plaques in a mouse model of AD and in the aged brain464, are present also in post-stroke tissue383. This astrocyte subpopulation, expressing high levels of Gfap, Vim and C3ar1, was reduced in mice treated with intranasal C3a. Notably, the expression profile of peri-infarct astrocytes was enriched in genes involved in the regulation of inflammatory response and responses to virus and wounding383. By contrast, the subpopulation of astrocytes expressing low levels of Gfap and enriched in genes involved in neural plasticity was reduced in the peri-infarct cortex, but only in mice treated with physiological saline383.

C3a treatment starting 1 week after stroke reduces astrocyte reactivity and enhances the expression of positive regulators of neural plasticity, increasing synaptic density and axonal sprouting. Figure created in BioRender.
Reduced peri-infarct astrocyte reactivity in C3a-treated mice was associated with upregulation of Igf1 and Thbs4, coding for insulin-like growth factor 1 (IGF-1) and thrombospondin-4 (THBS4), respectively, both of which are highly expressed in peri-infarct astrocytes383. IGF-1 stimulates axon outgrowth 465, and THBS4 promotes synaptogenesis of excitatory synapses466. The C3a-treated mice had also higher density of excitatory synapses and higher expression of the membrane phosphoprotein growth-associated protein 43467, which is an established marker of axonal sprouting and plasticity468,469. Treatment with C3a starting 7 days after stroke accelerated recovery of motor function383,467, stimulated global white matter reorganization and increased peri-infarct structural connectivity383. The increase in expression of positive regulators of neural plasticity provides a plausible mechanistic link between the modulation of astrocyte reactivity, enhanced neural plasticity in the peri-infarct motor cortex, and improved motor function recovery in C3a-treated mice.
In conclusion, GFAP-expressing reactive astrocytes appear as negative regulators of neuronal functioning in the chronic phase after stroke, and C3aR signaling modulates astrocyte reactivity in the post-stroke brain. C3a administered intranasally starting 1 week after stroke reduces astrocyte reactivity, with a positive effect on recovery of motor function. As pharmacological treatments to enhance rehabilitation and improve outcomes in the post-acute and chronic phase after stroke are lacking, clinical testing of C3aR agonists is warranted. Given the broad therapeutic window, the majority of stroke survivors, including those who received but were not helped by the acute interventions, would potentially benefit from such a treatment.
Connexins—suppressing hemichannels
Connexins (Cx) are a family of membrane channel proteins that, in vertebrates, assemble into connexons. These connexons can form intercellular gap junction channels or plasmalemmal pores, known as hemichannels470. Specific to astrocytes are connexins Cx43 (the most abundant) and Cx30470. In the CNS, gap junctions connect astrocytes and oligodendrocytes into ‘panglial’ syncytia, enabling long-distance functional integration of glial cells, supporting intercellular signaling via ions, metabolites and second messengers, and maintaining glial isopotentiality471,472,473. Nonjunctional or unpaired connexons, generally known as hemichannels, act as plasmalemmal channels, distinguished by permeability to relatively big (~1 kDa) molecules474. Connexins also exert numerous nonchannel functions: they regulate astrocyte morphology and interactions with synapses, influence gene expression, control glial reactivity and modulate mitochondrial function and ATP production475,476,477. In pathological settings, connexins are redistributed from gap junctions to hemichannels; the latter contribute to pathogenesis, and their pharmacological suppression provides beneficial outcomes.
Acute brain damage—trauma and ischemia
Acute trauma, ischemia and stroke are characterized by prominent increases in the presence and activity of Cx43 hemichannels in astrocytes, contributing to the secondary damage of the nervous tissue478. Increased hemichannel activity is associated with the release of glutamate and ATP, which can trigger excitotoxicity479. Furthermore, in the context of brain trauma, Cx43 appears to activate cytolytic P2X7 purinoceptors and downregulate astrocytic expression of EAAT1, both events promoting excitotoxicity480. In hypoxia and ischemia, opening of Cx43 hemichannels can lead to astrocytic depolarization and death481. In cerebral ischemia, Cx43 hemichannels do not contribute to an acute phase injury but rather propagate secondary damage in the aftermath481.
Neurodegeneration and AD
Increased presence and activity of astrocytic Cx43 hemichannels seems to be a common feature of all major neurodegenerative pathologies, including AD, PD, HD, ALS and MS482,483,484,485,486,487. Exposure of astrocytes to various β-amyloid peptides, and β-amyloid25–35 in particular, increased Cx43 expression and appears to redistribute connexins from gap junction assemblies to hemichannels486,488,489, the latter, in turn, mediating excessive release of glutamate and ATP resulting in excitotoxicity489,490,491. Expression of Cx43 is significantly upregulated in AD post-mortem brain tissue and in AD murine models of β-amyloidosis such as APPswe/PS1dE9 mice and 5xFAD mice489,492,493. Post-mortem studies also revealed increased Cx43 protein levels in astrocytes surrounding β-amyloid plaques494, although, paradoxically, levels of Gja1 (encoding Cx43) mRNA were decreased492, suggesting post-transcriptional dysregulation. Besides potentiating the expression of Cx43, exposure to β-amyloid opens Cx43 hemichannels, as shown by ethidium bromide uptake assay489,490. Activity of Cx43 hemichannels is high in reactive astrocytes surrounding β-amyloid plaques495. The opening of Cx43 hemichannels can be induced by cytosolic Ca2+ signals or by a reduction in cholesterol491. Notably, the gap junctional connectivity of astrocytes in the hippocampus of APPswe/PS1dE9 mice remains unaffected489. Finally, a profound decrease in astrocytic expression of Cx43 was found in the post-mortem samples of patients with advanced-stage PD; the degree of the Cx43 loss correlated with the severity of symptoms, including nonmotor symptoms such as depression and sleep irregularities209.
Neuropathic pain
In the spinal cord, astrocytic Cx43 expression is upregulated following nerve injury, chemotherapy or spinal cord injury496,497,498,499. An increase in the activity of astrocyte Cx43 hemichannels contributes to the central sensitization of pain. Genetic ablation of astrocytic Cx43 reduces neuronal hyperactivity and mechanical allodynia in rodent models of neuropathic pain500. Similarly, the knockdown of Cx43 by small interfering RNA intrathecal administration reduced mechanical allodynia in a spinal nerve ligation model501. Mechanistically, opening of Cx43 hemichannels leads to the release of ATP and glutamate, which promotes spinal neuronal hyperactivity, reactive astrogliosis and microgliosis498,502,503. Suppression of Cx43 hemichannels via intrathecal injection of blockers such as Gap26, Gap27 or Peptide 5 reduces the production of chemokines CXCL1 and CXCL12, as well as cytokines IL-1β and IL-6, in various neuropathic pain models, including nerve injury, chemotherapy-induced and bone cancer models496,498,504,505.
Therapeutic targeting of Cx43 hemichannels
Targeting astrocyte connexin hemichannels has disease-modifying effects in various neurological diseases (Fig. 10). Several Cx43 targeting drugs underwent clinical trials506. Antisense oligodeoxynucleotides (AsODNs) targeting Cx43 expression have proven effective in the treatment of skin wounds, corneal injuries, and retinal disorders506. Recently, a fiber-hydrogel scaffold-mediated Cx43-AsODN delivery system was tested for the treatment of spinal cord injury and was shown to suppress reactive gliosis and reduce neuronal loss507. A peptide mimetic, aCT1, which reduces Cx43–ZO1 interaction to preserve gap junction connectivity while inhibiting Cx43 hemichannels, has been tested in clinical trials for skin wounds, corneal injuries and retinal disorders506,508. However, the effect of these drugs in neurological diseases requires further testing in vivo.

In physiological conditions, Cx43 forms gap junction channels (GJCs) that mediate the formation of astrocytic network. However, in pathological conditions, such as trauma, ischemia, neurodegeneration and neuropathic pain, pathogenic insults and neuroinflammation trigger reactive astrogliosis, accompanied by the activation of Cx43 hemichannels (HCs), with its GJC function unaltered or downregulated. Cx43 hemichannels contribute to the release of ATP and glutamate. ATP can activate P2X receptors on astrocytes in an autocrine manner, suppress EAAT expression and increase the release of chemokines and cytokines. ATP can also activate P2X receptors on microglia, promoting reactive microgliosis. In addition, ATP and glutamate activate P2X and NMDA receptors on neurons, respectively. These processes downstream of Cx43 HC activation collectively contribute to neuronal excitotoxicity, degeneration and even cell death.
Several other peptides suppress Cx43 hemichannels and alleviate neurological symptoms. Intrathecally injected Cx43 blockers Peptide5, Gap26 and Gap27, for example, reduced neuropathic pain496,498,509. Similarly, Gap26 and Gap27 prevented increases in extracellular glutamate and caspase-3 and significantly reduced cerebral infarct volume480. Infusion of Peptide 5, a selective Cx43 hemichannel blocker, 90 min after a 30-min period of bilateral carotid artery occlusion, thus limited brain damage510.
All these peptide hemichannel inhibitors, however, cannot cross the blood–brain barrier, which limits their clinical potential. In addition, these peptides are mimetic peptides targeting the extracellular loop of Cx43, which have prominent off-target effects, interfering with the channel functions of Cx32, Cx37 and Cx40511. Gap26 and Gap27 could also inhibit gap junction channel function of Cx43512,513, possibly by blockage of the docking sites of opposing hemichannels. Gap19, a mimetic peptide of the Cx43 intracellular loop, inhibits hemichannel opening by suppressing the tail–loop interaction without impacting on the gap junction function514. As the target of Gap19 is intracellular, an addition of cell-permeable sequence TAT to Gap19 produced a TAT–Gap19 peptide, which improves the hemichannel inhibitory efficacy (IC50 ~7 μM versus 50 μM for Gap19) and makes this formulation to cross blood-brain barrier515. Another peptide, TAT–Cx43266–283, mimics the Cx43 C terminus and can suppress Cx43 hemichannels without affecting gap junction communication516,517. Both TAT–Gap19 and TAT–Cx43266–283 effectively alleviate symptoms in mouse models of epilepsy516,518. However, peptide drugs suffer from a major drawback of short half-life in the circulation, which limits their bioavailability and their therapeutic potential in chronic neurodegenerative diseases519. This problem was recently solved by using lipid nanoparticle to package TAT–Cx43266-283 (TAT–Cx43@LNP), which achieve long-term retention in the brain and effectively alleviate cognitive decline in a mouse model of AD517.
Alternative strategies utilize small molecules to target Cx43 hemichannels. Boldine, a natural alkaloid that blocks hemichannels, reduced the release of ATP and glutamate, and alleviates neuronal damage in APP/PS1 mice520. A carbenoxolone derivative INI-0602 with high blood–brain barrier penetration blocks hemichannels and alleviates memory deficits in APP/PS1 mice after intraperitoneal administration521. A recent study used computational screening to develop a small-molecule compound D4, which shows high efficiency in blocking Cx43 hemichannels (10 nM) without affecting gap junction function (up to 200 μM)522. D4 could be orally administrated, readily crosses the blood–brain barrier and effectively suppresses reactive gliosis in an epilepsy model523. However, D4 also shows off-target effects blocking other connexin channels, including Cx26, Cx30 and Cx45522,523.
MAO-B—reversing tonic inhibition and oxidative stress
Considering that reactive astrocytes contribute to various neurological disorders, including neurotrauma, AD, PD, stroke, metabolic disorders and epilepsy, targeting MAO-B and astrocytic GABA tonic inhibition may provide a broad-spectrum therapeutic strategy.
Alzheimer’s disease
In AD, reactive astrocytes cluster around β-amyloid plaques and exhibit increased MAO-B expression, resulting in heightened GABA synthesis and tonic inhibition of neuronal circuits524. Excessive astrocytic GABA release suppresses excitatory transmission, impairs synaptic plasticity and contributes to memory dysfunction524. In contrast to the healthy brain, where tonic inhibition fine-tunes neuronal excitability, in disease, excessive astrocytic GABA secretion suppresses excitatory transmission, reduces glutamatergic input and disrupts memory-related processes524,525,526,527. Under physiological conditions, hippocampal astrocytes contain low [GABA]i. However, in APP/PS1 mice, which develop AD-related β-amyloid plaques, periplaque reactive astrocytes exhibit significantly increased GABA immunoreactivity; notably, β-amyloid directly upregulates MAO-B expression, linking β-amyloid pathology with aberrant astrocytic regulation of neurotransmission510. In the APPNL-F mouse model of β-amyloidosis, increased tonic GABA tone was observed even before the development of plaques276. Elevated astrocytic GABA is associated with increased GFAP immunostaining characteristic of reactive astrocytes524,528. In reactive astrocytes, Best-1, the GABA-releasing channel, redistributes from the perisynaptic leaflets to the soma, probably reflecting a pathological rearrangement of gliotransmission that favors GABA over glutamate529. A positive correlation between GABA levels and proximity to senile plaques suggests localized induction of MAO-B activity, reinforcing its pathological significance524). Imaging studies using PET tracers such as [18F]SMBT-1 confirm that MAO-B is associated with reactive astrogliosis in AD and shows strong correlations with β-amyloid and tau pathology367,530. Besides GABA-related effects, H2O2 generated in the process of MAO-B-catalyzed putrescine conversion evokes oxidative stress and consequent neuronal damage531. Increased H2O2 amplifies reactive astro- and microgliosis, potentiates lipid peroxidation and perpetrates mitochondrial malfunction, further exacerbating AD progression346. Pharmacological occlusion of MAO-B with selective inhibitors reduces astrocyte reactivity, restores synaptic function and mitigates cognitive deficits in amyloidosis model mice532,533.
Excessive astrocytic GABA synthesis and release lead to abnormal synaptic transmission and cognitive deficits. Tonic GABA release in the dentate gyrus disrupts synaptic plasticity by persistently activating extrasynaptic GABAA and GABAB receptors, thereby diminishing excitatory neurotransmission and long-term potentiation, favoring synaptic depression and memory impairment534. Blocking MAO-B with deprenyl restores synaptic transmission and plasticity in APPNL-F mice276. Chronic synaptic inhibition reduces neuronal excitability, leading to synaptic depression and memory impairment535. Post-mortem human studies confirmed this mechanism, demonstrating elevated GFAP, MAO-B and GABA expression in reactive astrocytes within the temporal cortex, with a strong correlation between GFAP and MAO-B levels345.
Parkinson’s disease
MAO-B contributes to the pathogenesis of PD by boosting astrocytic GABA transmission and contributing to oxidative stress. Expression of MAO-B is upregulated in reactive astrocytes in the substantia nigra, leading to excessive GABA production and oxidative stress. This astrocyte-derived GABA mediates tonic inhibition, thus suppressing the excitability of neighboring dopaminergic neurons. As a result, the synthesis and release of dopamine is blocked, markedly reducing dopamine levels and worsening motor impairments348,536,537,538. Post-mortem analyses of PD brains consistently demonstrated elevated MAO-B activity in reactive astrocytes346. The loss of dopaminergic neurons is also exacerbated by oxidative stress triggered by H2O2 produced by MAO-B activity539. Neuronal death, in turn, amplifies secondary reactive gliosis, creating a feedforward loop exacerbating neurodegeneration in PD.
Neuropathic pain
Astrocytic MAO-B contributes to neuropathic pain by providing for an excessive GABA release in the spinal dorsal horn, thus altering chloride homeostasis and promoting neuronal hyperexcitability265,540. The pathogenesis of the chronic pain involves BDNF (microglia-derived)-dependent downregulation of neuronal KCC2 transporter leading to a depolarizing shift of Cl− reversal potential (ECl). Thus, opening of GABAA receptors generates Cl− efflux and neuronal hyperexcitability541. Increased astrocytic GABA synthesis, driven by upregulated MAO-B activity, further amplifies this pathological excitation by acting on extrasynaptic GABAA receptors containing the α5 subunit. Furthermore, MAO-B-dependent astrocytic GABA release contributes to metabolic dysregulation in neuropathic pain. [18F]FDG-microPET imaging of chronic pain animal models revealed increased glucose metabolism in the ipsilateral dorsal horn, correlating with heightened neuronal excitability and glial reactivity265. Beyond its role in neurotransmission, MAO-B contributes to reactive astrogliosis and neuroinflammation. Thus, MAO-B-mediated astrocytic GABA synthesis and its involvement in neuronal tonic excitation augments synaptic transmission and regional metabolism, contributing to the development of neuropathic pain.
Therapeutic targeting of astrocytic MAO-B
Given its prominent role in neurodegeneration and inflammation, astrocytic MAO-B represents a promising therapeutic target. Pharmacological inhibitors such as selegiline, rasagiline and newer reversible inhibitors such as KDS2010 are effective in mitigating MAO-B-mediated pathology531. These inhibitors reduce astrocytic reactivity, normalize GABAergic neurotransmission and limit oxidative stress, offering potential treatment strategies for several neurological disorders including AD, PD, neuropathic pain, stroke and others. Furthermore, gene-silencing approaches targeting astrocytic MAO-B have demonstrated neuroprotective effects in preclinical models, highlighting potential for future genetic therapies538.
Pharmacological inhibitors of MAO-B are classified into irreversible (selegiline and rasagiline) and reversible inhibitors (safinamide and KDS2010)531,542. While irreversible inhibitors were widely used in clinical settings due to their high potency (IC50 ~10 nM), their limited selectivity (selegiline, ~150-fold; rasagiline, ~52-fold over MAO-A) results in potential off-target effects, complicating their long-term efficacy348. Moreover, prolonged treatment with irreversible inhibitors can trigger compensatory mechanisms, including the upregulation of DAO-dependent GABA synthesis, thus undermining the therapeutic impact530,531. By contrast, highly selective reversible inhibitors, such as KDS2010 (~12,500-fold selectivity for MAO-B over MAO-A), demonstrated superior efficacy in mitigating aberrant astrocytic GABA release and oxidative stress without inducing compensatory pathways543. Preclinical studies demonstrated that KDS2010 restores dopaminergic function, alleviates motor deficits and reduces neuroinflammation in PD models, positioning it as a promising candidate for future disease-modifying interventions531. The ongoing development of highly selective MAO-B inhibitors and gene-silencing approaches targeting astrocytic MAO-B further underscores the potential of this enzyme as a therapeutic target in PD. Pharmacological inhibition of MAO-B using compounds such as selegiline or safinamide reduces oxidative stress, restores synaptic function and improves cognitive performance in AD models544.
Similarly, suppression of MAO-B activity is useful in the treatment of neuropathic pain. Pharmacological inhibition of MAO-B using KDS2010 restores GABAergic neurotransmission and alleviates mechanical allodynia and chronic pain symptoms540. Treatment with KDS2010 also reduces regional hypermetabolism, suggesting that astrocytic GABA synthesis plays a key role in sustaining pathological neuronal activity. The suppression of neuroinflammation and reduction of astrocyte-mediated excitatory signaling through MAO-B inhibition highlight its therapeutic potential for chronic pain management, reinforcing the importance of targeting astrocytic MAO-B as a novel strategy in neuropathic pain treatment540.
Urea cycle—restoring neuroprotection and limiting oxidative damage
Astrocytes use the urea cycle to clean up excessive β-amyloid; in the context of AD, the urea metabolism undergoes a metabolic switch545. The urea cycle, discovered by Hans Krebs and Kurt Henseleit546, converts toxic ammonium (obligatory product of amino acids catabolism) to urea. This cycle is highly expressed in hepatocytes, whereas all components of the cycle are also selectively expressed in astrocytes545. In the healthy brain, astrocytes process urea in a noncyclic manner; in AD, this enzymatic cascade is upregulated and switched to the urea cycle mode, resulting in both helpful and harmful outcomes545 (Fig. 11). The upregulated urea cycle removes β-amyloid and ammonia on one hand, while on the other, overproduction of the urea cycle metabolite ornithine leads to the overexpression of the enzyme ornithine decarboxylase 1 (ODC1), which produces excessive putrescine that subsequently degrades into GABA, H2O2 and ammonium (see previous section). The produced ammonium feeds back into the urea cycle, thus creating a vicious loop that exacerbates neurotoxicity. An increase in the expression of urea cycle enzymes and ODC1 was detected in astrocytes from patients with AD and mouse models of AD545.

Schematic of the astrocyte putrescine degradation pathway involved in the late steps of toxic protein degradation process in normal (left) and AD (right) conditions. Healthy cortical and hippocampal astrocytes show low levels of activity in putrescine degradation metabolism, while reactive astrocytes show high levels of putrescine and, thus, high activity of putrescine degradation enzymes, including MAO-B, which produces H2O2, NH3 and, ultimately, GABA.
Inhibition of ODC1 with difluoromethyl ornithine (DFMO) reduced the production of toxic byproducts and improved memory performance in APP/PS1 mice, a widely used transgenic model of AD. Suppression of ODC1 activity appears to redirect ornithine metabolism toward urea production rather than putrescine, and because urea can be efficiently eliminated from astrocytes via the bloodstream and excreted in urine, no adverse effects were observed under the reported treatment regimen. Notably, DFMO-treated APP/PS1 mice also exhibited a reduction in hippocampal β-amyloid plaque burden, suggesting enhanced clearance mechanisms547. With prolonged treatment, sustained inhibition of astrocytic ODC1 was associated with near-complete removal of β-amyloid deposits from the brain in this model. While these findings are encouraging, further studies are needed to evaluate the long-term safety, reproducibility and translational applicability across additional models and treatment paradigms. Besides reducing β-amyloid load, treatment with DFMO coerced astrocytes into a neuroprotective state. Incidentally, these ‘neuroprotective’ astrocytes had genetic similarities with the astrocytes after environmental enrichment and physical exercise, supporting regeneration of the nervous tissue548. The involvement of astrocytic autophagic plasticity upstream to the urea cycle was recently confirmed549. In summary, pharmacological inhibition of ODC1 prevents harmful effects downstream to the urea cycle while preserving its beneficial role in clearing toxic substances. At the same time, while DFMO shows promise in animal models, it has failed in a single-subject case study550 and causes side effects during long-term administration, prompting the need for better therapeutics.
Astrocytic α7 nicotinic acetylcholine receptors—containing β-amyloid deposition
The α7 nicotinic acetylcholine (α7nACh) receptors highly expressed in the human brain mediate cholinergic neurotransmission and neurobiological processes fundamental for memory consolidation and inflammatory responses551. Homomeric α7nAChR is assembled from five α7 subunits forming a cation channel with high permeability to Ca2+ with PCa/Pmonovalent ≈ 6 (ref. 552). Opening of α7nACh receptors thus generates Ca2+ influx modulating various signaling cascades and inflammatory responses essential for cell survival and memory consolidation. The α7nACh receptors are also expressed in astrocytes and are linked to generation of Ca2+ signals553. In the early 2000s, it was noted that α7nACh receptors bind β-amyloid with high (picomolar range) affinity554,555. In cortices and hippocampi of sporadic AD, expression of α7nACh decreases in neurons, while increasing in astrocytes556,557. Even higher increases in astrocytic α7nACh receptors were found in autosomal-dominant AD, with significant positive correlation between numbers of senile plaques and α7 nACh receptor expression558. A recent hypothesis postulated that overexpression of α7 nAChRs in astrocytes is an early modulator of β-amyloid pathology in the context of AD1,559 (Fig. 12). Because reactive astrogliosis in AD precedes β-amyloid deposition355,356, it was proposed that astrocytes are involved in the spreading of β-amyloid pathology. Soluble β-amyloid is suggested to form a complex with α7nACh receptors on neuronal membrane, leading to the release of this complex to be taken up by the astrocytes. Subsequently, β-amyloid is released to the extracellular space as an aggregate and forms senile plaques (Fig. 12, step 1). Uptake of the soluble α7ACh receptor/β-amyloid complex by astrocytes triggers Ca2+ influx, followed by glutamate secretion, which induces excitotoxicity and neuronal death (Fig. 12, step 2).

Soluble β-amyloid forms a complex with α7nACh receptors (step 1a,b) on the neuronal membrane followed by internalization of the complex (step 2). The neuron releases the α7nACh receptor/β-amyloid complex (step 3), which is taken up by reactive astrocyte (step 4) and undergoes lysis, and β-amyloid aggregates are secreted to the extracellular space and form senile plaques (steps 5 and 6). In addition, soluble β-amyloid interacts and forms a complex with α7nACh receptors on the astrocytic membranes (step 1), inducing α7nACh receptors overexpression (step 2b). High β-amyloid causes increased Ca2+ influx through α7nACh receptors into astrocytes (steps 2a and 3), which triggers a Ca2+-dependent glutamate release (steps 4 and 5), followed by hyperactivation of extrasynaptic NMDA receptors leading to glutamate excitotoxicity and neuronal death (step 6). Based on ref. 559.
To further understand the functional role of astrocytic α7nACh receptors in the healthy brain as well as in different disorders, it is essential to develop highly specialized molecular tools capable of detecting α7nACh receptors in the brain of living individuals. [18F]ASEM was developed as a first-generation PET radiotracer that binds to α7nACh receptors with nanomolar affinity560. The PET scans demonstrated increased binding in patients with MCI across all brain regions561. A second-generation tracer [11C] Kln83 with improved selectivity was subsequently designed and is now used in clinical settings562,563.
Aquaporin 4—manipulating astrocytic water fluxes to manage edema
The water channel Aquaporin-4 (AQP4) is abundantly expressed in astrocytes and ependymoglia; it is particularly enriched in the astrocytic endfoot membranes that abut brain capillaries564 (Fig. 13). As such, AQP4 is uniquely positioned to impact the exchange of water between blood and the brain. Attesting to such a role, specific deletion of the perivascular pool of AQP4 significantly reduces the size of experimentally induced edema565, mimicking the effect of a global AQP4 knockout566.

a Schematic illustration of AQP4 expression in brain. AQP4 (blue symbols) is expressed in astrocytes and ependymocytes, and is particularly enriched in astrocytic endfeet abutting on brain microvessels and in astrocytic processes underneath the pial surface. Lower levels of AQP4 are found in perisynaptic astrocytic membranes. Modified from ref. 571. b A 3D reconstruction based on serial electron micrographs indicating that capillaries are completely covered by a perivascular glial sheath. Four astrocytic endfeet (pve I–IV) are shown together with a pericyte (pe) that is sandwiched between the capillary and the endfeet. A cell process (pvcp), probably of microglial origin, is seen peripheral to the glial sheath. Reproduced, with permission from ref. 568.
Non-invasive MRI based on multiple echo time arterial spin labeling has confirmed that water flux between blood and brain tissue is reduced in animals following targeted deletion of AQP4567. That perivascular endfoot membranes and their AQP4 pools are rate limiting when it comes to water exchange across the brain–blood interface, is consistent with three-dimensional (3D) electron microscopic reconstructions showing that astrocyte endfeet provide a complete covering of brain microcapillaries568. The wealth of data supporting a role for AQP4 in mediating water flux across the brain–blood interface suggests the potential use of AQP4 inhibitors in the treatment of stroke and other conditions associated with brain edema. In theory, such inhibitors would curb edema formation, but also impact infarct size. Thus, deletion of AQP4 has been found to reduce infarct volume not only after middle cerebral artery occlusion (which is associated with a massive edema565), but also in experimental models where edema is much less of a confounding factor (distal middle cerebral artery occlusion569). The mechanisms underlying the specific effect on infarct volume remain to be identified, but one possibility is that the protection is mediated through increased astroglial activation in the border zone of the infarct569.
The development of clinically useful AQP4 inhibitors has proved difficult for several reasons570. Any attempt to inhibit AQP4 in a clinical setting must take into account the fact that AQP4 allows bidirectional transport of water571. Thus, the theoretical time window for treatment will close when the edema transits to its resolution phase and AQP4 switches from being an influx route for water to support water efflux572. In the latter phase, treatment with AQP4 inhibitors would make matters worse and slow down recovery rather than speeding it up. These complications notwithstanding, the potential upside of identifying clinically useful AQP4 inhibitors is promising and should inspire further studies and clinical trials.
As an alternative to the search for new inhibitors, one could look for signaling pathways that regulate AQP4 expression at the brain–blood interface or in the brain at large. Recently, it was shown that canonical BMPs—and BMP2 and 4 in particular—cause an upregulation of AQP4 expression in mature astrocytes and dysregulate the associated dystrophin complex by differentially affecting its individual members573. BMP signaling pathways could be considered as a target for putative treatment strategies. Another strategy relies on the redistribution of AQP4, which is at least in part regulated by calmodulin. Inhibition of calmodulin by trifluoperazine (a drug licensed to treat schizophrenia) suppressed AQP4 localization at the blood–spinal cord barrier. In a model of spinal cord injury, treatment with trifluoperazine resulted in attenuation of edema and acceleration of functional recovery574.
Any attempt to pharmacologically manipulate AQP4—directly, through inhibitors, or indirectly, through targeting signaling pathways—must be discussed in the context of the roles that AQP4 normally subserves. It is well known that the function of AQP4 goes far beyond that of a specific water channel. AQP4 is involved in cell volume control and K+ homeostasis (through interaction with other membrane proteins575,576) and appears to be pro-inflammatory in different experimental models of neurological disease577,578. It is also well documented that AQP4 serves as an autoantigen in neuromyelitis optica579. Most relevant, however, is the role that AQP4 appears to play in propelling or facilitating the glymphatic system of the brain. The glymphatic system mediates influx of CSF into the brain tissue from vascular spaces around arteries and efflux of interstitial fluid through perivenous spaces. It is well documented that this fluid flow contributes to the clearance of waste products from the brain580. Early studies showed that deletion of AQP4 reduces clearance of macromolecular substances from the brain after intrastriatal injections581, and a more recent study demonstrated reduced clearance of extracellular solutes after specific deletion of the perivascular pool of AQP4582. The perivascular AQP4 pool appeared to be primarily relevant for clearance of larger molecules such as 500-kDa dextran. Pharmacological inhibition of AQP4 by AER-271 suppressed glymphatic influx to brain tissue583, supporting the conclusions based on the genetic studies referred to above. Amyloid peptides are among the molecules whose clearance from the brain is sensitive to AQP4 deletion and suppression of the glymphatic system581. Potential mechanistic coupling between AQP4 and amyloid clearance is supported by early experimental studies showing that amyloid deposits are typically associated with vessels that lack perivascular AQP4584.
It follows from the discussion above that AQP4 has multifarious roles in the brain. Pharmacological manipulation with AQP4 function or expression holds promise in several clinical settings but might have side effects that deserve careful consideration. Most notably, any pharmacological intervention targeting AQP4 could affect functions as diverse as cell volume control, K+ homeostasis and clearance of waste products including peptides associated with neurodegenerative disease. After 30 years of research on AQP4, we have a solid grasp on the role of this water channel in brain physiology and pathophysiology. Recent breakthroughs have set the stage for new attempts to target AQP4 for new therapies.
Astrocytic autophagy plasticity—boosting degradation of pathological proteins
Neurons exposed to β-amyloid oligomers fail to cope with this proteotoxic stress, leading to a reduction in the expression of autophagy-related genes (MAP1LC3B/LC3B and SQSTM1/p62, two key autophagy-related genes) alongside a decrease in microtubule-associated protein 2 (MAP2). By contrast, astrocytes respond to β-amyloid oligomers by upregulating autophagy-related gene expression. Notably, the expression of MAP1LC3B/LC3B and SQSTM1/p62 is elevated in parallel with the increase in GFAP immunoreactivity. Thus, astrocytes respond to β-amyloid proteotoxic stress by undergoing plastic changes in both morphology and gene expression to clear β-amyloid oligomers through phagocytic autophagy585. Astrocytes orchestrate the autophagy response gradually, coordinating the sequential expression of autophagy components. This astrocytic autophagy plasticity facilitates β-amyloid clearance by modulating the expression of autophagy-related genes. Inhibition of autophagic function impairs mitochondrial health, increases oxidative stress and exacerbates astrocytic cell death. These findings suggest that autophagy plasticity plays a role in astrocyte survival under neurodegenerative conditions. Moreover, suppression of astrocytic autophagy plasticity increases the size of β-amyloid plaques and the number of hypertrophic GFAP-positive astrocytes in a mouse model of β-amyloidosis585. These pathological sequelae are associated with hippocampal neuronal damage and memory loss (Fig. 14). In summary, astrocytes—unlike neurons—exhibit adaptable autophagy plasticity to increase astrocyte survival and clear β-amyloid, thereby improving cognition. Therefore, boosting astrocytic autophagy plasticity may be therapeutically relevant.

Normal astrocytic autophagy plasticity is activated in response to AD-related stressors such as β-amyloid (Aβ) oligomers through upregulation of MAP1LC3B/LC3B and SQSTM1/p62, promoting phagophore formation, enhancing β-amyloid clearance, supporting astrocyte survival and contributing to improved cognitive function in AD mouse models. By contrast, interrupted astrocytic autophagy plasticity impairs mitochondrial function, increases oxidative stress and induces astrocytic cell death, leading to neuronal damage, exacerbated Aβ plaque accumulation and worsened memory deficits. Drawn based on ref. 585.
Aerobic glycolysis—restoring astrocytic metabolic excitability
The brain mass represents around 2% of the total human body mass. However, its metabolic demands are one order of magnitude higher—the consumption of bodily glucose by the brain amounts to 20% of total body energy expenditure. This consumption increases further during cognitive tasks and memory formation and learning586. Under normal conditions, neurons lack energy reserves in the form of glycogen or lipid droplets. Neuronal resilience to energy-demanding states is therefore limited and depends on energy substrates supplied by neuroglia, particularly astrocytes587,588,589,590. Astrocytic energy metabolism is regulated by noradrenergic innervation of the brain which mainly comes from locus coeruleus586. Noradrenergic neurons dwelling in the locus coeruleus are sensitive to various pathological insults and become altered or malfunctional in many neurological disorders, including attention-deficit/hyperactivity disorders, sleep-associated disturbances591, congenital central hypoventilation syndrome, depression and anxiety, and in neurodegenerative diseases, including AD and PD592,593. Recent clinical research has revealed that locus coeruleus neurons are among the first to degenerate in many, if not all, neurodegenerative diseases594. Thus, under conditions of locus coeruleus neurodegeneration, levels of noradrenaline in the CNS are reduced, as demonstrated in an animal model of ß-amyloidosis595, which affects energy provision to neuronal networks. Indeed, a hypoenergetic state is regularly recorded in patients with AD356.
Astrocytes are regulators of CNS energy metabolism596. They are anatomically linked to the blood–brain barrier597 and mediate the uptake of substrates from the systemic circulation into the CNS. Numerous transporters are found in astrocyte plasmalemma, including those that transport glucose and fatty acids, and astrocytes store energy in the form of lipid droplets586,598,599 and glycogen600,601, which is key in generating L-lactate in the process of aerobic glycolysis602.
Adrenergic regulation of astrocyte aerobic glycolysis
The term aerobic glycolysis was introduced by Otto Heinrich Warburg603, who revealed that glycolysis with L-lactate production in cancer tissue occurred even in the presence of oxygen, which is generally known as the Warburg effect604. Aerobic glycolysis is operational in astrocytes during neurodevelopment and even in adulthood in some areas of the CNS605. Aerobic glycolysis is inefficient in producing ATP, but it generates biosynthetic intermediates, an advantage for developing and dynamically growing tissues606. Astrocytes are morphologically plastic cells, especially during learning and memory formation416,607, and biosynthetic aerobic glycolysis intermediates are probably key to supporting these processes608.
The phenomenon of aerobic glycolysis, together with its product L-lactate, is linked to the astrocyte-to-neuron lactate shuttle609, a concept that considers L-lactate as an energy source for neurons. Although this shuttle was presented as a cornerstone of brain energy metabolism, it has been known for a long time that glycolysis-derived pyruvate has two fates in astrocytes: conversion to L-lactate and decarboxylation in the mitochondria610,611. About 25% of the oxidative metabolism of the brain accounts for the pyruvate metabolism612. Moreover, astrocytes are metabolically unique because they preferentially produce and release citrate, a key component of mitochondrial function613. Furthermore, citrate is an intermediate for sterol and fatty acid biosynthesis614,615 and is used in astrocytes for the production of cholesterol, supporting synaptogenesis616, lipogenesis and lipid droplet formation in astrocytes614,615. Noradrenergic stimulation of astrocytes affects lipid droplet formation598. Pathological conditions related to neurodegeneration affect lipid droplets homeostasis, probably through impaired second messenger adrenergic signaling617.
Impaired noradrenergic regulation of astrocyte energy provision in neurodegenerative disorders
Brain activity, including cognitive functions and locomotion, increases metabolism in astrocytes and neurons. Locomotion induces energy processes differently in astrocytes and neurons. In neurons, activation of locomotion triggers a prompt increase in mitochondrial respiration, but in astrocytes, respiration develops much slower618. Enhanced L-lactate availability, produced by astrocytes stimulated with noradrenaline, supports learning619. Imaging of 18F 2-deoxy glucose by the MRI–PET method in patients revealed reduced D-glucose metabolism in patients suffering from neurodegeneration356. In part, this hypometabolic state reflects reduced levels of noradrenaline595 in the CNS due to degeneration of locus coeruleus. In addition to noradrenaline stimulating astrocytes to provide energy in the form of L-lactate, other mechanisms may also contribute to excitation–energy coupling. Activation of the adenosine A2B receptor in astrocytes stimulated glucose metabolism and the release of L-lactate, which increases the extracellular pool of readily available energy substrates620.
When released from astrocytes, L-lactate may exit the CNS through systemic circulation621; what is remaining acts as a signaling molecule binding to L-lactate-sensitive receptors622,623,624. The expression of the canonical L-lactate hydroxycarboxylic acid receptor 1 (HCA1)625 is very low in the brain. HCA1, previously known as the orphan G protein-coupled receptor 81 (GPR81), is coupled to Gi protein and downregulates cAMP production. Exposure of astrocytes to extracellular L-lactate and selective HCA1 agonists, 3Cl-5OH-BA626 or Compound 2627, triggers responses similar to those mediated by β-adrenoceptors628,629,630, namely an increase in cAMP, cAMP-dependent PKA activity and L-lactate production. The latter was attenuated by adenylate cyclase pharmacological inhibition and is still present in HCA1-knockout astrocytes624. Thus, the effects of extracellular L-lactate and HCA1 agonists on cAMP signaling and metabolism in astrocytes are independent of HCA1 and are mediated by a yet unidentified Gs protein-coupled L-lactate receptor624,631. Upon noradrenergic stimulation L-lactate released from astrocytes can back-excite astrocytes to produce even more L-lactate during increased brain energy demand. This form of intercellular signaling was proposed to be the basis of metabolic excitability624 (Fig. 15), which contributes to the maintenance of the concentration gradient of the extracellular L-lactate pool favoring supplementation of neurons energy demand624,632. Astrocytic metabolic excitability is reduced in neurodegeneration owing to the reduced bioavailability of noradrenaline. By contrast, in a neurodevelopmental animal model of intellectual disability631, L-lactate sensing by astrocytes was reported to be increased, indicating the validity of the metabolic excitability model as a therapeutic target.

The schematic shows lactate turnover in astrocytes and its signalling function. Extracellular L-lactate controls cytosolic L-lactate synthesis in astrocytes by yet unidentified receptors coupled to adenylate cyclase activity and cytosolic cAMP increase in astrocytes. In the brain, L-lactate is formed in the cytoplasm of astrocytes (IN) and is released through monocarboxylate transporters (MCTs) 1 and 4 and/or lactate-permeable channels. Extracellularly (OUT), L-lactate can be transported to neighboring cells as an energy substrate. However, L-lactate can also act as a signaling molecule by binding to the L-lactate receptors of neighboring cells, stimulating adenylate cyclase activity (AC) and an increase in cAMP synthesis. Elevated cytosolic cAMP levels facilitate glycogen degradation by activating glycogen phosphorylase (GP) and glycolysis with L-lactate as the end product. In the absence of the L-lactate positive feedback mechanism (‘metabolic excitability’), the L-lactate tissue concentration gradient could be dissipated, reducing the availability of L-lactate as a metabolic fuel, when local energy demands, especially in the brain, are high. This model shares similarities with the ‘autocrine lactate loop’ acting (oppositely) on [cAMP]i through the GPR81 receptor in adipocytes675. GS, glycogen synthase; TCA, tricarboxylic acid cycle. Glucose denotes phosphorylated and free glucose. Reproduced, with permission, from ref. 624.
Targeting aerobic glycolysis in astrocytes
There are several strategies to contain astrocytic impairments associated with reduced levels of noradrenaline. Voluntary physical exercise improves memory by enhancing noradrenergic input633, an effect possibly mediated by β-adrenoceptors634,635. Pharmacological approaches to increase noradrenaline by using uptake inhibitors were not clinically effective636. Transplanting noradrenergic neurons into the CNS was shown to be effective in animal models637, but represents a challenging approach. Another possibility is to stimulate pathways mimicking the action of noradrenaline in producing L-lactate but independently of adrenergic receptors638. A possible receptor that enhances L-lactate production in astrocytes is an orphan GPR27639. Enhanced L-lactate production in astrocytes may engage the mechanism of metabolic excitability, maintaining the extracellular pool of L-lactate. A similar maintenance of the extracellular pool of L-lactate can be also mediated by the A2B adenosine receptors620. While the concept is in line with improving the key pathobiological defect in neurodegeneration (locus coeruleus degeneration, reduced noradrenergic innervation and decreased L-lactate production), further developments are needed to bring the proposal to a feasible clinical outcome.
Conclusions and further perspectives
Restoring brain function and prolonging cognitive longevity are the primary therapeutic challenges of the twenty-first century. Astrocytes—their function, protective capacity, malfunctions, and pathological changes—are fundamental to virtually every CNS disorder. Several astrocyte-specific molecules, most notably the EAAT1 and EAAT2 glutamate transporters, AQP4 water channels, Cx43 connexins, ezrin, and the intermediate filament proteins GFAP and vimentin, contribute to many of these pathologies. Targeting these molecules and developing astrocyte-specific therapeutic strategies may improve both disease-preventing and disease-modifying therapeutic outcomes in diseases of cognition.
References
Verkhratsky, A. & Nedergaard, M. The homeostatic astroglia emerges from evolutionary specialization of neural cells. Philos. Trans. R. Soc. Lond. B 371, 20150428 (2016).
Paolicelli, R. C. et al. Microglia states and nomenclature: a field at its crossroads. Neuron 110, 3458–3483 (2022).
Nave, K. A. & Werner, H. B. Myelination of the nervous system: mechanisms and functions. Annu. Rev. Cell Dev. Biol. 30, 503–533 (2014).
Verkhratsky, A. & Butt, A. M. Neuroglia: Function and Pathology (Elsevier, 2023).
Kettenmann, H. & Ransom, B. R. Neuroglia (OUP, 2013).
Niu, J., Verkhratsky, A., Butt, A. & Yi, C. Oligodendroglia and myelin: supporting the connectome. Adv. Neurobiol. 43, 1–37 (2025).
Sofroniew, M. V. & Vinters, H. V. Astrocytes: biology and pathology. Acta Neuropathol. 119, 7–35 (2010).
Semyanov, A. & Verkhratsky, A. Inclusive brain: from neuronal doctrine to the active milieu. Function 3, zqab069 (2022).
Verkhratsky, A. et al. Astrocytes in human central nervous system diseases: a frontier for new therapies. Signal Transduct. Target. Ther. 8, 396 (2023).
Virchow, R. Ueber das granulirte Ansehen der Wandungen der Gehirnventrikel. In Gesammelte Abhandlungen zur wissenschaftlichen Medicin (ed. Virchow, R.), p. 885–891 (Meidinger Sohn & Comp., 1856).
Virchow, R. Die Cellularpathologie in ihrer Begründung auf physiologische und pathologische Gewebelehre 20 Vorlesungen, gehalten während d. Monate Febr., März u. April 1858 im Patholog. Inst. zu Berlin (August Hirschwald, 1858).
Golgi, C. Sulla Struttura della Sostanza Grigia del Cervello. Gazz. Med. Itl. 33, 244–246 (1873).
Lenhossék, M. V. Der feinere Bau des Nervensystems im Lichte neuester Forschung 2nd edn (Fischer’s Medicinische Buchhandlung H. Kornfield, 1895).
Verkhratsky, A. & Nedergaard, M. Physiology of Astroglia. Physiol. Rev. 98, 239–389 (2018).
Semyanov, A. & Verkhratsky, A. Astrocytic processes: from tripartite synapses to the active milieu. Trends Neurosci. 44, 781–792 (2021).
Khakh, B. S. & Sofroniew, M. V. Diversity of astrocyte functions and phenotypes in neural circuits. Nat. Neurosci. 18, 942–952 (2015).
Miller, R. H. & Raff, M. C. Fibrous and protoplasmic astrocytes are biochemically and developmentally distinct. J. Neurosci. 4, 585–592 (1984).
Reichenbach, A. & Bringmann, A. Müller Cells in the Healthy and Diseased Retina (Springer, 2010).
Franze, K. et al. Muller cells are living optical fibers in the vertebrate retina. Proc. Natl Acad. Sci. USA 104, 8287–8292 (2007).
Rose, C. R. & Verkhratsky, A. Sodium homeostasis and signalling: the core and the hub of astrocyte function. Cell Calcium 117, 102817 (2024).
Aramideh, J. A., Vidal-Itriago, A. & Morsch, M. M. B. Cytokine signalling at the microglial penta-partite synapse. Int. J. Mol. Sci. 22, 13186 (2021).
Dityatev, A. & Rusakov, D. A. Molecular signals of plasticity at the tetrapartite synapse. Curr. Opin. Neurobiol. 21, 353–359 (2011).
Araque, A., Parpura, V., Sanzgiri, R. P. & Haydon, P. G. Tripartite synapses: glia, the unacknowledged partner. Trends Neurosci. 22, 208–215 (1999).
Verkhratsky, A. & Nedergaard, M. Astroglial cradle in the life of the synapse. Philos. Trans. R. Soc. Lond. B 369, 20130595 (2014).
Augusto-Oliveira, M. et al. Astroglia-specific contributions to the regulation of synapses, cognition and behaviour. Neurosci. Biobehav. Rev. 118, 331–357 (2020).
Laake, J. H., Slyngstad, T. A., Haug, F. M. & Ottersen, O. P. Glutamine from glial cells is essential for the maintenance of the nerve terminal pool of glutamate: immunogold evidence from hippocampal slice cultures. J. Neurochem. 65, 871–881 (1995).
Hertz, L. The glutamate–glutamine (GABA) cycle: importance of late postnatal development and potential reciprocal interactions between biosynthesis and degradation. Front. Endocrinol. 4, 59 (2013).
Verkhratsky, A., Arranz, A. M., Ciuba, K. & Pekowska, A. Evolution of neuroglia. Ann. N. Y. Acad. Sci. 1518, 120–130 (2022).
Moroz, L. L. et al. The ctenophore genome and the evolutionary origins of neural systems. Nature 510, 109–114 (2014).
Zalc, B., Goujet, D. & Colman, D. The origin of the myelination program in vertebrates. Curr. Biol. 18, R511–512 (2008).
Tang, Y., Illes, P. & Verkhratsky, A. Glial-neuronal sensory organs: evolutionary journey from Caenorhabditis elegans to mammals. Neurosci. Bull. 36, 561–564 (2020).
Edwards, T. N. & Meinertzhagen, I. A. The functional organisation of glia in the adult brain of Drosophila and other insects. Prog. Neurobiol. 90, 471–497 (2010).
Freeman, M. R. Drosophila central nervous system glia. Cold Spring Harb. Perspect. Biol. 7, a020552 (2015).
Dunski, E. & Pekowska, A. Keeping the balance: trade-offs between human brain evolution, autism, and schizophrenia. Front. Genet. 13, 1009390 (2022).
Zug, R. Developmental disorders caused by haploinsufficiency of transcriptional regulators: a perspective based on cell fate determination. Biol. Open 11, bio058896 (2022).
Oberheim, N. A. et al. Uniquely hominid features of adult human astrocytes. J. Neurosci. 29, 3276–3287 (2009).
Falcone, C. et al. Cortical interlaminar astrocytes across the therian mammal radiation. J. Comp. Neurol. 527, 1654–1674 (2019).
Ciani, C. et al. Varicose-projection astrocytes: a reactive phenotype associated with neuropathology. Preprint at bioRxiv https://doi.org/10.1101/2025.03.17.643488 (2025).
Pekowska, A., Verkhratsky, A. & Falcone, C. Evolution of neuroglia: from worm to man. Handb. Clin. Neurol. 209, 7–26 (2025).
O’Leary, L. A. et al. Characterization of vimentin-immunoreactive astrocytes in the human brain. Front. Neuroanat. 14, 31 (2020).
Kanton, S. et al. Organoid single-cell genomic atlas uncovers human-specific features of brain development. Nature 574, 418–422 (2019).
Ma, S. et al. Molecular and cellular evolution of the primate dorsolateral prefrontal cortex. Science 377, eabo7257 (2022).
Jorstad, N. L. et al. Comparative transcriptomics reveals human-specific cortical features. Science 382, eade9516 (2023).
Khrameeva, E. et al. Single-cell-resolution transcriptome map of human, chimpanzee, bonobo, and macaque brains. Genome Res. 30, 776–789 (2020).
Ciuba, K. et al. Molecular signature of primate astrocytes reveals pathways and regulatory changes contributing to human brain evolution. Cell Stem Cell 32, 426–444 (2025).
Golgi, C. Opera Omnia (Hoepli, 1903).
Schaeffer, S. & Iadecola, C. Revisiting the neurovascular unit. Nat. Neurol. 24, 1198–1209 (2021).
Sanmarco, L. M., Polonio, C. M., Wheeler, M. A. & Quintana, F. J. Functional immune cell–astrocyte interactions. J. Exp. Med. 218, (2021).
Mills, W. A. 3rd et al. Astrocyte plasticity in mice ensures continued endfoot coverage of cerebral blood vessels following injury and declines with age. Nat. Commun. 13, 1794 (2022).
Lawal, O., Ulloa Severino, F. P. & Eroglu, C. The role of astrocyte structural plasticity in regulating neural circuit function and behavior. Glia 70, 1467–1483 (2022).
Perez-Catalan, N. A., Doe, C. Q. & Ackerman, S. D. The role of astrocyte-mediated plasticity in neural circuit development and function. Neural Dev. 16, 1 (2021).
Allen, N. J. Astrocyte regulation of synaptic behavior. Annu. Rev. Cell Dev. Biol. 30, 439–463 (2014).
Verkhratsky, A., Matteoli, M., Parpura, V., Mothet, J. P. & Zorec, R. Astrocytes as secretory cells of the central nervous system: idiosyncrasies of vesicular secretion. EMBO J. 35, 239–257 (2016).
Loureiro, S. O. et al. Homocysteine induces cytoskeletal remodeling and production of reactive oxygen species in cultured cortical astrocytes. Brain Res. 1355, 151–164 (2010).
Zeisel, A. et al. Molecular architecture of the mouse nervous system. Cell 174, 999–1014 (2018).
Herrero-Navarro, A. et al. Astrocytes and neurons share region-specific transcriptional signatures that confer regional identity to neuronal reprogramming. Sci. Adv. 7, eabe8978 (2021).
Kelly, S. M. et al. Radial glial lineage progression and differential intermediate progenitor amplification underlie striatal compartments and circuit organization. Neuron 99, 345–361(2018).
La Manno, G. et al. Molecular diversity of midbrain development in mouse, human, and stem cells. Cell 167, 566–580 (2016).
Jorstad, N. L. et al. Transcriptomic cytoarchitecture reveals principles of human neocortex organization. Science 382, eadf6812 (2023).
Yacawych, W. T. et al. A single dorsal vagal complex circuit mediates the aversive and anorectic responses to GLP1R agonists. Preprint at bioRxiv https://doi.org/10.1101/2025.01.21.634167 (2025).
Zhong, W. et al. The neuropeptide landscape of human prefrontal cortex. Proc. Natl Acad. Sci. USA 119, e2123146119 (2022).
Romanov, R. A. et al. Molecular design of hypothalamus development. Nature 582, 246–252 (2020).
Lyu, P. et al. Common and divergent gene regulatory networks control injury-induced and developmental neurogenesis in zebrafish retina. Nat. Commun. 14, 8477 (2023).
Harkany, T. et al. Molecularly stratified hypothalamic astrocytes are cellular foci for obesity. Research Square https://doi.org/10.21203/rs.3.rs-3748581/v1 (2024).
Noh, K. et al. Cortical astrocytes modulate dominance behavior in male mice by regulating synaptic excitatory and inhibitory balance. Nat. Neurosci. 26, 1541–1554 (2023).
Kofuji, P. & Araque, A. Astrocytes and behavior. Annu. Rev. Neurosci. 44, 49–67 (2021).
Barnett, D. et al. Astrocytes as drivers and disruptors of behavior: new advances in basic mechanisms and therapeutic targeting. J. Neurosci. 43, 7463–7471 (2023).
Cho, W.-H. et al. Hippocampal astrocytes modulate anxiety-like behavior. Nat. Commun. 13, 6536 (2022).
Ding, F., O’Donnell, J., Xu, Q., Kang, N., Goldman, N. & Nedergaard, M. Changes in the composition of brain interstitial ions control the sleep-wake cycle. Science 352, 550–555 (2016).
Untiet, V. et al. Astrocytic chloride is brain state dependent and modulates inhibitory neurotransmission in mice. Nat. Commun. 14, 1871 (2023).
Larsen, B. R. et al. Contributions of the Na+/K+-ATPase, NKCC1, and Kir4.1 to hippocampal K+ clearance and volume responses. Glia 62, 608–622 (2014).
Untiet, V., Kovermann, P., Gerkau, N. J., Gensch, T., Rose, C. R. & Fahlke, C. Glutamate transporter-associated anion channels adjust intracellular chloride concentrations during glial maturation. Glia 65, 388–400 (2017).
Andersen, J. V., Schousboe, A. & Verkhratsky, A. Astrocyte energy and neurotransmitter metabolism in Alzheimer’s disease: Integration of the glutamate/GABA–glutamine cycle. Prog. Neurobiol. 217, 102331 (2022).
Vandenberg, R. J. & Ryan, R. M. Mechanisms of glutamate transport. Physiol. Rev. 93, 1621–1657 (2013).
Zhou, Y. & Danbolt, N. C. GABA and glutamate transporters in brain. Front. Endocrinol. 4, 165 (2013).
Srivastava, I., Vazquez-Juarez, E. & Lindskog, M. Reducing glutamate uptake in rat hippocampal slices enhances astrocytic membrane depolarization while down-regulating CA3–CA1 synaptic response. Front. Synaptic. Neurosci. 12, 37 (2020).
Vazquez-Juarez, E., Srivastava, I. & Lindskog, M. The effect of ketamine on synaptic mistuning induced by impaired glutamate reuptake. Neuropsychopharmacology 48, 1859–1868 (2023).
de Ceglia, R. et al. Specialized astrocytes mediate glutamatergic gliotransmission in the CNS. Nature 622, 120–129 (2023).
Vardjan, N., Parpura, V., Verkhratsky, A. & Zorec, R. Gliocrine system: astroglia as secretory cells of the CNS. Adv. Exp. Med. Biol. 1175, 93–115 (2019).
Bridges, R. J., Natale, N. R. & Patel, S. A. System xc- cystine/glutamate antiporter: an update on molecular pharmacology and roles within the CNS. Br. J. Pharmacol. 165, 20–34 (2012).
Woo, D. H. et al. TREK-1 and Best1 channels mediate fast and slow glutamate release in astrocytes upon GPCR activation. Cell 151, 25–40 (2012).
Kim, H., Choi, S., Lee, E., Koh, W. & Lee, C. J. Tonic NMDA receptor currents in the brain: regulation and cognitive functions. Biol. Psychiatry 96, 164–175 (2024).
Koh, W. et al. Astrocytes render memory flexible by releasing D-serine and regulating NMDA receptor tone in the hippocampus. Biol. Psychiatry 91, 740–752 (2022).
Sah, P., Hestrin, S. & Nicoll, R. A. Tonic activation of NMDA receptors by ambient glutamate enhances excitability of neurons. Science 246, 815–818 (1989).
Cuenod, M., Do, K. Q., Grandes, P., Morino, P. & Streit, P. Localization and release of homocysteic acid, an excitatory sulfur-containing amino acid. J. Histochem. Cytochem. 38, 1713–1715 (1990).
Do, K. Q., Benz, B., Sorg, O., Pellerin, L. & Magistretti, P. J. β-Adrenergic stimulation promotes homocysteic acid release from astrocyte cultures: evidence for a role of astrocytes in the modulation of synaptic transmission. J. Neurochem. 68, 2386–2394 (1997).
Koh, W., Kwak, H., Cheong, E. & Lee, C. J. GABA tone regulation and its cognitive functions in the brain. Nat. Rev. Neurosci. 24, 523–539 (2023).
Kwak, H. et al. Astrocytes control sensory acuity via tonic inhibition in the thalamus. Neuron 108, 691–706 (2020).
Oh, S. J. & Lee, C. J. Distribution and function of the Bestrophin-1 (Best1) channel in the brain. Exp. Neurobiol. 26, 113–121 (2017).
Di Palma, M., Koh, W., Lee, C. J. & Conti, F. A quantitative analysis of bestrophin 1 cellular localization in mouse cerebral cortex. Acta Physiol. 241, e14245 (2025).
Verkhratsky, A., Untiet, V. & Matchkov, V. V. Chloride fluxes and GABA release sustain inhibition in the CNS: The role for Bestrophin 1 anion channels. Acta Physiol. 241, e14254 (2025).
Melone, M., Ciappelloni, S. & Conti, F. A quantitative analysis of cellular and synaptic localization of GAT-1 and GAT-3 in rat neocortex. Brain Struct. Funct. 220, 885–897 (2015).
Barakat, L. & Bordey, A. GAT-1 and reversible GABA transport in Bergmann glia in slices. J. Neurophysiol. 88, 1407–1419 (2002).
Attwell, D., Barbour, B. & Szatkowski, M. Nonvesicular release of neurotransmitter. Neuron 11, 401–407 (1993).
Richerson, G. B. & Wu, Y. Dynamic equilibrium of neurotransmitter transporters: not just for reuptake anymore. J. Neurophysiol. 90, 1363–1374 (2003).
Yoon, B. E. et al. Glial GABA, synthesized by monoamine oxidase B, mediates tonic inhibition. J. Physiol. 592, 4951–4968 (2014).
Woo, J. et al. Control of motor coordination by astrocytic tonic GABA release through modulation of excitation/inhibition balance in cerebellum. Proc. Natl Acad. Sci. USA 115, 5004–5009 (2018).
Lee, J. M. et al. Generation of astrocyte-specific MAOB conditional knockout mouse with minimal tonic GABA inhibition. Exp. Neurobiol. 31, 158–172 (2022).
Wu, Y., Wang, W., Diez-Sampedro, A. & Richerson, G. B. Nonvesicular inhibitory neurotransmission via reversal of the GABA transporter GAT-1. Neuron 56, 851–865 (2007).
Kilb, W. & Kirischuk, S. GABA release from astrocytes in health and disease. Int. J. Mol. Sci. 23, 15859 (2022).
Unichenko, P., Myakhar, O. & Kirischuk, S. Intracellular Na+ concentration influences short-term plasticity of glutamate transporter-mediated currents in neocortical astrocytes. Glia 60, 605–614 (2012).
Heja, L. et al. Astrocytes convert network excitation to tonic inhibition of neurons. BMC Biol. 10, 26 (2012).
Eskandari, S., Willford, S. L. & Anderson, C. M. Revised ion/substrate coupling stoichiometry of GABA transporters. Adv. Neurobiol. 16, 85–116 (2017).
Savtchenko, L., Megalogeni, M., Rusakov, D. A., Walker, M. C. & Pavlov, I. Synaptic GABA release prevents GABA transporter type-1 reversal during excessive network activity. Nat. Commun. 6, 6597 (2015).
Bhandage, A. K., Kanatani, S. & Barragan, A. Toxoplasma-induced hypermigration of primary cortical microglia implicates GABAergic signaling. Front. Cell Infect. Microbiol. 9, 73 (2019).
Serrano-Regal, M. P. et al. Oligodendrocyte differentiation and myelination is potentiated via GABAB receptor activation. Neuroscience 439, 163–180 (2020).
Alfonsa, H. et al. Intracellular chloride regulation mediates local sleep pressure in the cortex. Nat. Neurosci. 26, 64–78 (2023).
Stern, Y., Barnes, C. A., Grady, C., Jones, R. N. & Raz, N. Brain reserve, cognitive reserve, compensation, and maintenance: operationalization, validity, and mechanisms of cognitive resilience. Neurobiol. Aging 83, 124–129 (2019).
Stern, Y. What is cognitive reserve? Theory and research application of the reserve concept. J. Int. Neuropsychol. Soc. 8, 448–460 (2002).
Kittner, S. J. et al. Methodological issues in screening for dementia: the problem of education adjustment. J.Chronic Dis. 39, 163–170 (1986).
Zhang, M. Y. et al. The prevalence of dementia and Alzheimer’s disease in Shanghai, China: impact of age, gender, and education. Ann. Neurol. 27, 428–437 (1990).
Ahangari, N., Fischer, C. E., Schweizer, T. A. & Munoz, D. G. Cognitive resilience and severe Alzheimer’s disease neuropathology. Aging Brain 3, 100065 (2023).
Verkhratsky, A. & Zorec, R. Neuroglia in cognitive reserve. Mol. Psychiatry 29, 3962–3967 (2024).
Yeh, C. Y., Wu, K. Y., Huang, G. J. & Verkhratsky, A. Radial stem astrocytes (aka neural stem cells): Identity, development, physio-pathology, and therapeutic potential. Acta Physiol. 238, e13967 (2023).
Gotz, M., Nakafuku, M. & Petrik, D. Neurogenesis in the developing and adult brain—similarities and key differences. Cold Spring Harb. Perspect. Biol. 8, a018853 (2016).
Ullian, E. M., Sapperstein, S. K., Christopherson, K. S. & Barres, B. A. Control of synapse number by glia. Science 291, 657–661 (2001).
Chung, W. S., Allen, N. J. & Eroglu, C. Astrocytes control synapse formation, function, and elimination. Cold Spring Harb. Perspect. Biol. 7, a020370 (2015).
Escartin, C. et al. Reactive astrocyte nomenclature, definitions, and future directions. Nat. Neurosci. 24, 312–325 (2021).
O’Shea, T. M., Burda, J. E. & Sofroniew, M. V. Cell biology of spinal cord injury and repair. J. Clin. Invest. 127, 3259–3270 (2017).
Skinnider, M. A. et al. Single-cell and spatial atlases of spinal cord injury in the Tabulae Paralytica. Nature 631, 150–163 (2024).
del Río-Hortega, P. & Penfield, W. G. Cerebral cicatrix: the reaction of neuroglia and microglia to brain wounds. Bull. John Hopkins Hosp. 41, 278–303 (1927).
Penfield, W. in Special Cytology. The Form and Functions of the Cell in Health and Disease (ed. Cowdry, E. V.) p. 1033–1068 (Paul B. Hoeber, 1928).
Penfield, W. & Buckley, R. C. Punctures of the brain. Arch. Neurol. Psychiat. 20, 1–13 (1928).
Al-Dalahmah, O. et al. Single-nucleus RNA-seq identifies Huntington disease astrocyte states. Acta Neuropathol. Commun. 8, 19 (2020).
Burda, J. E. et al. Divergent transcriptional regulation of astrocyte reactivity across disorders. Nature 606, 557–564 (2022).
Itoh, N. et al. Cell-specific and region-specific transcriptomics in the multiple sclerosis model: focus on astrocytes. Proc. Natl Acad. Sci. USA 115, E302–E309 (2018).
Kamphuis, W., Kooijman, L., Orre, M., Stassen, O., Pekny, M. & Hol, E. M. GFAP and vimentin deficiency alters gene expression in astrocytes and microglia in wild-type mice and changes the transcriptional response of reactive. Glia 63, 1036–1056 (2015).
Yu, X. et al. Context-specific striatal astrocyte molecular responses are phenotypically exploitable. Neuron 108, 1146–1162 (2020).
Burda, J. E., Bernstein, A. M. & Sofroniew, M. V. Astrocyte roles in traumatic brain injury. Exp. Neurol. 275, 305–315 (2016).
Sofroniew, M. V. Astrocyte barriers to neurotoxic inflammation. Nat. Rev. Neurosci. 16, 249–263 (2015).
Sofroniew, M. V. Astrocyte reactivity: subtypes, states, and functions in CNS innate immunity. Trends Immunol. 41, 758–770 (2020).
Shulyatnikova, T. & Verkhratsky, A. Astroglia in sepsis associated encephalopathy. Neurochem. Res. 45, 83–99 (2020).
Verkhratsky, A., Augusto-Oliveira, M., Pivoriunas, A., Popov, A., Brazhe, A. & Semyanov, A. Astroglial asthenia and loss of function, rather than reactivity, contribute to the ageing of the brain. Pflugers Arch. 473, 753–774 (2021).
Popov, A. et al. Astrocyte dystrophy in ageing brain parallels impaired synaptic plasticity. Aging Cell 20, e13334 (2021).
Popov, A. et al. Mitochondrial malfunction and atrophy of astrocytes in the aged human cerebral cortex. Nat. Commun. 14, 8380 (2023).
Diaz-Castro, B., Robel, S. & Mishra, A. Astrocyte endfeet in brain function and pathology: open questions. Annu. Rev. Neurosci. 46, 101–121 (2023).
Hazell, A. S. Astrocytes are a major target in thiamine deficiency and Wernicke’s encephalopathy. Neurochem. Int. 55, 129–135 (2009).
Hazell, A. S. et al. Loss of astrocytic glutamate transporters in Wernicke encephalopathy. Glia 58, 148–156 (2009).
Galland, F. et al. Hyperammonemia compromises glutamate metabolism and reduces BDNF in the rat hippocampus. NeuroToxicology 62, 46–55 (2017).
Vitale, M., Tottene, A., Zarin Zadeh, M., Brennan, K. C. & Pietrobon, D. Mechanisms of initiation of cortical spreading depression. J. Headache Pain 24, 105 (2023).
Binder, D. K. & Steinhauser, C. Astrocytes and epilepsy. Neurochem. Res. 46, 2687–2695 (2021).
Ueda, Y. et al. Collapse of extracellular glutamate regulation during epileptogenesis: down-regulation and functional failure of glutamate transporter function in rats with chronic seizures induced by kainic acid. J. Neurochem. 76, 892–900 (2001).
Banasr, M., Sanacora, G. & Esterlis, I. Macro- and microscale stress-associated alterations in brain structure: translational link with depression. Biol. Psychiatry 90, 118–127 (2021).
Rajkowska, G., Hughes, J., Stockmeier, C. A., Javier Miguel-Hidalgo, J. & Maciag, D. Coverage of blood vessels by astrocytic endfeet is reduced in major depressive disorder. Biol. Psychiatry 73, 613–621 (2013).
Aten, S. et al. Chronic stress impairs the structure and function of astrocyte networks in an animal model of depression. Neurochem. Res. 48, 1191–1210 (2023).
Lin, S.-S. et al. Electroacupuncture prevents astrocyte atrophy to alleviate depression. Cell Death Dis. 14, 343 (2023).
Lin, S.-S. et al. Astrocyte ezrin defines resilience to stress-induced depressive behaviours in mice. Natl Sci. Rev. 13, nwaf480 (2025).
Howland, D. S. et al. Focal loss of the glutamate transporter EAAT2 in a transgenic rat model of SOD1 mutant-mediated amyotrophic lateral sclerosis (ALS). Proc. Natl Acad. Sci. USA 99, 1604–1609 (2002).
Rothstein, J. D., Van Kammen, M., Levey, A. I., Martin, L. J. & Kuncl, R. W. Selective loss of glial glutamate transporter GLT-1 in amyotrophic lateral sclerosis. Ann. Neurol. 38, 73–84 (1995).
Diaz-Castro, B., Gangwani, M. R., Yu, X., Coppola, G. & Khakh, B. S. Astrocyte molecular signatures in Huntington’s disease. Sci. Transl. Med. 11, eaaw8546 (2019).
Tong, X. et al. Astrocyte Kir4.1 ion channel deficits contribute to neuronal dysfunction in Huntington’s disease model mice. Nat. Neurosci. 17, 694–703 (2014).
Jiang, R., Diaz-Castro, B., Looger, L. L. & Khakh, B. S. Dysfunctional calcium and glutamate signaling in striatal astrocytes from Huntington’s disease model mice. J. Neurosci. 36, 3453–3470 (2016).
Lievens, J. C. et al. Impaired glutamate uptake in the R6 Huntington’s disease transgenic mice. Neurobiol. Dis. 8, 807–821 (2001).
Verkhratsky, A., Marutle, A., Rodriguez-Arellano, J. J. & Nordberg, A. Glial asthenia and functional paralysis: a new perspective on neurodegeneration and Alzheimer’s disease. Neuroscientist 21, 552–568 (2015).
Bugiani, M. & Breur, M. Leukodystrophies due to astroyctic dysfunction. Brain Pathol. 28, 369–371 (2018).
Trias, E., Barbeito, L. & Yamanaka, K. Phenotypic heterogeneity of astrocytes in motor neuron disease. Clin. Exp. Neuroimmunol. 9, 225–234 (2018).
Wilhelmsson, U. et al. Injury leads to the appearance of cells with characteristics of both microglia and astrocytes in mouse and human brain. Cereb. Cortex 27, 3360–3377 (2017).
Balaban, D., Miyawaki, E. K., Bhattacharyya, S. & Torre, M. The phenomenon of clasmatodendrosis. Heliyon 7, e07605 (2021).
Faulkner, J. R., Herrmann, J. E., Woo, M. J., Tansey, K. E., Doan, N. B. & Sofroniew, M. V. Reactive astrocytes protect tissue and preserve function after spinal cord injury. J. Neurosci. 24, 2143–2155 (2004).
Rolls, A., Shechter, R. & Schwartz, M. The bright side of the glial scar in CNS repair. Nat. Rev. Neurosci. 10, 235–241 (2009).
Anderson, M.A. et al. Astrocyte scar formation aids central nervous system axon regeneration. Nature 532, 195–200 (2016).
Liu, K. et al. PTEN deletion enhances the regenerative ability of adult corticospinal neurons. Nat. Neurosci. 13, 1075–1081 (2010).
Mokalled, M. H., Patra, C., Dickson, A. L., Endo, T., Stainier, D. Y. & Poss, K. D. Injury-induced ctgfa directs glial bridging and spinal cord regeneration in zebrafish. Science 354, 630–634 (2016).
Dias, D. O. & Goritz, C. Fibrotic scarring following lesions to the central nervous system. Matrix. Biol. 68-69, 561–570 (2018).
Dias, D. O. et al. Pericyte-derived fibrotic scarring is conserved across diverse central nervous system lesions. Nat. Commun. 12, 5501 (2021).
Holl, D. et al. Distinct origin and region-dependent contribution of stromal fibroblasts to fibrosis following traumatic injury in mice. Nat. Neurosci. 27, 1285–1298 (2024).
Goritz, C., Dias, D. O., Tomilin, N., Barbacid, M., Shupliakov, O. & Frisen, J. A pericyte origin of spinal cord scar tissue. Science 333, 238–242 (2011).
Nogueira-Rodrigues, J. et al. Rewired glycosylation activity promotes scarless regeneration and functional recovery in spiny mice after complete spinal cord transection. Dev. Cell 57, 440–450 (2022).
Streeter, K. A., Sunshine, M. D., Brant, J. O., Sandoval, A. G. W., Maden, M. & Fuller, D. D. Molecular and histologic outcomes following spinal cord injury in spiny mice, Acomys cahirinus. J. Comp. Neurol. 528, 1535–1547 (2020).
Bundesen, L. Q., Scheel, T. A., Bregman, B. S. & Kromer, L. F. Ephrin-B2 and EphB2 regulation of astrocyte-meningeal fibroblast interactions in response to spinal cord lesions in adult rats. J. Neurosci. 23, 7789–7800 (2003).
Bardehle, S. et al. Live imaging of astrocyte responses to acute injury reveals selective juxtavascular proliferation. Nat. Neurosci. 16, 580–586 (2013).
Meletis, K. et al. Spinal cord injury reveals multilineage differentiation of ependymal cells. PLoS Biol. 6, e182 (2008).
Barnabe-Heider, F. et al. Origin of new glial cells in intact and injured adult spinal cord. Cell Stem Cell 7, 470–482 (2010).
Sabelstrom, H. et al. Resident neural stem cells restrict tissue damage and neuronal loss after spinal cord injury in mice. Science 342, 637–640 (2013).
Chen, M. et al. Leucine zipper-bearing kinase is a critical regulator of astrocyte reactivity in the adult mammalian CNS. Cell Rep. 22, 3587–3597 (2018).
Dias, D. O. et al. Reducing pericyte-derived scarring promotes recovery after spinal cord injury. Cell 173, 153–165(2018).
Verkhratsky, A., Zorec, R., Rodriguez-Arellano, J. J. & Parpura, V. Neuroglia in ageing. Adv. Exp. Med. Biol. 1175, 181–197 (2019).
Wruck, W. & Adjaye, J. Meta-analysis of human prefrontal cortex reveals activation of GFAP and decline of synaptic transmission in the aging brain. Acta Neuropathol. Commun. 8, 26 (2020).
Soreq, L. et al. Major shifts in glial regional identity are a transcriptional hallmark of human brain aging. Cell Rep. 18, 557–570 (2017).
Verkerke, M., Hol, E. M. & Middeldorp, J. Physiological and pathological ageing of astrocytes in the human brain. Neurochem. Res. 46, 2662–2675 (2021).
Pakkenberg, B. & Gundersen, H. J. Neocortical neuron number in humans: effect of sex and age. J. Comp. Neurol. 384, 312–320 (1997).
Verkhratsky, A. & Semyanov, A. Astrocytes in ageing. Subcell. Biochem. 103, 253–277 (2023).
Rodriguez-Callejas, J. D., Fuchs, E. & Perez-Cruz, C. Atrophic astrocytes in aged marmosets present tau hyperphosphorylation, RNA oxidation, and DNA fragmentation. Neurobiol. Aging 129, 121–136 (2023).
Wanner, I. B. et al. Glial scar borders are formed by newly proliferated, elongated astrocytes that interact to corral inflammatory and fibrotic cells via STAT3-dependent mechanisms after spinal cord injury. J. Neurosci. 33, 12870–12886 (2013).
Myer, D. J., Gurkoff, G. G., Lee, S. M., Hovda, D. A. & Sofroniew, M. V. Essential protective roles of reactive astrocytes in traumatic brain injury. Brain 129, 2761–2772 (2006).
Lee, J.-H. et al. H2O2-induced astrocytic collagen triggers neuronal death via fucosylation-dependent glial barrier formation upon ischemic stroke. Preprint at bioRxiv https://doi.org/10.1101/2025.05.01.651594 (2025).
Colón Ortiz, C. & Eroglu, C. Astrocyte signaling and interactions in multiple sclerosis. Curr. Opin. Cell Biol. 86, 102307 (2024).
Holl, D. & Goritz, C. Decoding fibrosis in the human central nervous system. Am. J. Physiol. Cell Physiol. 325, C1415–C1420 (2023).
Chen, T. et al. Astrocyte-microglia interaction drives evolving neuromyelitis optica lesion. J. Clin. Invest. 130, 4025–4038 (2020).
Zhang, L., Verkhratsky, A. & Shi, F. D. Astrocytes and microglia in multiple sclerosis and neuromyelitis optica. Handb. Clin. Neurol. 210, 133–145 (2025).
Karpuk, N., Burkovetskaya, M., Fritz, T., Angle, A. & Kielian, T. Neuroinflammation leads to region-dependent alterations in astrocyte gap junction communication and hemichannel activity. J. Neurosci. 31, 414–425 (2011).
Preman, P., Alfonso-Triguero, M., Alberdi, E., Verkhratsky, A. & Arranz, A. M. Astrocytes in Alzheimer’s disease: pathological significance and molecular pathways. Cells 10, 540 (2021).
Verkhratsky, A., Rodrigues, J. J., Pivoriunas, A., Zorec, R. & Semyanov, A. Astroglial atrophy in Alzheimer’s disease. Pflugers Arch. 471, 1247–1261 (2019).
Arranz, A. M. & De Strooper, B. The role of astroglia in Alzheimer’s disease: pathophysiology and clinical implications. Lancet Neurol. 18, 406–414 (2019).
Verkhratsky, A., Zorec, R., Rodríguez, J. J. & Parpura, V. Astroglia dynamics in ageing and Alzheimer’s disease. Curr. Opin. Pharmacol. 26, 74–79 (2016).
Condello, C., Yuan, P., Schain, A. & Grutzendler, J. Microglia constitute a barrier that prevents neurotoxic protofibrillar Abeta42 hotspots around plaques. Nat. Commun. 6, 6176 (2015).
Kraft, A. W. et al. Attenuating astrocyte activation accelerates plaque pathogenesis in APP/PS1 mice. FASEB J. 27, 187–198 (2013).
Dai, D. L., Li, M. & Lee, E. B. Human Alzheimer’s disease reactive astrocytes exhibit a loss of homeostastic gene expression. Acta Neuropathol. Commun. 11, 127 (2023).
Sokolova, D. et al. Astrocyte-derived MFG-E8 facilitates microglial synapse elimination in Alzheimer’s disease mouse models. Preprint at bioRxiv https://doi.org/10.1101/2024.08.31.606944 (2024).
Castegna, A. et al. Proteomic identification of oxidatively modified proteins in Alzheimer’s disease brain. Part I: creatine kinase BB, glutamine synthase, and ubiquitin carboxy-terminal hydrolase L-1. Free Radic. Biol. Med. 33, 562–571 (2002).
Zheng, T. et al. Characterization of reduced astrocyte creatine kinase levels in Alzheimer’s disease. Glia 72, 1590–1603 (2024).
Butterfield, D. A. & Mattson, M. P. Apolipoprotein E and oxidative stress in brain with relevance to Alzheimer’s disease. Neurobiol. Dis. 138, 104795 (2020).
Choi, J., Forster, M. J., McDonald, S. R., Weintraub, S. T., Carroll, C. A. & Gracy, R. W. Proteomic identification of specific oxidized proteins in ApoE-knockout mice: relevance to Alzheimer’s disease. Free Radic. Biol. Med. 36, 1155–1162 (2004).
Pamplona, R. et al. Proteins in human brain cortex are modified by oxidation, glycoxidation, and lipoxidation. Effects of Alzheimer disease and identification of lipoxidation targets. J. Biol. Chem. 280, 21522–21530 (2005).
Tong, J. et al. Low levels of astroglial markers in Parkinson’s disease: relationship to alpha-synuclein accumulation. Neurobiol. Dis. 82, 243–253 (2015).
Mirza, B., Hadberg, H., Thomsen, P. & Moos, T. The absence of reactive astrocytosis is indicative of a unique inflammatory process in Parkinson’s disease. Neuroscience 95, 425–432 (2000).
Kano, M. et al. Reduced astrocytic reactivity in human brains and midbrain organoids with PRKN mutations. NPJ Parkinsons Dis. 6, 33 (2020).
Ramos-Gonzalez, P., Mato, S., Chara, J. C., Verkhratsky, A., Matute, C. & Cavaliere, F. Astrocytic atrophy as a pathological feature of Parkinson’s disease with LRRK2 mutation. NPJ Parkinsons Dis. 7, 31 (2021).
Hastings, N., Rahman, S., Stempor, P. A., Wayland, M. T., Kuan, W.-L. & Kotter, M. R. N. Connexin 43 is downregulated in advanced Parkinson’s disease in multiple brain regions which correlates with symptoms. Sci. Rep. 15, 10250 (2025).
Morales, I., Sanchez, A., Puertas-Avendano, R., Rodriguez-Sabate, C., Perez-Barreto, A. & Rodriguez, M. Neuroglial transmitophagy and Parkinson’s disease. Glia 68, 2277–2299 (2020).
Kuter, K. Z., Cenci, M. A. & Carta, A. R. The role of glia in Parkinson’s disease: emerging concepts and therapeutic applications. Prog. Brain Res. 252, 131–168 (2020).
Lindstrom, V. et al. Extensive uptake of alpha-synuclein oligomers in astrocytes results in sustained intracellular deposits and mitochondrial damage. Mol. Cell. Neurosci. 82, 143–156 (2017).
Rostami, J. et al. Human astrocytes transfer aggregated alpha-synuclein via tunneling nanotubes. J. Neurosci. 37, 11835–11853 (2017).
Gusella, J. F. et al. A polymorphic DNA marker genetically linked to Huntington’s disease. Nature 306, 234–238 (1983).
Tabrizi, S. J., Flower, M. D., Ross, C. A. & Wild, E. J. Huntington disease: new insights into molecular pathogenesis and therapeutic opportunities. Nat. Rev. Neurol. 16, 529–546 (2020).
Osipovitch, M. et al. Human ESC-derived chimeric mouse models of huntington’s disease reveal cell-intrinsic defects in glial progenitor cell differentiation. Cell Stem Cell 24, 107–122(2019).
Octeau, J. C., Chai, H., Jiang, R., Bonanno, S. L., Martin, K. C. & Khakh, B. S. An optical neuron–astrocyte proximity assay at synaptic distance scales. Neuron 98, 49–66 (2018).
Hodges, A. et al. Regional and cellular gene expression changes in human Huntington’s disease brain. Hum. Mol. Genet. 15, 965–977 (2006).
Skotte, N. H. et al. Integrative characterization of the R6/2 mouse model of Huntington’s disease reveals dysfunctional astrocyte metabolism. Cell Rep. 23, 2211–2224 (2018).
Bradford, J., Shin, J. Y., Roberts, M., Wang, C. E., Li, X. J. & Li, S. Expression of mutant huntingtin in mouse brain astrocytes causes age-dependent neurological symptoms. Proc. Natl Acad. Sci. USA 106, 22480–22485 (2009).
Wood, T. E., Barry, J., Yang, Z., Cepeda, C., Levine, M. S. & Gray, M. Mutant huntingtin reduction in astrocytes slows disease progression in the BACHD conditional Huntington’s disease mouse model. Hum. Mol. Genet. 28, 487–500 (2019).
Le Ber, I. et al. Phenotype variability in progranulin mutation carriers: a clinical, neuropsychological, imaging and genetic study. Brain 131, 732–746 (2008).
Ghetti, B., Oblak, A. L., Boeve, B. F., Johnson, K. A., Dickerson, B. C. & Goedert, M. Invited review: frontotemporal dementia caused by microtubule-associated protein tau gene (MAPT) mutations: a chameleon for neuropathology and neuroimaging. Neuropathol. Appl. Neurobiol. 41, 24–46 (2015).
Todd, T. W. & Petrucelli, L. Insights into the pathogenic mechanisms of chromosome 9 open reading frame 72 (C9orf72) repeat expansions. J. Neurochem. 138, 145–162 (2016).
Gerrits, E. et al. Neurovascular dysfunction in GRN-associated frontotemporal dementia identified by single-nucleus RNA sequencing of human cerebral cortex. Nat. Neurosci. 25, 1034–1048 (2022).
Rajicic, A. et al. WDR49-positive astrocytes mark severity of neurodegeneration in frontotemporal lobar degeneration and Alzheimer’s disease. Glia 73, 948–968 (2025).
Heller, C. et al. Plasma glial fibrillary acidic protein is raised in progranulin-associated frontotemporal dementia. J. Neurol. Neurosurg. Psychiatry 91, 263–270 (2020).
Filiano, A. J. et al. Dissociation of frontotemporal dementia-related deficits and neuroinflammation in progranulin haploinsufficient mice. J. Neurosci. 33, 5352–5361 (2013).
Koppers, M. et al. C9orf72 ablation in mice does not cause motor neuron degeneration or motor deficits. Ann. Neurol. 78, 426–438 (2015).
Chew, J. et al. Neurodegeneration. C9ORF72 repeat expansions in mice cause TDP-43 pathology, neuronal loss, and behavioral deficits. Science 348, 1151–1154 (2015).
Hallmann, A. L. et al. Astrocyte pathology in a human neural stem cell model of frontotemporal dementia caused by mutant TAU protein. Sci. Rep. 7, 42991 (2017).
Umoh, M. E. et al. A proteomic network approach across the ALS–FTD disease spectrum resolves clinical phenotypes and genetic vulnerability in human brain. EMBO Mol. Med. 10, 48–62 (2018).
Yamanaka, K. & Komine, O. The multi-dimensional roles of astrocytes in ALS. Neurosci. Res. 126, 31–38 (2018).
Taha, D. M. et al. Astrocytes display cell autonomous and diverse early reactive states in familial amyotrophic lateral sclerosis. Brain 145, 481–489 (2022).
Valori, C. F., Sulmona, C., Brambilla, L. & Rossi, D. Astrocytes: dissecting their diverse roles in amyotrophic lateral sclerosis and frontotemporal dementia. Cells 12, 1450 (2023).
Diaz-Amarilla, P. et al. Phenotypically aberrant astrocytes that promote motoneuron damage in a model of inherited amyotrophic lateral sclerosis. Proc. Natl Acad. Sci. USA 108, 18126–18131 (2011).
Haidet-Phillips, A. M. et al. Astrocytes from familial and sporadic ALS patients are toxic to motor neurons. Nat. Biotechnol. 29, 824–828 (2011).
Re, D. B. et al. Necroptosis drives motor neuron death in models of both sporadic and familial ALS. Neuron 81, 1001–1008 (2014).
Qian, K. et al. Sporadic ALS astrocytes induce neuronal degeneration in vivo. Stem Cell Rep. 8, 843–855 (2017).
Wang, L., Gutmann, D. H. & Roos, R. P. Astrocyte loss of mutant SOD1 delays ALS disease onset and progression in G85R transgenic mice. Hum. Mol. Genet. 20, 286–293 (2011).
Yamanaka, K. et al. Astrocytes as determinants of disease progression in inherited amyotrophic lateral sclerosis. Nat. Neurosci. 11, 251–253 (2008).
Pramatarova, A., Laganiere, J., Roussel, J., Brisebois, K. & Rouleau, G. A. Neuron-specific expression of mutant superoxide dismutase 1 in transgenic mice does not lead to motor impairment. J. Neurosci. 21, 3369–3374 (2001).
Lino, M. M., Schneider, C. & Caroni, P. Accumulation of SOD1 mutants in postnatal motoneurons does not cause motoneuron pathology or motoneuron disease. J. Neurosci. 22, 4825–4832 (2002).
Papadeas, S. T., Kraig, S. E., O’Banion, C., Lepore, A. C. & Maragakis, N. J. Astrocytes carrying the superoxide dismutase 1 (SOD1G93A) mutation induce wild-type motor neuron degeneration in vivo. Proc. Natl Acad. Sci. USA 108, 17803–17808 (2011).
Lepore, A. C. et al. Focal transplantation-based astrocyte replacement is neuroprotective in a model of motor neuron disease. Nat. Neurosci. 11, 1294–1301 (2008).
Ziff, O. J., Clarke, B. E., Taha, D. M., Crerar, H., Luscombe, N. M. & Patani, R. Meta-analysis of human and mouse ALS astrocytes reveals multi-omic signatures of inflammatory reactive states. Genome Res. 32, 71–84 (2022).
Provenzano, F., Torazza, C., Bonifacino, T., Bonanno, G. & Milanese, M. The key role of astrocytes in amyotrophic lateral sclerosis and their commitment to glutamate excitotoxicity. Int. J. Mol. Sci. 24, 15430 (2023).
Rothstein, J. D., Martin, L. J. & Kuncl, R. W. Decreased glutamate transport by the brain and spinal cord in amyotrophic lateral sclerosis. N. Engl. J. Med. 326, 1464–1468 (1992).
Bruijn, L. I. et al. ALS-linked SOD1 mutant G85R mediates damage to astrocytes and promotes rapidly progressive disease with SOD1-containing inclusions. Neuron 18, 327–338 (1997).
Guo, H. et al. Increased expression of the glial glutamate transporter EAAT2 modulates excitotoxicity and delays the onset but not the outcome of ALS in mice. Hum. Mol. Genet. 12, 2519–2532 (2003).
Marchetto, M. C., Muotri, A. R., Mu, Y., Smith, A. M., Cezar, G. G. & Gage, F. H. Non-cell-autonomous effect of human SOD1 G37R astrocytes on motor neurons derived from human embryonic stem cells. Cell Stem Cell 3, 649–657 (2008).
Jimenez-Riani, M., Diaz-Amarilla, P., Isasi, E., Casanova, G., Barbeito, L. & Olivera-Bravo, S. Ultrastructural features of aberrant glial cells isolated from the spinal cord of paralytic rats expressing the amyotrophic lateral sclerosis-linked SOD1G93A mutation. Cell Tissue Res. 370, 391–401 (2017).
Kovacs, G. G. Astroglia and tau: new perspectives. Front. Aging Neurosci. 12, 96 (2020).
Yoshida, M. Astrocytic inclusions in progressive supranuclear palsy and corticobasal degeneration. Neuropathology 34, 555–570 (2014).
Kovacs, G. G. et al. Neuropathology of the hippocampus in FTLD-Tau with Pick bodies: a study of the BrainNet Europe Consortium. Neuropathol. Appl. Neurobiol. 39, 166–178 (2013).
Ferrer, I. et al. Familial globular glial tauopathy linked to MAPT mutations: molecular neuropathology and seeding capacity of a prototypical mixed neuronal and glial tauopathy. Acta Neuropathol. 139, 735–771 (2020).
Kovacs, G. G. et al. Aging-related tau astrogliopathy (ARTAG): harmonized evaluation strategy. Acta Neuropathol. 131, 87–102 (2016).
Fang, B. et al. Autoimmune glial fibrillary acidic protein astrocytopathy: a novel meningoencephalomyelitis. JAMA Neurol. 73, 1297–1307 (2016).
Gravier-Dumonceau, A. et al. Glial fibrillary acidic protein autoimmunity: a French cohort study. Neurology 98, e653–e668 (2022).
Flanagan, E. P. et al. Glial fibrillary acidic protein immunoglobulin G as biomarker of autoimmune astrocytopathy: analysis of 102 patients. Ann. Neurol. 81, 298–309 (2017).
Long, Y. et al. Autoimmune glial fibrillary acidic protein astrocytopathy in Chinese patients: a retrospective study. Eur. J. Neurol. 25, 477–483 (2018).
Wu, T. et al. Glial fibrillary acidic protein astrocytopathy based on a two-center Chinese cohort study. Ann. Clin. Transl. Neurol. 12, 813–1822 (2025).
Wan, D. et al. Novel meningoencephalomyelitis associated with vimentin IgG autoantibodies. JAMA Neurol. 82, 47–257 (2025).
Gwak, Y. S., Kang, J., Unabia, G. C. & Hulsebosch, C. E. Spatial and temporal activation of spinal glial cells: role of gliopathy in central neuropathic pain following spinal cord injury in rats. Exp. Neurol. 234, 362–372 (2012).
Ju, Y. H. et al. Tonic excitation by astrocytic GABA causes neuropathic pain by augmenting neuronal activity and glucose metabolism. Exp. Mol. Med. 56, 1193–1205 (2024).
Tsuda, M. et al. JAK-STAT3 pathway regulates spinal astrocyte proliferation and neuropathic pain maintenance in rats. Brain 134, 1127–1139 (2011).
DeLeo, J. A., Rutkowski, M. D., Stalder, A. K. & Campbell, I. L. Transgenic expression of TNF by astrocytes increases mechanical allodynia in a mouse neuropathy model. Neuroreport 11, 599–602 (2000).
Menetski, J. et al. Mice overexpressing chemokine ligand 2 (CCL2) in astrocytes display enhanced nociceptive responses. Neuroscience 149, 706–714 (2007).
Treder, M. S. Special section on gaze-independent brain-computer interfaces. J. Neural. Eng. 9, 040201 (2012).
Ji, R. R., Donnelly, C. R. & Nedergaard, M. Astrocytes in chronic pain and itch. Nat. Rev. Neurosci. 20, 667–685 (2019).
Ongur, D., Drevets, W. C. & Price, J. L. Glial reduction in the subgenual prefrontal cortex in mood disorders. Proc. Natl Acad. Sci. USA 95, 13290–13295 (1998).
Rajkowska, G. & Stockmeier, C. A. Astrocyte pathology in major depressive disorder: insights from human postmortem brain tissue. Curr. Drug Targets 14, 1225–1236 (2013).
Gomez-Galan, M., De Bundel, D., Van Eeckhaut, A., Smolders, I. & Lindskog, M. Dysfunctional astrocytic regulation of glutamate transmission in a rat model of depression. Mol. Psychiatry 18, 582–594 (2013).
Gomez-Galan, M. et al. Running opposes the effects of social isolation on synaptic plasticity and transmission in a rat model of depression. PLoS ONE 11, e0165071 (2016).
Agudelo, L. Z. et al. Skeletal muscle PGC-1alpha1 modulates kynurenine metabolism and mediates resilience to stress-induced depression. Cell 159, 33–45 (2014).
Portal, B. et al. Early astrocytic dysfunction is associated with mistuned synapses as well as anxiety and depressive-like behavior in the AppNL-F mouse model of Alzheimer’s disease. J. Alzheimers Dis. 100, 1017–1037 (2024).
Depaauw-Holt, L. R. et al. A divergent astrocytic response to stress alters activity patterns via distinct mechanisms in male and female mice. Nat. Commun. 16, 6372 (2025).
Srivastava, I. et al. Reactive astrocytes with reduced function of glutamate transporters in the App(NL-G-F) knock-in mice. ACS Chem. Neurosci. 16, 2035–2047 (2025).
Srivastava, I., Vazquez-Juarez, E., Henning, L., Gomez-Galan, M. & Lindskog, M. Blocking astrocytic GABA restores synaptic plasticity in prefrontal cortex of rat model of depression. Cells 9, 1705 (2020).
Banasr, M. & Duman, R. S. Glial loss in the prefrontal cortex is sufficient to induce depressive-like behaviors. Biol. Psychiatry 64, 863–870 (2008).
Codeluppi, S. A. et al. Prefrontal cortex astroglia modulate anhedonia-like behavior. Mol. Psychiatry 28, 4632–4641 (2023).
John, C. S., Sypek, E. I., Carlezon, W. A., Cohen, B. M., Ongur, D. & Bechtholt, A. J. Blockade of the GLT-1 transporter in the central nucleus of the amygdala induces both anxiety and depressive-like symptoms. Neuropsychopharmacology 40, 1700–1708 (2015).
Sun, J. D., Liu, Y., Yuan, Y. H., Li, J. & Chen, N. H. Gap junction dysfunction in the prefrontal cortex induces depressive-like behaviors in rats. Neuropsychopharmacology 37, 1305–1320 (2012).
Banasr, M. et al. Glial pathology in an animal model of depression: reversal of stress-induced cellular, metabolic and behavioral deficits by the glutamate-modulating drug riluzole. Mol. Psychiatry 15, 501–511 (2010).
Nasca, C., Bigio, B., Zelli, D., Nicoletti, F. & McEwen, B. S. Mind the gap: glucocorticoids modulate hippocampal glutamate tone underlying individual differences in stress susceptibility. Mol. Psychiatry 20, 755–763 (2015).
Nasca, C. et al. L-acetylcarnitine causes rapid antidepressant effects through the epigenetic induction of mGlu2 receptors. Proc. Natl Acad. Sci. USA 110, 4804–4809 (2013).
Kim, J. S., Schmid-Burgk, W., Claus, D. & Kornhuber, H. H. Increased serum glutamate in depressed patients. Arch. Psychiatr. Nervenkr. 232, 299–304 (1982).
Mineur, Y. S., Picciotto, M. R. & Sanacora, G. Antidepressant-like effects of ceftriaxone in male C57BL/6J mice. Biol. Psychiatry 61, 250–252 (2007).
Anacker, C. et al. Neuroanatomic differences associated with stress susceptibility and resilience. Biol. Psychiatry 79, 840–849 (2016).
Dong, L., Li, B., Verkhratsky, A. & Peng, L. Cell type-specific in vivo expression of genes encoding signalling molecules in the brain in response to chronic mild stress and chronic treatment with fluoxetine. Psychopharmacology 232, 2827–2835 (2015).
McEwen, B. S. In pursuit of resilience: stress, epigenetics, and brain plasticity. Ann. N. Y. Acad. Sci. 1373, 56–64 (2016).
Nestler, E. J. & Russo, S. J. Neurobiological basis of stress resilience. Neuron 112, 1911–1929 (2024).
Rajkowska, G. Postmortem studies in mood disorders indicate altered numbers of neurons and glial cells. Biol. Psychiatry 48, 766–777 (2000).
Harrison, P. J., Colbourne, L. & Harrison, C. H. The neuropathology of bipolar disorder: systematic review and meta-analysis. Mol. Psychiatry 25, 1787–1808 (2020).
Li, C. T., Yang, K. C. & Lin, W. C. Glutamatergic dysfunction and glutamatergic compounds for major psychiatric disorders: evidence from clinical neuroimaging studies. Front. Psychiatry 9, 767 (2018).
de Sousa, R. T. et al. Genetic studies on the tripartite glutamate synapse in the pathophysiology and therapeutics of mood disorders. Neuropsychopharmacology 42, 787–800 (2017).
Butt, A. M. & Rivera, A. D. Astrocytes in bipolar disorder. Adv. Neurobiol. 26, 95–113 (2021).
Feresten, A. H., Barakauskas, V., Ypsilanti, A., Barr, A. M. & Beasley, C. L. Increased expression of glial fibrillary acidic protein in prefrontal cortex in psychotic illness. Schizophr. Res. 150, 252–257 (2013).
Windrem, M. S. et al. Human iPSC glial mouse chimeras reveal glial contributions to schizophrenia. Cell Stem Cell 21, 195–208(2017).
Dietz, A. G., Goldman, S. A. & Nedergaard, M. Glial cells in schizophrenia: a unified hypothesis. Lancet Psychiatry 7, 272–281 (2020).
Toker, L., Mancarci, B. O., Tripathy, S. & Pavlidis, P. Transcriptomic evidence for alterations in astrocytes and parvalbumin interneurons in subjects with bipolar disorder and schizophrenia. Biol. Psychiatry 84, 787–796 (2018).
Yeung, R. K. et al. Gabrb2-knockout mice displayed schizophrenia-like and comorbid phenotypes with interneuron–astrocyte–microglia dysregulation. Transl. Psychiatry 8, 128 (2018).
Liu, Z. et al. Dysregulated glial differentiation in schizophrenia may be relieved by suppression of SMAD4- and REST-dependent signaling. Cell Rep. 27, 3832–3843 (2019).
Rivera, A. D. & Butt, A. M. Astrocytes are direct cellular targets of lithium treatment: novel roles for lysyl oxidase and peroxisome-proliferator activated receptor-gamma as astroglial targets of lithium. Transl. Psychiatry 9, 211 (2019).
Pietilainen, O. et al. Astrocytic cell adhesion genes linked to schizophrenia correlate with synaptic programs in neurons. Cell Rep. 42, 111988 (2023).
Ling, E. et al. A concerted neuron–astrocyte program declines in ageing and schizophrenia. Nature 627, 604–611 (2024).
Binder, D. K. Astrocytes: stars of the sacred disease. Epilepsy Curr. 18, 172–179 (2018).
Plata, A. et al. Astrocytic atrophy following status epilepticus parallels reduced Ca2+ activity and impaired synaptic plasticity in the rat hippocampus. Front. Mol. Neurosci. 11, 215 (2018).
Bedner, P. & Steinhauser, C. Altered Kir and gap junction channels in temporal lobe epilepsy. Neurochem. Int. 63, 682–687 (2013).
Chever, O., Djukic, B., McCarthy, K. D. & Amzica, F. Implication of Kir4.1 channel in excess potassium clearance: an in vivo study on anesthetized glial-conditional Kir4.1 knock-out mice. J. Neurosci. 30, 15769–15777 (2010).
Haj-Yasein, N. N. et al. Evidence that compromised K+ spatial buffering contributes to the epileptogenic effect of mutations in the human Kir4.1 gene (KCNJ10). Glia 59, 1635–1642 (2011).
Glass, M. & Dragunow, M. Neurochemical and morphological changes associated with human epilepsy. Brain Res. Brain Res. Rev. 21, 29–41 (1995).
Sarac, S., Afzal, S., Broholm, H., Madsen, F. F., Ploug, T. & Laursen, H. Excitatory amino acid transporters EAAT-1 and EAAT-2 in temporal lobe and hippocampus in intractable temporal lobe epilepsy. APMIS 117, 291–301 (2009).
Lopes, M. W. et al. Time-dependent modulation of AMPA receptor phosphorylation and mRNA expression of NMDA receptors and glial glutamate transporters in the rat hippocampus and cerebral cortex in a pilocarpine model of epilepsy. Exp. Brain Res. 226, 153–163 (2013).
Gorter, J. A. et al. Glutamate transporters alterations in the reorganizing dentate gyrus are associated with progressive seizure activity in chronic epileptic rats. J. Comp. Neurol. 442, 365–377 (2002).
Watanabe, T., Morimoto, K., Hirao, T., Suwaki, H., Watase, K. & Tanaka, K. Amygdala-kindled and pentylenetetrazole-induced seizures in glutamate transporter GLAST-deficient mice. Brain Res. 845, 92–96 (1999).
Ortinski, P. I. et al. Selective induction of astrocytic gliosis generates deficits in neuronal inhibition. Nat. Neurosci. 13, 584–591 (2010).
Aronica, E. et al. Upregulation of adenosine kinase in astrocytes in experimental and human temporal lobe epilepsy. Epilepsia 52, 1645–1655 (2011).
Amiry-Moghaddam, M. et al. Delayed K+ clearance associated with aquaporin-4 mislocalization: phenotypic defects in brains of alpha-syntrophin-null mice. Proc. Natl Acad. Sci. USA 100, 13615–13620 (2003).
Steinhauser, C., Seifert, G. & Bedner, P. Astrocyte dysfunction in temporal lobe epilepsy: K+ channels and gap junction coupling. Glia 60, 1192–1202 (2012).
Pannasch, U. et al. Astroglial networks scale synaptic activity and plasticity. Proc. Natl Acad. Sci. USA 108, 8467–8472 (2011).
Jen, J. C., Wan, J., Palos, T. P., Howard, B. D. & Baloh, R. W. Mutation in the glutamate transporter EAAT1 causes episodic ataxia, hemiplegia, and seizures. Neurology 65, 529–534 (2005).
Kovermann, P. et al. Increased glutamate transporter-associated anion currents cause glial apoptosis in episodic ataxia 6. Brain Commun. 2, fcaa022 (2020).
Nguyen, T. D. et al. Astrocytic NKCC1 inhibits seizures by buffering Cl- and antagonizing neuronal NKCC1 at GABAergic synapses. Epilepsia 64, 3389–3403 (2023).
Wallraff, A., Kohling, R., Heinemann, U., Theis, M., Willecke, K. & Steinhauser, C. The impact of astrocytic gap junctional coupling on potassium buffering in the hippocampus. J. Neurosci. 26, 5438–5447 (2006).
Egawa, K., Yamada, J., Furukawa, T., Yanagawa, Y. & Fukuda, A. Cl− homeodynamics in gap junction-coupled astrocytic networks on activation of GABAergic synapses. J. Physiol. 591, 3901–3917 (2013).
Pietrobon, D. & Conti, F. Astrocytic Na+, K+ ATPases in physiology and pathophysiology. Cell Calcium 118, 102851 (2024).
Pietrobon, D. & Moskowitz, M. A. Chaos and commotion in the wake of cortical spreading depression and spreading depolarizations. Nat. Rev. Neurosci. 15, 379–393 (2014).
Capuani, C. et al. Defective glutamate and K+ clearance by cortical astrocytes in familial hemiplegic migraine type 2. EMBO Mol. Med. 8, 967–986 (2016).
Brenner, M., Johnson, A. B., Boespflug-Tanguy, O., Rodriguez, D., Goldman, J. E. & Messing, A. Mutations in GFAP, encoding glial fibrillary acidic protein, are associated with Alexander disease. Nat. Genet. 27, 117–120 (2001).
Messing, A., Brenner, M., Feany, M. B., Nedergaard, M. & Goldman, J. E. Alexander disease. J. Neurosci. 32, 5017–5023 (2012).
Pekny, M. et al. Astrocytes: a central element in neurological diseases. Acta Neuropathol. 131, 323–345 (2016).
Pajares, M. A., Hernandez-Gerez, E., Pekny, M. & Perez-Sala, D. Alexander disease: the road ahead. Neural Regen. Res. 18, 2156–2160 (2023).
Matusova, Z. et al. Aberrant neurodevelopment in human iPS cell-derived models of Alexander disease. Glia 73, 57–79 (2025).
van der Knaap, M. S. et al. Mutations in each of the five subunits of translation initiation factor eIF2B can cause leukoencephalopathy with vanishing white matter. Ann. Neurol. 51, 264–270 (2002).
Bugiani, M., Vuong, C., Breur, M. & van der Knaap, M. S. Vanishing white matter: a leukodystrophy due to astrocytic dysfunction. Brain Pathol. 28, 408–421 (2018).
Bugiani, M. et al. Defective glial maturation in vanishing white matter disease. J. Neuropathol. Exp. Neurol. 70, 69–82 (2011).
van der Knaap, M. S. & Bugiani, M. Leukodystrophies: a proposed classification system based on pathological changes and pathogenetic mechanisms. Acta Neuropathol. 134, 351–382 (2017).
Brignone, M. S. et al. MLC1 protein: a likely link between leukodystrophies and brain channelopathies. Front. Cell. Neurosci. 9, 66 (2015).
Boor, I. et al. MLC1 is associated with the dystrophin–glycoprotein complex at astrocytic endfeet. Acta Neuropathol. 114, 403–410 (2007).
Lanciotti, A. et al. Megalencephalic leukoencephalopathy with subcortical cysts protein 1 functionally cooperates with the TRPV4 cation channel to activate the response of astrocytes to osmotic stress: dysregulation by pathological mutations. Hum. Mol. Genet. 21, 2166–2180 (2012).
Min, R. & van der Knaap, M. S. Genetic defects disrupting glial ion and water homeostasis in the brain. Brain Pathol. 28, 372–387 (2018).
Dubey, M. et al. Seizures and disturbed brain potassium dynamics in the leukodystrophy megalencephalic leukoencephalopathy with subcortical cysts. Ann. Neurol. 83, 636–649 (2018).
Alsaad, I. et al. Targeting MAO-B with small-molecule inhibitors: a decade of advances in anticancer research (2012–2024). Molecules 30, 126 (2024).
Jaisa-Aad, M., Munoz-Castro, C., Healey, M. A., Hyman, B. T. & Serrano-Pozo, A. Characterization of monoamine oxidase-B (MAO-B) as a biomarker of reactive astrogliosis in Alzheimer’s disease and related dementias. Acta Neuropathol. 147, 66 (2024).
Lopresti, B. J. et al. Kinetic modeling of the monoamine oxidase-B radioligand [18F]SMBT-1 in human brain with positron emission tomography. J. Cereb. Blood Flow Metab. 44, 1262–1276 (2024).
Wu, N., Zhang, X., Li, Y., Zhang, J. & Cui, M. Fluorinated coumarin derivatives as selective PET tracer for MAO-B imaging. J. Med. Chem. 68, 324–337 (2025).
Nam, M. H., Sa, M., Ju, Y. H., Park, M. G. & Lee, C. J. Revisiting the role of astrocytic MAOB in Parkinson’s disease. Int. J. Mol. Sci. 23, 4453 (2022).
Kumlien, E., Nilsson, A., Hagberg, G., Langstrom, B. & Bergstrom, M. PET with 11C-deuterium-deprenyl and 18F-FDG in focal epilepsy. Acta Neurol. Scand. 103, 360–366 (2001).
Johansson, A. et al. Evidence for astrocytosis in ALS demonstrated by [11C](L)-deprenyl-D2 PET. J. Neurol. Sci. 255, 17–22 (2007).
Engler, H. et al. Multitracer study with positron emission tomography in Creutzfeldt–Jakob disease. Eur. J. Nucl. Med. Mol. Imaging 30, 85–95 (2003).
Engler, H. et al. Imaging astrocytosis with PET in Creutzfeldt–Jakob disease: case report with histopathological findings. Int. J. Clin. Exp. Med. 5, 201–207 (2012).
Carter, S. F. et al. Evidence for astrocytosis in prodromal Alzheimer disease provided by 11C-deuterium-L-deprenyl: a multitracer PET paradigm combining 11C-Pittsburgh compound B and 18F-FDG. J. Nucl. Med. 53, 37–46 (2012).
Nordberg, A. Molecular imaging in Alzheimer’s disease: new perspectives on biomarkers for early diagnosis and drug development. Alzheimers Res. Ther. 3, 34 (2011).
Rodriguez-Vieitez, E. et al. Astrocytosis precedes amyloid plaque deposition in Alzheimer APPswe transgenic mouse brain: a correlative positron emission tomography and in vitro imaging study. Eur. J. Nucl. Med. Mol. Imaging 42, 1119–1132 (2015).
Rodriguez-Vieitez, E. et al. Diverging longitudinal changes in astrocytosis and amyloid PET in autosomal dominant Alzheimer’s disease. Brain 139, 922–936 (2016).
Scholl, M. et al. Early astrocytosis in autosomal dominant Alzheimer’s disease measured in vivo by multi-tracer positron emission tomography. Sci. Rep. 5, 16404 (2015).
Carter, S. F., Chiotis, K., Nordberg, A. & Rodriguez-Vieitez, E. Longitudinal association between astrocyte function and glucose metabolism in autosomal dominant Alzheimer’s disease. Eur. J. Nucl. Med. Mol. Imaging 46, 348–356 (2019).
Kumar, A., Fontana, I. C. & Nordberg, A. Reactive astrogliosis: A friend or foe in the pathogenesis of Alzheimer’s disease. J. Neurochem. 164, 309–324 (2023).
Fontana, I. C. et al. Astrocyte Signature in Alzheimer’s disease continuum through a multi-PET tracer imaging perspective. Cells 12, 1469 (2023).
Tyacke, R. J. et al. Evaluation and initial in vitro and ex vivo characterization of the potential positron emission tomography ligand, BU99008 (2-(4,5-dihydro-1H-imidazol-2-yl)-1- methyl-1H-indole), for the imidazoline(2) binding site. Synapse 66, 542–551 (2012).
Tyacke, R. J. et al. Evaluation of 11C-BU99008, a PET ligand for the imidazoline2 binding site in human brain. J. Nucl. Med. 59, 1597–1602 (2018).
Calsolaro, V. et al. Astrocyte reactivity with late-onset cognitive impairment assessed in vivo using (11)C-BU99008 PET and its relationship with amyloid load. Mol. Psychiatry 26, 5848–5855 (2021).
Livingston, N. R. et al. Relationship between astrocyte reactivity, using novel (11)C-BU99008 PET, and glucose metabolism, grey matter volume and amyloid load in cognitively impaired individuals. Mol. Psychiatry 27, 2019–2029 (2022).
Wilson, H. et al. Imidazoline 2 binding sites reflecting astroglia pathology in Parkinson’s disease: an in vivo11C-BU99008 PET study. Brain 142, 3116–3128 (2019).
Harada, R. et al. 18F-SMBT-1: a selective and reversible PET tracer for monoamine oxidase-B imaging. J. Nucl. Med. 62, 253–258 (2021).
Villemagne, V. L. et al. Assessing reactive astrogliosis with 18F-SMBT-1 across the Alzheimer disease spectrum. J. Nucl. Med. 63, 1560–1569 (2022).
Moriguchi, S. et al. Monoamine oxidase B total distribution volume in the prefrontal cortex of major depressive disorder: an [11C]SL25.1188 positron emission tomography study. JAMA Psychiatry 76, 634–641 (2019).
Nisha Aji, K. et al. Evidence of altered monoamine oxidase B, an astroglia marker, in early psychosis and high-risk state. Mol. Psychiatry 30, 2049–2058 (2024).
Gill, T. et al. Imaging of astrocytes in posttraumatic stress disorder: A PET study with the monoamine oxidase B radioligand [11C]SL25.1188. Eur. Neuropsychopharmacol. 54, 54–61 (2022).
Koshimori, Y. et al. Astrogliosis marker 11C-SL25.1188 PET in traumatic brain injury with persistent symptoms. Brain 146, 4469–4475 (2023).
Braga, J. et al. Astrogliosis marker [11C]SL25.1188 after COVID-19 with ongoing depressive and cognitive symptoms. Biol. Psychiatry 15, 816–824 (2024).
Nam, M. H. et al. Visualizing reactive astrocyte-neuron interaction in Alzheimer’s disease using 11C-acetate and 18F-FDG. Brain 146, 2957–2974 (2023).
Takata, K. et al. 11C-acetate PET imaging in patients with multiple sclerosis. PLoS ONE 9, e111598 (2014).
Kumar, A. et al. Astroglial tracer BU99008 detects multiple binding sites in Alzheimer’s disease brain. Mol. Psychiatry 26, 5833–5847 (2021).
Fontana, I. C., Kumar, A., Okamura, N. & Nordberg, A. Multitracer approach to understanding the complexity of reactive astrogliosis in Alzheimer’s brains. ACS Chem. Neurosci. 15, 328–336 (2024).
Pekny, M. & Pekna, M. Astrocyte reactivity and reactive astrogliosis: costs and benefits. Physiol. Rev. 94, 1077–1098 (2014).
Hol, E. M. & Pekny, M. Glial fibrillary acidic protein (GFAP) and the astrocyte intermediate filament system in diseases of the central nervous system. Curr. Opin. Cell Biol. 32, 121–130 (2015).
Hagemann, T. L. et al. Antisense suppression of glial fibrillary acidic protein as a treatment for Alexander disease. Ann. Neurol. 83, 27–39 (2018).
Hagemann, T. L. et al. Antisense therapy in a rat model of Alexander disease reverses GFAP pathology, white matter deficits, and motor impairment. Sci. Transl. Med. 13, eabg4711 (2021).
Wilhelmsson, U. et al. Absence of glial fibrillary acidic protein and vimentin prevents hypertrophy of astrocytic processes and improves post-traumatic regeneration. J. Neurosci. 24, 5016–5021 (2004).
Lepekhin, E. A., Eliasson, C., Berthold, C. H., Berezin, V., Bock, E. & Pekny, M. Intermediate filaments regulate astrocyte motility. J. Neurochem. 79, 617–625 (2001).
Stokowska, A. et al. Complement C3a treatment accelerates recovery after stroke via modulation of astrocyte reactivity and cortical connectivity. J. Clin. Invest. 133, e162253 (2023).
Eliasson, C. et al. Intermediate filament protein partnership in astrocytes. J. Biol. Chem. 274, 23996–24006 (1999).
Pekny, M. et al. Abnormal reaction to central nervous system injury in mice lacking glial fibrillary acidic protein and vimentin. J. Cell Biol. 145, 503–514 (1999).
Pekny, M., Wilhelmsson, U., Tatlisumak, T. & Pekna, M. Astrocyte activation and reactive gliosis—a new target in stroke? Neurosci. Lett. 689, 45–55 (2019).
Nakazawa, T. et al. Attenuated glial reactions and photoreceptor degeneration after retinal detachment in mice deficient in glial fibrillary acidic protein and vimentin. Invest. Ophthalmol. Vis. Sci. 48, 2760–2768 (2007).
Sihlbom, C., Wilhelmsson, U., Li, L., Nilsson, C. L. & Pekny, M. 14-3-3 expression in denervated hippocampus after entorhinal cortex lesion assessed by culture-derived isotope tags in quantitative proteomics. J. Proteome Res. 6, 3491–3500 (2007).
Lu, Y. B. et al. Reactive glial cells: increased stiffness correlates with increased intermediate filament expression. FASEB J. 25, 624–631 (2011).
Lebkuechner, I., Wilhelmsson, U., Mollerstrom, E., Pekna, M. & Pekny, M. Heterogeneity of Notch signaling in astrocytes and the effects of GFAP and vimentin deficiency. J. Neurochem. 135, 234–248 (2015).
Wilhelmsson, U. et al. Astrocytes negatively regulate neurogenesis through the Jagged1-mediated Notch pathway. Stem Cells 30, 2320–2329 (2012).
Potokar, M. et al. Cytoskeleton and vesicle mobility in astrocytes. Traffic 8, 12–20 (2007).
Potokar, M. et al. Intermediate filaments attenuate stimulation-dependent mobility of endosomes/lysosomes in astrocytes. Glia 58, 1208–1219 (2010).
Vardjan, N. et al. IFN-γ-induced increase in the mobility of MHC class II compartments in astrocytes depends on intermediate filaments. J. Neuroinflammation 9, 144 (2012).
Lasic, E. et al. Nestin affects fusion pore dynamics in mouse astrocytes. Acta Physiol. 228, e13399 (2020).
Wilhelmsson, U. et al. Nestin regulates neurogenesis in mice through Notch signaling from astrocytes to neural stem cells. Cereb. Cortex 29, 4050–4066 (2019).
Lundkvist, A., Reichenbach, A., Betsholtz, C., Carmeliet, P., Wolburg, H. & Pekny, M. Under stress, the absence of intermediate filaments from Muller cells in the retina has structural and functional consequences. J. Cell Sci. 117, 3481–3488 (2004).
Verardo, M. R. et al. Abnormal reactivity of muller cells after retinal detachment in mice deficient in GFAP and vimentin. Invest. Ophthalmol. Vis. Sci. 49, 3659–3665 (2008).
de Pablo, Y., Nilsson, M., Pekna, M. & Pekny, M. Intermediate filaments are important for astrocyte response to oxidative stress induced by oxygen-glucose deprivation and reperfusion. Histochem. Cell Biol. 140, 81–91 (2013).
Ding, M., Eliasson, C., Betsholtz, C., Hamberger, A. & Pekny, M. Altered taurine release following hypotonic stress in astrocytes from mice deficient for GFAP and vimentin. Brain Res. Mol. Brain Res. 62, 77–81 (1998).
Li, L. et al. Protective role of reactive astrocytes in brain ischemia. J. Cereb Blood Flow Metab. 28, 468–481 (2008).
Wunderlich, K. A. et al. Retinal functional alterations in mice lacking intermediate filament proteins glial fibrillary acidic protein and vimentin. FASEB J. 29, 4815–4828 (2015).
Pekny, M. et al. The impact of genetic removal of GFAP and/or vimentin on glutamine levels and transport of glucose and ascorbate in astrocytes. Neurochem. Res. 24, 1357–1362 (1999).
Liu, Z. et al. Beneficial effects of gfap/vimentin reactive astrocytes for axonal remodeling and motor behavioral recovery in mice after stroke. Glia 62, 2022–2033 (2014).
Jarlestedt, K. et al. Attenuation of reactive gliosis does not affect infarct volume in neonatal hypoxic-ischemic brain injury in mice. PLoS ONE 5, e10397 (2010).
Macauley, S. L., Pekny, M. & Sands, M. S. The role of attenuated astrocyte activation in infantile neuronal ceroid lipofuscinosis. J. Neurosci. 31, 15575–15585 (2011).
Berg, A., Zelano, J., Pekna, M., Wilhelmsson, U., Pekny, M. & Cullheim, S. Axonal regeneration after sciatic nerve lesion is delayed but complete in GFAP- and vimentin-deficient mice. PLoS ONE 8, e79395 (2013).
Sofroniew, M. V. Dissecting spinal cord regeneration. Nature 557, 343–350 (2018).
Aswendt, M. et al. Reactive astrocytes prevent maladaptive plasticity after ischemic stroke. Prog. Neurobiol. 209, 102199 (2022).
Larsson, A., Wilhelmsson, U., Pekna, M. & Pekny, M. Increased cell proliferation and neurogenesis in the hippocampal dentate gyrus of old GFAP(−/−)Vim(−/−) mice. Neurochem. Res. 29, 2069–2073 (2004).
Wilhelmsson, U. et al. The role of GFAP and vimentin in learning and memory. Biol. Chem. 400, 1147–1156 (2019).
Cho, K. S. et al. Re-establishing the regenerative potential of central nervous system axons in postnatal mice. J. Cell Sci. 118, 863–872 (2005).
Menet, V., Prieto, M., Privat, A. & Gimenez y Ribotta, M. Axonal plasticity and functional recovery after spinal cord injury in mice deficient in both glial fibrillary acidic protein and vimentin genes. Proc. Natl Acad. Sci. USA 100, 8999–9004 (2003).
Kinouchi, R. et al. Robust neural integration from retinal transplants in mice deficient in GFAP and vimentin. Nat. Neurosci. 6, 863–868 (2003).
Widestrand, A. et al. Increased neurogenesis and astrogenesis from neural progenitor cells grafted in the hippocampus of GFAP−/− Vim−/− mice. Stem Cells 25, 2619–2627 (2007).
Henneberger, C. et al. LTP induction boosts glutamate spillover by driving withdrawal of perisynaptic astroglia. Neuron 108, 919–936 (2020).
Fehon, R. G., McClatchey, A. I. & Bretscher, A. Organizing the cell cortex: the role of ERM proteins. Nat. Rev. Mol. Cell Biol. 11, 276–287 (2010).
Pore, D. & Gupta, N. The ezrin–radixin–moesin family of proteins in the regulation of B-cell immune response. Crit. Rev. Immunol. 35, 15–31 (2015).
Adyshev, D. M. et al. Ezrin/radixin/moesin proteins differentially regulate endothelial hyperpermeability after thrombin. Am. J. Physiol. Lung Cell. Mol. Physiol. 305, L240–L255 (2013).
Tsai, F.-C. et al. Ezrin enrichment on curved membranes requires a specific conformation or interaction with a curvature-sensitive partner. eLife 7, e37262 (2018).
Derouiche, A. & Geiger, K. D. Perspectives for Ezrin and Radixin in Astrocytes: Kinases, Functions and Pathology. Int. J. Mol. Sci. 2, 776 (2019).
Schacke, S. et al. Ezrin deficiency triggers glial fibrillary acidic protein upregulation and a distinct reactive astrocyte phenotype. Glia 70, 2309–2329 (2022).
Lavialle, M., Aumann, G., Anlauf, E., Pröls, F., Arpin, M. & Derouiche, A. Structural plasticity of perisynaptic astrocyte processes involves ezrin and metabotropic glutamate receptors. Proc. Natl Acad. Sci. USA 108, 12915–12919 (2011).
Chen, L. et al. Ezrin-mediated astrocyte-synapse signaling regulates cognitive function via astrocyte morphological changes in fine processes in male mice. Brain Behav. Immun. 124, 177–191 (2025).
Badia-Soteras, A. et al. Retraction of astrocyte leaflets from the synapse enhances fear memory. Biol. Psychiatry 94, 226–238 (2023).
Sullivan, S. M. et al. Cytoskeletal anchoring of GLAST determines susceptibility to brain damage: an identified role for GFAP. J. Biol. Chem. 282, 29414–29423 (2007).
Nijboer, C. H., Heijnen, C. J., Degos, V., Willemen, H. L. D. M., Gressens, P. & Kavelaars, A. Astrocyte GRK2 as a novel regulator of glutamate transport and brain damage. Neurobiol. Dis. 54, 206–215 (2013).
Zhou, B. et al. Astroglial dysfunctions drive aberrant synaptogenesis and social behavioral deficits in mice with neonatal exposure to lengthy general anesthesia. PLoS Biol. 17, e3000086 (2019).
Qureshi, T. et al. Slc38a1 Conveys astroglia-derived glutamine into GABAergic interneurons for neurotransmitter GABA synthesis. Cells 9, 1686 (2020).
Hertz, L. Functional interactions between neurons and astrocytes I. Turnover and metabolism of putative amino acid transmitters. Prog. Neurobiol. 13, 277–323 (1979).
Lehre, K. P. & Danbolt, N. C. The number of glutamate transporter subtype molecules at glutamatergic synapses: chemical and stereological quantification in young adult rat brain. J. Neurosci. 18, 8751–8757 (1998).
Chaudhry, F. A., Lehre, K. P., van Lookeren Campagne, M., Ottersen, O. P., Danbolt, N. C. & Storm-Mathisen, J. Glutamate transporters in glial plasma membranes: highly differentiated localizations revealed by quantitative ultrastructural immunocytochemistry. Neuron 15, 711–720 (1995).
Radulescu, A. R. et al. Estimating the glutamate transporter surface density in distinct sub-cellular compartments of mouse hippocampal astrocytes. PLoS Comput. Biol. 18, e1009845 (2022).
Scalise, M., Pochini, L., Galluccio, M. & Indiveri, C. Glutamine transport. From energy supply to sensing and beyond. Biochim. Biophys. Acta 1857, 1147–1157 (2016).
Broer, S. The SLC38 family of sodium-amino acid co-transporters. Pflugers Arch. 466, 155–172 (2014).
Verkhratsky, A. & Rose, C. R. Na+-dependent transporters: the backbone of astroglial homeostatic function. Cell Calcium 85, 102136 (2020).
Todd, A. C., Marx, M. C., Hulme, S. R., Broer, S. & Billups, B. SNAT3-mediated glutamine transport in perisynaptic astrocytes in situ is regulated by intracellular sodium. Glia 65, 900–916 (2017).
Li, B., Xia, M., Zorec, R., Parpura, V. & Verkhratsky, A. Astrocytes in heavy metal neurotoxicity and neurodegeneration. Brain Res. 1752, 147234 (2021).
Kobayashi, E. et al. Activated forms of astrocytes with higher GLT-1 expression are associated with cognitive normal subjects with Alzheimer pathology in human brain. Sci. Rep. 8, 1712 (2018).
Rothstein, J. D. et al. β-lactam antibiotics offer neuroprotection by increasing glutamate transporter expression. Nature 433, 73–77 (2005).
Hamidi, N., Nozad, A., Sheikhkanloui Milan, H., Salari, A. A. & Amani, M. Effect of ceftriaxone on paired-pulse response and long-term potentiation of hippocampal dentate gyrus neurons in rats with Alzheimer-like disease. Life Sci. 238, 116969 (2019).
Zumkehr, J. et al. Ceftriaxone ameliorates tau pathology and cognitive decline via restoration of glial glutamate transporter in a mouse model of Alzheimer’s disease. Neurobiol. Aging 36, 2260–2271 (2015).
Lin, C. L., Kong, Q., Cuny, G. D. & Glicksman, M. A. Glutamate transporter EAAT2: a new target for the treatment of neurodegenerative diseases. Future Med. Chem. 4, 1689–1700 (2012).
Takahashi, K. et al. Restored glial glutamate transporter EAAT2 function as a potential therapeutic approach for Alzheimer’s disease. J. Exp. Med. 212, 319–332 (2015).
Scimemi, A., Meabon, J. S., Woltjer, R. L., Sullivan, J. M., Diamond, J. S. & Cook, D. G. Amyloid-β1-42 slows clearance of synaptically released glutamate by mislocalizing astrocytic GLT-1. J. Neurosci. 33, 5312–5318 (2013).
Fontana, A. C. Current approaches to enhance glutamate transporter function and expression. J. Neurochem. 134, 982–1007 (2015).
Miller, R. G., Mitchell, J. D., Lyon, M. & Moore, D. H. Riluzole for amyotrophic lateral sclerosis (ALS)/motor neuron disease (MND). Cochrane Database Syst. Rev. CD001447 (2002).
Carbone, M., Duty, S. & Rattray, M. Riluzole elevates GLT-1 activity and levels in striatal astrocytes. Neurochem. Int. 60, 31–38 (2012).
Pekna, M. & Pekny, M. The complement system: a powerful modulator and effector of astrocyte function in the healthy and diseased central nervous system. Cells 10, 1812 (2021).
Davoust, N., Jones, J., Stahel, P. F., Ames, R. S. & Barnum, S. R. Receptor for the C3a anaphylatoxin is expressed by neurons and glial cells. Glia 26, 201–211 (1999).
van Beek, J. et al. Expression of receptors for complement anaphylatoxins C3a and C5a following permanent focal cerebral ischemia in the mouse. Exp. Neurol. 161, 373–382 (2000).
Ember, J. A., Jagels, M. A. & Hugli, T. in The Human Complement System in Health and Disease (eds Volanakis J. E. & Frank, M. M.) (Marcel Dekker, 1998).
Hannedouche, S. et al. Identification of the C3a receptor (C3AR1) as the target of the VGF-derived peptide TLQP-21 in rodent cells. J. Biol. Chem. 288, 27434–27443 (2013).
Trani, E. et al. Isolation and characterization of VGF peptides in rat brain. Role of PC1/3 and PC2 in the maturation of VGF precursor. J. Neurochem. 81, 565–574 (2002).
Sayah, S., Jauneau, A. C., Patte, C., Tonon, M. C., Vaudry, H. & Fontaine, M. Two different transduction pathways are activated by C3a and C5a anaphylatoxins on astrocytes. Brain Res. Mol. Brain Res. 112, 53–60 (2003).
Shinjyo, N., Ståhlberg, A., Dragunow, M., Pekny, M. & Pekna, M. Complement-derived anaphylatoxin C3a regulates in vitro differentiation and migration of neural progenitor cells in vitro. Stem Cells 27, 2824–2832 (2009).
McCarthy, J. D., Cao, Q., Winsor, N., Van Limbergen, J. & Stadnyk, A. W. The anaphylatoxin C3a primes model colonic epithelial cells for expression of inflammatory mediators through Gαi. Mol. Immunol. 103, 125–132 (2018).
Shinjyo, N., de Pablo, Y., Pekny, M. & Pekna, M. Complement peptide C3a promotes astrocyte survival in response to ischemic stress. Mol. Neurobiol. 53, 3076–3087 (2016).
Sayah, S., Ischenko, A., Zhakhov, A., Bonnard, A. S. & Fontaine, M. Expression of cytokines by human astrocytomas following stimulation by C3a and C5a anaphylatoxins: specific increase in interleukin-6 mRNA expression. J. Neurochem. 72, 2426–2436 (1999).
Jauneau, A. C., Ischenko, A., Chan, P. & Fontaine, M. Complement component anaphylatoxins upregulate chemokine expression by human astrocytes. FEBS Lett. 537, 17–22 (2003).
Jauneau, A.-C. et al. Interleukin-1β and anaphylatoxins exert a synergistic effect on NGF expression by astrocytes. J. Neuroinflammation 3, 8 (2006).
van Beek, J. et al. Complement anaphylatoxin C3a is selectively protective against NMDA-induced neuronal cell death. NeuroReport 12, 289–293 (2001).
Pekna, M., Siqin, S., de Pablo, Y., Stokowska, A., Torinsson Naluai Å & Pekny, M. Astrocyte responses to complement peptide C3a are highly context-dependent. Neurochem. Res. 48, 1233–1241 (2022).
Habib, N. et al. Disease-associated astrocytes in Alzheimer’s disease and aging. Nat. Neurosci. 23, 701–706 (2020).
Ozdinler, P. H. & Macklis, J. D. IGF-I specifically enhances axon outgrowth of corticospinal motor neurons. Nat. Neurosci. 9, 1371–1381 (2006).
Crosby, N. D., Zaucke, F., Kras, J. V., Dong, L., Luo, Z. D. & Winkelstein, B. A. Thrombospondin-4 and excitatory synaptogenesis promote spinal sensitization after painful mechanical joint injury. Exp. Neurol. 264, 111–120 (2015).
Stokowska, A. et al. Complement peptide C3a stimulates neural plasticity after experimental brain ischemia. Brain 140, 353–369 (2017).
Benowitz, L. I. & Routtenberg, A. GAP-43: an intrinsic determinant of neuronal development and plasticity. Trends Neurosci. 20, 84–91 (1997).
Lin, L. H., Bock, S., Carpenter, K., Rose, M. & Norden, J. J. Synthesis and transport of GAP-43 in entorhinal cortex neurons and perforant pathway during lesion-induced sprouting and reactive synaptogenesis. Brain Res. Mol. Brain Res. 14, 147–153 (1992).
Giaume, C., Naus, C. C., Saez, J. C. & Leybaert, L. Glial connexins and pannexins in the healthy and diseased brain. Physiol. Rev. 101, 93–145 (2021).
Rouach, N., Koulakoff, A., Abudara, V., Willecke, K. & Giaume, C. Astroglial metabolic networks sustain hippocampal synaptic transmission. Science 322, 1551–1555 (2008).
Giaume, C. & Liu, X. From a glial syncytium to a more restricted and specific glial networking. J. Physiol.106, 34–39 (2012).
Kiyoshi, C. M. et al. Syncytial isopotentiality: a system-wide electrical feature of astrocytic networks in the brain. Glia 66, 2756–2769 (2018).
Sáez, J. C. & Leybaert, L. Hunting for connexin hemichannels. FEBS Lett. 588, 1205–1211 (2014).
Martins-Marques, T., Ribeiro-Rodrigues, T., Batista-Almeida, D., Aasen, T., Kwak, B. R. & Girao, H. Biological functions of Connexin43 beyond intercellular communication. Trends Cell Biol. 29, 835–847 (2019).
Pannasch, U. et al. Connexin 30 sets synaptic strength by controlling astroglial synapse invasion. Nat. Neurosci. 17, 549–558 (2014).
Boulay, A. C. et al. Connexin 43 controls the astrocyte immunoregulatory phenotype. Brain Sci. 8, 50 (2018).
Tichauer, J. E. & Rovegno, M. Role of astrocytes connexins - pannexins in acute brain injury. Neurotherapeutics 22, e00523 (2025).
Prieto-Villalobos, J. et al. Astroglial hemichannels and pannexons: the hidden link between maternal inflammation and neurological disorders. Int. J.Mol. Sci. 22, 9503 (2021).
Sun, L. et al. A novel cognitive impairment mechanism that astrocytic p-connexin 43 promotes neuronic autophagy via activation of P2X7R and down-regulation of GLT-1 expression in the hippocampus following traumatic brain injury in rats. Behav. Brain Res. 291, 315–324 (2015).
Orellana, J. A. et al. Hypoxia in high glucose followed by reoxygenation in normal glucose reduces the viability of cortical astrocytes through increased permeability of connexin 43 hemichannels. Glia 58, 329–343 (2010).
Ahmadian, E., Eftekhari, A., Samiei, M., Maleki Dizaj, S. & Vinken, M. The role and therapeutic potential of connexins, pannexins and their channels in Parkinson’s disease. Cell. Signal. 58, 111–118 (2019).
Huang, X. et al. Astroglial Connexins In Neurodegenerative Diseases. Front. Mol. Neurosci. 14, 657514 (2021).
Vis, J. C., Nicholson, L. F., Faull, R. L., Evans, W. H., Severs, N. J. & Green, C. R. Connexin expression in Huntington’s diseased human brain. Cell Biol. Int. 22, 837–847 (1998).
Almad, A. A. et al. Connexin 43 in astrocytes contributes to motor neuron toxicity in amyotrophic lateral sclerosis. Glia 64, 1154–1169 (2016).
Denaro, S., D’Aprile, S., Vicario, N. & Parenti, R. Mechanistic insights into connexin-mediated neuroglia crosstalk in neurodegenerative diseases. Front. Cell. Neurosci. 19, 1532960 (2025).
Kawasaki, A. et al. Modulation of connexin 43 in rotenone-induced model of Parkinson’s disease. Neuroscience 160, 61–68 (2009).
Maulik, M., Vasan, L., Bose, A., Dutta Chowdhury, S., Sengupta, N. & Das Sarma, J. Amyloid-β regulates gap junction protein connexin 43 trafficking in cultured primary astrocytes. J. Biol. Chem. 295, 15097–15111 (2020).
Yi, C. et al. Astroglial connexin43 contributes to neuronal suffering in a mouse model of Alzheimer’s disease. Cell Death Differ. 23, 1691–1701 (2016).
Orellana, J. A. et al. ATP and glutamate released via astroglial connexin 43 hemichannels mediate neuronal death through activation of pannexin 1 hemichannels. J. Neurochem. 118, 826–840 (2011).
Ye, B. et al. Dual pathways mediate β-amyloid stimulated glutathione release from astrocytes. Glia 63, 2208–2219 (2015).
Angeli, S. et al. Altered expression of glial gap junction proteins Cx43, Cx30, and Cx47 in the 5XFAD model of Alzheimer’s disease. Front. Neurosci. 14, 582934 (2020).
Mei, X., Ezan, P., Giaume, C. & Koulakoff, A. Astroglial connexin immunoreactivity is specifically altered at β-amyloid plaques in β-amyloid precursor protein/presenilin1 mice. Neuroscience 171, 92–105 (2010).
Nagy, J. I., Li, W., Hertzberg, E. L. & Marotta, C. A. Elevated connexin43 immunoreactivity at sites of amyloid plaques in Alzheimer’s disease. Brain Res. 717, 173–178 (1995).
Asghari, K., Niknam, Z., Mohammadpour-Asl, S. & Chodari, L. Cellular junction dynamics and Alzheimer’s disease: a comprehensive review. Mol. Biol. Rep. 51, 273 (2024).
Tonkin, R. S. et al. Attenuation of mechanical pain hypersensitivity by treatment with Peptide5, a connexin-43 mimetic peptide, involves inhibition of NLRP3 inflammasome in nerve-injured mice. Exp. Neurol. 300, 1–12 (2018).
Vicario, N. et al. Simultaneous activation of mu and delta opioid receptors reduces allodynia and astrocytic Connexin 43 in an animal model of neuropathic pain. Mol. Neurobiol. 56, 7338–7354 (2019).
Chen, G., Park, C. K., Xie, R. G., Berta, T., Nedergaard, M. & Ji, R. R. Connexin-43 induces chemokine release from spinal cord astrocytes to maintain late-phase neuropathic pain in mice. Brain 137, 2193–2209 (2014).
Wu, X. F., Liu, W. T., Liu, Y. P., Huang, Z. J., Zhang, Y. K. & Song, X. J. Reopening of ATP-sensitive potassium channels reduces neuropathic pain and regulates astroglial gap junctions in the rat spinal cord. Pain 152, 2605–2615 (2011).
Chen, M. J. et al. Astrocytic CX43 hemichannels and gap junctions play a crucial role in development of chronic neuropathic pain following spinal cord injury. Glia 60, 1660–1670 (2012).
Xu, Q. et al. Suppression of spinal connexin 43 expression attenuates mechanical hypersensitivity in rats after an L5 spinal nerve injury. Neurosci. Lett. 566, 194–199 (2014).
Huang, C. et al. Critical role of connexin 43 in secondary expansion of traumatic spinal cord injury. J. Neurosci. 32, 3333–3338 (2012).
Luo, L. L., Wang, J. W., Yin, X. L., Chen, X. Y., Zhang, X. F. & Ye, Z. C. Astrocytic connexin 43 deletion ameliorates SNI-induced neuropathic pain by reducing microglia activation. Biochem. Biophys. Res. Commun. 638, 192–199 (2023).
Li, G. Z. et al. CaMKII and Ca(V)3.2 T-type calcium channel mediate Connexin-43-dependent inflammation by activating astrocytes in vincristine-induced neuropathic pain. Cell Biol. Toxicol. 39, 679–702 (2023).
Hang, L. H. et al. Connexin 43 mediates CXCL12 production from spinal dorsal horn to maintain bone cancer pain in rats. Neurochem. Res. 41, 1200–1208 (2016).
Su, Y., Verkhratsky, A. & Yi, C. Targeting connexins: possible game changer in managing neuropathic pain? Trends Mol. Med. 30, 642–659 (2024).
Chin, J. S., Milbreta, U., Becker, D. L. & Chew, S. Y. Targeting connexin 43 expression via scaffold mediated delivery of antisense oligodeoxynucleotide preserves neurons, enhances axonal extension, reduces astrocyte and microglial activation after spinal cord injury. J. Tissue Eng. 14, 20417314221145789 (2023).
Rhett, J. M., Jourdan, J. & Gourdie, R. G. Connexin 43 connexon to gap junction transition is regulated by zonula occludens-1. Mol. Biol. Cell 22, 1516–1528 (2011).
Kaji, K., Shinoda, M., Honda, K., Unno, S., Shimizu, N. & Iwata, K. Connexin 43 contributes to ectopic orofacial pain following inferior alveolar nerve injury. Mol. Pain 12, 1744806916633704 (2016).
Davidson, J. O., Green, C. R., Nicholson, L. F., Bennet, L. & Gunn, A. J. Connexin hemichannel blockade is neuroprotective after, but not during, global cerebral ischemia in near-term fetal sheep. Exp. Neurol. 248, 301–308 (2013).
Evans, W. H. & Leybaert, L. Mimetic peptides as blockers of connexin channel-facilitated intercellular communication. Cell Commun. Adhes. 14, 265–273 (2007).
Desplantez, T., Verma, V., Leybaert, L., Evans, W. H. & Weingart, R. Gap26, a connexin mimetic peptide, inhibits currents carried by connexin43 hemichannels and gap junction channels. Pharmacol. Res. 65, 546–552 (2012).
Pollok, S. et al. Connexin 43 mimetic peptide Gap27 reveals potential differences in the role of Cx43 in wound repair between diabetic and non-diabetic cells. J. Cell. Mol. Med. 15, 861–873 (2011).
Wang, N. et al. Selective inhibition of Cx43 hemichannels by Gap19 and its impact on myocardial ischemia/reperfusion injury. Basic Res. Cardiol. 108, 309 (2013).
Abudara, V. et al. The connexin43 mimetic peptide Gap19 inhibits hemichannels without altering gap junctional communication in astrocytes. Front. Cell. Neurosci. 8, 306 (2014).
Gangoso, E. et al. A c-Src inhibitor peptide based on Connexin43 exerts neuroprotective effects through the inhibition of glial hemichannel activity. Front. Mol. Neurosci. 10, 418 (2017).
Su, Y. et al. Connexin43 hemichannel blockade turns microglia neuroprotective and mitigates cognitive deficits in a mouse model of amyloidosis. Nat. Commun. 16, 5621 (2025).
Walrave, L. et al. Inhibition of astroglial connexin43 hemichannels with TAT-Gap19 exerts anticonvulsant effects in rodents. Glia 66, 1788–1804 (2018).
Mathur, D., Singh, S., Mehta, A., Agrawal, P. & Raghava, G. P. S. In silico approaches for predicting the half-life of natural and modified peptides in blood. PLoS ONE 13, e0196829 (2018).
Yi, C. et al. Inhibition of glial hemichannels by boldine treatment reduces neuronal suffering in a murine model of Alzheimer’s disease. Glia 65, 1607–1625 (2017).
Takeuchi, H. et al. Blockade of gap junction hemichannel suppresses disease progression in mouse models of amyotrophic lateral sclerosis and Alzheimer’s disease. PLoS ONE 6, e21108 (2011).
Cisterna, B. A. et al. Active acetylcholine receptors prevent the atrophy of skeletal muscles and favor reinnervation. Nat. Commun. 11, 1073 (2020).
Guo, A. et al. Inhibition of connexin hemichannels alleviates neuroinflammation and hyperexcitability in temporal lobe epilepsy. Proc. Natl Acad. Sci. USA 119, e2213162119 (2022).
Jo, S. et al. GABA from reactive astrocytes impairs memory in mouse models of Alzheimer’s disease. Nat. Med. 20, 886–896 (2014).
Targa Dias Anastacio, H., Matosin, N. & Ooi, L. Neuronal hyperexcitability in Alzheimer’s disease: what are the drivers behind this aberrant phenotype? Transl. Psychiatry 12, 257 (2022).
Mitew, S., Kirkcaldie, M. T., Dickson, T. C. & Vickers, J. C. Altered synapses and gliotransmission in Alzheimer’s disease and AD model mice. Neurobiol. Aging 34, 2341–2351 (2013).
Yarishkin, O., Lee, J., Jo, S., Hwang, E. M. & Lee, C. J. Disinhibitory action of astrocytic GABA at the perforant path to dentate gyrus granule neuron synapse reverses to inhibitory in Alzheimer’s disease model. Exp. Neurobiol. 24, 211–218 (2015).
Chun, H. et al. Astrocytic proBDNF and tonic GABA distinguish active versus reactive astrocytes in hippocampus. Exp. Neurobiol. 27, 155–170 (2018).
Oh, S. J. et al. Direct interaction with 14-3-3γ promotes surface expression of Best1 channel in astrocyte. Mol. Brain 10, 51 (2017).
Villemagne, V. L. et al. First-in-humans evaluation of 18F-SMBT-1, a novel 18F-labeled monoamine oxidase-B PET tracer for imaging reactive astrogliosis. J. Nucl. Med. 63, 1551–1559 (2022).
Nam, M. H., Na, H., Lee, J. C. & Yun, M. A key mediator and imaging target in Alzheimer’s disease: unlocking the role of reactive astrogliosis through MAOB. Nucl. Med. Mol. Imaging. 58, 177–184 (2024).
Heo, J. J. et al. Suffruticosol B from Paeonia lactiflora ameliorates Alzheimer’s disease pathology by inhibiting MAO-B activity. Phytother. Res. 39, 593–603 (2024).
Kong, Y. et al. Relationship between reactive astrocytes, by [18F]SMBT-1 imaging, with amyloid-β, tau, glucose metabolism, and TSPO in mouse models of Alzheimer’s disease. Mol. Neurobiol. 61, 8387–8401 (2024).
Wu, Z., Guo, Z., Gearing, M. & Chen, G. Tonic inhibition in dentate gyrus impairs long-term potentiation and memory in an Alzheimer’s [corrected] disease model. Nat. Commun. 5, 4159 (2014).
Chamberlain, S. E., Sadowski, J. H., Teles-Grilo Ruivo, L. M., Atherton, L. A. & Mellor, J. R. Long-term depression of synaptic kainate receptors reduces excitability by relieving inhibition of the slow afterhyperpolarization. J. Neurosci. 33, 9536–9545 (2013).
Heo, J. Y. et al. Aberrant tonic inhibition of dopaminergic neuronal activity causes motor symptoms in animal models of Parkinson’s disease. Curr. Biol. 30, 276–291(2020).
Guzman-Sastoque, P. et al. Assessment of CRISPRa-mediated gdnf overexpression in an In vitro Parkinson’s disease model. Front. Bioeng. Biotechnol. 12, 1420183 (2024).
Liu, L. L. et al. Loss of DJ-1 function contributes to Parkinson’s disease pathogenesis in mice via RACK1-mediated PKC activation and MAO-B upregulation. Acta Pharmacol. Sin. 44, 1948–1961 (2023).
Bao, L. et al. Mitochondria are the source of hydrogen peroxide for dynamic brain-cell signaling. J. Neurosci. 29, 9002–9010 (2009).
Pham, T. L. et al. MAO-B inhibitor, KDS2010, alleviates spinal nerve ligation-induced neuropathic pain in rats through competitively blocking the BDNF/TrkB/NR2B signaling. J. Pain 23, 2092–2109 (2022).
Mapplebeck, J. C. S. et al. Chloride dysregulation through downregulation of KCC2 mediates neuropathic pain in both sexes. Cell Rep. 28, 590–596 (2019).
Nam, M. H. et al. KDS2010, a newly developed reversible MAO-B Inhibitor, as an effective therapeutic candidate for Parkinson’s disease. Neurotherapeutics 18, 1729–1747 (2021).
Park, J. H. et al. Newly developed reversible MAO-B inhibitor circumvents the shortcomings of irreversible inhibitors in Alzheimer’s disease. Sci. Adv. 5, eaav0316 (2019).
Alborghetti, M., Bianchini, E., De Carolis, L., Galli, S., Pontieri, F. E. & Rinaldi, D. Type-B monoamine oxidase inhibitors in neurological diseases: clinical applications based on preclinical findings. Neural Regen. Res. 19, 16–21 (2024).
Ju, Y. H. et al. Astrocytic urea cycle detoxifies Aβ-derived ammonia while impairing memory in Alzheimer’s disease. Cell Metab. 34, 1104–1120 (2022).
Krebs, H. A. & Henseleit, K. Untersuchungen über die Harnstoffbildung im Tierkörper. Hoppe Seylers Z. Physiol. Chem. 210, 33–66 (1932).
Bhalla, M. & Lee, C. J. Long-term inhibition of ODC1 in APP/PS1 mice rescues amyloid pathology and switches astrocytes from a reactive to active state. Mol. Brain 17, 3 (2024).
Dalleau, S., Baradat, M., Gueraud, F. & Huc, L. Cell death and diseases related to oxidative stress: 4-hydroxynonenal (HNE) in the balance. Cell Death Differ. 20, 1615–1630 (2013).
Kim, S. et al. Astrocytic autophagy plasticity modulates Aβ clearance and cognitive function in Alzheimer’s disease. Mol. Neurodegener. 19, 55 (2024).
Alber, J., McGarry, K., Noto, R. B. & Snyder, P. J. Use of eflornithine (DFMO) in the treatment of early Alzheimer’s disease: a compassionate use, single-case study. Front. Aging Neurosci. 10, 60 (2018).
Paterson, D. & Nordberg, A. Neuronal nicotinic receptors in the human brain. Prog. Neurobiol. 61, 75–111 (2000).
Pankratov, Y., Lalo, U., Krishtal, O. A. & Verkhratsky, A. P2X receptors and synaptic plasticity. Neuroscience 158, 137–148 (2009).
Shen, J. X. & Yakel, J. L. Functional α7 nicotinic ACh receptors on astrocytes in rat hippocampal CA1 slices. J. Mol. Neurosci. 48, 14–21 (2012).
Wang, H. Y., Lee, D. H., Davis, C. B. & Shank, R. P. Amyloid peptide Aβ1–42 binds selectively and with picomolar affinity to α7 nicotinic acetylcholine receptors. J. Neurochem. 75, 1155–1161 (2000).
Wang, H. Y., Lee, D. H., D’Andrea, M. R., Peterson, P. A., Shank, R. P. & Reitz, A. B. β-Amyloid1–42 binds to α7 nicotinic acetylcholine receptor with high affinity. Implications for Alzheimer’s disease pathology. J. Biol. Chem. 275, 5626–5632 (2000).
Teaktong, T. et al. Alzheimer’s disease is associated with a selective increase in α7 nicotinic acetylcholine receptor immunoreactivity in astrocytes. Glia 41, 207–211 (2003).
Xiu, J. et al. Lovastatin stimulates up-regulation of α7 nicotinic receptors in cultured neurons without cholesterol dependency, a mechanism involving production of the alpha-form of secreted amyloid precursor protein. J. Neurosci. Res. 82, 531–541 (2005).
Yu, W. F., Guan, Z. Z., Bogdanovic, N. & Nordberg, A. High selective expression of α7 nicotinic receptors on astrocytes in the brains of patients with sporadic Alzheimer’s disease and patients carrying Swedish APP 670/671 mutation: a possible association with neuritic plaques. Exp. Neurol. 192, 215–225 (2005).
Fontana, I. C., Kumar, A. & Nordberg, A. The role of astrocytic α7 nicotinic acetylcholine receptors in Alzheimer disease. Nat. Rev. Neurol. 19, 278–288 (2023).
Horti, A. G. et al. 18F-ASEM, a radiolabeled antagonist for imaging the alpha7-nicotinic acetylcholine receptor with PET. J. Nucl. Med. 55, 672–677 (2014).
Coughlin, J. M. et al. High availability of the α7-nicotinic acetylcholine receptor in brains of individuals with mild cognitive impairment: a pilot study using (18)F-ASEM PET. J. Nucl. Med. 61, 423–426 (2020).
Jia, Z. et al. Good manufacturing practice validation and radiation dosimetry for the clinical application of a novel α7-nACh R radioligand: [11C]KIn83. Molecules 30, 1356 (2025).
Nag, S. et al. Development of 11C-labeled ASEM analogues for the detection of neuronal nicotinic acetylcholine receptors (α7-nAChR). ACS Chem. Neurosci. 13, 352–362 (2022).
Nielsen, S., Nagelhus, E. A., Amiry-Moghaddam, M., Bourque, C., Agre, P. & Ottersen, O. P. Specialized membrane domains for water transport in glial cells: high-resolution immunogold cytochemistry of aquaporin-4 in rat brain. J. Neurosci. 17, 171–180 (1997).
Amiry-Moghaddam, M. et al. An α-syntrophin-dependent pool of AQP4 in astroglial end-feet confers bidirectional water flow between blood and brain. Proc. Natl Acad. Sci. USA 100, 2106–2111 (2003).
Manley, G. T. et al. Aquaporin-4 deletion in mice reduces brain edema after acute water intoxication and ischemic stroke. Nat. Med. 6, 159–163 (2000).
Ohene, Y. et al. Non-invasive MRI of brain clearance pathways using multiple echo time arterial spin labelling: an aquaporin-4 study. Neuroimage 188, 515–523 (2019).
Mathiisen, T. M., Lehre, K. P., Danbolt, N. C. & Ottersen, O. P. The perivascular astroglial sheath provides a complete covering of the brain microvessels: an electron microscopic 3D reconstruction. Glia 58, 1094–1103 (2010).
Skauli, N. et al. Aquaporin-4 deletion leads to reduced infarct volume and increased peri-infarct astrocyte reactivity in a mouse model of cortical stroke. J. Physiol. 602, 3151–3168 (2024).
Tradtrantip, L., Jin, B. J., Yao, X., Anderson, M. O. & Verkman, A. S. Aquaporin-targeted therapeutics: state-of-the-field. Adv. Exp. Med. Biol. 969, 239–250 (2017).
Nagelhus, E. A. & Ottersen, O. P. Physiological roles of aquaporin-4 in brain. Physiol. Rev. 93, 1543–1562 (2013).
Zeynalov, E. et al. The perivascular pool of aquaporin-4 mediates the effect of osmotherapy in postischemic cerebral edema. Crit. Care Med. 36, 2634–2640 (2008).
Skauli, N., Savchenko, E., Ottersen, O. P., Roybon, L. & Amiry-Moghaddam, M. Canonical bone morphogenetic protein signaling regulates expression of aquaporin-4 and its anchoring complex in mouse astrocytes. Front. Cell. Neurosci. 16, 878154 (2022).
Kitchen, P. et al. Targeting aquaporin-4 subcellular localization to treat central nervous system edema. Cell 181, 784–799 (2020).
Benfenati, V. et al. An aquaporin-4/transient receptor potential vanilloid 4 (AQP4/TRPV4) complex is essential for cell-volume control in astrocytes. Proc. Natl Acad. Sci. USA 108, 2563–2568 (2011).
Amiry-Moghaddam, M. et al. Delayed K+ clearance associated with aquaporin-4 mislocalization: phenotypic defects in brains of α-syntrophin-null mice. Proc. Natl Acad. Sci. USA 100, 13615–13620 (2003).
Li, L., Zhang, H., Varrin-Doyer, M., Zamvil, S. S. & Verkman, A. S. Proinflammatory role of aquaporin-4 in autoimmune neuroinflammation. FASEB J. 25, 1556–1566 (2011).
Prydz, A. et al. Pro-inflammatory role of AQP4 in mice subjected to intrastriatal injections of the parkinsonogenic toxin MPP. Cells 9, 2418 (2020).
Afzali, A. M. et al. B cells orchestrate tolerance to the neuromyelitis optica autoantigen AQP4. Nature 627, 407–415 (2024).
Hablitz, L. M. & Nedergaard, M. The glymphatic system: a novel component of fundamental neurobiology. J. Neurosci. 41, 7698–7711 (2021).
Iliff, J. J. et al. A paravascular pathway facilitates CSF flow through the brain parenchyma and the clearance of interstitial solutes, including amyloid b. Sci. Transl. Med. 4, 147ra111 (2012).
Bojarskaite, L. et al. Role of aquaporin-4 polarization in extracellular solute clearance. Fluids Barriers CNS 21, 28 (2024).
Giannetto, M. J. et al. Glymphatic fluid transport is suppressed by the aquaporin-4 inhibitor AER-271. Glia 72, 982–998 (2024).
Yang, J. et al. Loss of astrocyte polarization in the tg-ArcSwe mouse model of Alzheimer’s disease. J. Alzheimers Dis. 27, 711–722 (2011).
Kim, S. et al. Astrocytic autophagy plasticity modulates Abeta clearance and cognitive function in Alzheimer’s disease. Mol. Neurodegener. 19, 55 (2024).
Zorec, R. & Vardjan, N. Adrenergic regulation of astroglial aerobic glycolysis and lipid metabolism: Towards a noradrenergic hypothesis of neurodegeneration. Neurobiol. Dis. 182, 106132 (2023).
Fünfschilling, U. et al. Glycolytic oligodendrocytes maintain myelin and long-term axonal integrity. Nature 485, 517–521 (2012).
Saab, A. S., Tzvetanova, I. D. & Nave, K. A. The role of myelin and oligodendrocytes in axonal energy metabolism. Curr. Opin. Neurobiol. 23, 1065–1072 (2013).
Rinholm, J. E. & Bergersen, L. H. White matter lactate – does it matter? Neuroscience 276, 109–116 (2014).
Ioannou, M. S. et al. Neuron–astrocyte metabolic coupling protects against activity-induced fatty acid toxicity. Cell 177, 1522–1535 (2019).
Luthi, A. & Nedergaard, M. Anything but small: microarousals stand at the crossroad between noradrenaline signaling and key sleep functions. Neuron 113, 509–523 (2025).
Slater, C. & Wang, Q. Alzheimer’s disease: an evolving understanding of noradrenergic involvement and the promising future of electroceutical therapies. Clin. Transl. Med. 11, e397 (2021).
Robertson, I. H. A noradrenergic theory of cognitive reserve: implications for Alzheimer’s disease. Neurobiol. Aging 34, 298–308 (2013).
Bueicheku, E. et al. Spatiotemporal patterns of locus coeruleus integrity predict cortical tau and cognition. Nat. Aging 4, 625–637 (2024).
Abjorsbraten, K. S. et al. Impaired astrocytic Ca2+ signaling in awake-behaving Alzheimer’s disease transgenic mice. eLife 11, e75055 (2022).
Dienel, G. A. Does shuttling of glycogen-derived lactate from astrocytes to neurons take place during neurotransmission and memory consolidation? J. Neurosci. Res. 97, 863–882 (2019).
Verkhratsky, A. & Pivoriunas, A. Astroglia support, regulate and reinforce brain barriers. Neurobiol. Dis. 179, 106054 (2023).
Smolič, T. et al. Astrocytes in stress accumulate lipid droplets. Glia 69, 1540–1562 (2021).
Barber, C. N. & Raben, D. M. Lipid metabolism crosstalk in the brain: glia and neurons. Front. Cell. Neurosci. 13, 212 (2019).
Bak, L. K., Walls, A. B., Schousboe, A. & Waagepetersen, H. S. Astrocytic glycogen metabolism in the healthy and diseased brain. J. Biol. Chem. 293, 7108–7116 (2018).
Rothman, D. L. et al. Glucose sparing by glycogenolysis (GSG) determines the relationship between brain metabolism and neurotransmission. J. Cereb. Blood Flow Metab. 42, 844–860 (2022).
Fink, K., Velebit, J., Vardjan, N., Zorec, R. & Kreft, M. Noradrenaline-induced l-lactate production requires d-glucose entry and transit through the glycogen shunt in single-cultured rat astrocytes. J. Neurosci. Res. 99, 1084–1098 (2021).
Warburg, O. On the origin of cancer cells. Science 123, 309–314 (1956).
Vander Heiden, M. G., Cantley, L. C. & Thompson, C. B. Understanding the Warburg effect: the metabolic requirements of cell proliferation. Science 324, 1029–1033 (2009).
Goyal, M. S., Hawrylycz, M., Miller, J. A., Snyder, A. Z. & Raichle, M. E. Aerobic glycolysis in the human brain is associated with development and neotenous gene expression. Cell Metab. 19, 49–57 (2014).
Tech, K. & Gershon, T. R. Energy metabolism in neurodevelopment and medulloblastoma. Transl. Pediatr. 4, 12–19 (2015).
Ostroff, L. E., Manzur, M. K., Cain, C. K. & Ledoux, J. E. Synapses lacking astrocyte appear in the amygdala during consolidation of Pavlovian threat conditioning. J. Comp. Neurol. 522, 2152–2163 (2014).
Zorec, R., Parpura, V. & Verkhratsky, A. Astroglial vesicular network: evolutionary trends, physiology and pathophysiology. Acta Physiol. 222, 12915 (2018).
Magistretti, P. J. & Allaman, I. Lactate in the brain: from metabolic end-product to signalling molecule. Nat. Rev. Neurosci. 19, 235–249 (2018).
Sonnewald, U. Glutamate synthesis has to be matched by its degradation—where do all the carbons go? J. Neurochem. 131, 399–406 (2014).
Dienel, G. A. & McKenna, M. C. A dogma-breaking concept: glutamate oxidation in astrocytes is the source of lactate during aerobic glycolysis in resting subjects. J. Neurochem. 131, 395–398 (2014).
Hertz, L. Astrocytic energy metabolism and glutamate formation-relevance for 13C-NMR spectroscopy and importance of cytosolic/mitochondrial trafficking. Magn. Reson. Imaging. 29, 1319–1329 (2011).
Sonnewald, U., Westergaard, N., Krane, J., Unsgard, G., Petersen, S. B. & Schousboe, A. First direct demonstration of preferential release of citrate from astrocytes using [13C]NMR spectroscopy of cultured neurons and astrocytes. Neurosci. Lett. 128, 235–239 (1991).
Srere, P. A. The molecular physiology of citrate. Curr. Top. Cell. Regul. 33, 261–275 (1992).
Westergaard, N., Waagepetersen, H. S., Belhage, B. & Schousboe, A. Citrate, a ubiquitous key metabolite with regulatory function in the CNS. Neurochem. Res. 42, 1583–1588 (2017).
Mauch, D. H. et al. CNS synaptogenesis promoted by glia-derived cholesterol. Science 294, 1354–1357 (2001).
Velebit, J. et al. Astrocytes with TDP-43 inclusions exhibit reduced noradrenergic cAMP and Ca. Sci. Rep. 10, 6003 (2020).
Tiaglik, A. et al. Cerebral perfusion and metabolic response of astrocytes and neurons during locomotion. Preprint at bioRxiv https://doi.org/10.1101/2024.12.23.630097 (2024).
Gao, V. et al. Astrocytic beta2-adrenergic receptors mediate hippocampal long-term memory consolidation. Proc. Natl Acad. Sci. USA 113, 8526–8531 (2016).
Theparambil, S. M. et al. Adenosine signalling to astrocytes coordinates brain metabolism and function. Nature 632, 139–146 (2024).
Dienel, G. A. & Cruz, N. F. Aerobic glycolysis during brain activation: adrenergic regulation and influence of norepinephrine on astrocytic metabolism. J. Neurochem. 138, 14–52 (2016).
Briquet, M. et al. Activation of lactate receptor HCAR1 down-modulates neuronal activity in rodent and human brain tissue. J. Cereb. Blood Flow Metab. 42, 1650–1665 (2022).
de Castro Abrantes, H. et al. The lactate receptor HCAR1 modulates neuronal network activity through the activation of G. J. Neurosci. 39, 4422–4433 (2019).
Vardjan, N. et al. Enhancement of astroglial aerobic glycolysis by extracellular lactate-mediated increase in cAMP. Front. Mol. Neurosci. 11, 148 (2018).
Bergersen, L. H. & Gjedde, A. Is lactate a volume transmitter of metabolic states of the brain? Front. Neuroenerg. 4, 5 (2012).
Dvorak, C. A. et al. Identification of hydroxybenzoic acids as selective lactate receptor (GPR81) agonists with antilipolytic effects. ACS Med. Chem. Lett. 3, 637–639 (2012).
Sakurai, T. et al. Identification of a novel GPR81-selective agonist that suppresses lipolysis in mice without cutaneous flushing. Eur. J. Pharmacol. 727, 1–7 (2014).
Vardjan, N., Kreft, M. & Zorec, R. Dynamics of β-adrenergic/cAMP signaling and morphological changes in cultured astrocytes. Glia 62, 566–579 (2014).
Horvat, A., Zorec, R. & Vardjan, N. Adrenergic stimulation of single rat astrocytes results in distinct temporal changes in intracellular Ca(2+) and cAMP-dependent PKA responses. Cell Calcium 59, 156–163 (2016).
Horvat, A. & Vardjan, N. Astroglial cAMP signalling in space and time. Neurosci. Lett. 689, 5–10 (2019).
D’Adamo, P. et al. Inhibiting glycolysis rescues memory impairment in an intellectual disability Gdi1-null mouse. Metabolism 116, 154463 (2021).
Verkhratsky, A., Semyanov, A. & Zorec, R. Physiology of astroglial excitability. Function 1, zqaa016 (2020).
Segal, S. K., Stark, S. M., Kattan, D., Stark, C. E. & Yassa, M. A. Norepinephrine-mediated emotional arousal facilitates subsequent pattern separation. Neurobiol. Learn. Mem. 97, 465–469 (2012).
Ebrahimi, S., Rashidy-Pour, A., Vafaei, A. A. & Akhavan, M. M. Central beta-adrenergic receptors play an important role in the enhancing effect of voluntary exercise on learning and memory in rat. Behav. Brain Res. 208, 189–193 (2010).
Van Hoomissen, J. D., Holmes, P. V., Zellner, A. S., Poudevigne, A. & Dishman, R. K. Effects of beta-adrenoreceptor blockade during chronic exercise on contextual fear conditioning and mRNA for galanin and brain-derived neurotrophic factor. Behav. Neurosci. 118, 1378–1390 (2004).
Feinstein, D. L., Kalinin, S. & Braun, D. Causes, consequences, and cures for neuroinflammation mediated via the locus coeruleus: noradrenergic signaling system. J. Neurochem. 139, 154–178 (2016).
Coradazzi, M., Gulino, R., Garozzo, S. & Leanza, G. Selective lesion of the developing central noradrenergic system: short- and long-term effects and reinnervation by noradrenergic-rich tissue grafts. J. Neurochem. 114, 761–771 (2010).
Zorec, R., Parpura, V. & Verkhratsky, A. Preventing neurodegeneration by adrenergic astroglial excitation. FEBS J. 285, 3645–3656 (2018).
Dolanc, D. et al. The activation of GPR27 increases cytosolic L-lactate in 3T3 embryonic cells and astrocytes. Cells 11, 1009 (2022).
Labarta-Bajo, L. & Allen, N. J. Astrocytes in aging. Neuron. 113, 109–126 (2025).
O’Shea, T. M. et al. Derivation and transcriptional reprogramming of border-forming wound repair astrocytes after spinal cord injury or stroke in mice. Nat. Neurosci. 27, 1505–1521 (2024).
George, K. K., Heithoff, B. P., Shandra, O. & Robel, S. Mild traumatic brain injury/concussion initiates an atypical astrocyte response caused by blood–brain barrier dysfunction. J. Neurotrauma. 39, 211–226 (2022).
Shandra, O. et al. Repetitive diffuse mild traumatic brain injury causes an atypical astrocyte response and spontaneous recurrent seizures. J. Neurosci. 39, 1944–1963 (2019).
Boulenguez, P. et al. Down-regulation of the potassium-chloride cotransporter KCC2 contributes to spasticity after spinal cord injury. Nat. Med. 16, 302–307 (2010).
Tsuda, M. et al. P2X4 receptors induced in spinal microglia gate tactile allodynia after nerve injury. Nature 424, 778–783 (2003).
Coull, J. A. et al. Trans-synaptic shift in anion gradient in spinal lamina I neurons as a mechanism of neuropathic pain. Nature 424, 938–942 (2003).
Xu, J., Yan, Z., Bang, S., Velmeshev, D. & Ji, R.-R. GPR37L1 identifies spinal cord astrocytes and protects neuropathic pain after nerve injury. Neuron (2025).
Mitani, A. & Tanaka, K. Functional changes of glial glutamate transporter GLT-1 during ischemia: an in vivo study in the hippocampal CA1 of normal mice and mutant mice lacking GLT-1. J. Neurosci. 23, 7176–7182 (2003).
Harvey, B. K. et al. Targeted over-expression of glutamate transporter 1 (GLT-1) reduces ischemic brain injury in a rat model of stroke. PLoS ONE 6, e22135 (2011).
Hu, Y. Y., Xu, J., Zhang, M., Wang, D., Li, L. & Li, W. B. Ceftriaxone modulates uptake activity of glial glutamate transporter-1 against global brain ischemia in rats. J. Neurochem. 132, 194–205 (2015).
Zhang, L. Y. et al. The mechanism of GLT-1 mediating cerebral ischemic injury depends on the activation of p38 MAPK. Brain Res. Bull. 147, 1–13 (2019).
Jiang, S. et al. APOE from patient-derived astrocytic extracellular vesicles alleviates neuromyelitis optica spectrum disorder in a mouse model. Sci. Transl. Med. 16, eadg5116 (2024).
Asavapanumas, N., Tradtrantip, L. & Verkman, A. S. Targeting the complement system in neuromyelitis optica spectrum disorder. Expert Opin. Biol. Ther. 21, 1073–1086 (2021).
Ponath, G., Park, C. & Pitt, D. The role of astrocytes in multiple sclerosis. Front. Immunol. 9, 217 (2018).
Absinta, M. et al. A lymphocyte–microglia–astrocyte axis in chronic active multiple sclerosis. Nature 597, 709–714 (2021).
Olabarria, M., Noristani, H. N., Verkhratsky, A. & Rodriguez, J. J. Concomitant astroglial atrophy and astrogliosis in a triple transgenic animal model of Alzheimer’s disease. Glia 58, 831–838 (2010).
Rodriguez, J. J., Terzieva, S., Olabarria, M., Lanza, R. G. & Verkhratsky, A. Enriched environment and physical activity reverse astrogliodegeneration in the hippocampus of AD transgenic mice. Cell Death Dis. 4, e678 (2013).
Chun, H. et al. Severe reactive astrocytes precipitate pathological hallmarks of Alzheimer’s disease via H(2)O(2)(−) production. Nat. Neurosci. 23, 1555–1566 (2020).
Verkhratsky, A. et al. Neuroglial advances: new roles for established players. J. Neurochem. 169, e70080 (2025).
Almad, A. A. et al. Cx43 hemichannels contribute to astrocyte-mediated toxicity in sporadic and familial ALS. Proc. Natl Acad. Sci. USA 119, e2107391119 (2022).
Wilton, D. K. & Stevens, B. The contribution of glial cells to Huntington’s disease pathogenesis. Neurobiol. Dis. 143, 104963 (2020).
Bernard, R. et al. Altered expression of glutamate signaling, growth factor, and glia genes in the locus coeruleus of patients with major depression. Mol. Psychiatry 16, 634–646 (2011).
Li, B., Zhang, D. & Verkhratsky, A. Astrocytes in post-traumatic stress disorder. Neurosci. Bull. 38, 953–965 (2022).
Eid, T. et al. Loss of glutamine synthetase in the human epileptogenic hippocampus: possible mechanism for raised extracellular glutamate in mesial temporal lobe epilepsy. Lancet 363, 28–37 (2004).
Untiet, V., Nedergaard, M. & Verkhratsky, A. Astrocyte chloride, excitatory–inhibitory balance and epilepsy. Neural Regen. Res. 19, 1887 (2024).
Pietrobon, D. & Moskowitz, M. A. Pathophysiology of migraine. Annu. Rev. Physiol. 75, 365–391 (2013).
Messing, A., Head, M. W., Galles, K., Galbreath, E. J., Goldman, J. E. & Brenner, M. Fatal encephalopathy with astrocyte inclusions in GFAP transgenic mice. Am. J. Pathol. 152, 391 (1998).
Tian, R. et al. Alexander disease mutant glial fibrillary acidic protein compromises glutamate transport in astrocytes. J. Neuropathol. Exp. Neurol. 69, 335–345 (2010).
van der Knaap, M. S., Bugiani, M. & Abbink, T. E. M. Vanishing white matter. Handb. Clin. Neurol. 204, 77–94 (2024).
Leferink, P. S. et al. Astrocyte subtype vulnerability in stem cell models of vanishing white matter. Ann. Neurol. 86, 780–792 (2019).
Dooves, S. et al. Astrocytes are central in the pathomechanisms of vanishing white matter. J. Clin. Invest. 126, 1512–1524 (2016).
Mohamed, M. A. et al. Astrogliosis in aging and Parkinson’s disease dementia: a new clinical study with 11C-BU99008 PET. Brain Commun. 4, fcac199 (2022).
Retzius, G. Biologishe Untersuchungen. Neue Folge, Vol VI (Von Gustav Fischer, 1894).
Grosche, J., Matyash, V., Moller, T., Verkhratsky, A., Reichenbach, A. & Kettenmann, H. Microdomains for neuron–glia interaction: parallel fiber signaling to Bergmann glial cells. Nat. Neurosci. 2, 139–143 (1999).
Ahmed, K. et al. An autocrine lactate loop mediates insulin-dependent inhibition of lipolysis through GPR81. Cell Metab. 11, 311–319 (2010).
Acknowledgements
A.V. was supported by 2023 Key Support Project of the Liaoning Provincial Department of Science and Technology to Support the High-Quality Development of China Medical University ([2023]61-7), by Slovenian Research Agency grant J4-60077, by the Science and Technology Planning Project of Guangdong Province (2021B1212040006), and by the Sanming Project of Medicine in Shenzhen (SZSM202411023 and SZSM202411013). C.J.L. and S.L. were supported by the Institute for Basic Science (IBS), Center for Cognition and Sociality (IBS-R001-D2); C.G. was supported by the Swedish Research Council, Swedish Brain Foundation, Anna-Stina och John Mattsons Minnesstiftelse för sonen Johan, Ming Wai Lau Centre for Reparative Medicine, the Strategic Network for Stem Cells and Regenerative Medicine (STRATREGEN) at Karolinska Institutet, Wings for Life Foundation, and the InnoHK initiative of the Innovation and Technology Commission of the Hong Kong Special Administrative Region Government. T.H. was supported by the Swedish Research Council (2023-03058), Novo Nordisk Foundation (NNF23OC0084476); Hjärnfonden (FO2022-0300); the European Research Council (FOODFORLIFE, 2020-AdG-101021016) and the Austrian Science Fund (FWF; 10.55776/COE16). M.L. was supported by the Swedish Research Council, Olle Engkvist Foundation, The Swedish Alzheimer foundation. J.M. was supported by Swedish research council grant (VR-2023-02656), Hjärnfonden grant (FO2022-0234) and Alzheimerfonden grant (AF-994908). MHN was supported by an NRF Grant (RS-2024-00397737) and Korea Dementia Research Grant (RS-2024-00345328) through the Korea Dementia Research Center (KDRC). O.P.O. was supported by Research Council of Norway. M. Pekna has been supported by grants from Swedish Research Council (2024-02814), the Swedish state under the agreement between the Swedish government and the county councils, the ALF agreement (1005387), The Swedish Brain Foundation (FO2023-0157) and The Swedish Stroke Foundation. M. Pekny has been supported by grants from Swedish Research Council (2017-02255, 2019-00284, 2020-01148 and 2024-03159), the Swedish state under the agreement between the Swedish government and the county councils, the ALF agreement (965939 and 1005449) The Swedish Brain Foundation (FO2018-0252, FO2021-0082 and FO2024-0365), Hagströmer’s Foundation Millennium, The Swedish Stroke Foundation, Amlöv’s Foundation, Peter Eriksson Foundation, E. Jakobsson’s Foundation, La Caixa Foundation project Astromad and European Joint Program on Rare Diseases (EJPRD) project Alexander. A.P. was funded by the Dioscuri Grant (Dioscuri is a program initiated by the Max Planck Society (MPG), jointly managed with the National Science Centre in Poland (NCN) and mutually 486 funded by the Polish Ministry of Education and Science and the German Federal Ministry of Education and Research (BMBF)), by the OPUS17 (UMO-2019/33/B/NZ2/02437), OPUS22 (UMO-488 2021/43/B/NZ2/02934) and Sonata Bis 11 (UMO-2021/42/E/NZ2/00392) grants from the NCN, by EMBO Installation Grant. H.R. was supported by National Research Foundation of Korea (NRF) Grant (2022R1A2C3013138) and Korea Dementia Research Project Grant (RS-2023-KH137130) through the Korea Dementia Research Center (KDRC). C.H.S. was supported by NRF, RS-2021-NR061245 and KAIST startup grant, G04240046. V.U. was supported by the Novo Nordisk Foundation (NNF24OC0088143), Independent Research Fund Denmark (3101-00282B) and Lundbeck Foundation (114955). T.J.V. was supported by the Medical Research Council (MR/Z504518/1). Post-mortem brain tissue (for Fig. 2e) was obtained from the Oxford Brain Bank, which is supported by the Medical Research Council, Brains for Dementia Research (Alzheimer’s Society and Alzheimer Research UK), Autistica UK and the NIHR Oxford Biomedical Research Centre. C.Y. was supported by grants from the National Natural Science Foundation of China (32170980), Guangdong Basic and Applied Basic Research Foundation (2022B1515020012) and Shenzhen Fundamental Research Program (RCJC20231211090018040 and ZDSYS20220606100801003). R.Z. acknowledges support from the Slovenian Research and Innovation Agency (P3-0310; J3-2523; J3-50104; J3-3062; J4-60077; MR+, I0-0034, I0-0048; I0-0022; MRIC-Carl Zeiss Reference Centre for Laser Confocal Microscopy P3-0083) and EU grants (COST action CA18133; ERNEST; EU Interreg Italia-Slovenia Immunocluster-2, EU Interreg Italia-Slovenia COHERENCE). E.C. was supported by the Samsung Science and Technology Foundation under project number SSTF-BA2201-12, and by the National Research Foundation of Korea (NRF) grant funded by the Korean government (MSIT) (2021R1A2C3007164 and 2022M3E5E8016325). A.N. was supported by the Swedish Foundation for Strategic Research (SSF; RB13-0192), the Swedish Research Council (2017-06087, 2023-02649), the Swedish Brain Foundation (Hjärnfonden), the Swedish Alzheimer Foundation (Alzheimerfonden), Karolinska Institutet private bequests, and ALF Region Stockholm.
Funding
Open access funding provided by Karolinska Institute.
Author information
Authors and Affiliations
Contributions
A.V., C.J.L., E.C. and A.N. developed the concept and wrote the draft. H.C., C.G., T.H., J.L., S.L., M.L., W.K., J.M., M.H.N., O.P.O., M. Pekna, M. Pekny, A.P., H.R., C.H.S., E.O.T., V.U., T.J.V., W.Y., C.Y., R.Z., and M.Y. contributed to further writing, discussion and editing. All authors have read and approved the article.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Verkhratsky, A., Lee, C.J., Chun, H. et al. Curing the brain: in search for new astrocyte-specific therapies. Exp Mol Med (2026). https://doi.org/10.1038/s12276-026-01712-4
Received:
Revised:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s12276-026-01712-4
