
Abstract
Sarcomas are driven by diverse pathogenic mechanisms, including gene rearrangements in a subset of cases. Rare soft tissue sarcomas containing KMT2A fusions have recently been reported, characterized by a predilection for young adults, sclerosing epithelioid fibrosarcoma-like morphology, and an often aggressive course. Nonetheless, clinicopathologic and molecular descriptions of KMT2A-rearranged sarcomas remain limited. In this study, we identified by targeted next-generation RNA sequencing an index patient with KMT2A fusion-positive soft tissue sarcoma. In addition, we systematically searched for KMT2A structural variants in a comprehensive genomic profiling database of 14,680 sarcomas interrogated by targeted next-generation DNA and/or RNA sequencing. We characterized the clinicopathologic and molecular features of KMT2A fusion-positive sarcomas, including KMT2A breakpoints, rearrangement partners, and concurrent genetic alterations. Collectively, we identified a cohort of 34 sarcomas with KMT2A fusions (0.2%), and YAP1 was the predominant partner (n = 16 [47%]). Notably, a complex rearrangement with YAP1 consistent with YAP1–KMT2A–YAP1 fusion was detected in most cases, with preservation of KMT2A CxxC-binding domain in the YAP1–KMT2A–YAP1 fusion and concurrent deletions of corresponding exons in KMT2A. The tumors often affected younger adults (age 20–66 [median 40] years) and histologically showed variably monomorphic epithelioid-to-spindle shaped cells embedded in a dense collagenous stroma. Ultrastructural evidence of fibroblastic differentiation was noted in one tumor examined. Our cohort also included two sarcomas with VIM–KMT2A fusions, each harboring concurrent mutations in CTNNB1, SMARCB1, and ARID1A and characterized histologically by sheets of spindle-to-round blue cells. The remaining 16 KMT2A-rearranged sarcomas in our cohort exhibited diverse histologic subtypes, each with unique novel fusion partners. In summary, KMT2A-fusion-positive sarcomas most commonly exhibit sclerosing epithelioid fibrosarcoma-like morphology and complex YAP1–KMT2A–YAP1 fusions. Cases also include rare spindle-to-round cell sarcomas with VIM–KMT2A fusions and tumors of diverse histologic subtypes with unique KMT2A fusions to non-YAP1 non-VIM partners.
Similar content being viewed by others

Introduction
Sarcomas are mesenchymal tumors driven by diverse pathogenic mechanisms, including gene rearrangements in a subset of cases [1]. A fraction of sarcomas remain unclassified or difficult to classify based on histologic and immunohistochemical profile, with no putative driver mutation identified [2]. Recently, oncogenic rearrangements involving Lysine methyltransferase 2A (KMT2A), a frequent fusion partner in some acute leukemias, have been reported in solid tumors, including types B2 and B3 thymomas and soft tissue sarcomas [3,4,5,6,7]. Yoshida et al. first described two soft tissue sarcomas with KMT2A rearrangements, one each to the partners yes-associated protein 1 (YAP1) and vimentin (VIM) [3]. Two subsequent studies described YAP1 as a recurrent fusion partner in KMT2A-rearranged sarcomas that showed histologic features reminiscent of sclerosing epithelioid fibrosarcoma, and in some cases, low-grade fibromyxoid sarcoma [4, 6]. Nonetheless, clinicopathologic and molecular descriptions of KMT2A-rearranged sarcomas remain limited.
In this study, we described an index patient with a sarcoma that showed sclerosing epithelioid fibrosarcoma-like morphology and harbored rearrangements in KMT2A and YAP1. Prompted by this case and the recent literature, we queried KMT2A structural alterations in a comprehensive genomic profiling database of sarcomas and identified 33 additional KMT2A-rearranged sarcomas, including 15 additional tumors with similar sclerosing epithelioid fibrosarcoma-like morphology and complex rearrangements with YAP1, two cases with spindle-to-round cytomorphology and VIM–KMT2A fusions, and 16 sarcomas of diverse histologic subtypes each with unique novel fusion partners with KMT2A. We characterized the clinicopathologic and molecular features of KMT2A fusion-positive sarcomas, including KMT2A breakpoints, rearrangement partners, and concurrent genomic alterations.
Materials and methods
Characterization of index patient sarcoma
This part of the study was approved by the Partners Institutional Review Board (Protocol No. 2016P001180). Clinicopathologic and immunophenotypic features were reviewed. Electron microscopy was performed using a FEI Morgagni transmission electron microscope, with images captured with Advanced Microscopy Techniques 2K digital CCD camera, as previously described [8].
Next-generation sequencing-based anchored multiplex PCR was performed for targeted RNA fusion transcript detection as described [9], and targeted sequencing of genomic DNA was performed using Illumina (San Diego, CA) for detection of single nucleotide variants, insertion/deletion (indel), and copy number alterations (lists of gene targets in Supplementary Table 1). Briefly, RNA and DNA were first extracted from formalin-fixed paraffin-embedded tissue (Formapure RNA Isolation, Agencourt AMPure; Beckman Coulter Life Sciences, Indianapolis, IL). In the RNA-based assays, double-stranded cDNA was synthesized, end repaired, adenylated, and ligated with half-functional adapters; two hemi-nested PCR reactions using custom primers from ArcherDx (Boulder, CO) were performed. A laboratory-developed algorithm was used for fusion detection and annotation, with alignment to reference human genome (hg19) using bwa-mem [10].
Comprehensive genomic analysis and cohort
Comprehensive genomic profiling was performed in a Clinical Laboratory Improvement Amendments-certified, College of American Pathologists-accredited laboratory (Foundation Medicine, Inc., Cambridge, MA, USA). Approval for this study, including a waiver of informed consent and a HIPAA waiver of authorization, was obtained from the Western Institutional Review Board (Protocol No. 20152817). The presence of tumor was confirmed on the hematoxylin and eosin-stained slides prior to nucleic acid extraction and sequencing. DNA and RNA were extracted from 40 μm sections of formalin-fixed paraffin-embedded tissue. Comprehensive genomic profiling was performed using FoundationOne Heme® (F1H), a clinical grade, high-throughput, hybridization capture-based NGS assay for targeted sequencing of all exons of 406 genes, and RNA sequencing of 265 genes. F1H methods used have been previously reported in detail [11,12,13,14]. Sequences were analyzed for genomic alterations including short variant alterations, copy number alterations, and gene rearrangements as described [11,12,13,14]. Filtering of gene rearrangement candidates was performed by mapping quality (MQ > 30) and distribution of alignment positions (s.d. >10), along with manual review by expert analysts. Tumor mutational burden (mutations/Mb) was determined on 1.2 Mbp of sequenced DNA [13]. For sequence alignment of transcripts, the reference sequences for KMT2A and YAP1 used were NM_005933 and NM_006106, respectively; to be consistent with prior literature, the nomenclature was converted to NM_001197104 and NM_001130145, respectively.
After exclusion of hematologic-related myeloid/monocytic sarcoma, this cohort of non-hematologic soft tissue sarcomas harboring KMT2A fusions comprised 33 cases. Clinicopathological data including patient age, gender, tumor site, and stage (based on AJCC 8th edition) [15] were extracted from the accompanying pathology reports, with histologic review of representative hematoxylin and eosin-stained sections of all tumors by IC, YPH, LRM, EAW, and GPN.
Results
KMT2A–YAP1 fusion in the index patient with aggressive soft tissue sarcoma showing sclerosing epithelioid fibrosarcoma-like histology
A 42-year-old woman with no prior medical history presented with a palpable abdominal mass and associated discomfort. Abdominopelvic computed tomography demonstrated a 13.0 cm mass in the left upper quadrant with multiple peritoneal metastases (Fig. 1a). After the diagnostic biopsy (see below), the patient received palliative radiation and died of disease 7 months after presentation.

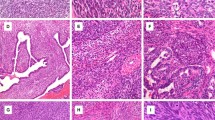
a Abdominopelvic computed tomography shows a 13.0 cm mass in the left upper quadrant. b Histology shows tumor cells infiltrating around fibroadipose tissue, with fibrotic stroma (H&E, 40×). c A variably epithelioid-to-spindly region of tumor cells in a sclerotic stroma with gaping vessels and infiltration of eosinophils (H&E, 200×). d Tumor cells infiltrate among individual adipocytes and form round aggregates, replacing residual adipocytes surrounded by sclerosis (H&E, 400×). e Epithelioid-to-spindle tumor cells contain moderate amounts of eosinophilic cytoplasm, large nuclei, vesicular chromatin, and often prominent nucleoli (H&E, 400×). f A focal region of tumor cells shows haphazard arrangement and feathery collagenous stroma (H&E, 400×). g By immunohistochemistry, tumor cells show diffuse nuclear cyclin D1 staining (400×). h, i Ultrastructural analysis shows an extracellular collagenous matrix, abundant dilated endoplasmic reticulum (h, 8900×), and small aggregates of filaments (i, 14000×).
The biopsy demonstrated an infiltrative sarcoma with spindled-to-epithelioid cytomorphology within a dense hyalinized-to-sclerotic background, histologically reminiscent of sclerosing epithelioid fibrosarcoma. Variable cytologic and architectural patterns were seen, including varying tumor cell density and deceptively bland-appearing pauci-cellular areas (Fig. 1b–f). By immunohistochemistry, tumor cells demonstrated strong nuclear cyclin D1 expression (Fig. 1g) and multifocal smooth muscle actin staining but were negative for MUC4, ERG, CD34, S-100 protein, desmin, STAT6, CD45, MDM2, CDK4, ALK, and ROS1. By electron microscopy, tumor cells showed evidence of fibroblastic differentiation, with prominent dilated rough endoplasmic reticulum, aggregates of filaments, and cohesion as joined by small junctions (Fig. 1h, i). Cytogenetic analysis performed on a fresh tumor sample showed a normal karyotype in 15 metaphases.
Using an RNA-based fusion assay (Supplementary Table 1d), we identified an in-frame oncogenic fusion between KMT2A exon 6 (5′) and YAP1 exon 9 (3′). Due to the primer design, this assay could not assess breakpoints that contain KMT2A as a 3′ partner. Additional RNA-based fusion assays (using non-KMT2A gene targets; Supplementary Table 1a, b) and targeted DNA-based next-generation sequencing (Supplementary Table 1c) identified no additional pathologic alterations, including EWSR1 or FUS rearrangements.
Soft tissue sarcomas with complex rearrangements between YAP1 and KMT2A
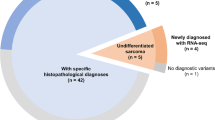
To assess the overall frequency of KMT2A structural alterations in sarcomas, we queried a genomic profiling database of 14,680 sarcomas, in which there was coverage of KMT2A by DNA and/or RNA-based sequencing methods. We identified 33 (0.2%) soft tissue sarcomas beyond the index case that harbored KMT2A rearrangements. This cohort included 15 additional soft tissue sarcomas with complex rearrangements between YAP1 and KMT2A (Table 1), two soft tissue sarcomas with VIM–KMT2A (Supplementary Table 2, patients #S1–S2), and 16 soft tissue sarcomas with KMT2A fusions to unique non-YAP1 non-VIM partners (Supplementary Table 2, patients #S3–S18).
Collectively, among the 16 patients with YAP1–KMT2A fusion-positive sarcomas, 56% were female, and ages ranged from 20 to 66 (median 40) years. Primary tumor locations included abdomen/peritoneum/retroperitoneum (36%), hip/back/buttock (29%), upper or lower extremities (21%), and chest/scalp (14%). This cohort included 13 patients with stage IV (81%), one patient with stage III (6%), and two patients with stage II disease (13%). Of sixteen tumors, the submitting pathologic diagnoses included “sclerosing epithelioid fibrosarcoma” in five, “sclerosing fibrosarcoma” in one, and descriptive diagnoses that included “sarcoma,” “favor sarcoma,” or “spindle cell neoplasm” in the remainder ten cases (including the descriptor “epithelioid” in six cases and “fibrosarcoma” in one case).
Histology of this cohort (available for 15 of 16 tumors) demonstrated features resembling sclerosing epithelioid fibrosarcoma in all tumors, with variably monomorphic epithelioid-to-spindle cells embedded in a collagenous stroma (Fig. 2a–c). Characteristic features of low-grade fibromyxoid sarcoma, such as arcade-like vessels and variably prominent myxoid background, were absent. The index case was remarkable for a small region with feathery-appearing stromal collagen and showed staghorn hemangiopericytoma-like vasculature multifocally. Accompanying pathology reports specified that immunohistochemistry for MUC4 was negative in all four tumors tested, and that break-apart FISH for EWSR1 rearrangements was negative in all four tested cases.

a–c Representative histology of three YAP1–KMT2A fusion-positive sarcomas with variably monomorphic epithelioid-to-spindle cells, morphologically reminiscent of sclerosing epithelioid fibrosarcoma (a H&E, 200×; b, c H&E, 400×). d, e Schematic of complex YAP1–KMT2A–YAP1 rearrangement, with variant I (d) and variant II (e) fusions along with co-occurring KMT2A deletions.
Fusions between YAP1 and KMT2A were detected by RNA-based assays in all 16 sarcomas, six of which were additionally confirmed by orthogonal DNA-based assays (Table 1). In each of the 15 sarcomas in which both the 5′ and 3′ breakpoints of KMT2A could be evaluated, two sets of breakpoints involving KMT2A and YAP1 were identified. In 13 tumors (patients #2–14), the first sets of breakpoints corresponded to fusion of YAP1 exon 5 to KMT2A exon 4, and the second sets of breakpoints corresponded to fusion of KMT2A exon 6 to YAP1 exon 9 (variant I), indicative of insertion of KMT2A exons 4–6 into YAP1 and loss of YAP1 exons 6–8. Of these, eight tumors had separate KMT2A deletions of exons 4–6 detected at the DNA and/or RNA level (Table 1).
In the remaining two tumors (patients #15–16), the first sets of breakpoints corresponded to fusion of YAP1 exon 3 to KMT2A exon 5, and the second sets of breakpoints corresponded to fusion of KMT2A exon 6 to YAP1 exon 5 (variant II), indicative of insertion of KMT2A exons 5–6 into YAP1 between exons 3 and 5. In one of these two tumors, separate KMT2A deletion of exon 5–6 was detected.
No gene amplification of KMT2A was identified. Concurrent genomic alterations included homozygous loss of CDKN2A in eight tumors, homozygous loss of RB1 in two tumors, and MDM2 amplification in two tumors (Table 1). Median tumor mutational burden (TMB) was 1.2 mutations/Mb (range <0.8–5.7).
Soft tissue sarcomas with VIM–KMT2A fusion
Two soft tissue sarcomas with VIM–KMT2A fusion were identified (Supplementary Table 2, patients #S1–S2); both occurred in men, aged 43 and 46 years, and involved the lower extremities. Histologically, both were round and spindle cell sarcomas, characterized by monomorphic spindled-to-round cells with moderate amounts of eosinophilic cytoplasm (Fig. 3a, b). VIM was identified as the 5′ fusion partner to KMT2A, with breakpoints near exon 4 and exon 2 respectively (Fig. 3c). Both tumors harbored concurrent mutations in CTNNB1, SMARCB1, and ARID1A (Supplementary Table 2). TMB was 0.8 and 1.6 mutations/Mb.

a, b Representative histology of two VIM–KMT2A sarcomas with spindle-to-round cell morphology (a H&E, 200×; b H&E, 400×). c Schematic of VIM–KMT2A rearrangement.
Diverse soft tissue sarcomas with KMT2A fusion to non-YAP1 Non-VIM partners
Sixteen tumors of diverse histologic subtypes were identified with KMT2A fusion to nonrecurrent partners (Supplementary Table 2, patients #S3–S18). These tumors affected patients across a wide age range (6–70 [median 57] years) and encompassed diverse histologic subtypes, including leiomyosarcoma, atypical lipomatous tumor, and undifferentiated pleomorphic sarcoma (Fig. 4a–d), among others. None of these 16 tumors demonstrated histologic features of sclerosing epithelioid fibrosarcoma or low-grade fibromyxoid sarcoma. KMT2A was the 3′ fusion partner in eight tumors and the 5′ fusion partner in seven tumors, with no reciprocal transcripts identified in any of the cases. The remaining tumor (patient #S7) showed separate fusion partners to KMT2A 5′ and 3′ fragments. Pathogenic TP53 mutation was the most common co-occurring mutation, present in seven (44%) tumors in this group. Median TMB was 3.2 mutations/Mb (range <0.8–8.1).

a Giant-cell rich pleomorphic sarcoma (case #S5; H&E, 200×). b Atypical lipomatous tumor (case #S10; H&E, 200×). c Undifferentiated pleomorphic sarcoma (case #S13; H&E, 200×). d High-grade endometrial stromal sarcoma (case #S17; H&E, 200×).
Discussion
In this study, we described 34 soft tissue sarcomas with KMT2A rearrangements. YAP1 is a commonly recurrent partner to KMT2A, and YAP1–KMT2A fusion-positive sarcomas exhibit sclerosing epithelioid fibrosarcoma-like histomorphology. We found that YAP1–KMT2A rearrangements are complex in configuration, showing two reciprocally oriented breakpoints and often concurrent KMT2A exon deletions, altogether in keeping with a complex YAP1–KMT2A–YAP1 fusion. We also described the features of VIM–KMT2A sarcomas as well as sarcomas of diverse histologic subtypes that harbored unique nonrecurrent KMT2A fusions of unclear significance.
Sarcomas containing complex rearrangements of YAP1 and KMT2A
This study augments the clinicopathologic descriptions of YAP1–KMT2A fusion-positive sarcomas [3, 4, 6]. YAP1–KMT2A fusion-positive sarcomas may behave aggressively, given the rapid decline in the index patient and the advanced tumor stage in most cases. A limitation of our study is that the queried database is enriched for aggressive cases and advanced disease where genetic testing is desired for therapeutic target identification. From 11 patients with available follow-up in the three prior studies, three showed no recurrence after initial resection, five recurred locally or with unresectable tumors, and three harbored metastatic disease; altogether, three of 11 reported patients died of disease [3, 4, 6].
YAP1–KMT2A fusion-positive sarcomas contained histologic patterns reminiscent of sclerosing epithelioid fibrosarcoma. While low-grade fibromyxoid sarcoma-like histology was noted in three of seven cases in one study [6], this was not observed in other studies [3, 4]. Our histologic review, limited to a representative section in most cases, precluded full assessment of such “hybrid” features. However, none of the tumors received a histologic diagnosis of low-grade fibromyxoid sarcoma rendered by the submitting pathologist. Immunohistochemical expression of cyclin D1, as noted in the index case, might represent a diagnostic clue. The lack of MUC4 expression in the four tested tumors herein was also noted previously in all 13 sequencing-confirmed cases [3, 4, 6]. We additionally provided the first description of the ultrastructural features of YAP1–KMT2A fusion-positive sarcoma, which appeared similar to those reported in sclerosing epithelioid fibrosarcoma [16, 17]. Sclerosing epithelioid fibrosarcoma is characterized by MUC4 expression and recurrent EWSR1–CREB3L1 or FUS–CREB3L2 fusion in most cases [18]. While MUC4 status may be a useful discriminator between EWSR1/FUS-rearranged sclerosing epithelioid fibrosarcoma and YAP1–KMT2A fusion-positive tumors, additional studies to determine the performance of this immunohistochemical marker would be needed for definitive assessment. Furthermore, identification of YAP1–KMT2A fusion-positive soft tissue sarcomas with sclerosing epithelioid sarcoma-like histologic and ultrastructural features raises the question of whether these sarcomas should be classified as a distinct entity related to or within the spectrum of sclerosing epithelioid fibrosarcoma.
Detection of the complex YAP1–KMT2A rearrangement can be challenging. In prior studies, concurrent KMT2A and YAP1 rearrangements were noted in only one tumor tested by both break-apart fluorescence in situ hybridization (FISH) probes [3]; negative FISH results for KMT2A and/or YAP1 were common in cases where the fusion was confirmed by sequencing [4]. FISH testing is thus not sensitive for identifying YAP1–KMT2A fusion-positive sarcoma. Other detection methods include targeted next-generation DNA-based or RNA-based sequencing assays. While KMT2A is frequently included on the sequencing panels for hematolymphoid neoplasms, it is less often covered on solid tumor sequencing panels due to the low prevalence of KMT2A structural alterations in solid tumors, including sarcomas. Nonetheless, our findings suggest that cases with sclerosing epithelioid fibrosarcoma-like histology that lack EWSR1 and FUS rearrangements may be selected for additional testing that includes KMT2A or YAP1 coverage.
While prior studies have generally assumed the expression of two separate reciprocal transcripts and hypothesized the YAP1–KMT2A fusion alone to be pathogenic [3, 4, 6], published data show frequently discrepant sets of breakpoints between the YAP1–KMT2A transcripts and the KMT2A–YAP1 transcripts (see below), suggesting a complex rearrangement. Based on our analysis, we hypothesize a novel YAP1–KMT2A–YAP1 configuration (Fig. 2d, e), which may be accounted for by a “cut-and-paste” structural mechanism in some cases [19].
This complex YAP1–KMT2A–YAP1 fusion configuration is supported by multiple lines of reasoning and evidence. First, YAP1 is located 16 Mb centromeric to KMT2A on the same plus strand on chromosome 11q; reciprocal translocations between YAP1 and KMT2A could not be accounted for by a single intrachromosomal inversion event. Deletion between 5′ YAP and 3′ KMT2A could also generate an in-frame fusion but would not account for the two sets of fusion breakpoints observed. We detected two sets of reciprocally oriented breakpoints involving KMT2A and YAP1 in every fully evaluated case, along with frequent concurrent exon deletion of KMT2A involving the same exons implicated in the complex fusion. In fact, a review of all previously published YAP1–KMT2A fusion-positive sarcomas with reported breakpoints confirmed our observations, with KMT2A exons 5–6 (or exons 4–6) present in all predicted full-length transcripts and shared between sets of reciprocals (Table 1; patients #17–29), except in patient case #19; this lone exception demonstrated an out-of-frame 5′ KMT2A transcript lacking exon 6, similar to a low-level splice variant seen in the index case absent of KMT2A exon 6. Otherwise, the inclusion of KMT2A exons 5–6 (which encodes the CxxC-binding domain) within a YAP1–KMT2A–YAP1 fusion characterized all eight prior cases reporting both breakpoints, similar to current cases [3, 4, 6].
Second, given that the KMT2A CxxC-binding domain is integral to HOX gene regulation, it is notable that no increase in HOXA-related gene expression was detected by RNA sequencing in three YAP1–KMT2A fusion-positive sarcomas in a prior study [4]. KMT2A (formerly MLL) belongs to the polymerase associated factor complex (PAFc) [20], which modifies chromatins and regulates target genes such as HOX [21,22,23]. In ~10% of acute leukemias in which KMT2A is fused to one of >80 fusion partners [20, 24], KMT2A fusion breakpoints are located primarily within exons 8–14 (the so-called “breakpoint cluster region”) [20, 22, 25], preserving both the CxxC-binding domain in exons 5–6 and the adjacent repression domain 2 (RD2) largely encoded by exons 7–8. The latter is required for interaction with PAFc and engagement of transcription machinery at downstream targets critical to leukemogenesis, including HOXA cluster genes [22, 25]. The lack of HOXA-related gene expression in those three YAP1–KMT2A fusion-positive sarcomas argues against a simple YAP1–KMT2A fusion that preserves these domains, but rather a complex YAP1–KMT2A–YAP1 fusion that retains the CxxC-binding domain but lacks the RD2 domain. Given the importance of the CxxC-binding domain in the pathogenesis of acute leukemias, the consistent retention of the CxxC-binding domain in YAP1–KMT2A–YAP1 fusion suggests that it plays a key role in the pathogenesis of these sarcomas [25, 26].
In addition to KMT2A exons 5–6, the YAP1–KMT2A–YAP1 construct retains at least YAP1 exons 1–3 and exon 9, with the former encoding the TEAD-binding domain. This domain is responsible for the downstream activation of TEAD transcription factors documented in other YAP1-related oncogenic fusions [27]. YAP1 exon 9 contains a PDZ-binding motif that has been demonstrated to play a role in some forms of YAP1-mediated oncogenesis, which is required for TEAD-associated transcription of the cell proliferation gene connective tissue growth factor (CCN2/CTGF) [28]. Interestingly, CCN2 encodes for a matricellular protein that is ubiquitously overexpressed in fibrotic diseases, and overexpression in fibroblasts is sufficient to cause fibrosis in tissues of multiple types [29].
Sarcomas with VIM–KMT2A rearrangements and other nonrecurrent partners to KMT2A
Among the remaining KMT2A rearrangements in this study, only the VIM–KMT2A fusion was recurrent (n = 2) and was associated with small round-to-spindle histology. The only previously published VIM–KMT2A fusion-positive sarcoma [3] showed similar histology and expressed NKX2.2 and EMA, both of which were also expressed in one of our cases herein and may represent potential diagnostic adjuncts. Collectively, these three VIM–KMT2A sarcomas presented in young adult males in the lower extremities. VIM, located on chromosome 10p13, encodes vimentin and has been described in fusions with FOS in a subset of epithelioid hemangioma [30, 31]. The initial study on a VIM–KMT2A sarcoma demonstrated high expression of KMT2A and the downstream target HOXA genes [3], likely driven by the highly active VIM promoter in the VIM–KMT2A fusion, with retention of both CxxC-binding and RD2 domains in KMT2A that allow PAFc-associated chromatin modification of HOX-related genes.
In the remaining sarcomas with KMT2A fusions to non-YAP1 non-VIM partners, none of the gene partners were recurrent or previously described, and these sarcomas harbored diverse histologic diagnoses. While leiomyosarcoma was the most common diagnosis, there was no clear unifying theme, and none showed sclerosing epithelioid fibrosarcoma-like histology as in YAP1–KMT2A fusion-positive sarcomas or small round blue cell-like histology in VIM–KMT2A fusion-positive sarcomas. The rarity of nonrecurrent KMT2A fusions in histologically diverse sets of sarcomas underscores the importance of correlating molecular results with clinicopathologic parameters when reaching the diagnosis. Given their rarity and the frequent presence of diverse co-occurring mutations, these nonrecurrent KMT2A fusions may represent sequelae of genomic instability rather than bona fide oncogenic drivers. The fusions have not been corroborated by orthogonal techniques, and the significance of these nonrecurrent fusions should be interpreted with caution.
In conclusion, in a comprehensive genomic profiling database of 14,680 sarcomas, KMT2A rearrangements were observed in 0.2% of cases. Most KMT2A fusions involved complex rearrangements with YAP1, consistent with a YAP1–KMT2A–YAP1 fusion configuration. YAP1–KMT2A fusion-positive sarcomas primarily affected young adults and showed a sclerosing epithelioid fibrosarcoma-like histology. VIM–KMT2A sarcomas also showed a predilection for young adults, however with a spindle-to-round cell morphology. Other unique and nonrecurrent KMT2A fusions occurred in sarcomas of diverse histologic subtypes with unclear significance. Identification of sarcomas with pathogenic KMT2A fusions raises a possibility of targeted therapies which are actively being pursued in KMT2A-rearranged leukemias [32, 33]. Comprehensive genomic profiling of sarcomas may aid the characterization of recurrent complex alterations and enable precise tumor classification in conjunction with their clinicopathologic contexts.
References
Fletcher C, Bridge J, Hogendoorn P, Mertens F, editors. WHO classification of tumours of soft tissue and bone. Pathology and genetics of tumours of soft tissue and bone. 4th ed. Lyon: IARC Press; 2013.
Mastrangelo G, Coindre JM, Ducimetière F, Dei Tos AP, Fadda E, Blay JY, et al. Incidence of soft tissue sarcoma and beyond: a population-based prospective study in 3 European regions. Cancer. 2012;118:5339–48.
Yoshida A, Arai Y, Tanzawa Y, Wakai S, Hama N, Kawai A, et al. KMT2A (MLL) fusions in aggressive sarcomas in young adults. Histopathology. 2019;75:508–19.
Kao YC, Lee JC, Zhang L, Sung YS, Swanson D, Hsieh TH, et al. Recurrent YAP1 and KMT2A gene rearrangements in a subset of MUC4-negative sclerosing epithelioid fibrosarcoma. Am J Surg Pathol. 2019;44:368–77.
Watson S, Perrin V, Guillemot D, Reynaud S, Coindre JM, Karanian M, et al. Transcriptomic definition of molecular subgroups of small round cell sarcomas. J Pathol. 2018;245:29–40.
Puls F, Agaimy A, Flucke U, Ploegmakers M, Stoehr R, Kindblom L, et al. Recurrent fusions between YAP1 and KMT2A in morphologically distinct neoplasms within the spectrum of low-grade fibromyxoid sarcoma and sclerosing epithelioid fibrosarcoma. Am J Surg Pathol. 2020;44:594–606.
Massoth LR, Hung YP, Dias-santagata D, Onozato M, Shah N, et al. Pan-Cancer landscape analysis reveals recurrent KMT2A-MAML2 gene fusion in aggressive histologic subtypes of thymoma. JCO Precis Oncol. 2020;4:109–15.
Massoth LR, Selig MK, Little BP, Chebib I, Kradin RL. Multiple calcifying fibrous pseudotumors of the pleura: ultrastructural analysis provides insight on mechanism of dissemination. Ultrastruct Pathol. 2019;43:154–61.
Zheng Z, Liebers M, Zhelyazkova B, Cao Y, Panditi D, Lynch KD, et al. Anchored multiplex PCR for targeted next-generation sequencing. Nat Med. 2014;20:1479–84.
Li H, Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics. 2009;25:1754–60.
Frampton GM, Fichtenholtz A, Otto GA, Wang K, Downing SR, He J, et al. Development and validation of a clinical cancer genomic profiling test based on massively parallel DNA sequencing. Nat Biotechnol. 2013;31:1023–31.
Sun JX, He Y, Sanford E, Montesion M, Frampton GM, Vignot S, et al. A computational approach to distinguish somatic vs. germline origin of genomic alterations from deep sequencing of cancer specimens without a matched normal. PLoS Pathog. 2018;14:e1005965.
Chalmers ZR, Connelly CF, Fabrizio D, Gay L, Ali SM, Ennis R, et al. Analysis of 100,000 human cancer genomes reveals the landscape of tumor mutational burden. Genome Med. 2017;9:34.
He J, Abdel-Wahab O, Nahas MK, Wang K, Rampal RK, Intlekofer AM, et al. Integrated genomic DNA/RNA profiling of hematologic malignancies in the clinical setting. Blood. 2016;127:3004–14.
Amin MB, Edge S, Greene F, Byrd DR, Brookland RK, Washington MK, et al. AJCC cancer staging system. 8th ed. New York: Springer; 2017.
Jiao YF, Nakamura SI, Sugai T, Uesugi N, Habano W, Ogata M, et al. Overexpression of MDM2 in a sclerosing epithelioid fibrosarcoma: genetic, immunohistochemical and ultrastructural study of a case. Pathol Int. 2002;52:135–40.
Donner LR, Clawson K, Dobin SM. Sclerosing epithelioid fibrosarcoma: a cytogenetic, immunohistochemical, and ultrastructural study of an unusual histological variant. Cancer Genet Cytogenet. 2000;119:127–31.
Arbajian E, Puls F, Magnusson L, Thway K, Fisher C, Sumathi VP, et al. Recurrent EWSR1–CREB3L1 gene fusions in sclerosing epithelioid fibrosarcoma. Am J Surg Pathol. 2014;38:801–8.
Li Y, Roberts ND, Wala JA, Shapira O, Schumacher SE, Kumar K, et al. Patterns of somatic structural variation in human cancer genomes. Nature. 2020;578:112–21.
Winters AC, Bernt KM. MLL-rearranged leukemias—an update on science and clinical approaches. Front Pediatr. 2017;5:11–3.
Milne TA, Briggs SD, Brock HW, Martin ME, Gibbs D, Allis CD, et al. MLL targets SET domain methyltransferase activity to Hox gene promoters. Mol Cell. 2002;10:1107–17.
Tan J, Muntean AG, Hess JL. PAFc, a key player in MLL-rearranged leukemogenesis abstract. Oncotarget. 2010;1:461–5.
Butler LH, Slany R, Cui X, Cleary ML, Mason DY. The HRX proto-oncogene product is widely expressed in human tissues and localizes to nuclear structures. Blood. 1997;89:3361–70.
Meyer C, Burmeister T, Gröger D, Tsaur G, Fechina L, Renneville A, et al. The MLL recombinome of acute leukemias in 2017. Leukemia. 2018;32:273–84.
Muntean AG, Tan J, Sitwala K, Huang Y, Bronstein J, Connelly JA, et al. The PAF complex synergizes with MLL fusion proteins at HOX loci to promote leukemogenesis. Cancer Cell. 2010;17:609–21.
Ayton PM, Chen EH, Cleary ML. Binding to nonmethylated CpG DNA is essential for target recognition, transactivation, and myeloid transformation by an MLL oncoprotein. Mol Cell Biol. 2004;24:10470–8.
Sekine S, Kiyono T, Ryo E, Ogawa R, Wakai S, Ichikawa H, et al. Recurrent YAP1-MAML2 and YAP1-NUTM1 fusions in poroma and porocarcinoma. J Clin Investig. 2019;130:3827–32.
Shimomura T, Miyamura N, Hata S, Miura R, Hirayama J, Nishina H. The PDZ-binding motif of Yes-associated protein is required for its co-activation of TEAD-mediated CTGF transcription and oncogenic cell transforming activity. Biochem Biophys Res Commun. 2014;443:917–23.
Jun J, Lau L. Taking aim at the extracellular matrix. Nat Rev Drug Discov. 2011;1:945–63.
van Ijzendoorn DGP, de Jong D, Romagosa C, Picci P, Benassi MS, Gambarotti M, et al. Fusion events lead to truncation of FOS in epithelioid hemangioma of bone. Genes Chromosom Cancer. 2015;54:565–74.
Huang SC, Zhang L, Sung YS, Chen CL, Krausz T, Dickson BC, et al. Frequent FOS gene rearrangements in epithelioid hemangioma: a molecular study of 58 cases with morphologic reappraisal. Am J Surg Pathol. 2015;39:1313–21.
Somers K, Wen VW, Middlemiss SMC, Osborne B, Forgham H, Jung MS, et al. A novel small molecule that kills a subset of MLL-rearranged leukemia cells by inducing mitochondrial dysfunction. Oncogene. 2019;38:3824–42.
Chan AKN, Chen CW. Rewiring the epigenetic networks in MLL-rearranged leukemias: epigenetic dysregulation and pharmacological interventions. Front Cell Dev Biol. 2019;7:81.
Acknowledgements
The authors thank Dr. Sheng Xiao for helpful input on cytogenetic analysis; Drs. Tasos Gogakos and John Iafrate for helpful discussions on molecular analysis.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
DCP, ESS, BMA, JAV, JSR, and EAW are employees of Foundation Medicine, Inc., a wholly owned subsidiary of Roche Holdings, Inc. and Roche Finance Ltd; these employees have equity interest in an affiliate of these Roche entities. KJW receives research funding from Novo Nordisk for studies unrelated to cancer.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Massoth, L.R., Hung, Y.P., Nardi, V. et al. Pan-sarcoma genomic analysis of KMT2A rearrangements reveals distinct subtypes defined by YAP1–KMT2A–YAP1 and VIM–KMT2A fusions. Mod Pathol 33, 2307–2317 (2020). https://doi.org/10.1038/s41379-020-0582-4
Received:
Revised:
Accepted:
Published:
Version of record:
Issue date:
DOI: https://doi.org/10.1038/s41379-020-0582-4
This article is cited by
-
KMT2A-Mediated transcriptional regulation in stemness and cancer: molecular mechanisms and therapeutic opportunities
Medical Oncology (2025)
-
YAP1::KMT2A-rearranged sarcomas harbor a unique methylation profile and are distinct from sclerosing epithelioid fibrosarcoma and low-grade fibromyxoid sarcoma
Virchows Archiv (2025)
-
Clinical Utility of an RNA-based Gene Fusion Assay in Sarcoma for Diagnosis and Management: Experience in an Australian Laboratory
Molecular Diagnosis & Therapy (2025)
-
Introduction and impact of routine whole genome sequencing in the diagnosis and management of sarcoma
British Journal of Cancer (2024)
-
KMT2A-rearranged sarcoma with unusual fusion gene CBX6::KMT2A::PYGO1
Virchows Archiv (2023)
