
Abstract
Background
Genome-wide association studies (GWAS) have identified numerous body mass index (BMI) loci. However, most underlying mechanisms from risk locus to BMI remain unknown. Leveraging omics data through integrative analyses could provide more comprehensive views of biological pathways on BMI.
Methods
We analyzed genotype and blood gene expression data from up to 5619 samples in the Framingham Heart Study (FHS). Using 3992 single-nucleotide polymorphisms (SNPs) at 97 BMI loci and 1408 transcripts within 1 Mb, we performed separate association analyses of transcript with BMI and SNP with transcript (PBMI and PSNP, respectively) and then a correlated meta-analysis between the full summary data sets (PMETA). Transcripts were prioritized if we identified transcripts that met Bonferroni-corrected significance within each omic, showed stronger associations in the correlated meta-analysis than each omic, and had corresponding SNPs in the SNP-transcript-BMI association that were at least nominally associated with BMI in FHS data. We tested for generalization of identified association in a Hispanic ancestry sample of blood gene expression data and other samples in hypothalamus, nucleus accumbens, liver, and visceral adipose tissue (VAT) with significant threshold: PMETA < 0.05 & PMETA < PSNP & PMETA < PBMI.
Results
Among 308 significant SNP-transcript-BMI associations, we identified seven genes (NT5C2, GSTM3, SNAPC3, SPNS1, TMEM245, YPEL3, and ZNF646) in five association regions. We generalized results for SNAPC3 and YPEL3 in Hispanic ancestry sample, for YPEL3 in the nucleus accumbens, ZNF646 and GSTM3 in VAT, and NT5C2, SNAPC3, TMEM245, YPEL3, and ZNF646 in liver.
Conclusion
The identified genes help link the genetic variation at obesity-risk loci to biological mechanisms and health outcomes, thus translating GWAS findings to function.
Similar content being viewed by others


Introduction
Obesity is an enormous global public health burden. Since obesity is a major risk factor for numerous health outcomes, including cardiometabolic diseases [1], the rapid increase in the global obesity burden requires immediate public health action and a better understanding of obesity pathogenicity to prevent it. Decades of research, including genome-wide association studies (GWAS), have demonstrated the fundamental role of genetic susceptibility in obesity risk [2,3,4,5,6]. Each GWAS-identified locus potentially provides novel biologic insight; yet identifying the functional variants, genes, and underlying pathways at these loci has limited translation for precision medicine.
A major barrier to precision medicine for obesity has been the identification of functional genes underlying GWAS findings. Of the thousands of genomic regions associated with obesity-related traits by GWAS, over 90% are in non-coding, potentially regulatory regions of the genome [7]. Previous work mapping body mass index (BMI)-related genes implicates the involvement of synaptic function and glutamate receptor signaling, which impinge on key hypothalamic circuits that respond to changes in feeding and fasting and are regulated by key obesity-related molecules such as BDNF and MC4R [8, 9]. These pathways overlap with a proposed mechanism of action of topiramate, a component of one new FDA-approved weight-loss drug [10, 11]. However, our understanding of the fundamental mechanisms underlying genetic risk for obesity is limited and controversial even for FTO, with the most prominent effects on BMI [12].
Transcriptomics lie along pathways linking genetic susceptibility to obesity and is emerging as powerful disease biomarkers [13, 14] that may provide targetable “mechanistic bridges” linking GWAS findings with obesity risk. Large-scale characterization and integration of OMICs have been challenging because the comprehensive collection of molecular data has, until very recently, been either unavailable or cost-prohibitive in the context of a single study. However, OMICs scans in the same individuals in which obesity-associated loci discoveries were made are now available [15, 16], thereby facilitating comprehensive and efficient integration with genetic data to illuminate the underlying genes and mechanistic pathways of obesity-associated loci. Thus, studies that integrate GWAS with transcriptomics may lead to breakthroughs that reveal the genes contributing to obesity, identify individuals or groups that could benefit from aggressive prevention or treatment [17], or the repurposing of therapeutics [18].
Whole blood tissue is a key metabolic tissue critical to understanding obesity and further precision medicine. Blood is a sentinel tissue and a system integrator of tissue and organ-level perturbation in its physiological role; so all major metabolic perturbations may lead to adaptive responses in blood [19]. Additionally, blood is an easily accessible and minimally invasive tissue source clinically, making it an ideal starting point for research on molecular assessments of whole-blood OMICs contributing to the development of precision prevention, diagnosis, and treatment. Previous studies have shown that BMI-associated genes are highly enriched in brain tissue, including the hypothalamus and other regions of the brain involved in memory, appetite regulation, and metabolism [6]. Similarly, obesity-related genes involved in energy homeostasis (i.e. SEC16B [20], HMGCR [21], etc) are also expressed in the liver [22], a metabolically active organ [23], and human and mouse studies have shown associations between BMI genes and non-alcoholic fatty liver disease [24]. Therefore, our study used samples from whole blood tissue in the discovery stage, providing a strong foundation for generalizing to other potentially relevant tissues.
In this study, we analyzed GWAS data and transcriptomic data generated in whole blood in 5619 participants from the Framingham Heart Study (FHS) to identify potential causal genes through which known loci operate on obesity phenotypes (BMI). We used a correlated meta-analysis procedure to efficiently screen loci for potential candidate genes that are jointly associated with BMI and SNPs in linkage disequilibrium (LD) with established BMI-associated GWAS SNPs and follow-up promising associations in other obesity-relevant tissues.
Methods
Study sample
We included participants from both the Offspring cohort and the third generation (Gen3) cohort of the FHS. The Offspring cohort of FHS began in 1971 and consisted of children of the Original cohort and spouses of these children [25]. Gen3 cohort comprised children from the offspring families enrolled in 2002 [26]. The time intervals between clinical examinations for Offspring and Gen3 cohorts were approximately 4–6 years.
Since the timing of the blood sample taken for RNA collection was close to the eighth clinical examination (Exam 8) for the Offspring cohort and the second clinical examination (Exam 2) for the Gen3 cohort, our study was restricted to subjects with available blood sample, genotype data, and BMI information in either Exam 8 of the Offspring study or Exam 2 of the Gen3 study.
Data description
FHS participants were genotyped using the Affymetrix GeneChip Human Mapping 500 K Array Set and another Affymetrix 50 K gene-centric array. The genotype imputation was performed using the Michigan Imputation Server with HRC reference panel release 1.1 April 2016 (HRC r1.1).
Fasting peripheral whole blood samples (2.5 ml) were collected from FHS participants at the eighth clinical examination (Exam 8) of the Offspring cohort and the second clinical examination (Exam 2) of the Gen3 cohort. The details of RNA collection and expression data cleaning have been previously described [27]. In our study, we used the expression data that have been adjusted using technical covariates and blood count [28, 29].
Height and weight were measured at Exam 8 of the Offspring cohort and Exam 2 of the Gen3 cohort. BMI was then calculated by weight (kg)/height(m)2.
SNP-transcript association and transcript-BMI association
We analyzed 3992 SNPs that are in LD (r2 > 0.8) with 97 previously reported BMI variants from GIANT BMI GWAS paper [6] and the 1408 transcripts with a start position within 1 Mb of these variants. These 97 loci were originally selected as they have been well replicated in more recent GWAS studies [5, 30], have generalized across multiple global populations [30,31,32], are in well-imputed regions of the genomes, and many still lack strong candidate genes; thus, causal genes at these loci are also still under investigation.
We performed two kinds of association modeling. The first was a SNP-transcript association model, with the transcript as the outcome, and the SNP genotype as the predictor, adjusting for covariates including age at expression data collection, sex, and cohort identifier. We performed this first model for every SNP-transcript pair, using a linear mixed effects model to account for relatedness. The second model assessed the association between transcript and BMI, with expression of the transcript as the outcome, and BMI as the predictor, adjusting for age at expression data collection, sex, cohort identifier, and familial relatedness. We performed the second model for each transcript separately. In this manuscript, we will denote the p value of the SNP from the first model as PSNP and the p value of BMI from the second model as PBMI.
Correlated meta-analysis and causal inference test
We used the correlated meta-analysis model of Province and Borecki [33] to account for the potential dependence between the SNP-transcript and transcript-BMI associations, thus correcting for type I error, while still maintaining power for discovery by empirically estimating the null distribution from the test statistics This correlated meta-analysis model estimated the degree of correlation between SNP-transcript and transcript-BMI associations, and corrected for the inflation of type-I error that would be observed in a traditional meta-analysis (that assumes the two associations are statistically independent). Our model used a tetrachoric correlation, which was less sensitive to contamination from the alternative hypothesis than the Pearson correlation, thus preventing over-correction of the correlation.
In our analysis, for every SNP, we estimated the covariance matrix \(\Sigma\) between two association results (\({Z}_{\mathrm{SNP}}={\Phi }^{-1}({P}_{\mathrm{SNP}})\) and \({Z}_{\mathrm{BMI}}={\Phi }^{-1}({P}_{\mathrm{BMI}})\)) using tetrachoric correlation, and then we calculated \({Z}_{\mathrm{meta}}={(Z}_{\mathrm{SNP}}+{Z}_{\mathrm{BMI}})\, \sim N(0,\mathrm{sum}(\Sigma ))\) and \({P}_{\mathrm{meta}}=1-\Phi ({Z}_{\mathrm{meta}})\) for each SNP-transcript pair.
After performing the correlated meta-analysis, we further screened the results to identify transcripts that met Bonferroni-corrected significance for each omic and were more significant in the correlated meta-analysis than in each omic. Thus, we included five criteria: Pmeta < PSNP, Pmeta < PBMI, PSNP < (0.05/1408) = 3.6 × 10−5, PBMI < 3.6 × 10−5, and the SNPs in the identified SNP-transcript pairs should have at least nominal association (p < 0.05) with BMI in FHS. The first two criteria ensured that both the SNP-transcript and transcript-BMI associations contributed to the meta-analysis. The third and fourth criteria guaranteed the Bonferroni-corrected significance of each association. The last criterion restricted the SNPs to those at least nominally associated with BMI in FHS data. The analysis code is available upon request.
We also performed causal inference test (CIT) analysis on the signals identified in the correlated meta-analysis. CIT is a hypothesis testing approach to identify potential mediators of the effects of genetic variants on traits of interest [34, 35]. Considering a transcript as a potential mediator of an effect of SNP on BMI, CIT could simultaneously test four conditions contributing to causal relationship evaluation among SNP, transcript, and BMI, and produce an omnibus test p-value. The omnibus null hypothesis is that the null hypothesis is true in at least one of the four components. We also performed CIT in the reverse direction, where we considered BMI as a potential mediator of an effect of SNP on a transcript. For each transcript we identified from the correlated meta-analysis, we analyzed the most significant SNP from its SNP-transcript associations in CIT analysis to avoid multicollinearity in the model.
Biological interrogation and functional annotation
Regulatory variants are more likely to drive correlated signals of gene expression and SNP association than coding variants. To characterize candidate regulatory variants, we used chromatin marks and other epigenomic data that define regulatory elements or link regulatory elements to gene transcription start sites. We focused on data sets for liver, and component cell types, especially preadipocytes, adipocytes, and hepatocytes. We compared them to other tissues because tissue-restricted regulatory elements may be more likely to be relevant and functional. The resources we considered include accessible chromatin based on the assay for transposase-accessible chromatin (ATAC-seq) or DNase hypersensitivity from brain, blood, and liver; histone mark and transcription factor ChIP-seq and chromatin states from ENCODE [36] used for visual inspection and to assess variant overlap with potential candidate cis regulatory elements (cCREs). Additional resources used for variant annotation as described in Supplementary Note 1 include GeneCards [37], OMIM [38], and GTEx [39].
Generalization: Cameron County Hispanic Cohort (CCHC)
The CCHC was established on the Texas-Mexico border in 2004 [40]. This randomly ascertained community cohort currently comprises over 5000 people and is approximately 60% female. All CCHC individuals were genotyped using the Illumina MEGAEX array [41].
RNA sequencing of CCHC participants was conducted using stored whole blood with sufficient quantity and quality. Sample collection and transcriptome profiling were described in detail previously [42]. We implemented a negative binomial model in DESeq2 [43] to identify BMI genes with covariate adjustment for sex, age, 10 PEER factors [44], and filtered results using default thresholds (N = 934).
We performed eQTL mapping using the GTEx v8 pipeline [39]. We identified eQTLs in cis (within 1 Mb) for each gene using FastQTL [45] with adjustment for sex, RNA-seq batch, 5 genetic principal components (PCs), and 10 PEER factors.
Generalization on liver tissue and visceral adipose tissue (VAT): MyCode Bariatric Surgery Program (BSP)
The MyCode™Community Health Initiative (MyCode) study is a healthcare-based population study in central and northeastern Pennsylvania with ~2 million patients [46, 47]. We leveraged existing transcriptomic profiling in the Geisinger Health System’s (GHS) Bariatric Surgery Program (BSP) study (N = 2224) to generalize observed joint associations from whole blood in FHS to liver (N = 2224) and VAT (N = 657) tissues. Liver tissue sample collection and transcriptome profiling were described in detail previously [48, 49]. VAT samples were collected during the same procedure as described for the liver on a subset of subjects in the BSP, following similar storage procedures, as described previously [50]. Association analyses were performed using FastQTL [45], adjusting for sex, age, self-identified race/ethnicity, the first three genomic PCs to control for ancestry, and 60 PEER factors [44].
Generalization on brain tissue
Analyses of hypothalamus (N = 131) and nucleus accumbens (N = 198) were conducted on samples from three cohorts: the Framingham Heart Study (FHS), the Religious Orders Study (ROS), and the Rush Memory and Aging Project (MAP). Details of RNA sequencing of hypothalamus and nucleus accumbens and the transcript-BMI association analysis were described previously [51]. In brief, total RNA were isolated using QIAzol Lysis Reagent (Qiagen, Valencia CA) and purified using miRNeasy MinElute Cleanup columns. Clipped large RNA sequencing reads shorter than 50 nucleotides were removed, and clipped small RNA sequencing reads shorter than 15 or longer than 23 were removed. For quality control, the nucleotide trimming tool sickle [52] v 1.33 was applied with Phred quality threshold of 20, removing low quality ends of reads and once again applying the lower bound read length filter. Differential expression testing of last measured BMI was performed using linear regression with LIMMA. We restricted our analysis to samples with RIN > 3 and BRAAK score ≤4. Covariates considered were sex, cohort, age at death, and sequencing batch. For the eQTL analysis, we used FastQTL and adjusted for covariates: 5 first genetic PCs, PEER factors according to the GTEx recommendations (15 PEER factors for hypothalamus and 30 PEER factors for nucleus accumbens), sex, age at death, cohort, and sequencing batch. We further performed meta-analysis using p values of SNP-transcript and transcript-BMI associations via Fisher’s method [53], producing a meta-analyzed p value.
Results
Sample characteristics
The characteristics of samples included in the discovery correlated meta-analysis, and generalization analyses are shown in Supplementary Table 1. The age distribution was similar for FHS whole blood, CCHC whole blood, and BSP liver analyses, with a mean ranging from 47 to 58, and the brain analyses had relatively older subjects with a mean age of 88. All study samples had a larger proportion of females compared to males. The BSP sample had a relatively higher BMI compared to other study samples. FHS and BSP are dominantly European ancestry, while CCHC was 100% Hispanic/Latino.
Correlated Meta-analysis
Figure 1 shows the general workflow of our entire study. The models and filtering criteria of each step have been included in the “Methods” section. Separate results for suggestive SNP-transcript (PSNP < (0.05/1408) = 3.6 × 10−5) and transcript-BMI (PBMI < 3.6 × 10−5) signals in FHS analysis are provided in Supplementary Tables 2 and 3. The SNP-transcript analysis identified 3424 SNP-transcript pairs, including 1208 unique SNPs (corresponding to 31 GIANT BMI GWAS loci) and 74 unique transcripts. Genes, most frequently implicated with over 100 SNP-transcript analysis, were AS3MT, DMXL2, NT5C2, PRKAG3, RQCD1, SNAPC3, TMOD2, TTLL4, USP37, and VIL1. The most significant SNP-transcript pair was rs8049439-TUFM from 16p11.2 with PSNP = 6.07 × 10−203. We did not observe inflation in the SNP-transcript analysis (Q-Q plot as Supplementary Fig. 1). The transcript-BMI analysis identified 306 transcripts with PBMI < 3.6 × 10−5, corresponding to 78 GIANT BMI GWAS SNPs. The most significant transcript was AHSP at 16p11.2 with PBMI = 1.60 × 10−106.

a Step 1 included single omics associations for SNP to gene expression (PSNP) and gene expression to BMI (PBMI). b Step 2 included the correlated meta-analysis to account for the interdependence between PSNP and PBMI. c Identifying all SNP—Gene—BMI combinations that met our filtering criteria, which included correlated meta-analysis results that are more significant than individual omics associations. d All significant SNP—Gene—BMI combinations were followed by generalization in blood, liver, adipose, brain tissues and causal inference testing (CIT) analysis.
In the FHS correlated meta-analysis, we found 308 SNP-transcript-BMI associations corresponding to seven unique genes (NT5C2, YPEL3, ZNF646, SPNS1, GSTM3, SNAPC3, and TMEM245) potentially involved in transcriptional pathways from SNP to BMI (Table 1). 115 variants were involved in the SNP-transcript-BMI associations for NT5C2, including the reported BMI variant rs11191560. YPEL3, ZNF646 and SPNS1 were in the same region (16p11.2), and we observed 10, 46 and 91 SNP-transcript-BMI associations for YPEL3, ZNF646 and SPNS1 respectively, including three reported BMI variants rs4787491, rs9925964 and rs3888190. At the TMEM245 locus, we pinpointed the SNP-transcript-BMI association to the reported BMI variant rs6477694. GSTM3 was located at 1p13.3, with 4 SNP-transcript-BMI associations detected, including previously reported BMI signal rs17024393. SNAPC3, located at 9p22.3, had 41 SNP-transcript-BMI associations identified. In the directional causal analysis using CIT model, YPEL3 reached nominal significance (p value = 0.011), indicating its role as a potential mediator between genetic variants and BMI.
Generalization to Hispanic/Latino participants and other obesity-relevant tissues
We tested for generalization of the above seven genes using CCHC blood gene expression data. Among the identified 308 SNP-transcript-BMI associations, 37 SNP-transcript-BMI associations corresponding to SNAPC3 and 10 SNP-transcript-BMI associations corresponding to YPEL3 remained significant (Pmeta< 0.05 & Pmeta < PSNP & Pmeta < PBMI) (Supplementary Table 4). Regional association plots for each gene show annotation information (Fig. 2 and Supplementary Figs. 2–6). Of note, the top PMETA SNP for SNAPC3 and YPEL3 are within or proximal to putative candidate cis-Regulatory Elements (cCREs) based on ENCODE [36] regulatory data on blood, brain, liver, and VAT tissues (Fig. 2).

Annotation for potential candidate cis-regulatory elements from ENCODE are included for each reported SNP in the region. A SNAPC3, B YPEL3.
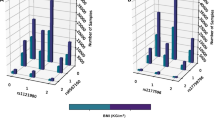
We also tested for generalization using gene expression in brain tissues. Hypothalamus tissue showed no significant SNP-transcript-BMI association. In contrast, the 10 SNP-transcript-BMI associations corresponding to YPEL3 were significant in the generalization analysis on nucleus accumbens. Additionally, we were able to generalize signals in liver tissue for NT5C2, SNAPC3, TMEM245, YPEL3, and ZNF646, including 103, 40, 1, 10, and 15 SNP-transcript-BMI associations, respectively (Supplementary Table 4). Finally, we observed significant generalization in VAT for GSTM3 and ZNF646, including 4 and 46 SNP-transcript-BMI associations, respectively. While the direction of effect was consistent for both brain tissues, even for non-significant associations (Table 2, Fig. 3 and Supplementary Table 4). The direction of effect was not always consistent across tissue types; however, consistency of direction of effect across various tissues may not be expected. Further work may be needed to clarify expectations of directional consistency across tissues with respect to BMI ~ Gene and SNP ~ associations.

Results are provided for discovery sample (FHS, blue), generalization in blood (CCHC, red), and generalization to hypothalamus (Hypo, green), nucleus accumbens (Accum, purple), liver (brown) and VAT (yellow) tissues. We provide individual effect estimates and p values for each OMIC and meta-analysis. Filled diamonds indicate significant associations in the meta-analysis (Note: FHS is noted as NULL, as all are significant).
Biological interrogation and functional annotation
Previous studies of gene function and bioinformatics characteristics (see Methods) of the significant genes highlight nearby signatures of gene regulation (Supplementary Note 1, Fig. 2 and Supplementary Figs. 2–6). Top SNPs from our correlated meta-analysis often coincide with regulatory elements, particularly in relevant tissues, while known index SNPs more often fell outside of these proposed regulatory elements. For example, our top SNP in YPEL3 overlaps a likely regulatory element in three tissue types (brain, liver, and blood), but the previously known index variant overlaps a probable regulatory element in blood. Further, there is stronger evidence supporting the likely regulatory function by cCREs overlap for our lead SNP (Fig. 2B; i.e. promoter−like signature (PLS) for our lead SNP vs. DNase−only for known index SNP in blood). Similar evidence is observed for our top correlated SNP for NT5C2 and GSTM3. For ZNF646, both the known and top correlated meta SNP are overlapping cCREs in multiple tissues. However, other loci do not provide evidence for cCREs overlap for either index SNP.
Seven genes were identified as significant in our correlated meta-analysis, many with potential relevance to metabolic and neurodevelopmental phenotypes (see Supplementary Note 1 for additional details). For example, NT5C2, a purine-metabolizing enzyme, is ubiquitously expressed and has been associated with reduced adiposity, obesity, and obesity-related depression [54,55,56,57,58]. Functional studies suggest its variants may influence BMI through miRNA-mediated regulation [59]. YPEL3, involved in glial development and apoptosis, has been linked to both BMI and schizophrenia, and mouse knockdown models showing altered fat composition, according to the International Mouse Phenotyping Consortium (IMPC) [60,61,62]. GSTM3, a detoxification enzyme, is highly polymorphic and associated with several cardiometabolic phenotypes, including hyperinsulinemia, type 2 diabetes, hypertension, and polycystic ovary syndrome (PCOS) [63,64,65]. Lastly, SNAPC3, a component of the small nuclear RNA transcription complex, is associated with schizophrenia and early-life growth trajectories via epigenetic regulation [66, 67]. These findings highlight the diverse biological pathways potentially contributing to metabolic and neuropsychiatric traits.
Discussion
This study incorporated a correlated meta-analysis method to perform integrative analysis using genotype, gene expression, and phenotype (BMI) data. From the discovery analysis using the FHS whole blood data, we identified seven genes (NT5C2, YPEL3, ZNF646, SPNS1, GSTM3, SNAPC3, and TMEM245) that potentially lie along the pathway linking genetic variation to elevated BMI. Among those seven genes, YPEL3 and SNAPC3 associations were validated in whole blood in the CCHC study. In the analyses of tissues other than blood, NT5C2, SNAPC3, TMEM245, YPEL3, and ZNF646 associations generalized in the liver tissue, ZNF646 and GSTM3 in VAT, and YPEL3 in the nucleus accumbens.
Our literature search provides further details on potential roles for identified genes for obesity (Supplementary Note 1). YPEL3 is located at 16p11.2, a gene dense region well-known for a microdeletion associated with neurocognitive developmental delay and predisposition to obesity [68,69,70]. Literature has reported that this region’s deletion event is related to a highly-penetrant form of obesity [71, 72], and is age- and gender-dependent [73, 74]. Within this region, SH2B1 has received much attention as the likely causal gene underlying the mosaic effects of the 16p11.2 deletion and is thought to regulate body weight and glucose metabolism [75, 76]; and as a result, YPEL3 has rarely been considered in previous studies. One of the previous studies that considered YPEL3 [61] identified it as a pleiotropic gene jointly influencing BMI and risk of schizophrenia. In contrast, another study [77] asserted that the association between YPEL3 and schizophrenia is due to its correlation with expression of INO80E, another possible candidate gene for BMI and risk of schizophrenia in the 16p11.2 region. Other model organism studies have shown alterations in YPEL3 results in altered obesity phenotypes. For example, YPEL3 knockdown in Drosophila melanogaster resulted in significant changes in body fat percentage [62]. Despite the controversial findings of YPEL3 in the literature, several pieces of evidence support a role of YPEL3 in BMI. First, the gene is highly expressed in whole blood and brain, similar to well-known BMI-related genes (Supplementary Note 1). Also, YPEL3 was the sole candidate gene in this region identified by the current analysis and it showed significant causal SNP-transcript-BMI relationship in the CIT analysis. Further, the blood expression results were validated in an independent study of Hispanic participants, and the results generalized to both brain and liver tissues. Combined, this evidence suggests that more attention is warranted on this gene in the future.
NT5C2 is located at 10q24.32, which has been reported as a highlight locus of autism spectrum disorder, brain arterial diameters, and schizophrenia [78,79,80]. NT5C2 deletion was found to be protective in mice fed a high fat diet (HFD) [54]. A previous study in zebrafish found NT5C2 as a potential causal gene in this region for blood pressure [81]. Notably, variation in this gene is also associated with lower visceral and subcutaneous fat [57], obesity, and the concurrence of obesity and depression [58] (Supplementary Note 1). Further, animal studies of NT5C2 knock-outs show changes in body weight gain, insulin resistance on high-fat diet, and white adipose tissue mass [54, 56]. Kumar et al. found that rs11191548 decreased miRNA binding efficiency, which may explain the functional role of NT5C2 influencing BMI [59]. Yet, our significant findings linking SNP variation to NT5C2 gene expression with BMI in liver tissue is novel and a role for this gene in other tissues warrants further exploration. Literature shows strong support for YPEL3 and NT5C2 as likely candidate genes underlying the association with BMI in these two regions. However, existing knowledge that may offer a role for the other genes in the pathway to BMI is sparse.
While support for other genes identified herein is limited in the literature, SNAPC3, which validated in CCHC, and TMEM245, which generalized to liver tissue, have connections to obesity-related traits. For example, similar to both genes mentioned above, SNAPC3 variants have also been associated with schizophrenia [66]. Also, DNA methylation in SNAPC3 has been reported to mediate the association between breastfeeding and early-life growth trajectories [67]. The expression level of TMEM245 has been associated with atrial fibrillation [82], and schizophrenia-associated variants have been reported within this gene [83].
In recent years, there has been growing interest in developing integrative approaches that utilize various OMICs data to uncover underlying biological mechanisms of obesity. Smemo et al. [12] found that obesity-associated variants within FTO were functionally connected with IRX3 and IRX5 expression. Voisin et al. [84] and Tang et al. [85] evaluated the association and the interaction between obesity-associated SNPs and DNA methylation changes. Kogelman et al. [86] detected co-expression patterns among eQTLs, integrated with protein data, and detected several obesity candidate genes, such as ENPP1, CTSL, and ABHD12B. More recently, integrative analyses on multiple obesity and neuro-related phenotypes provided further gene lists that potentially affected relevant phenotypes jointly [61, 77]. Also, a recent study colocalized splice junction quantitative trait loci (sQTLs) measured in subcutaneous adipose tissue with 24 BMI GWAS loci, including with YPEL3 [87], and another study has reported 162 BMI signals with a colocalized adipose eQTL [88].
When individual-level data is available, combining multiple OMICs datasets to perform further analysis is preferred [86, 89]. Yet, few integrative studies using summary-level data exist [61, 90], limiting cross study analyses. Thus, among all the integrative OMICs analyses, the correlation between OMICs is often ignored [90, 91]. In our study, we leveraged the correlated meta-analysis framework proposed [33], which is a robust approach to integrate “suspected” correlated SNP-transcript association and transcript-BMI association. This approach is useful for performing statistical integration and has been incorporated into many colocalization and polygenic pleiotropy detection methods [92, 93]. By performing correlated meta-analysis using summary level data, we ensured the correlation between summary statistics of OMICs scans were considered. Given the complex and potentially bidirectional relationship between gene expression levels and BMI, this approach is well-suited as it does not require an assumption about the direction of causation. Instead, this computationally simple, fast, and scalable approach can serve as a tool for refining or prioritizing known signals with diverse types of data, with the possibility of conducting follow-up analyses such as CIT to explore causal direction post-hoc. Indeed, we did identify evidence of potential causal relationships from SNP to BMI through expression of YPEL3 in our CIT analyses. However, interpreting null findings at other genes is complex given the power requirements for causal inferences, relationships among genes for polygenic traits, the complex pathways that connect genetic variation to phenotypic expression, horizontal pleiotropy/epistasis, and limitations of the CIT to model bidirectional, direct and indirect effects. Thus, other mediating factors should be explored in future investigations.
There are potential limitations to our study. First, the data sets used in this analysis are limited in demographic variation. For example, there is a limited age range for brain-related tissues (mean age > 85 years for both tissues), as these tissues are available posthumously. Also, there is limited race/ethnic diversity in the discovery and liver and VAT generalization cohorts, all with primarily White/European Americans. These concerns are somewhat mitigated by the attempt to generalize associations from FHS to Hispanic/Latino participants of the CCHC. Another potential limitation is the differences in gene expression data collection between our cohort studies (i.e. array-based vs. RNAseq), which may limit the potential generalizations. Additionally, we used population-level cohort data for our analyses and thus expression data are measured under circumstances that may not be optimal for detecting relevant differences in gene expression125, and has the potential to influence both the strength and direction of gene expression. For example, SNP-associated gene expression changes in response to environmental exposures (i.e. high-fat foods, medications, etc). Last, we were unable to conduct in vitro or in vivo functional validation of the candidate genes and/or SNPs identified through our analyses.
Yet, compared to other integrative studies, our study has several strengths. To our knowledge, our study is the first one that takes the correlation between OMICs scans into the integrative analysis of BMI. We not only have a discovery study using whole blood samples from European ancestry, but validate these joint associations in an independent study of Hispanic participants, and generalize our findings to other tissues, including liver, VAT, and nucleus accumbens. Yet, our study has some limitations. First, the traditional meta-analysis instead of the correlated meta-analysis was used in the generalization analyses due to data sparsity. Also, we only included two types of OMICs data in our analyses, genetics and gene expression data. However, these analyses gave us a comprehensive view of how our findings can be interpreted across ancestry and tissue type. And, our work offers a framework for future investigations incorporating additional OMICs data, such as DNA methylation or protein data, as well as additional tissues, that can also be adopted for other traits of interest.
Conclusion
Our study aimed to narrow in on causal genes that underly known obesity susceptibility loci. Specifically, we were interested in genetic variation that may be operating on variation in BMI through alterations in gene expression. Our integrative, multi-omics approach identified seven candidate genes within five genomic regions for BMI. Among these seven, we find the strongest support for YPEL3, NT5C2, and SNAPC3, through generalization across ethnicities, generalization across BMI-relevant tissues, and/or existing literature with a connection to BMI-related traits or gene functions. This deep dive into the etiology of obesity risk loci gets us one step forward to connecting genetic variation to biological mechanisms and health outcomes, and thus translating GWAS findings to function so that obesity precision treatment and prevention can begin.
Data availability
The individual level data are available on dbGAP (dbGAP ID: phs000007.v2.p1 and phs002611.v3.p16 for FHS and phs003894.v1.p1 for CCHC). MyCode data can be accessed by Geisinger investigators. There are restrictions to the sharing of MyCode DiscovEHR genetic datasets related to agreements between Geisinger and the Regeneron Genetics Center.
References
Collaboration PS, Whitlock G, Lewington S, Sherliker P, Clarke R, Emberson J, et al. Body-mass index and cause-specific mortality in 900 000 adults: collaborative analyses of 57 prospective studies. Lancet. 2009;373:1083–96.
Shungin D, Winkler TW, Croteau-Chonka DC, Ferreira T, Locke AE, Mägi R, et al. New genetic loci link adipose and insulin biology to body fat distribution. Nature. 2015;518:187–96.
Berndt SI, Gustafsson S, Mägi R, Ganna A, Wheeler E, Feitosa MF, et al. Genome-wide meta-analysis identifies 11 new loci for anthropometric traits and provides insights into genetic architecture. Nat Genet. 2013;45:501–12.
Pulit SL, Stoneman C, Morris AP, Wood AR, Glastonbury CA, Tyrrell J, et al. Meta-analysis of genome-wide association studies for body fat distribution in 694 649 individuals of European ancestry. Hum Mol Genet. 2019;28:166–74.
Yengo L, Sidorenko J, Kemper KE, Zheng Z, Wood AR, Weedon MN, et al. Meta-analysis of genome-wide association studies for height and body mass index in ∼700000 individuals of European ancestry. Hum Mol Genet. 2018;27:3641–9.
Locke AE, Kahali B, Berndt SI, Justice AE, Pers TH, Day FR, et al. Genetic studies of body mass index yield new insights for obesity biology. Nature. 2015;518:197–206.
Fall T, Ingelsson E. Genome-wide association studies of obesity and metabolic syndrome. Mol Cell Endocrinol. 2014;382:740–57.
Melo CV, Mele M, Curcio M, Comprido D, Silva CG, Duarte CB. BDNF regulates the expression and distribution of vesicular glutamate transporters in cultured hippocampal neurons. PLoS ONE. 2013;8:e53793.
Shen Y, Fu WY, Cheng EY, Fu AK, Ip NY. Melanocortin-4 receptor regulates hippocampal synaptic plasticity through a protein kinase A-dependent mechanism. J Neurosci. 2013;33:464–72.
Gibbs JW 3rd, Sombati S, DeLorenzo RJ, Coulter DA. Cellular actions of topiramate: blockade of kainate-evoked inward currents in cultured hippocampal neurons. Epilepsia. 2000;41:S10–16.
Poulsen CF, Simeone TA, Maar TE, Smith-Swintosky V, White HS, Schousboe A. Modulation by topiramate of AMPA and kainate mediated calcium influx in cultured cerebral cortical, hippocampal and cerebellar neurons. Neurochem Res. 2004;29:275–82.
Smemo S, Tena JJ, Kim KH, Gamazon ER, Sakabe NJ, Gomez-Marin C, et al. Obesity-associated variants within FTO form long-range functional connections with IRX3. Nature. 2014;507:371–5.
Wang TJ, Larson MG, Vasan RS, Cheng S, Rhee EP, McCabe E, et al. Metabolite profiles and the risk of developing diabetes. Nat Med. 2011;17:448–53.
Kraus WE, Pieper CF, Huffman KM, Thompson DK, Kraus VB, Morey MC, et al. Association of plasma small-molecule intermediate metabolites with age and body mass index across six diverse study populations. J Gerontol Ser A Biol Sci Med Sci. 2016;71:1507–13.
Justice AE, Winkler TW, Feitosa MF, Graff M, Fisher VA, Young K, et al. Genome-wide meta-analysis of 241,258 adults accounting for smoking behaviour identifies novel loci for obesity traits. Nat Commun. 2017;8:14977.
Mendelson MM, Marioni RE, Joehanes R, Liu C, Hedman ÅK, Aslibekyan S, et al. Association of body mass index with DNA methylation and gene expression in blood cells and relations to cardiometabolic disease: a Mendelian randomization approach. PLoS Med. 2017;14:1–30.
Howard G, Moy CS, Howard VJ, McClure LA, Kleindorfer DO, Kissela BM, et al. Where to focus efforts to reduce the Black-White disparity in stroke mortality: incidence versus case fatality?. Stroke. 2016;47:1893–8.
Tardif JC, Rheaume E, Lemieux Perreault LP, Gregoire JC, Feroz Zada Y, Asselin G, et al. Pharmacogenomic determinants of the cardiovascular effects of dalcetrapib. Circ Cardiovasc Genet. 2015;8:372–82.
Ghosh S, Dent R, Harper ME, Gorman SA, Stuart JS, McPherson R. Gene expression profiling in whole blood identifies distinct biological pathways associated with obesity. BMC Med Genomics. 2010;3:56.
Shi R, Lu W, Tian Y, Wang B. Intestinal SEC16B modulates obesity by regulating chylomicron metabolism. Mol Metab. 2023;70:101693.
Duan Y, Gong K, Xu S, Zhang F, Meng X, Han J. Regulation of cholesterol homeostasis in health and diseases: from mechanisms to targeted therapeutics. Signal Transduct Target Ther. 2022;7:265.
Speliotes EK, Willer CJ, Berndt SI, Monda KL, Thorleifsson G, Jackson AU, et al. Association analyses of 249,796 individuals reveal 18 new loci associated with body mass index. Nat Genet. 2010;42:937–48.
Rui L. Energy metabolism in the liver. Compr Physiol. 2014;4:177–97.
Sokouti B. Significant roles of potential genes and their mutations in nonalcoholic fatty liver disease. Clin Exp Hepatol. 2023;9:95–105.
Feinleib M, Kannel WB, Garrison RJ, McNamara PM, Castelli WP. The Framingham offspring study. Design and preliminary data. Prev Med. 1975;4:518–25.
Splansky GL, Corey D, Yang Q, Atwood LD, Cupples LA, Benjamin EJ, et al. The Third Generation Cohort of the National Heart, Lung, and Blood Institute’s Framingham Heart Study: design, recruitment, and initial examination. Am J Epidemiol. 2007;165:1328–35.
Wang L, Perez J, Heard-Costa N, Chu AY, Joehanes R, Munson PJ, et al. Integrating genetic, transcriptional, and biological information provides insights into obesity. Int J Obes. 2019;43:457–67.
Joehanes R, Zhang X, Huan T, Yao C, Ying SX, Nguyen QT, et al. Integrated genome-wide analysis of expression quantitative trait loci aids interpretation of genomic association studies. Genome Biol. 2017;18:16.
Zhang X, Joehanes R, Chen BH, Huan T, Ying S, Munson PJ, et al. Identification of common genetic variants controlling transcript isoform variation in human whole blood. Nat Genet. 2015;47:345–52.
Huang J, Huffman JE, Huang Y, Do Valle I, Assimes TL, Raghavan S, et al. Genomics and phenomics of body mass index reveals a complex disease network. Nat Commun. 2022;13:7973.
Ng MCY, Graff M, Lu Y, Justice AE, Mudgal P, Liu C-T, et al. Discovery and fine-mapping of adiposity loci using high density imputation of genome-wide association studies in individuals of African ancestry: African Ancestry Anthropometry Genetics Consortium. PLoS Genet. 2017;13:e1006719.
Akiyama M, Okada Y, Kanai M, Takahashi A, Momozawa Y, Ikeda M, et al. Genome-wide association study identifies 112 new loci for body mass index in the Japanese population. Nat Genet. 2017;49:1458–67.
Province MA, Borecki IB. A correlated meta-analysis strategy for data mining ‘OMIC’ scans. Proceedings of the Pacific symposium on biocomputing 2013. Singapore; 2013, pp. 236–46.
Millstein J, Zhang B, Zhu J, Schadt EE. Disentangling molecular relationships with a causal inference test. BMC Genet. 2009;10:23.
Millstein J, Chen GK, Breton CV. cit: hypothesis testing software for mediation analysis in genomic applications. Bioinformatics. 2016;32:2364–5.
Consortium EP. An integrated encyclopedia of DNA elements in the human genome. Nature. 2012;489:57–74.
Stelzer G, Rosen N, Plaschkes I, Zimmerman S, Twik M, Fishilevich S, et al. The GeneCards Suite: from gene data mining to disease genome sequence analyses. Curr Protoc Bioinform. 2016;54:1 30 31–1 30 33.
Amberger JS, Bocchini CA, Scott AF, Hamosh A. OMIM.org: leveraging knowledge across phenotype-gene relationships. Nucleic Acids Res. 2019;47:D1038–43.
Consortium Gte. The GTEx Consortium atlas of genetic regulatory effects across human tissues. Science. 2020;369:1318–30.
Chen HH, Petty LE, North KE, McCormick JB, Fisher-Hoch SP, Gamazon ER, et al. Novel diabetes gene discovery through comprehensive characterization and integrative analysis of longitudinal gene expression changes. Hum Mol Genet. 2022;31:3191–205.
Anwar MY, Baldassari AR, Polikowsky HG, Sitlani CM, Highland HM, Chami N, et al. Genetic pleiotropy underpinning adiposity and inflammation in self-identified Hispanic/Latino populations. BMC Med Genomics. 2022;15:192.
Sabotta CM, Kwan SY, Petty LE, Below JE, Joon A, Wei P, et al. Genetic variants associated with circulating liver injury markers in Mexican Americans, a population at risk for non-alcoholic fatty liver disease. Front Genet. 2022;13:995488.
Love MI, Huber W, Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014;15:550.
Stegle O, Parts L, Piipari M, Winn J, Durbin R. Using probabilistic estimation of expression residuals (PEER) to obtain increased power and interpretability of gene expression analyses. Nat Protoc. 2012;7:500–7.
Ongen H, Buil A, Brown AA, Dermitzakis ET, Delaneau O. Fast and efficient QTL mapper for thousands of molecular phenotypes. Bioinformatics. 2016;32:1479–85.
Carey DJ, Fetterolf SN, Davis FD, Faucett WA, Kirchner HL, Mirshahi U, et al. The Geisinger MyCode community health initiative: an electronic health record-linked biobank for precision medicine research. Genet Med. 2016;18:906–13.
Dewey FE, Murray MF, Overton JD, Habegger L, Leader JB, Fetterolf SN, et al. Distribution and clinical impact of functional variants in 50,726 whole-exome sequences from the DiscovEHR study. Science. 2016;354. https://doi.org/10.1126/science.aaf6814.
Leti F, Malenica I, Doshi M, Courtright A, Van Keuren-Jensen K, Legendre C, et al. High-throughput sequencing reveals altered expression of hepatic microRNAs in nonalcoholic fatty liver disease-related fibrosis. Transl Res. 2015;166:304–14.
Gerhard GS, Benotti P, Wood GC, Chu X, Argyropoulos G, Petrick A, et al. Identification of novel clinical factors associated with hepatic fat accumulation in extreme obesity. J Obes. 2014;2014:368210.
Gerhard GS, Styer AM, Strodel WE, Roesch SL, Yavorek A, Carey DJ, et al. Gene expression profiling in subcutaneous, visceral and epigastric adipose tissues of patients with extreme obesity. Int J Obes. 2014;38:371–8.
Christian W, Julie AS, Thor DS, Joli B, Adam L, Ann M, et al. RNA-sequencing of human post-mortem hypothalamus and nucleus accumbens identifies expression profiles associated with obesity. bioRxiv. 2022. https://doi.org/10.1101/2022.01.08.473382.
Joshi N, Fass J. Sickle: a sliding-window, adaptive, quality-based trimming tool for FastQ files (Version 1.33) [Software]. 2011.
Fisher RA. Statistical methods for research workers. 4th ed. Edinburgh. Oliver and Boyd; 1932.
Johanns M, Kviklyte S, Chuang SJ, Corbeels K, Jacobs R, Herinckx G, et al. Genetic deletion of soluble 5’-nucleotidase II reduces body weight gain and insulin resistance induced by a high-fat diet. Mol Genet Metab. 2019;126:377–87.
Kulkarni SS, Karlsson HK, Szekeres F, Chibalin AV, Krook A, Zierath JR. Suppression of 5’-nucleotidase enzymes promotes AMP-activated protein kinase (AMPK) phosphorylation and metabolism in human and mouse skeletal muscle. J Biol Chem. 2011;286:34567–74.
Camici M, Garcia-Gil M, Allegrini S, Pesi R, Tozzi MG. Evidence for a cross-talk between cytosolic 5’-nucleotidases and AMP-activated protein kinase. Front Pharmacol. 2020;11:609849.
Hotta K, Kitamoto A, Kitamoto T, Mizusawa S, Teranishi H, Matsuo T, et al. Genetic variations in the CYP17A1 and NT5C2 genes are associated with a reduction in visceral and subcutaneous fat areas in Japanese women. J Hum Genet. 2012;57:46–51.
Samaan Z, Lee YK, Gerstein HC, Engert JC, Bosch J, Mohan V, et al. Obesity genes and risk of major depressive disorder in a multiethnic population: a cross-sectional study. J Clin Psychiatry. 2015;76:e1611–1618.
Kumar P, Traurig M, Baier LJ. Identification and functional validation of genetic variants in potential miRNA target sites of established BMI genes. Int J Obes. 2020;44:1191–5.
Blanco-Sánchez B, Clément A, Stednitz SJ, Kyle J, Peirce JL, McFadden M, et al. yippee like 3 (ypel3) is a novel gene required for myelinating and perineurial glia development. PLoS Genet. 2020;16:e1008841.
Liu H, Sun Y, Zhang X, Li S, Hu D, Xiao L, et al. Integrated analysis of summary statistics to identify pleiotropic genes and pathways for the comorbidity of schizophrenia and cardiometabolic disease. Front Psychiatry. 2020;11. https://www.frontiersin.org/articles/10.3389/fpsyt.2020.00256.
Baranski TJ, Kraja AT, Fink JL, Feitosa M, Lenzini PA, Borecki IB, et al. A high throughput, functional screen of human body mass index GWAS loci using tissue-specific RNAi Drosophila melanogaster crosses. PLoS Genet. 2018;14:e1007222.
Pass GJ, Becker W, Kluge R, Linnartz K, Plum L, Giesen K, et al. Effect of hyperinsulinemia and type 2 diabetes-like hyperglycemia on expression of hepatic cytochrome p450 and glutathione s-transferase isoforms in a New Zealand obese-derived mouse backcross population. J Pharmacol Exp Ther. 2002;302:442–50.
Delles C, Padmanabhan S, Lee WK, Miller WH, McBride MW, McClure JD, et al. Glutathione S-transferase variants and hypertension. J Hypertens. 2008;26:1343–52.
Cortón M, Botella-Carretero JI, López JA, Camafeita E, San Millán JL, Escobar-Morreale HF, et al. Proteomic analysis of human omental adipose tissue in the polycystic ovary syndrome using two-dimensional difference gel electrophoresis and mass spectrometry. Hum Reprod. 2008;23:651–61.
Goes FS, McGrath J, Avramopoulos D, Wolyniec P, Pirooznia M, Ruczinski I, et al. Genome-wide association study of schizophrenia in Ashkenazi Jews. Am J Med Genet B Neuropsychiatr Genet. 2015;168:649–59.
Briollais L, Rustand D, Allard C, Wu Y, Xu J, Rajan SG, et al. DNA methylation mediates the association between breastfeeding and early-life growth trajectories. Clin Epigenetics. 2021;13:231.
Christian SL, Brune CW, Sudi J, Kumar RA, Liu S, Karamohamed S, et al. Novel submicroscopic chromosomal abnormalities detected in autism spectrum disorder. Biol Psychiatry. 2008;63:1111–7.
Stoppel LJ, Kazdoba TM, Schaffler MD, Preza AR, Heynen A, Crawley JN, et al. R-Baclofen reverses cognitive deficits and improves social interactions in two lines of 16p11.2 deletion mice. Neuropsychopharmacology. 2018;43:513–24.
Bochukova EG, Huang N, Keogh J, Henning E, Purmann C, Blaszczyk K, et al. Large, rare chromosomal deletions associated with severe early-onset obesity. Nature. 2010;463:666–70.
Walters RG, Jacquemont S, Valsesia A, de Smith AJ, Martinet D, Andersson J, et al. A new highly penetrant form of obesity due to deletions on chromosome 16p11.2. Nature. 2010;463:671–5.
Maillard AM, Hippolyte L, Rodriguez-Herreros B, Chawner SJ, Dremmel D, Aguera Z, et al. 16p11.2 Locus modulates response to satiety before the onset of obesity. Int J Obes 2016;40:870–6.
Yu Y, Zhu H, Miller DT, Gusella JF, Platt OS, Wu BL, et al. Age- and gender-dependent obesity in individuals with 16p11.2 deletion. J Genet Genomics. 2011;38:403–9.
Perrone L, Marzuillo P, Grandone A, del Giudice EM. Chromosome 16p11.2 deletions: another piece in the genetic puzzle of childhood obesity. Ital J Pediatr. 2010;36:43.
Rui L. SH2B1 regulation of energy balance, body weight, and glucose metabolism. World J Diab. 2014;5:511–26.
Jiang L, Su H, Wu X, Shen H, Kim MH, Li Y, et al. Leptin receptor-expressing neuron Sh2b1 supports sympathetic nervous system and protects against obesity and metabolic disease. Nat Commun. 2020;11:1517.
Vysotskiy M, Zhong X, Miller-Fleming TW, Zhou D, Cox NJ, Weiss LA. Integration of genetic, transcriptomic, and clinical data provides insight into 16p11.2 and 22q11.2 CNV genes. Genome Med. 2021;13:172.
Consortium ASDWG of TPG. Meta-analysis of GWAS of over 16,000 individuals with autism spectrum disorder highlights a novel locus at 10q24.32 and a significant overlap with schizophrenia. Mol Autism. 2017;8:21.
Liu M, Khasiyev F, Sariya S, Spagnolo-Allende A, Sanchez DL, Andrews H, et al. Chromosome 10q24.32 variants associate with brain arterial diameters in diverse populations: a genome-wide association study. medRxiv. 2023. https://doi.org/10.1101/2023.01.31.23285251.
Wang J, Liu J, Li S, Li X, Yang J, Dang X, et al. Genetic regulatory and biological implications of the 10q24.32 schizophrenia risk locus. Brain. 2023;146:1403–19.
Vishnolia KK, Hoene C, Tarhbalouti K, Revenstorff J, Aherrahrou Z, Erdmann J. Studies in zebrafish demonstrate that CNNM2 and NT5C2 are most likely the causal genes at the blood pressure-associated locus on human chromosome 10q24.32. Front Cardiovasc Med. 2020;7:135.
Zhang PP, Sun J, Li W. Genome-wide profiling reveals atrial fibrillation-related circular RNAs in atrial appendages. Gene. 2020;728:144286.
Xu C, Aragam N, Li X, Villla EC, Wang L, Briones D, et al. BCL9 and C9orf5 are associated with negative symptoms in schizophrenia: meta-analysis of two genome-wide association studies. PLoS ONE. 2013;8:e51674.
Voisin S, Almen MS, Zheleznyakova GY, Lundberg L, Zarei S, Castillo S, et al. Many obesity-associated SNPs strongly associate with DNA methylation changes at proximal promoters and enhancers. Genome Med. 2015;7:103.
Tang Y, Jin B, Zhou L, Lu W. MeQTL analysis of childhood obesity links epigenetics with a risk SNP rs17782313 near MC4R from meta-analysis. Oncotarget. 2017;8:2800–6.
Kogelman LJ, Zhernakova DV, Westra HJ, Cirera S, Fredholm M, Franke L, et al. An integrative systems genetics approach reveals potential causal genes and pathways related to obesity. Genome Med. 2015;7:105.
Brotman SM, Raulerson CK, Vadlamudi S, Currin KW, Shen Q, Parsons VA, et al. Subcutaneous adipose tissue splice quantitative trait loci reveal differences in isoform usage associated with cardiometabolic traits. Am J Hum Genet. 2022;109:66–80.
Brotman SM, El-Sayed Moustafa JS, Guan L, Broadaway KA, Wang D, Jackson AU, et al. Adipose tissue eQTL meta-analysis reveals the contribution of allelic heterogeneity to gene expression regulation and cardiometabolic traits. bioRxiv. 2023. https://doi.org/10.1101/2023.10.26.563798.
Zhang X, Zhou Z, Xu H, Liu CT. Integrative clustering methods for multi-omics data. Wiley Interdiscip Rev Comput Stat. 2022;14. https://doi.org/10.1002/wics.1553.
Wang B, Lunetta KL, Dupuis J, Lubitz SA, Trinquart L, Yao L, et al. Integrative omics approach to identifying genes associated with atrial fibrillation. Circ Res. 2020;126:350–60.
Clark C, Dayon L, Masoodi M, Bowman GL, Popp J. An integrative multi-omics approach reveals new central nervous system pathway alterations in Alzheimer’s disease. Alzheimers Res Ther. 2021;13:71.
Wang F, Panjwani N, Wang C, Sun L, Strug LJ. A flexible summary statistics-based colocalization method with application to the mucin cystic fibrosis lung disease modifier locus. Am J Hum Genet. 2022;109:253–69.
Foley CN, Staley JR, Breen PG, Sun BB, Kirk PDW, Burgess S, et al. A fast and efficient colocalization algorithm for identifying shared genetic risk factors across multiple traits. Nat Commun. 2021;12:764.
Acknowledgements
Individual Acknowledgements: SG, ID, GC, CTL, KEN, AEJ, CDS, TM, KLM were funded in part by NIH R01 DK122503. MYA was funded by NIH NIDDK 3R01DK122503 – 02W1. MG was supported in part by NIH R01HL163262. JIR was funded in part by National Center for Minority Health Disparity (NCMHD), MD000170P20, McCormick. In part, KEN was funded by NIH R01HL142302, R01HL151152, R01HD057194, R01HG010297, and R01HL143885. AD was in part supported by NIH 1R01DK099269. Study Acknowledgements: The Framingham Heart Study (FHS): The FHS is funded by National Institutes of Health contract N01-HC-25195. The laboratory work for this investigation was funded by the Division of Intramural Research, National Heart, Lung, and Blood Institute, National Institutes of Health, Bethesda, MD. The analytical component of this project was funded by the Division of Intramural Research, National Heart, Lung, and Blood Institute, and the Center for Information Technology, National Institutes of Health, Bethesda, MD. The visualization tools and data resources for this project were funded by the National Center for Biotechnology Information, National Institutes of Health, Bethesda, MD. The FHS acknowledges the support of contracts NO1-HC-25195, HHSN268201500001I and 75N92019D00031 from the National Heart, Lung and Blood Institute and grant supplement R01 HL092577-06S1 for this research. We also acknowledge the dedication of the FHS study participants without whom this research would not be possible. This manuscript does not necessarily reflect the opinions or views of the NHLBI, NIH or DHHS. The Framingham Brain Donation program is funded by R01 AG054076 and previously by AG008122 with additional support for the brain bank by NIA grants RF1AG054156 and P30AG13846. Cameron County Hispanic Community (CCHC) Cohort: We thank our cohort team including the CRU, data and lab staff of the CCHC team. We thank Valley Baptist Medical Center, Brownsville, for providing the space for our Clinical Research Unit. CCHC study and team members were funded by: National Institutes of Health, National Center for Minority Health Disparity, MD000170P20, UL1 TR000371, National Heart, Lung, and Blood Institute, 1R01HL142302, 2R01HL142302, National Center for Advancing Translational Sciences, (NCATS), Clinical and Translational Science Awards (CTSA), UL1 9/30/06-6/30/11, UL1TR0003716/27/12-/31/17, UL1TR0031677/1/19-6/30/2. MyCode Bariatric Surgery Cohort: We thank all the participants of the MyCode Community Health Initiative Study (MyCode) and the MyCode Research Team. We thank the members of the Geisinger-Regeneron DiscovEHR Collaboration who have been critical in the generation of the genetic and transcriptomic data used in this study. This study was funded in part by NIH HL142302, NIH 2R01HL142302, National Center for Advancing Translational Sciences, UL1 9/30/06-6/30/11, UL1TR0003716/27/12-/31/17, UL1TR0031677/1/19-(NCATS), Clinical and Translational Science Awards (CTSA). Religious Orders Study (ROS) and the Rush Memory and Aging Project (MAP): Funding for ROS and MAP was provided by National Institute of Aging (NIA) grants P30AG10161, P30AG72975, R01AG15819, R01AG17917, U01AG46152, and U01AG61356.
Author information
Authors and Affiliations
Contributions
Participate in project concept and design: LAC, KEN, AEJ, CTL. Participate in parent study concept and design: JBM, JIR, LAC, KEN. Participate in phenotype data acquisition and/or QC: HX, ID, SPFH, RJFL, NHC, JBM, TM, CDS, LAC, AD, KEN, AEJ, CTL. Participate in genotype or gene expression data acquisition and/or QC: HX, ID, MYA, HHC, LEP, MG, GC, HL, NHC, SPFH, AD, RJFL, JBM, TM, CDS, LAC, KEN, AEJ, CTL. Participate in data analysis and interpretation: HX, SG, ID, MYA, RJFL, KLM, KEN, AEJ, CTL. Drafted the manuscript and revised according to co-author suggestions: HX, SG, SPFH, KEN, AEJ, CTL. All authors critically reviewed the manuscript, suggested revisions as needed, and approved the final version.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Ethics approval and consent to participate
All participants provided written informed consent. This study was approved by each corresponding institutional committee: the Institutional Review Board of the Boston University Medical Campus, the Committee for the Protection of Human Subjects (CPHS) at the University of Texas Health Sciences Center at Houston, and the Geisinger Institutional Review Board. All experiments were performed in accordance with relevant guidelines and regulations.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Xu, H., Gupta, S., Dinsmore, I. et al. Integrating genetic and transcriptomic data to identify genes underlying obesity risk loci. Int J Obes 49, 2346–2357 (2025). https://doi.org/10.1038/s41366-025-01898-z
Received:
Revised:
Accepted:
Published:
Version of record:
Issue date:
DOI: https://doi.org/10.1038/s41366-025-01898-z
