
Abstract
Alzheimer’s disease is increasingly viewed as a breakdown of balanced excitation–inhibition (E/I) homeostasis layered atop classical proteinopathy. Restoring circuit-level neural excitation and inhibition is rapidly becoming a tractable therapeutic strategy, guiding trials of circuit-modulating drugs such as levetiracetam. To date, however, findings across species and modalities remain fragmented, and it is unclear how to contextualize AD-focused E/I findings across scales and methodologies. Synthesizing over 150 studies of E/I homeostasis in AD, we organize the results into several prevailing themes: excitatory/inhibitory effects of amyloid and tau, whether hyperexcitation precedes amyloid plaque deposition, progressive oscillatory slowing (a shift of aggregate neural signal frequencies towards lower frequencies) as AD worsens, early preclinical hyperexcitation peaking in MCI and transitioning to hypoexcitation in AD, sex differences in E/I trajectories, APOE4 as a mediating factor, the contribution of neuroinflammation and metabolic dysfunction to E/I imbalance, and E/I-focused trials/experiments, particularly involving levetiracetam. These dominant themes are interpreted in a framework of multidimensional E/I homeostasis, rather than a single-axis imbalance. To support this integration, we first outline the microscale, mesoscale, and macroscale techniques used to assess E/I in AD, ranging from patch clamping and extracellular recordings to EEG/MEG and fMRI. By charting these multiscale E/I shifts, our synthesis offers a unifying framework to guide future experimental work and accelerate the design of biomarker-driven trials of E/I-targeted therapies in Alzheimer’s disease.
Similar content being viewed by others

Introduction
Overview of Alzheimer’s Disease (AD) with links to sex differences and E/I balance
Alzheimer’s disease (AD) is a progressive, non-curable neurodegenerative disorder affecting 1 in 9 people age 65 and older in the U.S, with a two-fold greater prevalence in females [1, 2]. AD is the leading cause of dementia, representing a growing economic and caregiving burden, and a condition in desperate need of disease-modifying therapies. The clinical picture includes progressive memory loss, executive dysfunction, and behavioral changes [3], while the National Institute on Aging defines AD biologically by its core pathological features: amyloid plaques and tau tangles [4].
Current treatments such as acetylcholinesterase inhibitors (e.g., rivastigmine, donepezil), NMDA antagonists (e.g., memantine), and recent amyloid-targeting monoclonal antibodies offer limited or contested benefits [5, 6]. Meanwhile, major risk factors such as female sex and the genetic APOE4 allele (which confers 4x with one allele, 12x with two) point to unresolved mechanisms that remain unaddressed.
One promising framework reconceptualizes AD through the lens of excitation/inhibition (E/I) balance, which is a dynamic equilibrium that maintains neural circuits near a critical point for optimal information transfer and emergent behaviors like traveling cortical waves [7,8,9,10,11]. Rather than viewing AD solely as a proteinopathy, this perspective frames the disease as progressive destabilization of E/I homeostasis, where disrupted circuit dynamics both result from and contribute to pathological changes.
Epilepsy, in which seizures reflect runaway excitation, provides both a clinical model and an epidemiological tracer of E/I dysregulation. A meta-analysis by Chen et al. of 27 studies involving cohorts (~1000–3000 participants per study) found seizures in ~3% of AD cases, representing a 1.86-fold increased seizure risk. Early- and late-onset AD carried 5-year hazard ratios (compares how quickly an event happens in one group versus another over time) of 4.06 and 1.52, respectively, for epileptic events, both representing increased risk over time. Vice-versa, individuals with epilepsy had AD/dementia prevalences of 15%/17%, and a 2.83-fold increased risk of future dementia, representing a bidirectional risk increase between AD/dementia and epilepsy [12].
This meta-analysis stratifies risk not only for AD but also for related dementias (FTD—frontotemporal dementia, DLB—dementia with Lewy bodies, VD—vascular dementia). These findings highlight a central challenge: the specificity of E/I alterations remains unclear, raising the question of a shared, global E/I biomarker versus disorder-specific signatures across dementia subtypes and psychiatric illness. Notably, E/I changes are also reported in neurodevelopmental (e.g., autism, Down syndrome) [13,14,15,16,17,18,19] and neuropsychiatric disorders (e.g., schizophrenia, depression), and aging [17, 18, 20].
The challenge of specificity, across aging and neuropsychiatric disorders with E/I changes and within putative AD subtypes, calls for a rigorous classification framework. This framework should map E/I readouts across scales (single cell, local circuits, whole-brain networks) and modalities (patch clamp, calcium imaging, magnetic resonance spectroscopy, fMRI), aligning measurements to shared constructs. Doing so would reduce the conflation of superficially similar phenotypes that reflect distinct alterations, enabling disease- and subtype-specific profiles that can be linked to mechanistic and therapeutic targets. Despite this caveat, the aforementioned meta-analysis strengthens the E/I-imbalance hypothesis and highlights candidate, therapeutically actionable targets in AD. Importantly, E/I modulation may be clinically accessible through noninvasive electrophysiological treatments or existing medications that target glutamatergic (excitatory) or GABAergic (inhibitory) signaling, such as currently approved antiepileptic agents (e.g., levetiracetam, brivaracetam, lamotrigine, topiramate).
At present, the traditional biomarker models of AD, such as the current National Institute of Aging–Alzheimer’s Association’s (NIA-AA) revised criteria for diagnosis and staging of AD [4], do not explicitly include E/I dysregulation in the AD trajectory. However, these frameworks acknowledge that E/I-related observations, such as synaptic dysfunction, map onto currently established imaging biomarkers of neurodegeneration as a later downstream consequence of amyloid and tau accumulation. Yet, emerging evidence has been integrated into the NIA-AA’s framework by some authors, suggesting a more reciprocal relationship: E/I imbalances may not only follow the pathology but also precede and promote it, forming a self-reinforcing positive feedback loop that shifts the paradigm from linear causality to dynamic systems interaction [21,22,23,24,25,26]. Therefore, it is also worth noting that the NIA-AA’s criteria have undergone several revisions since 2011, having incorporated several new biomarker categories, and will likely evolve with biomarkers related to E/I brain dynamics.
Purpose and scope of this review chapter
This review integrates otherwise siloed evidence on excitation–inhibition (E/I) dysregulation in Alzheimer’s disease into a single multiscale framework spanning molecules, cells, circuits, and whole-brain imaging. While prior reviews often focus on a single aspect of E/I imbalance, such as epilepsy risk, EEG slowing, amyloid or tau mechanisms, or mouse circuit phenotypes, we map how these phenomena fit within a dynamic model of homeostatic E/I control across disease stages. We use this scaffold to highlight points of convergence and controversy, including:
-
Excitatory/inhibitory roles of amyloid and tau
-
Timing of hyperexcitation relative to plaque/tangle formation
-
Oscillatory slowing across disease progression
-
Sex differences in E/I mechanisms
-
Role of APOE4 as a modulator
-
Neuroinflammatory and metabolic mechanisms
-
Therapeutic implications of levetiracetam and related agents
Sex-specific factors receive special attention, as they increasingly appear to affect prognosis and therapeutic response, yet remain underrepresented in existing literature. We acknowledge that there exist several other subtopics that are equally important to cover as they intricately connect E/I homeostasis and Alzheimer’s disease, either observationally or through mechanistic studies. These important topics include but are not limited to sleep, glymphatic clearance, and neuropeptides and neurotransmitter systems underlying E/I homeostasis, but remain out of scope for this selective review [27,28,29,30,31,32].
Measuring excitation/inhibition in Alzheimer’s Disease (AD)
The phrase “excitation/inhibition (E/I) balance” is convenient shorthand, yet the underlying biology defies reduction to a single number. A cortical network can tip toward E/I dyshomeostasis in multiple distinct ways (Fig. 1): neurons may fire in tighter synchrony without changes to intrinsic excitability, local microcircuits may amplify while long-range hubs disconnect, or the same tissue may behave very differently during rest versus task engagement. Each case reflects a unique axis of homeostatic disruption:
-
1.
Population synchrony—from highly asynchronous spike trains to synchronized bursts;
-
2.
Intrinsic cell state—from hypo-excitable membranes to neurons that fire with minimal depolarization;
-
3.
Spatial scale—from local regions of interest (ROIs) to whole-brain network coupling;
-
4.
Behavioral context—from resting-state to task-evoked dynamics.

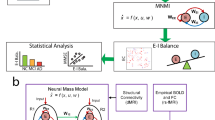
A theoretical example of two similar individuals with altered dyshomeostasis, that can separately be interpreted as a “hyperexcitation phenotype,” but through two distinct combinations of microscale, mesoscale, and macroscale observations (blue and red). Thus, a multidimensional E/I framework is more optimal to describe these two distinct phenotypes that don’t fit neatly into a binary hyper- vs hypo-phenotype and considers population and network dynamics. Four axes to describe E/I homeostasis through this multidimensional E/I framework are represented as sliding toggles including population synchrony (asynchrony versus synchronous), intrinsic cell state (hypoexcitable versus hyperexcitable), spatial scale (local vs global), and behavioral context (rest versus task). Hp = Hippocampus.
Frere et al. formalized a similar framework in which the brain continually steers activity back toward a stable operating region [33]. Figure 1 illustrates how two hyperexcitable phenotypes can emerge from distinct combinations of microscale and macroscale phenomena, although without this framework, both are often superficially reduced to a hyperexcitable phenotype, which, unfortunately, inappropriately simplifies a multidimensional system with cellular, local, and network dynamics.
To capture these dynamics, we categorize methodologies into three nested tiers (Fig. 2) that trade mechanistic precision for behavioral relevance and fall on a continuum from direct-to-indirect measurements (e.g., real-time ionic currents to delayed hemodynamic or transcriptional surrogates):
-
Microscale tools (e.g., patch clamp, single-cell calcium imaging, opto/chemogenetics, transcriptomics/proteomics) probe the ion channel and synaptic properties of individual neurons.
-
Mesoscale approaches such as field recordings (e.g., multielectrode arrays, laminar probes), bulk calcium imaging, and regional magnetic resonance spectroscopy (MRS) capture emergent microcircuit behavior or mean-field signals across a small population.
-
Macroscale techniques (e.g., EEG, MEG, fMRI, PET, and whole-brain modeling) extend the lens to centimeters of cortex and network-level dynamics relevant to clinical symptoms.

A representative sampling of published E/I methodologies in AD ranging different spatial scales: microscale (left, single cell and smaller) to mesoscale (middle, multiple cells to small ROIs) to macroscale (right, large ROIs to whole brain). Of note, several methodologies are not constrained by one scale (e.g., calcium imaging measures signal at cellular resolution but in a population of neurons). Likewise, computational modeling approaches span all three scales.
Microscale methodologies: molecules, synapses, and single cells
At the single-cell level, patch clamp electrophysiology, in which a glass pipette forms a giga-ohm seal with the membrane, is the gold standard for assessing excitability and synaptic input. Depending on the mode, the experimenter can leave the membrane voltage free while injecting a defined current (current clamp) or fix voltage and measure the ionic currents needed to maintain it (voltage clamp). In AD models, current-clamp recordings yield metrics such as resting membrane potential, input resistance, rheobase, and action potential shape, all of which reflect how close a neuron is to firing and how easily it does. Burst properties, like interictal burst duration, offer a real-time proxy for local circuit synchrony and the balance between recurrent excitation and inhibition.
Voltage-clamp recordings allow measurement of excitatory postsynaptic currents (EPSCs) and inhibitory postsynaptic currents (IPSCs)—either spontaneous, miniature, or evoked, depending on the stimulus and ion-channel blockade. The EPSC/IPSC ratio serves as a widely used readout of synaptic E/I balance. Short- and long-term synaptic plasticity are quantified via the paired-pulse ratio (PPR) and long-term potentiation/depression (LTP/LTD), indicating how circuits adjust their E/I set point over milliseconds to hours.
Calcium imaging detects intracellular Ca²⁺ influx, a downstream marker of spiking, and bridges single-cell and population-level data. Genetically encoded calcium indicators enable the visualization of spike-like transients, with key metrics including transient amplitude, decay constant (τ), event rate, and area under the ΔF/F₀ curve. When hundreds of neurons are recorded, correlation matrices, population sparseness, and neural activity overlap [34] metrics quantify circuit coordination and selectivity.
Optogenetic and chemogenetic tools (e.g., DREADDs) enable causal testing of E/I hypotheses by activating or silencing targeted neurons and recording the downstream impact with calcium imaging or electrophysiology. For example, in vivo stimulation of excitatory neurons has been shown to trigger tau release, linking hyperactivity to tau propagation [35].
Molecular profiling includes single-cell RNA-seq, proteomics, and immunohistochemistry, and reveals altered glutamatergic and GABAergic markers. Composite indices like tE/I (transcriptomic E/I) and pE/I (proteomic E/I) have revealed elevated excitatory drive in AD cortices [36]. More specific ratios (e.g., Gaba α1/α3, GluA1/GluA3, NKCC1/KCC2) indicate shifts in synaptic function and chloride homeostasis that affect inhibitory efficacy [37].
Markers of GABAergic interneurons further contextualize E/I shifts. Commonly profiled subtypes include parvalbumin (PV), somatostatin (SOM), neuropeptide Y (NPY), and calretinin (CR) [38, 39]. Each of these with distinct physiological roles and vulnerabilities in AD. However, marker expression alone may not fully predict function, as some subtypes (e.g., CR+) exhibit mixed or excitatory roles, depending on location [40].
Finally, morphological features such as the length of axon initial segment (AIS), which helps determine the spike threshold, provide structural correlates of long-term E/I remodeling. Similarly, the AIS and dendritic structure and spine density, which are frequently reduced in AD, complement functional metrics by capturing slower-timescale plasticity and degeneration [41, 42].
Mesoscale methodologies: microcircuits to sub-regions
Microscale tools scale up naturally when data are collected from dozens to thousands of neurons simultaneously. Advances such as two-photon or wide-field calcium imaging now enable tracking of hundreds [43] to thousands [44] of cells in vivo, while bulk transcriptomics and proteomics provide region-averaged molecular snapshots. Even multi-neuron patch clamps are possible, though less scalable. These approaches bridge single-cell excitability with emergent microcircuit dynamics.
The core mesoscale workhorse is an extracellular field recording, performed in vivo or in ex vivo slices using metal or glass electrodes or multielectrode arrays. High-pass filtering (>300 Hz) yields multi-unit activity (MUA), a population-level estimate of spike rates and inter-spike intervals. Low-pass filtering (<300 Hz) captures the local field potential (LFP), which reflects the summed excitatory and inhibitory transmembrane currents from nearby neurons. Arrays enable simultaneous recordings across brain regions, while slice perfusion systems support high-resolution ex vivo experiments [45].
LFPs support a broad range of E/I-relevant metrics. During evoked activity:
-
fEPSP/fIPSP slope measures baseline glutamatergic drive and inhibitory tone, respectively
-
Paired-pulse ratio (PPR) reflects short-term plasticity
-
Post-tetanic potentiation captures adaptation over tens of seconds to minutes
-
Long-term potentiation and depression (LTP, LTD) index longer-lasting changes in synaptic strength over hours to days
During spontaneous activity, power spectral analysis reveals shifts toward slower or faster rhythms, spike-phase coupling quantifies how tightly action potentials synchronize to oscillatory phases, and burst metrics (e.g., duration, frequency, coastline burst index) capture episodes of runaway excitation.
Magnetic resonance spectroscopy (MRS), an MRI-based technique, noninvasively estimates regional neurotransmitter concentrations, particularly glutamate and GABA, by analyzing their distinct spectroscopic signatures in predefined regions of interest (ROIs). While whole-brain MRS is theoretically possible and emerging, scan time limitations have historically confined measurements to select areas.
Together, these mesoscale methodologies illuminate how neural ensembles and their molecular context regulate E/I balance over time, which may be invisible at the single-cell level.
Macroscale methodologies: whole-brain
The two most direct macroscale electrophysiological methods, measuring the combined activity of millions of neurons across cortex-wide and subcortical networks, are electroencephalography (EEG) and magnetoencephalography (MEG). Both offer millisecond precision and do not rely on neurovascular coupling like in fMRI, albeit with relatively worse spatial resolution and ability to detect subcortical activity [46].
A primary use of EEG/MEG is power spectral analysis, which decomposes signals into canonical frequency bands: delta, theta, alpha, beta, and gamma. AD is often marked by:
-
Oscillatory slowing (increased delta/theta, reduced alpha/beta)
-
Altered aperiodic slope (1/f) and offset of the power spectrum, which provide information on tonic network drive.
Tools like the FOOOF algorithm (Fitting Oscillations and One-Over-F) [47] can decompose the power spectrum into these aperiodic and periodic components, yielding single-number indices such as:
-
Spectral Power Ratio (SPR): (alpha + beta)/(delta + theta), reflecting whether rhythms have shifted towards slower frequencies
-
Pathological Oscillatory Slowing Index (POSI): a normalized marker correlated with amyloid burden and cognitive decline in of slowing validated in AD cohorts [48]
Event-based metrics illuminate phasic hyperexcitability: subclinical epileptiform discharges, interictal spikes, and overt seizures. Rodent studies commonly score seizure severity on an eight-point motor scale [49,50,51]. Connectivity measures (e.g., phase-locking value, coherence) between ROIs and complexity metrics (e.g., Lempel-Ziv complexity) assess synchrony and information richness. Hyper- and hypoconnectivity in the default mode, frontoparietal, and hippocampal networks often align with AD disease progression [52].
Functional MRI (fMRI) complements EEG/MEG by offering whole-brain spatial coverage, including deep structures, albeit at lower temporal resolution. fMRI measures the blood-oxygen-level-dependent (BOLD) signal, an indirect proxy of synaptic input and spiking-related energy demand.
Frequency-based fMRI analyses use measures like:
-
fALFF (fractional amplitude of low-frequency fluctuations): the power below 0.1 Hz relative to the total spectrum, indexing baseline activity [53]
-
Signal variance during rest or peak BOLD amplitude during task-based designs, both reflecting regional excitation
More commonly, fMRI is used for functional connectivity analyses, with correlation matrices derived from ROI time series.
Finally, biophysical and phenomenological models offer new ways to bridge microscale mechanisms and macroscale observations. These frameworks often integrate multimodal data (e.g., fMRI, MEG, DWI) and simulate how cellular-level changes may manifest in whole-brain dynamics. Some generate novel disease biomarkers, while others help constrain competing theories of E/I disruption in AD. A brief overview of these models is included throughout this review [22, 24, 54,55,56,57,58,59,60,61,62,63,64,65,66,67,68,69] and readers are referred to Yang et al. [70], for a more detailed summary.
Prominent themes of excitation/inhibition imbalances in Alzheimer’s Disease (AD)
Amyloid-dependent hyperexcitation and tau-dependent destabilization
Setting the stage
Across experimental and computational studies of AD, a consistent yet nuanced picture of excitation-inhibition homeostatic disruption from amyloid and tau has emerged: one capturing large-scale human network phenomena and the other dissecting cellular and local circuit mechanisms in animal models. In humans, amyloid and tau pathology consistently alter regional and network activity, oscillatory frequency, and connectivity (most notably within the hippocampus and default mode network) and vice-versa are altered by the brain’s E/I homeostatic landscape. Amyloid’s effects dominate early in disease, driving hyperconnectivity and hyperactivity, whereas tau’s influence emerges later, spreading along functionally defined networks as symptoms advance. In contrast, animal models provide a mechanistic resolution. Amyloid-β (Aβ) in both plaque and soluble forms induces local hyperexcitation in a distance- and cell-type-dependent manner, while tau produces more variable effects ranging from circuit silencing to early hyperactivity followed by hypoactivity. Like human findings, both amyloid and tau are also shaped by the brain’s changing E/I patterns. When amyloid and tau are co-expressed, tau mirrors findings from human studies, often attenuating Aβ-driven excitation, though not universally. Overall, Aβ and tau appear to perturb distinct axes of a multidimensional E/I homeostatic space (Fig. 1), with their effects shaped by solubility, mutation type, region, age, and measurement scale.
Evidence in humans: amyloid, tau, and mixed pathology
The following sub-section details the human evidence underlying this macroscale picture of E/I homeostatic disruption in AD, specifically as it relates to amyloid and tau. Neuroimaging studies using fMRI, EEG, and PET have begun to delineate how Aβ and tau pathology alter cortical activity, functional coupling, and network synchrony. Collectively, these findings demonstrate that amyloid and tau perturb AD-related E/I homeostasis at the systems level, providing a critical foundation for animal studies aimed at elucidating their cellular mechanisms.
Direct measures of activity or inferred causal regional coupling with corresponding amyloid PET measurements offer some of the most direct evidence for altered E/I homeostasis. Using dynamic causal modeling (DCM), which infers directional excitatory or inhibitory influences between regions, task-based fMRI in cognitively normal older adults showed that higher global Aβ reversed the normally inhibitory medial temporal lobe to default mode network (DMN) connection, producing DMN hyperactivity. Entorhinal tau amplified this excitatory drive and predicted faster local tau accumulation, suggesting a feed-forward cascade linking amyloid, hyperexcitability, and tau spread [71]. Longitudinal resting-state fMRI further revealed that regional Aβ accumulation correlated with rising DMN and frontoparietal coupling [72], consistent with early compensatory hyperexcitability (see more in “Early/Preclinical Hyperexcitation”), while Aβ-positive MCI (mild cognitive impairment) individuals showed increased hippocampal activation during memory encoding [73]. In one custom simulation model based on rs-fMRI and DWI-based connectivity, global amyloid burden did not shift simulated E/I balance in cognitively normal adults, pointing to spatial distribution rather than total load, being a key feature of correlating amyloid burden with E/I alterations [62].
However, Canario et al. demonstrated in AD participants that microglial activation (measured by PET tracer 11C-PK11195) in AD participants increased posterior cingulate activity independent of local Aβ, indicating that amyloid’s effects depend on its inflammatory context (Canario et al.) [74].
Additionally, functional connectivity (FC) analyses show that amyloid and tau pathology disrupt the large-scale coordination of neural dynamics. Global Aβ mediates the link between FC disruption and tau deposition, supporting a cascading network failure that bridges molecular and systems pathology [75]. A dynamic mean-field model integrating fMRI and DWI further indicates that Aβ drives network instability during mild cognitive impairment, while tau predominates in later stages [65]. Across modalities, Aβ-positive individuals show reduced within-DMN but increased cross-network coupling [76], pattern complemented in EEG by elevated frontal theta frequency band, reduced beta band, and altered directed connectivity [77,78,79]. EEG-based machine learning algorithms have also shown substantial sensitivity and specificity in predicting amyloid positivity in MCI [79]. Together, these findings suggest that amyloid and progressively reorganize network synchrony and destabilize E/I homeostasis at the population level. Several reviews have synthesized the EEG/MEG/fMRI literature, framing Alzheimer’s disease as a “disconnection syndrome” marked by widespread functional decoupling across modalities. While these network alterations are robust, their variability across studies and methods has so far prevented the identification of a single, clinically useful connectivity phenotype [80, 81].
Additional evidence linking tau to E/I homeostasis comes from studies showing that tau accumulation follows the brain’s functional architecture in an activity-dependent manner. Regions with stronger resting-state connectivity show more correlated tau-PET change, and areas more connected to tau epicenters accumulate tau faster, with local Aβ amplifying this effect. The same functionally co-active circuits exhibiting altered intrinsic activity appear to serve as conduits for tau spread [82,83,84]. Complementary MEG findings show that higher CSF p-tau is associated with reduced connectivity and lower alpha/beta power, indicating global hypoactivity in MCI [85]. Computational modeling further supports tau-based hypoactivity even in the presence of amyloid: neural mass models fit to MEG and PET data suggest that amyloid prolongs inhibitory time constants, driving hyperexcitation, whereas tau prolongs excitatory time constants, promoting hypoexcitation [66]. Together, these empirical and computational findings converge on a view of amyloid and tau as opposing but interdependent forces that reshape, and are shaped by, the brain’s E/I homeostatic landscape.
The large-scale E/I alterations observed in humans find clear mechanistic counterparts in animal models. Whereas human imaging defines the network-level expression of amyloid- and tau-driven dysregulation, animal studies expose the underlying circuit and cellular events: amyloid-β-driven hyperexcitation through glutamatergic overdrive, synaptic dysfunction, interneuron loss, and astrocytic reuptake impairment, alongside tau-dependent transitions from hyper- to hypo-excitable states.
Amyloid in isolation—animal models
At the cellular level, amyloid-β (Aβ) consistently disrupts E/I homeostasis by increasing local glutamatergic excitability and weakening inhibition, effects most prominent near plaques. Multiple studies confirm Aβ-associated hyperexcitation in close proximity to amyloid plaques. In a foundational in vivo calcium imaging study, Busche et al. [86] showed that ~20% of cortical layer 2/3 neurons within 60 μm of plaques became hyperactive, while ~30% of more distant neurons were silenced [86]. This spatial gradient suggests localized excitotoxicity, likely driven by glutamatergic overdrive and interneuron impairment. In contrast, Algamal et al. reported that somatostatin (SOM) interneurons, rather than pyramidal cells, became hyperactive near plaques, implicating paradoxical interneuron disinhibition as a contributing mechanism [87].
Soluble Aβ oligomers reproduce the hyperexcitation phenotype but extend this mechanism: dysfunction of inhibitory interneurons, not merely disinhibition, drives and is a result of local and global hyperexcitation. For example, in APP mice, pre-plaque Aβ reduced firing thresholds and increased excitability [88]. Other studies found that soluble Aβ suppressed theta and gamma oscillations, likely via SOM and parvalbumin (PV) interneuron dysfunction [89]. Immunohistochemistry in 5XFAD mice confirmed reduced densities of CR and PV interneurons in the hippocampus, pointing to structural degradation of inhibition [90].
Microscale electrophysiology converges on the same endpoint of loss of inhibition. In CD1 mice, intracerebral Aβ injections increased pyramidal firing and spontaneous EPSCs by suppressing inhibitory transmission, identified by decreased amplitude and frequency of miniature IPSCs. These effects were modulated by D1 dopamine receptor signaling in fast-spiking interneurons [91]. Additional work implicates ion channelopathies: Nav1.1 haploinsufficiency in interneurons reduced IPSCs, increased pyramidal firing, and induced EEG hypersynchrony in hAPPJ20 mice and human AD tissue. Restoring Nav1.1 rescued inhibition and improved memory [92]. In addition, local field recordings show that Aβ oligomers selectively impair theta and gamma oscillations, sparing other bands, suggesting targeted interneuron disruption. Voltage-clamp data confirm that Aβ enhances GABA release from SOM and PV cells, leading to synaptic depression. Remarkably, optogenetic stimulation of these interneurons restores rhythmic precision, again pointing to inhibitory dysfunction as a primary driver of Aβ-induced desynchronization [89].
Synaptic dysfunction has also been implicated as a mechanism for hyperactivity and subsequent network failure from amyloid. Niraula et al. demonstrated in the hAPP mouse visual cortex with synaptic labeling and calcium imaging that an increased apical dendrite E/I synapse ratio (PSD-95/gephyrin) favors neuronal hyperactivity in amyloidosis and also subsequently disrupts functional connectivity and network architecture (decreased node degree) [34].
Glial mechanisms, particularly astrocytic dysfunction, further substantiate a hyperexcitation phenotype, but via glutamatergic excitability rather than reduced inhibitory tone. Zott et al. showed that Aβ disrupted GLT-1-mediated glutamate uptake in astrocytes, elevating extracellular glutamate and driving neuronal hyperactivity [93]. These effects were amplified in already-active neurons, suggesting that Aβ exaggerates preexisting excitability rather than generating new activity [93].
Taken together, these findings support a unifying model in which synaptic dysfunction, dendritic and axonal morphological changes, and inhibitory interneuron impairment underlie both local and global hyperexcitability. Paradoxical disinhibition and glutamatergic overdrive from astrocytic dysfunction can further amplify this phenotype and blur the timescale at which the core pattern of interneuron impairment is observed, which should be considered in future studies.
Tau in isolation—animal models
Compared to amyloid, tau pathology produces more heterogeneous effects on neural excitability, ranging from silencing and inhibition to hyperexcitation and epileptiform activity. These differences appear to reflect not only tau form (soluble vs aggregated), but also disease stage, brain region, mutation type, and network context. Taken together, the evidence supports a multidimensional and dynamic model, in which tau drives early circuit instability, often through both 1) hypoexcitatory changes, such as neuronal silencing and hyperinhibition and 2) hyperexcitatory changes, such as increased resting membrane potentials and dendritic morphological changes that can later transition into functional disconnection and network instability.
Tau in isolation—evidence for hypoexcitation and silencing
Several models implicate tau in neural hypoactivity. In Tg4510 mice, Busche et al. used calcium imaging to show increased neuronal silencing and decreased event rate, consistent with hypoexcitation. The same was observed in rTg21221, a non-tangle model, indicating that tau aggregation is not required. Suppressing tau expression reversed all effects [94]. In EC-Tau mice, Fu et al. reported reduced firing of entorhinal grid cells, alongside increased inhibitory neuron firing: a hyperinhibition + hypoexcitation signature associated with spatial memory deficits [95]. Hatch et al., using current clamp and morphometry in P301L tau mice, found that although membrane depolarization was present, a distal relocation of the axon initial segment (AIS) increased spike threshold and reduced firing, yielding a functionally hypoexcitable phenotype [41]. These four different tau-based models support a framework of tau-induced hypoexcitation through various mechanisms: neuronal silencing, hyperinhibition, and axonal morphological changes, which need not be mutually exclusive.
Tau in isolation—evidence for hyperexcitation
In contrast, the observed hypoexcitation from tau, several other models demonstrate a propensity for hyperexcitation and increased neuronal activity. In rTg4510 mice, Crimins et al. showed increased resting membrane and sag potentials, as well as higher firing rates that preceded neurofibrillary tangle (NFT) deposition, indicating early hyperexcitability [42]. Decker et al., using A152T mice, found increased calcium signals, fEPSPs, dendritic spine density, and epileptiform bursts, all of which were normalized by enhancing astrocytic glutamate uptake, implicating glial dysfunction in tau-induced excitability [96]. Two studies showed that reducing tau via antisense oligonucleotides or genetic knockdown made mice less susceptible to chemically induced seizures, suggesting that tau enhances network hyperexcitability through network population instability [51, 97]. These studies support several observed phenotypes of hyperexcitation such as increased action potentials and firing dynamics, potentiation, and dendritic morphological changes. One possible mechanism of bridging these studies with those that demonstrate a hypoexcitation phenotype is reframing these E/I phenotypes with a multidimensional view proposed in Fig. 1: tau-induced microscale hypoexcitation and hyperexcitation create mesoscale and macroscale network instability that can more easily destabilize transiently into global hyperexcitation phenotypes (e.g., seizures).
Tau in isolation—mixed mechanisms
A subset of models exhibits both hypo- and hyperactive features simultaneously, underscoring the need for a multidimensional E/I framework to explain their intertwined mechanisms. Some models reveal both hypo- and hyperactive features. Maeda et al. found that hTau-A152T and hTau-WT mice both exhibited increased fEPSPs and lower seizure thresholds (more unstable). However, only hTau-WT showed a lower paired-pulse ratio (PPR) and more epileptic spikes, suggesting different genetic mechanisms yield a similar pro-excitatory endpoint [98]. Separately, Shimojo et al., using PET imaging and synaptic histology on rTg4510 mice, found that inhibitory synapse loss (VGAT, GABAAR, GAD67) preceded excitatory loss, suggesting early disinhibition as a driving mechanism [99]. As a direct correlate of human studies, tau pathology also appears to follow activity-dependent spread along network connections, rather than simple proximity. Wu et al. showed that higher neuronal activation enhanced tau spread, and that induced hyperactivity stimulated tau release in vitro and in vivo [35]. Together, these findings suggest a bidirectional feedback between activity and tau, where increased activation accelerates tau spread, and accumulated tau disrupts excitability in opposing directions, silencing some neurons while overdriving others.
Mixed pathology—tau often masks amyloid effects
In humans, Alzheimer’s disease nearly always involves co-pathology, or a combination of amyloid, tau, and other neurodegenerative processes. This makes it crucial to examine how E/I homeostasis is altered in the presence of both amyloid and tau in animal models as well. A recurring theme across several models is that tau expression often dampens or alters the hyperexcitation typically induced by amyloid, although this effect is not universal.
Experimental support for tau-mediated masking comes from several animal studies. In entorhinal cortex slices from transgenic mice, APP expression alone increased spontaneous field potentials and epileptiform activity, but adding human tau (hAPP/hTau) normalized the hyperexcitability [100]. In Busche et al., wide-field calcium imaging showed that soluble tau expression suppressed Aβ-induced hyperactivity, even in the absence of neurofibrillary tangles. Suppressing tau failed to rescue firing abnormalities in co-pathology models (e.g., rTG4510 × APP/PS1), suggesting a nonlinear interaction rather than simple additive effects [94]. Similarly, the loss of dendritic Kv4.2, a potassium channel linked to amyloid hyperexcitability, was prevented by tau expression, indicating a possible protective mechanism [101].
However, other models report the opposite, namely tau exacerbating or failing to rescue amyloid-driven excitability. In a separate study, hAPP mice with tau showed heightened EPSCs and seizure susceptibility, and tau reduction normalized both, seemingly contradicting the protective hypothesis [50]. Viewed through a multidimensional E/I framework (Fig. 1), tau’s dual hyper- and hypo-excitable actions can both counterbalance amyloid-induced hyperactivity and heighten network fragility, paralleling human evidence of tau-linked E/I instability.
Importantly, causality between E/I alterations and protein pathologies may be bidirectional. In a co-pathology mouse model, Rodriguez et al. showed that chemogenetic attenuation of entorhinal hyperactivity reduced both Aβ burden and tau spread into the hippocampus. Similar to how network hyperactivity can provide a conduit for tau spread, this also suggests that altered E/I homeostasis can drive proteinopathy, not just reflect it [102].
Conclusion
The tendency to interpret seizure activity or spectral slowing as proxies for hyperexcitability risks oversimplifying E/I homeostasis as a one-dimensional axis (see “Measuring Excitation/Inhibition in Alzheimer’s Disease”). Tau may simultaneously increase local epileptiform events while reducing global connectivity or network power. These apparent contradictions are better understood within a multidimensional E/I framework, where different combinations of excitatory and inhibitory changes can all lead to instability. While general trends emerge such as amyloid-driven hyperexcitation (often proximity-dependent) and tau-linked hypoexcitation or masking, the effects of both proteins are modulated by spatial distribution, genetic background, stage of disease, and co-pathology. Future research should explicitly frame these observations within multiscale and multidimensional models to avoid over-reliance on binary E/I interpretations.
Early/preclinical hyperexcitation
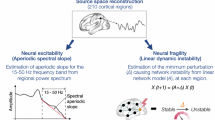
Growing evidence suggests that network hyperactivity emerges in the earliest stages of AD, often before detectable amyloid pathology. This aligns with updated biomarker models, such as the revised “Jack curves” (Fig. 3), which propose a transient surge in excitation–inhibition (E/I) imbalance peaking around mild cognitive impairment (MCI), followed by hypoactivity as neurodegeneration progresses. Reports of altered “neuronal activity,” “excitation,” or “synchronization” likely reflect this dynamic trajectory, not a fixed gain or loss of function [21,22,23,24,25,26]. Mapping E/I changes across spatial and temporal scales may clarify when early hyperactivity shifts from compensation to pathology.

Schematic representation of excitation/inhibition (E/I) and oscillatory peak frequency during the course of Alzheimer’s disease (preclinical, MCI, and dementia). This representation has been consolidated and adapted from various authors: Corriveau-Lecavalier et al., Burns et al., Koelewijn et al., Van Nifterick et al., Maestu et al., Toniolo et al. [21,22,23,24,25,26].
Compelling support comes from APP transgenic models. In one study, in vivo local field potential recordings revealed a collapse in gamma-band power prior to plaque formation or detectable cognitive deficits. Patch-clamp data from the same animals showed reduced EPSCs and IPSCs in pyramidal neurons and weakened excitatory drive onto fast-spiking interneurons, indicating a breakdown in coordinated firing that emerged before deposition [103].
Independent evidence comes from the APOE4 allele in cognitively normal and amyloid- and tau-negative mice and humans. APOE4 drives network-scale hypermetabolism in vivo: aged APOE4 mice exhibited elevated cerebral blood volume on fMRI, increased broadband LFP power, and higher excitatory (not inhibitory) firing rates, all in the absence of detectable Aβ or tau [104]. In an fMRI study of cognitively intact adults, APOE3/4 carriers showed the strongest task activation in medial frontal and parietal cortices compared with APOE3/3 peers, despite identical behavioral test performance [105]. Several other fMRI studies have shown similar findings with APOE4 carriers showing higher resting-state default mode network (DMN) activation [106], weaker precuneus-DMN connectivity at rest [107], and memory-task hyperactivation in hippocampal and prefrontal cortices with greater activation predicting 2-year memory decline [108].
MEG studies of APOE4 carriers converge, showing resting-state hyperconnectivity in alpha-beta bands and focal gamma-band increases. These patterns that reversed in AD, and consistent with a transition from early overconnectivity to disconnection [23]. A separate EEG study also showed greater alpha loss and theta-delta bursts during hyperventilation in cognitively healthy APOE4 relatives of AD patients [109]. Complementing these MEG and EEG observations of APOE4, in vivo MRS combined with amyloid PET showed that posterior-cingulate Glu and GABA levels increased with amyloid load, with steeper trajectories in APOE4 carriers—these neurochemical changes predicted faster memory decline [110].
Statistical physics models also support the idea that APOE4 influences early hyperexcitation: cross-sectional and longitudinal analyses reveal elevated E/I ratios in female APOE4 carriers, with early hyperexcitation linked to enhanced cognition, suggesting a possible sex-specific compensatory window [22, 58].
These results suggest that APOE4 alone may shift the E/I set point toward hyperexcitability, independent of proteinopathy.
Some of the strongest support for early hyperexcitation comes not only from these APOE-focused functional imaging studies, but in conjunction with other fMRI studies focusing on other AD risk factors and stages, such as age, autosomal dominant genetic risk (PSEN1), and differentiating MCI from AD. For example, in a hippocampal pattern separation task, cognitively normal older adults showed increased CA3–dentate gyrus activation despite normal performance, suggesting that circuit overactivation may arise even in the absence of clinical or genetic risk [111]. Longitudinal data of aging also show that higher baseline hippocampal activation predicts steeper cognitive decline [112]. In two separate studies of presymptomatic PSEN1-E280A mutation carriers, participants exhibited elevated hippocampal and parahippocampal BOLD activation and impaired default mode deactivation during a memory task despite identical performance. In EEG, PSEN1 mutation carriers display elevated alpha-to-theta ratios in the precuneus that correlated with age and cognitive performance [113].
Magnetic resonance spectroscopy (MRS) provides biochemical support for early E/I imbalance. In MCI, one study found elevated glutamate (Glx)/Cr and reduced GABA/Cr ratios, correlating with worse cognitive performance [114]. Another reported lower GABA+/Cr in the cingulate cortex, showing good diagnostic accuracy for MCI [115]. However, findings are mixed, with one study observed concurrent Glu and GABA reductions, suggesting more complex dynamics [116]. Despite variability, MRS data overall indicate that neurotransmitter shifts may precede structural degeneration.
Several studies go further by modulating E/I balance directly to evaluate causal influence on amyloid deposition. In APP/PS1 mice, parvalbumin (PV) interneurons became hyperexcitable around 16 weeks. This coincided with increased inhibitory transmission onto CA1 pyramidal neurons (increased PV AP frequency and sIPSC frequency). Chemogenetic silencing of PV cells via DREADDs normalized inhibitory tone, rescued spatial memory, and reduced soluble Aβ and plaque load [117].
Taken together, findings across fMRI, electrophysiology, modeling, and spectroscopy support early presymptomatic network hyperactivity that often emerges before plaque deposition, especially in genetically at-risk individuals. While methodological variability and individual differences remain, a consistent trajectory is taking shape: early excitation rises, often as a compensatory or stress response, before giving way to disconnection and hypoactivity as pathology progresses. This pattern holds across modalities and disease models, suggesting that E/I imbalance is a dynamic, evolving process shaped by age, genotype, and circuit resilience.
Late hypoexcitation and oscillatory slowing
Hyperexcitation appears to peak around the MCI stage, followed by a transition toward hypoexcitation in Alzheimer’s dementia, likely reflecting progressive neurodegeneration (see Fig. 3). This shift is accompanied by a well-documented electrophysiological phenomenon: oscillatory slowing, where spectral power shifts from higher-frequency bands (alpha, beta, gamma) to lower-frequency ones (theta, delta). Most evident in EEG, MEG, and field potentials, this slowing serves as a macroscale signal of altered excitation/inhibition (E/I) dynamics in late-stage disease.
Longitudinal and multimodal studies support this trajectory. In a task-based fMRI study, individuals with MCI showed elevated hippocampal activation at baseline, which declined over 2 years, while healthy controls remained stable [112]. MRS data similarly revealed stepwise reductions in Glx/Cr ratios from controls to MCI to AD, indicating progressive excitatory loss in the anterior cingulate and hippocampus [118].
Yet the notion of late-stage hypoexcitation is complicated by evidence of persistent seizure-like activity. One study found epileptiform discharges in over 50% of AD patients compared to 25% of controls; individuals with subclinical spiking declined more rapidly [119]. Foramen ovale recordings revealed hippocampal seizures in patients with normal scalp EEGs [120]. Several studies report that epileptiform activity can accelerate cognitive decline by up to 7 years [121, 122]. These findings suggest that AD brains may hover near a critical threshold, fluctuating between tonic hypoactivity and phasic hyperexcitability.
Molecular and cellular data also challenge a purely hypoactive model. Synaptic membrane transplants from AD parietal cortex into Xenopus oocytes revealed elevated E/I ratios across electrophysiological and transcriptomic domains [36]. Scaduto et al. reported higher AMPAR/GABA current ratios in the AD temporal cortex, correlating with amyloid/tau burden and cognitive decline [123].
Still, oscillatory slowing remains a robust late-stage marker. Studies consistently show lower peak frequencies, increased theta-delta power, and reduced thalamic alpha activity [61]. Resting-state MEG reveals widespread power loss, while early-onset AD cohorts show theta increases and hippocampal alpha reductions linked to clinical decline [124]. An early-onset cohort showed increased theta and reduced hippocampal alpha, correlating with cognitive decline [125]. Further, a Duffing–Van der Pol oscillator model of EEG showed a shift from alpha to theta dominance [63] while an activity-dependent connection-loss model also produced the same slowing [55].
Modeling studies also link oscillatory slowing to protein burden. Ranasinghe et al. found that tau correlated with longer excitation time constants, while amyloid predicted inhibition delays [66]. Jansen–Rit local circuit simulations required patient-specific PET maps to reproduce alpha–theta slowing; uniform PET burden erased the effect, indicating that the spatial deposition of amyloid matters [57]. Wiesman et al. introduced a Pathological Oscillatory Slowing Index and showed it tracked amyloid burden and predicted deficits in attention, language, and processing speed [48]. Some studies extend this slowing trajectory into asymptomatic at-risk populations. Cross-sectional MEG of participants across the dementia spectrum (cognitively healthy, MCI, mild AD, moderate AD, and severe AD) using the Kuramoto simulation model charted a stepwise rise in theta coupling and a fall in alpha coupling from healthy aging through severe dementia [67]. APOE4 carriers and PSEN1 mutation carriers exhibit altered alpha and theta power, even without symptoms [109, 113].
Other studies, especially in animal models, challenge the slowing–hypoexcitation link. Gamma power decreases preceded plaque formation without increases in slow rhythms [103]. In hAPP models, Aβ-driven hyperactivity in CA1 suppressed both theta and gamma; optogenetic activation of SST and PV interneurons restored both, implicating inhibitory dysfunction over reduced excitation [89]. Chemogenetic suppression of entorhinal hyperactivity reduced theta and limited amyloid/tau spread [102]. Longitudinal MEG data revealed that theta–beta hypersynchrony increased in stable MCI but declined in progressive cases, suggesting divergent trajectories [52]. Further, Van Nifterick et al. demonstrated in simulations that hyperactive neurons can cause spectral slowing by increasing inter-burst intervals. Hence, slowing may not necessarily reflect hypoexcitation [24].
In summary, oscillatory slowing is reliably observed in AD but cannot be simplistically interpreted as evidence of hypoexcitation. The coexistence of subclinical seizures, region-specific hyperactivity, and model-based contradictions argues for a more nuanced view - one that recognizes tonic/phasic dynamics, band-specific desynchronization, and regionally heterogeneous circuit failure.
Mechanism: APOE4 mediates excitation/inhibition
As discussed in “Early/Preclinical Hyperexcitation,” APOE4 is implicated at nearly every phase of the excitation-inhibition (E/I) trajectory, with many human and mice studies linking this genetic risk to altered E/I, especially before cognitive symptoms, and its effect mediated by sex [22, 23, 58, 62, 104, 105, 107]. Here, we expand upon this observational evidence linking APOE4 to altered E/I activity by focusing on studies probing cellular and molecular mechanisms.
One proposed mechanism involves astrocytic calcium dysregulation. Larramona-Arcas et al. found that ATP stimulation triggered prolonged calcium transients in APOE4 astrocytes, an effect specific to males and resistant to extracellular calcium chelation, thus implicating lysosomal release. Re-expression of APOE3 reversed the phenotype, and lipidomics revealed altered lysosomal membranes, pointing to gliotransmission-mediated excitability [126].
A second mechanism involves selective interneuron loss. Leung et al. reported that female APOE4 knock-in mice exhibit age-related reductions in GAD67+ and somatostatin+ interneurons in the hippocampus, correlating with spatial memory deficits. Males retained these populations, indicating a sex-specific vulnerability. Related to this, Li et al. demonstrated that GABAergic support of adult neurogenesis is compromised in APOE4 mice. Newborn dentate neurons received weaker inhibition, had shorter dendrites, and exhibited reduced branching and synaptic activity. Treatment with the GABA potentiator pentobarbital rescued dendritic growth and synaptic maturation, directly linking impaired inhibition to developmental arrest [127]. A fourth pathway involves modulation of synaptic plasticity by the renin–angiotensin system. Scheinman et al. found that APOE4–FAD mice had smaller baseline synaptic responses but exaggerated post-tetanic potentiation and long-term potentiation (LTP) in the Schaffer collaterals, indicating latent hyperexcitability. Angiotensin II suppressed these plasticity changes without affecting baseline transmission, suggesting that renin–angiotensin signaling modulates APOE4-linked excitability [128].
Hence, APOE4 may disrupt E/I balance through multiple, interacting pathways:
-
1.
Lysosomal calcium dysregulation in astrocytes
-
2.
Selective interneuron loss with sex-specific effects
-
3.
Impaired GABAergic support of neurogenesis
-
4.
Exaggerated synaptic plasticity modulated by renin–angiotensin signaling
Understanding how APOE4 reshapes circuit dynamics in vulnerable subgroups defined by sex, age, or cell-type specificity may help guide precision prevention strategies. Targeting early excitability changes, especially in preclinical carriers, could offer new windows for intervention and risk stratification in AD.
In summary, APOE4 appears to tilt the E/I balance via at least four mechanisms: lysosomal Ca²⁺ dysregulation in astrocytes, selective interneuron loss, impaired GABAergic support of neurogenesis, and dysregulated angiotensin signaling. The resulting region- and sex-specific hyperexcitability may precede or exacerbate downstream Alzheimer’s pathology.
Sex differences in AD-related excitation/inhibition imbalances
AD presents a notable sex disparity: women have nearly twice the lifetime risk of dementia compared to men [1, 2]. While longer lifespan plays a role, emerging evidence implicates sex-specific neurobiological mechanisms; particularly in excitation–inhibition (E/I) regulation as a central contributor. Yet most studies still fail to stratify by sex, missing key opportunities to uncover mechanisms and optimize interventions.
Divergent interneuron vulnerability and astrocytic excitability
Studies in APOE knock-in mice reveal marked sex differences in interneuron development and loss. Leung et al. found that male mice had fewer GAD67+, NPY+, and PV+ interneurons at 1 month. Over time, female APOE4 carriers exhibited pronounced loss of GAD67+ and SOM+ interneurons, while males showed modest gains. These were changes that predicted spatial memory deficits in females [40]. Astrocyte excitability also shows sex bias. Larramona-Arcas et al. reported that male APOE4 astrocytes exhibited exaggerated ATP-evoked Ca²⁺ transients, resistant to extracellular calcium chelation and linked to lysosomal release. Re-expression of APOE3 reversed the effect, suggesting a cell-autonomous, amyloid-independent gain of gliotransmission [126]. Sex also shapes intracellular signaling. Jain et al. found that estradiol potentiated EPSC amplitude in both sexes, but females required both PKA and MAPK/Src/ROCK signaling and dual calcium sources (L-type Ca²⁺ influx and internal Ca²⁺ release), while males could bypass PKA with a single calcium trigger [129]. These results suggest divergent molecular routes to similar excitatory outcomes.
Hormonal signaling, imaging, and network modeling
Hormone-linked E/I dynamics extend to human studies of estrogen receptors (ERs), which have genomic and rapid non-genomic effects on coordinating cerebral blood flow, metabolism, inflammation, and membrane depolarization. PET imaging of ERs by Mosconi et al. showed that menopause increased ER binding in the pituitary, posterior cingulate, caudate, and frontal cortex, distinguishing pre- from post-menopausal women and correlating with mood and cognitive symptoms [130].
Three computational modeling studies in humans using a statistical-physics framework applied to fMRI–DWI data also support sex-specific E/I dynamics in AD. In cognitively intact middle-aged adults, APOE4 carriers had higher whole-brain excitation only in females cross-sectionally, and this correlated with better spatial working memory [58]. In a follow-up study, the E/I ratio showed a less-steep age-related decrease in women with APOE4, driving a regional rise in right hippocampal excitation that predicted better executive scores, not observed in men [62]. A longitudinal study of 106 participants found that only female APOE4 carriers exhibited progressive increases in whole-brain E/I over several years [22].
Broader neurobiology supports a sex-specific circuit vulnerability. Tabatadze et al. proposed that perimenopause induces transient hypermetabolism, followed by a sharp decline, leaving energy-hungry PV interneurons disproportionately exposed to oxidative stress. These cells express β-estrogen receptors and lose excitability as estradiol declines [131]. These cells, anchoring gamma oscillations and comprise half of cortical interneurons, are critical to E/I balance, yet few AD studies stratify them by sex [131]. Terstege et al. further propose PV cells as sex-specific AD targets [132]. Last, Oliva et al. show small but widespread sex-based gene expression differences across 11 brain regions, reinforcing the biological rationale for stratification [133].
Together, these findings suggest that sex hormones shape receptor profiles, signaling cascades, metabolic resilience, and immune tone that govern E/I homeostasis from early development through menopause. Female circuits may rely more heavily on estradiol-sensitive PV interneurons, which become vulnerable during abrupt hormonal and metabolic shifts; male networks may follow a slower, androgen-biased trajectory. These asymmetries may help explain sex-specific patterns of hyperexcitation, oscillatory slowing, and APOE4 effects. Future E/I studies must explicitly stratify by sex and evaluate sex-specific interventions to reduce persistent disparities in AD risk, progression, and treatment response.
Metabolic, inflammatory, and oxidative stress exacerbate E/I imbalances in AD
Thus far, APOE effects, sex differences, amyloid and tau, early hyperexcitation, and later hypoexcitation and oscillatory slowing all seem distinctly disconnected with no unifying mechanism. While there may exist many varied mechanisms, neuroinflammation as a result of metabolic and oxidative stress is a popular and well-known framework that is obligatory to mention due to its systemic effects and numerous documentation in AD, aging, and general neuroscience studies of altered electrophysiology. In this section, we briefly describe select observations of altered GABA/glutamate, diabetes, identified genes, and glial involvement as it relate to metabolism, neuroinflammation, and oxidation in AD.
Metabolic dysregulation, oxidative stress, and neuroinflammation converge on the same endpoint: disrupted glutamatergic–GABAergic balance and network hyperexcitability [134, 135]. Aging, the primary risk factor for dementia, is marked by loss of redox homeostasis [136]. Reactive oxygen species (ROS) can directly perturb GABAergic and glutamatergic signaling, shifting excitation–inhibition (E–I) balance [137, 138]. Environmental and systemic metabolic stressors amplify this chemistry. Air pollution induces oxidative stress and inflammation, with postmortem brains of exposed individuals showing elevated pro-inflammatory cytokines and glial activation [139].
Dysregulated glucose metabolism is increasingly recognized as a major risk factor for AD, prompting some researchers to refer to AD as a form of “type 3 diabetes.” [140, 141] Diabetes, as a systemic disorder of metabolic homeostasis, may therefore represent an additional metabolic insult in AD: insulin regulates many synaptic proteins and enzymes and specifically modulates GABA and glutamate receptor expression [142, 143]. Consistent with that, altered cortical GABA and glutamate levels have been reported in type 1 and type 2 diabetes [144,145,146].
Neuroinflammation itself provides a unifying theme for these processes, correlating with impaired neurogenesis, oxidative stress, and classical AD pathologies (β-amyloid, tau) while disturbing GABAergic function and promoting E–I imbalance [15].
GWAS studies have also identified immune-related loci in late-onset AD risk: CD33, TREM2, ABCA7, and BIN1. BIN1 knockout mice show more inflammation [147] and neuronal hyperexcitability [148]. Inflammatory cytokines then provide a mechanistic bridge to excitability. Chronic glial activation promotes seizures [149]. Blocking the IL-1β receptor lowers seizure susceptibility, whereas exogenous IL-1β worsens it [150], and inhibiting IL-1β synthesis is anticonvulsive [151]. The proconvulsive effect of IL-1β is prevented by an NR2B antagonist, implicating NMDA receptors in this pathway [152]. TNF-α increases AMPA receptor surface expression and miniature EPSC frequency in hippocampal neurons [153], drives astrocytic glutamate release and NMDA receptor–mediated excitation in the dentate gyrus [154], and its reduction (including anti-TNF therapy) is protective against epilepsy in animals and patients [155, 156]. These same cytokine-driven shifts toward glutamatergic dominance and reduced inhibition are highly relevant for AD-related hyperexcitability.
Glial cells, such as microglia and astrocytes, are also intimately involved. Microglia carry several of the previously mentioned AD GWAS hits (ABCA7, CD33, TREM2); TREM2 promotes microglial phagocytosis, and its loss aggravates tau and Aβ pathology [157]. Microglia refine circuits through synaptic pruning [158], sense and modulate neuronal firing [159], and their depletion heightens seizure severity [160]. Increased microglia–neuron contact can boost synaptic activity and synchronize networks, but inflammatory activation (e.g., LPS) abolishes their regulatory control [161]. As briefly mentioned in “Amyloid-Dependent Hyperexcitation and Tau-Dependent Destabilization”, Canario et al. demonstrated with a microglial PET tracer in AD participants that microglial activation was sufficient to increase posterior cingulate activity independent of local Aβ [74]. In addition to microglia, astrocytes stabilize ionic and neurotransmitter milieus: they clear extracellular K+ during repolarization [162], and defective buffering raises K+, facilitating seizure initiation [163]. They also remove glutamate via EAAT1/2, transporters downregulated in AD brains [164,165,166], while AD mouse models show elevated astrocytic glutamate release [167]. Reactive astrocytosis (astrogliosis), common in AD [168], is linked to reduced IPSCs in nearby neurons and network hyperexcitability [169].
Together, metabolic insults, oxidative stress, and immune-glial dysregulation form a feedback loop that pushes cortical and hippocampal circuits toward excess excitation or inadequate inhibition. Framing these diverse findings around E–I balance highlights tractable intervention points: improving systemic metabolic health, dampening maladaptive inflammatory cascades (e.g., IL-1β or TNF-α pathways), and restoring glial homeostatic functions may all help re-center networks in AD.
Restoring E/I balance: opportunities and challenges in therapeutic targeting
Network hyperexcitability may be one of the earliest and most tractable circuit abnormalities in AD. In fact, direct modulation of excitability alters amyloid pathology. Chemogenetic silencing of PV interneurons in APP mice reduced soluble Aβ and restored memory, while low-dose levetiracetam improved synaptic plasticity and cognition without reducing plaque burden [117]. In contrast, prolonged optogenetic stimulation in another study exacerbated Aβ deposition and triggered seizures, highlighting the delicate balance of excitability in early intervention [170].
As a consequence of the many-cited studies within this review, electrophysiology-based treatment approaches to AD have received attention within various pharmacological philosophies. For instance, with the emergence of nonpharmacological and noninvasive techniques of sensory stimulation [171], transcranial direct current stimulation (tDCS) [172], transcranial alternating current stimulation (tACS) [173], and transcranial magnetic stimulation (TMS) [174], it is theoretically possible to delay, stop, or prevent AD. Additionally, there exist the invasive technique of deep brain stimulation that has gained promising use in Parkinson’s disease. However, all these approaches still suffer from small numbers of studies and heterogeneity of results, thus more high-quality studies are needed with more defined E/I based biomarkers in addition to primary endpoints of cognitive abilities (e.g., attention, memory).
In addition to non-pharmacological approaches, there exists some promising new agents as well as several agents already developed for use in AD, such as reducing overactivity of the excitatory NMDA receptor. For example, memantine, a low-affinity NMDA antagonist, is an established treatment for moderate-to-severe AD, which preferentially blocks calcium influx during excessive stimulation, without disrupting normal synaptic activity. Due to the beneficial results of memantine, there are other NMDA/glutamate targeting strategies which are still under investigation such as ifenprodil, riluzole, and AVP-786. In the same strategy of tamping down glutamatergic NMDA receptors, restoring inhibitory GABAergic tone has been proposed to counteract the neuronal overactivity in early AD. Many of these glutamatergic and GABAergic strategies, however, suffer from side effects, but investigations are still ongoing [26].
We point the curious reader to cited studies on sensory stimulation [171], tDCS [172], tACS [173], TMS [174], and DBS [175], as well as Toniolo et al.’s review discussing potential new approaches in modulating brain hyperexcitability in AD such as those modulating glutamatergic and GABAergic tone with an in-depth review of the mechanisms behind these approaches [26]. In this selective review, we focus our attention on the pharmacological approaches, specifically one promising candidate antiepileptic drug, levetiracetam (LEV) whose many studies have not yet been cohesively synthesized in one source.
Among viable therapeutic candidates in humans, levetiracetam (LEV) stands out for its CNS penetration, tolerability, and measurable effects on cognition and neural activity. While its mechanism is not fully understood, it likely modulates calcium-dependent neurotransmitter release via synaptic vesicle protein SV2A [176]. Here, we synthesize findings from human and animal studies, emphasizing how dose, physiology, genotype, and pathology influence outcomes.
Human studies: low-dose window and variable outcomes
In amnestic MCI, two small fMRI studies by Bakker et al. demonstrated that low-dose levetiracetam (62.5–125 mg BID) suppressed dentate gyrus overactivity and boosted decreased entorhinal activity and modestly improved pattern separation memory, while 250 mg provided no benefit, suggesting a narrow therapeutic window [177, 178]. This dosing insight informed AGB101, an extended-release 220 mg formulation designed for sustained low plasma levels. In the 78-week phase-2 HOPE4MCI trial (n = 164, amyloid-positive MCI), AGB101 slowed clinical decline and reduced entorhinal atrophy, but only in APOE4 non-carriers [179, 180].
In probable AD with seizures, Cumbo and Ligori showed that levetiracetam achieved better seizure control and fewer side effects than phenobarbital or lamotrigine. It also produced modest cognitive improvements (MMSE, ADAS-Cog), while phenobarbital was mildly detrimental [181,182,]. However, in mild AD patients without confirmed hyperexcitability, results were more mixed: the ILiAD pilot RCT (n = 8) showed no benefit [182, 183], and the 2021 phase-2a trial by Vossel et al. found no improvement on primary cognitive outcomes. Still, in the ~38% with baseline epileptiform activity, executive and spatial scores improved, reinforcing that baseline hyperexcitability may be a prerequisite for benefit [184].
Animal studies
In multiple AD mouse models, levetiracetam rescued a variety of hyperexcitability-linked deficits with mixed impact on protein pathologies:
-
hAPP model: Hall et al.: Restored Kv4.2 potassium channels, normalized spikes, rescued calbindin, and improved behavior deficits caused by Aβ [101].
-
APP knock-ins: Reduced cortical Aβ₄₂ and upregulated synaptic gene modules [185].
-
hAPP-J20 model: Improved memory and LTP, but did not reduce plaques, suggesting functional restoration without disease modification [186].
-
APP23/MAPT model: low-dose levetiracetam restored synaptic markers and cognition, but high doses were neurotoxic, causing apoptosis and eliminating benefit [187].
-
Tau-only model: no cognitive or synaptic benefit was seen, highlighting that levetiracetam may only work when amyloid-linked hyperexcitability is present [188].
Across preclinical and clinical studies, low dose levetiracetam shows promise for normalizing E/I balance and supporting cognitive function, but only in specific biological contexts:
-
Likely beneficial only when baseline hyperexcitability exists, best detected via extended EEG or MEG
-
Effects may depend on genotype (e.g., APOE4 carriers respond less) and pathology mix (amyloid vs. tau-driven)
-
Higher doses eliminate benefit and may introduce toxicity
-
No study has yet stratified by sex, and very few by APOE genotype—critical gaps for future trials
Overall, while levetiracetam is not a cure, it exemplifies a promising circuit-level intervention that may delay symptom progression if carefully matched to a patient’s electrophysiological profile.
Conclusions and future research priorities
AD research has undergone a paradigm shift, from viewing excitation–inhibition (E/I) imbalance as a late-stage byproduct to recognizing it as a dynamic, multiscale driver of disease onset and progression. E/I disturbances are now understood to emerge early, interact bidirectionally with amyloid and tau pathology, vary meaningfully by sex and genotype, and represent a potentially druggable target throughout the clinical continuum. Evidence from molecular to whole-brain levels suggests a trajectory of early hyperexcitability that peaks near the stage of mild cognitive impairment, followed by progressive hypoactivity and oscillatory slowing as neurodegeneration advances.
Yet this arc is not uniform. Regional variation, differential susceptibility among neuronal populations (e.g., GABAergic interneurons vs. glutamatergic pyramidal cells), and individual modifiers like APOE4 and sex introduce substantial heterogeneity. Technical variability such as differences in animal models, measurement scale, and analytical approach further complicates interpretation. Notably, APOE4 and female sex often accelerate the early hyperexcitable phase, while tau pathology appears to counteract or mask amyloid-induced excitation, creating complex, region- and scale-specific “tug-of-war” signatures. Some studies suggest amyloid exerts proximity-dependent excitatory effects, while tau’s impact may propagate along network connections. Additionally, low-dose levetiracetam provides proof-of-concept that E/I modulation can normalize network dynamics and improve cognition, but most reliably when baseline hyperactivity is present.
These findings challenge the notion of a single, static E/I “imbalance”, instead pointing to a spectrum of stable and unstable regimes that evolve over time and shape distinct clinical outcomes. Moving forward, research must shift from binary models to longitudinal, multidimensional mappings of E/I homeostasis across development, aging, and disease.
On the measurement front, future studies should attempt to integrate more than one methodology scale. These data should be embedded in biophysical or computational frameworks capable of bridging scales and simulating interventions in silico. Such models not only infer the multiscale drivers of E/I disruption but allow causal hypotheses to be tested virtually, accelerating translational insight. Research into E/I homeostasis in AD will greatly improve not just by focused studies on AD specific pathologies such as amyloid and tau, but also with development of general neuroscience models that can map observations between scales and methodologies.
Therapeutically, trials must adopt physiology-guided designs that stratify participants by sex, APOE genotype, and electrophysiological phenotype, ideally using long-duration EEG or model-derived metrics to identify optimal responders. Mechanistically, promising targets include presynaptic calcium stores, dendritic Kv4.2 potassium channels, interneuron subtype resilience, astrocytic lysosomal signaling, and renin–angiotensin modulation.
For easier visualization, Table 1 offers a preliminary organization of future research directions with example concrete next steps organized by a broader general topic: methodologies, clinical research, clinical trials, and basic science / preclinical studies.
Ultimately, restoring balanced, context-appropriate neural activity may emerge as a unifying therapeutic aim that is positioned upstream of protein aggregation, downstream of genetic risk, and well within reach of existing or emerging interventions. Realizing this vision will require multiscale biomarkers that track individual trajectories in real time, precision therapeutics tailored to neural dynamics rather than disease stage, and inclusive recruitment strategies that embed sex and genotype considerations into trial design from the outset. The central challenge of the next decade is to translate these mechanistic insights into predictive, preventative, and personalized strategies that redefine the trajectory of Alzheimer’s disease.
References
Beam CR, Kaneshiro C, Jang JY, Reynolds CA, Pedersen NL, Gatz M. Differences between women and men in incidence rates of dementia and Alzheimer’s disease. J Alzheimers Dis. 2018;64:1077–83. https://doi.org/10.3233/JAD-180141.
Podcasy JL, Epperson CN. Considering sex and gender in Alzheimer disease and other dementias. Dialog Clin Neurosci. 2016;18:437–46.
2024 Alzheimer’s disease facts and figures. Alzheimer’s & Dementia. 2024;20:3708–821. https://doi.org/10.1002/alz.13809.
Jack CR Jr, Andrews JS, Beach TG, Buracchio T, Dunn B, Graf A, et al. Revised criteria for diagnosis and staging of Alzheimer’s disease: Alzheimer’s Association Workgroup. Alzheimer’s Dement. 2024;20:5143–69. https://doi.org/10.1002/alz.13859.
van Dyck CH, Swanson CJ, Aisen P, Bateman RJ, Chen C, Gee M, et al. Lecanemab in early Alzheimer’s disease. N Engl J Med. 2023;388:9–21. https://doi.org/10.1056/NEJMoa2212948.
Kurkinen M. Lecanemab (Leqembi) is not the right drug for patients with Alzheimer’s disease. Adv Clin Exp Med. 2023;32:943–7. https://doi.org/10.17219/acem/171379.
Muller L, Chavane F, Reynolds J, Sejnowski TJ. Cortical travelling waves: mechanisms and computational principles. Nat Rev Neurosci. 2018;19:255–68. https://doi.org/10.1038/nrn.2018.20.
Tian Y, Tan Z, Hou H, Li G, Cheng A, Qiu Y, et al. Theoretical foundations of studying criticality in the brain. Netw Neurosci. 2022;6:1148–85. https://doi.org/10.1162/netn_a_00269.
Barak O, Tsodyks M. Working models of working memory. Curr Opin Neurobiol. 2014;25:20–4. https://doi.org/10.1016/j.conb.2013.10.008.
Lim S, Goldman MS. Balanced cortical microcircuitry for maintaining information in working memory. Nat Neurosci. 2013;16:1306–14. https://doi.org/10.1038/nn.3492.
Koolschijn RS, Shpektor A, Clarke WT, Ip IB, Dupret D, Emir UE, et al. Memory recall involves a transient break in excitatory-inhibitory balance. eLife. 2021;10. https://doi.org/10.7554/eLife.70071.
Chen L, Yang W, Yang F, Yu Y, Xu T, Wang D, et al. The crosstalk between epilepsy and dementia: a systematic review and meta-analysis. Epilepsy Behav. 2024;152:109640. https://doi.org/10.1016/j.yebeh.2024.109640.
Canitano R. Palumbi R. Excitation/inhibition modulators in autism spectrum disorder: current clinical research. Front Neurosci. 2021;15. https://doi.org/10.3389/fnins.2021.753274.
Bruining H, Hardstone R, Juarez-Martinez EL, Sprengers J, Avramiea AE, Simpraga S, et al. Measurement of excitation-inhibition ratio in autism spectrum disorder using critical brain dynamics. Sci Rep. 2020;10:9195. https://doi.org/10.1038/s41598-020-65500-4.
El-Ansary A, Al-Ayadhi L. GABAergic/glutamatergic imbalance relative to excessive neuroinflammation in autism spectrum disorders. J Neuroinflamm. 2014;11:189. https://doi.org/10.1186/s12974-014-0189-0.
Rubenstein JLR, Merzenich MM. Model of autism: increased ratio of excitation/inhibition in key neural systems. Genes, Brain Behav. 2003;2:255–67. https://doi.org/10.1034/j.1601-183X.2003.00037.x.
Uscătescu LC, Hyatt CJ, Dunn J, Kronbichler M, Calhoun V, Corbera S, et al. Using the excitation/inhibition ratio to optimize the classification of autism spectrum disorder and schizophrenia. medRxiv. Transl Psychiatry. 2025;15:234. https://doi.org/10.1038/s41398-025-03455-8.
Heinze K, Barron HC, Howes EK, Ramaswami M, Broome MR. Excitation and inhibition imbalance – a new therapeutic target for autism and psychosis? Br J Psychiatry: J Ment Sci. 2021;218:295. https://doi.org/10.1192/bjp.2020.163.
Abukhaled Y, Hatab K, Awadhalla M, Hamdan H. Understanding the genetic mechanisms and cognitive impairments in Down syndrome: towards a holistic approach. J Neurol. 2024;271:87–104. https://doi.org/10.1007/s00415-023-11890-0.
Selten M, Bokhoven HV, Kasri NN. Inhibitory control of the excitatory/inhibitory balance in psychiatric disorders. F1000Research. 2018;7:23. https://doi.org/10.12688/f1000research.12155.1
Corriveau-Lecavalier N, Adams JN, Fischer L, Molloy EN, Maass A. Cerebral hyperactivation across the Alzheimer’s disease pathological cascade. Brain Commun. 2024;6:fcae376. https://doi.org/10.1093/braincomms/fcae376.
Burns AP, Fortel I, Zhan L, Lazarov O, Mackin RS, Demos AP, et al. Longitudinal excitation-inhibition balance altered by sex and APOE-ε4. Commun Biol. 2025;8:1–9. https://doi.org/10.1038/s42003-025-07876-5.
Koelewijn L, Lancaster TM, Linden D, Dima DC, Routley BC, Magazzini L, et al. Oscillatory hyperactivity and hyperconnectivity in young APOE-ɛ4 carriers and hypoconnectivity in Alzheimer’s disease. Elife. 2019;8:e36011. https://doi.org/10.7554/eLife.36011.
van Nifterick AM, Gouw AA, van Kesteren RE, Scheltens P, Stam CJ, de Haan W. A multiscale brain network model links Alzheimer’s disease-mediated neuronal hyperactivity to large-scale oscillatory slowing. Alzheimer’s Res Ther. 2022;14:101. https://doi.org/10.1186/s13195-022-01041-4.
Maestú F, de Haan W, Busche MA, DeFelipe J. Neuronal excitation/inhibition imbalance: core element of a translational perspective on Alzheimer pathophysiology. Ageing Res Rev. 2021;69:101372. https://doi.org/10.1016/j.arr.2021.101372.
Toniolo S, Sen A, Husain M. Modulation of brain hyperexcitability: potential new therapeutic approaches in Alzheimer’s disease. Int J Mol Sci. 2020;21:9318. https://doi.org/10.3390/ijms21239318.
Bridi MCD, Zong FJ, Min X, Luo N, Tran T, Qiu J, et al. Daily oscillation of the excitation-inhibition balance in visual cortical circuits. Neuron. 2020;105:621–.e4. https://doi.org/10.1016/j.neuron.2019.11.011.
Hanke JM, Schindler KA, Seiler A. On the relationships between epilepsy, sleep, and Alzheimer’s disease: a narrative review. Epilepsy Behav. 2022;129:108609. https://doi.org/10.1016/j.yebeh.2022.108609.
Silva I, Silva J, Ferreira R, Trigo D. Glymphatic system, AQP4, and their implications in Alzheimer’s disease. Neurol Res Pract. 2021;3:5. https://doi.org/10.1186/s42466-021-00102-7.
Sun Q, Peng S, Xu Q, Weikop P, Hussain R, Song W, et al. Enhancing glymphatic fluid transport by pan-adrenergic inhibition suppresses epileptogenesis in male mice. Nat Commun. 2024;15:9600. https://doi.org/10.1038/s41467-024-53430-y.
Voumvourakis KI, Sideri E, Papadimitropoulos GN, Tsantzali I, Hewlett P, Kitsos D, et al. The dynamic relationship between the glymphatic system, aging, memory, and sleep. Biomedicines. 2023;11:2092. https://doi.org/10.3390/biomedicines11082092.
Zhu TT, Yang JJ, Hashimoto K. Noradrenergic modulation of glymphatic clearance: implications for neuropsychiatric disorders and mortality. Mol Psychiatry. 2025;30:4432–4. https://doi.org/10.1038/s41380-025-03051-8.
Frere S, Slutsky I. Alzheimer’s disease: from firing instability to homeostasis network collapse. Neuron. 2018;97:32–58. https://doi.org/10.1016/j.neuron.2017.11.028.
Niraula S, Doderer JJ, Indulkar S, Berry KP, Hauser WL, L'Esperance OJ, et al. Excitation-inhibition imbalance disrupts visual familiarity in amyloid and non-pathology conditions. Cell Rep. 2023;42:111946. https://doi.org/10.1016/j.celrep.2022.111946.
Wu JW, Hussaini SA, Bastille IM, Rodriguez GA, Mrejeru A, Rilett K, et al. Neuronal activity enhances tau propagation and tau pathology in vivo. Nat Neurosci. 2016;19:1085–92. https://doi.org/10.1038/nn.4328.
Lauterborn JC, Scaduto P, Cox CD, Schulmann A, Lynch G, Gall CM, et al. Increased excitatory to inhibitory synaptic ratio in parietal cortex samples from individuals with Alzheimer’s disease. Nat Commun. 2021;12:2603. https://doi.org/10.1038/s41467-021-22742-8.
Barbour AJ, Gourmaud S, Lancaster E, Li X, Stewart D, Irwin D, et al. Seizures exacerbate excitatory: inhibitory imbalance in Alzheimer’s disease and 5XFAD mice. Brain. 2024;147:2169–84. https://doi.org/10.1093/brain/awae126.
Fishell G, Kepecs A. Interneuron types as attractors and controllers. Annu Rev Neurosci. 2020; 43:1–30. https://doi.org/10.1146/annurev-neuro-070918-050421.
Kepecs A, Fishell G. Interneuron cell types: fit to form and formed to fit. Nature. 2014;505:318–26. https://doi.org/10.1038/nature12983.
Leung L, Andrews-Zwilling Y, Yoon SY, Jain S, Ring K, Dai J, et al. Apolipoprotein E4 causes age- and sex-dependent impairments of hilar GABAergic interneurons and learning and memory deficits in mice. PLOS ONE. 2012;7:e53569. https://doi.org/10.1371/journal.pone.0053569.
Hatch RJ, Wei Y, Xia D, Götz J. Hyperphosphorylated tau causes reduced hippocampal CA1 excitability by relocating the axon initial segment. Acta Neuropathol. 2017;133:717–30. https://doi.org/10.1007/s00401-017-1674-1.
Crimins JL, Rocher AB, Luebke JI. Electrophysiological changes precede morphological changes to frontal cortical pyramidal neurons in the rTg4510 mouse model of progressive tauopathy. Acta Neuropathol. 2012;124:777–95. https://doi.org/10.1007/s00401-012-1038-9.
Sofroniew NJ, Flickinger D, King J, Svoboda K. A large field of view two-photon mesoscope with subcellular resolution for in vivo imaging. eLife. 2016;5:e14472. https://doi.org/10.7554/eLife.14472.
Nietz AK, Popa LS, Streng ML, Carter RE, Kodandaramaiah SB, Ebner TJ. Wide-field calcium imaging of neuronal network dynamics in vivo. Biology. 2022;11:1601. https://doi.org/10.3390/biology11111601.
Steinmetz NA, Aydin C, Lebedeva A, Okun M, Pachitariu M, Bauza M, et al. Neuropixels 2.0: a miniaturized high-density probe for stable, long-term brain recordings. Science. 2021;372:eabf4588. https://doi.org/10.1126/science.abf4588.
Baillet S. Magnetoencephalography for brain electrophysiology and imaging. Nat Neurosci. 2017;20:327–39. https://doi.org/10.1038/nn.4504.
Kopčanová M, Tait L, Donoghue T, Stothart G, Smith L, Sandoval AAF, et al. Resting-state EEG signatures of Alzheimer’s disease are driven by periodic but not aperiodic changes. Neurobiol Dis. 2024;190:106380. https://doi.org/10.1016/j.nbd.2023.106380.
Wiesman AI, Murman DL, Losh RA, Schantell M, Christopher-Hayes NJ, Johnson HJ, et al. Spatially resolved neural slowing predicts impairment and amyloid burden in Alzheimer’s disease. Brain. 2022;145:2177–89. https://doi.org/10.1093/brain/awab430.
Palop JJ, Chin J, Roberson ED, Wang J, Thwin MT, Bien-Ly N, et al. Aberrant excitatory neuronal activity and compensatory remodeling of inhibitory hippocampal circuits in mouse models of Alzheimer’s disease. Neuron. 2007;55:697–711. https://doi.org/10.1016/j.neuron.2007.07.025.
Roberson ED, Halabisky B, Yoo JW, Yao J, Chin J, Yan F, et al. Amyloid-β/Fyn–induced synaptic, network, and cognitive impairments depend on tau levels in multiple mouse models of Alzheimer’s disease. J Neurosci. 2011;31:700–11. https://doi.org/10.1523/JNEUROSCI.4152-10.2011.
DeVos SL, Goncharoff DK, Chen G, Kebodeaux CS, Yamada K, Stewart FR, et al. Antisense reduction of tau in adult mice protects against seizures. J Neurosci. 2013;33:12887–97. https://doi.org/10.1523/JNEUROSCI.2107-13.2013.
Pusil S, López ME, Cuesta P, Bruña R, Pereda E, Maestú F. Hypersynchronization in mild cognitive impairment: the ‘X’ model. Brain. 2019;142:3936–50. https://doi.org/10.1093/brain/awz320.
Zou QH, Zhu CZ, Yang Y, Zuo XN, Long XY, Cao QJ, et al. An improved approach to detection of amplitude of low-frequency fluctuation (ALFF) for resting-state fMRI: fractional ALFF. J Neurosci Methods. 2008;172:137–41. https://doi.org/10.1016/j.jneumeth.2008.04.012.
Das S, Puthankattil SD. Functional connectivity and complexity in the phenomenological model of mild cognitive-impaired Alzheimer’s disease. Front Comput Neurosci. 2022;16. https://doi.org/10.3389/fncom.2022.877912.
de Haan W, Mott K, van Straaten ECW, Scheltens P, Stam CJ. Activity-dependent degeneration explains hub vulnerability in Alzheimer’s disease. PLOS Comput Biol. 2012;8:e1002582. https://doi.org/10.1371/journal.pcbi.1002582.
de Haan W, van Straaten ECW, Gouw AA, Stam CJ. Altering neuronal excitability to preserve network connectivity in a computational model of Alzheimer’s disease. PLOS Comput Biol. 2017;13:e1005707. https://doi.org/10.1371/journal.pcbi.1005707.
Stefanovski L, Triebkorn P, Spiegler A, Diaz-Cortes MA, Solodkin A, Jirsa V, et al. Linking molecular pathways and large-scale computational modeling to assess candidate disease mechanisms and pharmacodynamics in Alzheimer’s Disease. Front Comput Neurosci. 2019;13:54. https://doi.org/10.3389/fncom.2019.00054.
Fortel I, Korthauer LE, Morrissey Z, Zhan L, Ajilore O, Wolfson O, et al. Connectome signatures of hyperexcitation in cognitively intact middle-aged female APOE-ε4 carriers. Cereb Cortex. 2020;30:6350–62. https://doi.org/10.1093/cercor/bhaa190.
Alderson TH, Bokde ALW, Kelso JAS, Maguire L, Coyle D. Metastable neural dynamics in Alzheimer’s disease are disrupted by lesions to the structural connectome. NeuroImage. 2018;183:438–55. https://doi.org/10.1016/j.neuroimage.2018.08.033.
Arbabyazd L, Shen K, Wang Z, Hofmann-Apitius M, Ritter P, McIntosh AR, et al. Virtual connectomic datasets in Alzheimer’s disease and aging using whole-brain network dynamics modelling. eNeuro. 2021;8. https://doi.org/10.1523/ENEURO.0475-20.2021.
Cakir Y. Hybrid modeling of alpha rhythm and the amplitude of low-frequency fluctuations abnormalities in the thalamocortical region and basal ganglia in Alzheimer’s disease. Eur J Neurosci. 2020;52:2944–61. https://doi.org/10.1111/ejn.14666.
Fortel I, Zhan L, Ajilore O, Wu Y, Mackin S, Leow A. Disrupted excitation-inhibition balance in cognitively normal individuals at risk of Alzheimer’s disease. JAD. 2023;95:1–19. https://doi.org/10.3233/JAD-230035.
Ghorbanian P, Ramakrishnan S, Ashrafiuon H. Stochastic coupled oscillator model of EEG for Alzheimer’s disease. Annu Int Conf IEEE Eng Med Biol Soc. 2014;2014:706–9. https://doi.org/10.1109/EMBC.2014.6943688.
Li G, Hsu LM, Wu Y, Bozoki AC, Shih YYI, Yap PT. Revealing excitation-inhibition imbalance in Alzheimer’s disease using multiscale neural model inversion of resting-state functional MRI. Commun Med. 2025;5:1–21. https://doi.org/10.1038/s43856-025-00736-7.
Patow G, Stefanovski L, Ritter P, Deco G, Kobeleva X. for the Alzheimer’s Disease Neuroimaging Initiative. Whole-brain modeling of the differential influences of amyloid-beta and tau in Alzheimer’s disease. Alzheimer’s Res Ther. 2023;15:210. https://doi.org/10.1186/s13195-023-01349-9.
Ranasinghe KG, Verma P, Cai C, Xie X, Kudo K, Gao X, et al. Altered excitatory and inhibitory neuronal subpopulation parameters are distinctly associated with tau and amyloid in Alzheimer’s disease. eLife. 2022;11:e77850. https://doi.org/10.7554/eLife.77850.
Ruiz-Gómez SJ, Hornero R, Poza J, Maturana-Candelas A, Pinto N, Gómez C. Computational modeling of the effects of EEG volume conduction on functional connectivity metrics. Application to Alzheimer’s disease continuum. J Neural Eng. 2019;16:066019. https://doi.org/10.1088/1741-2552/ab4024.
Sanz Perl Y, Fittipaldi S, Gonzalez Campo C, Moguilner S, Cruzat J, Fraile-Vazquez ME, et al. Model-based whole-brain perturbational landscape of neurodegenerative diseases. eLife. 2023;12:e83970. https://doi.org/10.7554/eLife.83970.
Triebkorn P, Stefanovski L, Dhindsa K, Diaz-Cortes MA, Bey P, Bülau K, et al. Brain simulation augments machine-learning-based classification of dementia. Alzheimers Dement. 2022;8:e12303. https://doi.org/10.1002/trc2.12303.
Yang L, Lu J, Li D, Xiang J, Yan T, Sun J, et al. Alzheimer’s disease: insights from large-scale brain dynamics models. Brain Sci. 2023;13:1133. https://doi.org/10.3390/brainsci13081133.
Giorgio J, Adams JN, Maass A, Jagust WJ, Breakspear M. Amyloid induced hyperexcitability in default mode network drives medial temporal hyperactivity and early tau accumulation. Neuron. 2024;112:676–686.e4. https://doi.org/10.1016/j.neuron.2023.11.014.
Moffat G, Zhukovsky P, Coughlan G, Voineskos AN. Unravelling the relationship between amyloid accumulation and brain network function in normal aging and very mild cognitive decline: a longitudinal analysis. Brain Commun. 2022;4:fcac282. https://doi.org/10.1093/braincomms/fcac282.
Huijbers W, Mormino EC, Schultz AP, Wigman S, Ward AM, Larvie M, et al. Amyloid-β deposition in mild cognitive impairment is associated with increased hippocampal activity, atrophy and clinical progression. Brain. 2015;138:1023–35. https://doi.org/10.1093/brain/awv007.
Canário N, Jorge L, Martins R, Santana I, Castelo-Branco M. Dual PET-fMRI reveals a link between neuroinflammation, amyloid binding and compensatory task-related brain activity in Alzheimer’s disease. Commun Biol. 2022;5:804. https://doi.org/10.1038/s42003-022-03761-7.
Jones DT, Graff-Radford J, Lowe VJ, Wiste HJ, Gunter JL, Senjem ML, et al. Tau, amyloid, and cascading network failure across the Alzheimer’s disease spectrum. Cortex. 2017;97:143–59. https://doi.org/10.1016/j.cortex.2017.09.018.
Elman JA, Madison CM, Baker SL, Vogel JW, Marks SM, Crowley S, et al. Effects of beta-amyloid on resting state functional connectivity within and between networks reflect known patterns of regional vulnerability. Cereb Cortex. 2016;26:695–707. https://doi.org/10.1093/cercor/bhu259.
Al-Ezzi A, Arechavala RJ, Butler R, Nolty A, Kang JJ, Shimojo S, et al. Disrupted brain functional connectivity as early signature in cognitively healthy individuals with pathological CSF amyloid/tau. Commun Biol. 2024;7:1037. https://doi.org/10.1038/s42003-024-06673-w.
Park J, Kim WJ, Jung HW, Kim JJ, Park JY. Relationship between regional relative theta power and amyloid deposition in mild cognitive impairment: an exploratory study. Front Neurosci. 2025;19. https://doi.org/10.3389/fnins.2025.1510878.
Kim NH, Park U, Yang DW, Choi SH, Youn YC, Kang SW. PET-validated EEG-machine learning algorithm predicts brain amyloid pathology in pre-dementia Alzheimer’s disease. Sci Rep. 2023;13:10299. https://doi.org/10.1038/s41598-023-36713-0.
Engels MMA, van der Flier WM, Stam CJ, Hillebrand A, Scheltens PH, van Straaten ECW. Alzheimer’s disease: the state of the art in resting-state magnetoencephalography. Clin Neurophysiol. 2017;128:1426–37. https://doi.org/10.1016/j.clinph.2017.05.012.
Eyler LT, Elman JA, Hatton SN, Gough S, Mischel AK, Hagler DJ, et al. Resting state abnormalities of the default mode network in mild cognitive impairment: a systematic review and meta-analysis. J Alzheimers Dis. 2019;70:107–20. https://doi.org/10.3233/JAD-180847.
Franzmeier N, Brendel M, Beyer L, Slemann L, Kovacs GG, Arzberger T, et al. Tau deposition patterns are associated with functional connectivity in primary tauopathies. Nat Commun. 2022;13:1362. https://doi.org/10.1038/s41467-022-28896-3.
Franzmeier N, Neitzel J, Rubinski A, Smith R, Strandberg O, Ossenkoppele R, et al. Functional brain architecture is associated with the rate of tau accumulation in Alzheimer’s disease. Nat Commun. 2020;11:347. https://doi.org/10.1038/s41467-019-14159-1.
Nabizadeh F, Alzheimer’s Disease Neuroimaging Initiative (ADNI). Disruption in functional networks mediated tau spreading in Alzheimer’s disease. Brain Commun. 2024;6:fcae198. https://doi.org/10.1093/braincomms/fcae198.
Canuet L, Pusil S, López ME, Bajo R, Pineda-Pardo JÁ, Cuesta P, et al. Network disruption and cerebrospinal fluid amyloid-beta and phospho-tau levels in mild cognitive impairment. J Neurosci. 2015;35:10325–30. https://doi.org/10.1523/JNEUROSCI.0704-15.2015.
Busche MA, Eichhoff G, Adelsberger H, Abramowski D, Wiederhold KH, Haass C, et al. Clusters of Hyperactive Neurons Near Amyloid Plaques in a Mouse Model of Alzheimer’s Disease. Science. 2008;321:1686–9. https://doi.org/10.1126/science.1162844.
Algamal M, Russ AN, Miller MR, Hou SS, Maci M, Munting LP, et al. Reduced excitatory neuron activity and interneuron-type-specific deficits in a mouse model of Alzheimer’s disease. Commun Biol. 2022;5:1–9. https://doi.org/10.1038/s42003-022-04268-x.
Busche MA, Chen X, Henning HA, Reichwald J, Staufenbiel M, Sakmann B, et al. Critical role of soluble amyloid-β for early hippocampal hyperactivity in a mouse model of Alzheimer’s disease. Proc Natl Acad Sci. 2012;109:8740–5. https://doi.org/10.1073/pnas.1206171109.
Chung H, Park K, Jang HJ, Kohl MM, Kwag J. Dissociation of somatostatin and parvalbumin interneurons circuit dysfunctions underlying hippocampal theta and gamma oscillations impaired by amyloid β oligomers in vivo. Brain Struct Funct. 2020;225:935–54. https://doi.org/10.1007/s00429-020-02044-3.
Giesers NK, Wirths O. Loss of hippocampal calretinin and parvalbumin interneurons in the 5XFAD mouse model of Alzheimer’s Disease. ASN Neuro. 2020;12: 1759091420925356. https://doi.org/10.1177/1759091420925356.
Ren SQ, Yao W, Yan JZ, Jin C, Yin JJ, Yuan J, et al. Amyloid β causes excitation/inhibition imbalance through dopamine receptor 1-dependent disruption of fast-spiking GABAergic input in anterior cingulate cortex. Sci Rep. 2018;8:302. https://doi.org/10.1038/s41598-017-18729-5.
Verret L, Mann EO, Hang GB, Barth AM, Cobos I, Ho K, et al. Inhibitory interneuron deficit links altered network activity and cognitive dysfunction in Alzheimer's model. Cell. 2012;149:708–21. https://doi.org/10.1016/j.cell.2012.02.046.
Zott B, Simon MM, Hong W, Unger F, Chen-Engerer HJ, Frosch MP, et al. A vicious cycle of β amyloid–dependent neuronal hyperactivation. Science. 2019;365:559–65. https://doi.org/10.1126/science.aay0198.
Busche MA. Tau suppresses neuronal activity in vivo, even before tangles form. Brain. 2019;142:843–6. https://doi.org/10.1093/brain/awz060.
Fu H, Rodriguez GA, Herman M, Emrani S, Nahmani E, Barrett G, et al. Tau pathology induces excitatory neuron loss, grid cell dysfunction, and spatial memory deficits reminiscent of early Alzheimer’s disease. Neuron. 2017;93:533–541.e5. https://doi.org/10.1016/j.neuron.2016.12.023.
Decker JM, Krüger L, Sydow A, Dennissen FJ, Siskova Z, Mandelkow E, et al. The Tau/A152T mutation, a risk factor for frontotemporal-spectrum disorders, leads to NR2B receptor-mediated excitotoxicity. EMBO Rep. 2016;17:552–69. https://doi.org/10.15252/embr.201541439.
Holth JK, Bomben VC, Reed JG, Inoue T, Younkin L, Younkin SG, et al. Tau loss attenuates neuronal network hyperexcitability in mouse and drosophila genetic models of epilepsy. J Neurosci. 2013;33:1651–9. https://doi.org/10.1523/JNEUROSCI.3191-12.2013.
Maeda S, Djukic B, Taneja P, Yu GQ, Lo I, Davis A, et al. Expression of A152T human tau causes age-dependent neuronal dysfunction and loss in transgenic mice. EMBO Rep. 2016;17:530–51. https://doi.org/10.15252/embr.201541438.
Shimojo M, Takuwa H, Takado Y, Tokunaga M, Tsukamoto S, Minatohara K, et al. Selective disruption of inhibitory synapses leading to neuronal hyperexcitability at an early stage of tau pathogenesis in a mouse model. J Neurosci. 2020;40:3491–501. https://doi.org/10.1523/JNEUROSCI.2880-19.2020.
Angulo SL, Orman R, Neymotin SA, Liu L, Buitrago L, et al. Tau and amyloid-related pathologies in the entorhinal cortex have divergent effects in the hippocampal circuit. Neurobiol Dis. 2017;108:261–76. https://doi.org/10.1016/j.nbd.2017.08.015.
Hall AM, Throesch BT, Buckingham SC, Markwardt SJ, Peng Y, Wang Q, et al. Tau-dependent Kv4.2 depletion and dendritic hyperexcitability in a mouse model of Alzheimer’s disease. J Neurosci. 2015;35:6221–30. https://doi.org/10.1523/JNEUROSCI.2552-14.2015.
Rodriguez GA, Barrett GM, Duff KE, Hussaini SA. Chemogenetic attenuation of neuronal activity in the entorhinal cortex reduces Aβ and tau pathology in the hippocampus. PLoS Biol. 2020;18:3000851. https://doi.org/10.1371/journal.pbio.3000851.
Arroyo-García LE, Isla AG, Andrade-Talavera Y, Balleza-Tapia H, Loera-Valencia R, Alvarez-Jimenez L, et al. Impaired spike-gamma coupling of area CA3 fast-spiking interneurons as the earliest functional impairment in the AppNL-G-F mouse model of Alzheimer’s disease. Mol Psychiatry. 2021;26:5557–67. https://doi.org/10.1038/s41380-021-01257-0.
Nuriel T, Angulo SL, Khan U, Ashok A, Chen Q, Figueroa HY, et al. Neuronal hyperactivity due to loss of inhibitory tone in APOE4 mice lacking Alzheimer’s disease-like pathology. Nat Commun. 2017;8:1464. https://doi.org/10.1038/s41467-017-01444-0.
Wishart HA, Saykin AJ, Rabin LA, Santulli RB, Flashman LA, Guerin SJ, et al. Increased brain activation during working memory in cognitively intact adults with the APOE ε4 allele. AJP. 2006;163:1603–10. https://doi.org/10.1176/ajp.2006.163.9.1603.
Filippini N, MacIntosh BJ, Hough MG, Goodwin GM, Frisoni GB, Smith SM, et al. Distinct patterns of brain activity in young carriers of the APOE-ε4 allele. Proc Natl Acad Sci. 2009;106:7209–14. https://doi.org/10.1073/pnas.0811879106.
Sheline YI, Morris JC, Snyder AZ, Price JL, Yan Z, D'Angelo G, et al. APOE4 allele disrupts resting state fMRI connectivity in the absence of amyloid plaques or decreased CSF Aβ42. J Neurosci. 2010;30:17035–40. https://doi.org/10.1523/JNEUROSCI.3987-10.2010.
Bookheimer SY, Strojwas MH, Cohen MS, Saunders AM, Pericak-Vance MA, Mazziotta JC, et al. Patterns of brain activation in people at risk for Alzheimer’s Disease. N Engl J Med. 2000;343:450–6. https://doi.org/10.1056/NEJM200008173430701.
Ponomareva NV, Korovaitseva GI, Rogaev EI. EEG alterations in non-demented individuals related to apolipoprotein E genotype and to risk of Alzheimer's disease. Neurobiol Aging. 2008;29:819–27. https://doi.org/10.1016/j.neurobiolaging.2006.12.019.
Schreiner SJ, Van Bergen JMG, Gietl AF, Buck A, Hock C, Pruessmann KP, et al. Gray matter gamma-hydroxy-butyric acid and glutamate reflect beta-amyloid burden at old age. Alzheimer's Dement (Amst. 2024;16:12587. https://doi.org/10.1002/dad2.12587.
Yassa MA, Lacy JW, Stark SM, Albert MS, Gallagher M, Stark CEL. Pattern separation deficits associated with increased hippocampal CA3 and dentate gyrus activity in nondemented older adults. Hippocampus. 2011;21:968–79. https://doi.org/10.1002/hipo.20808.
O'Brien JL, O'Keefe KM, LaViolette PS, DeLuca AN, Blacker D, Dickerson BC. Longitudinal fMRI in elderly reveals loss of hippocampal activation with clinical decline. Neurology. 2010;74:1969–76. https://doi.org/10.1212/WNL.0b013e3181e3966e.
Ochoa JF, Alonso JF, Duque JE, Tobón CA, Baena A, Lopera F, et al. Precuneus failures in subjects of the PSEN1 E280A family at risk of developing alzheimer’s disease detected using quantitative electroencephalography. J Alzheimers Dis. 2017;58:1229–44. https://doi.org/10.3233/JAD-161291.
Velu L, Pellerin L, Julian A, Paccalin M, Giraud C, Fayolle P, et al. Early rise of glutamate-glutamine levels in mild cognitive impairment: evidence for emerging excitotoxicity. J Neuroradiol. 2024;51:168–75. https://doi.org/10.1016/j.neurad.2023.09.003.
Fu X, Qin M, Liu X, Cheng L, Zhang L, Zhang X, et al. Decreased GABA levels of the anterior and posterior cingulate cortex are associated with executive dysfunction in mild cognitive impairment. Front Neurosci. 2023;17:1220122. https://doi.org/10.3389/fnins.2023.1220122.
Oeltzschner G, Wijtenburg SA, Mikkelsen M, Edden RAE, Barker PB, Joo JH, et al. Neurometabolites and associations with cognitive deficits in mild cognitive impairment: a magnetic resonance spectroscopy study at 7 Tesla. Neurobiol Aging. 2019;73:211–8. https://doi.org/10.1016/j.neurobiolaging.2018.09.027.
Hijazi S, Heistek TS, Scheltens P, Neumann U, Shimshek DR, Mansvelder HD, et al. Early restoration of parvalbumin interneuron activity prevents memory loss and network hyperexcitability in a mouse model of Alzheimer’s disease. Mol Psychiatry. 2020;25:3380–98. https://doi.org/10.1038/s41380-019-0483-4.
Huang D, Liu D, Yin J, Qian T, Shrestha S, Ni H. Glutamate-glutamine and GABA in brain of normal aged and patients with cognitive impairment. Eur Radio. 2017;27:2698–705. https://doi.org/10.1007/s00330-016-4669-8.
Horvath AA, Papp A, Zsuffa J, Szucs A, Luckl J, Radai F, et al. Subclinical epileptiform activity accelerates the progression of Alzheimer’s disease: a long-term EEG study. Clin Neurophysiol. 2021;132:1982–9. https://doi.org/10.1016/j.clinph.2021.03.050.
Lam AD, Deck G, Goldman A, Eskandar EN, Noebels J, Cole AJ. Silent hippocampal seizures and spikes identified by foramen ovale electrodes in Alzheimer’s disease. Nat Med. 2017;23:678–80. https://doi.org/10.1038/nm.4330.
Vossel KA, Beagle AJ, Rabinovici GD, Shu H, Lee SE, Naasan G, et al. Seizures and epileptiform activity in the early stages of Alzheimer disease. JAMA Neurol. 2013;70:1158–66. https://doi.org/10.1001/jamaneurol.2013.136.
Vossel KA, Ranasinghe KG, Beagle AJ, Mizuiri D, Honma SM, Dowling AF, et al. Incidence and impact of subclinical epileptiform activity in Alzheimer’s disease. Ann Neurol. 2016;80:858–70. https://doi.org/10.1002/ana.24794.
Scaduto P, Lauterborn JC, Cox CD, Fracassi A, Zeppillo T, Gutierrez BA, et al. Functional excitatory to inhibitory synaptic imbalance and loss of cognitive performance in people with Alzheimer’s disease neuropathologic change. Acta Neuropathol. 2023;145:303–24. https://doi.org/10.1007/s00401-022-02526-0.
Engels MM, Hillebrand A, van der Flier WM, Stam CJ, Scheltens P, van Straaten EC. Slowing of hippocampal activity correlates with cognitive decline in early onset Alzheimer’s disease. An MEG study with virtual electrodes. Front Hum Neurosci. 2016;10:238. https://doi.org/10.3389/fnhum.2016.00238.
de Haan W, Stam CJ, Jones BF, Zuiderwijk IM, van Dijk BW, Scheltens P. Resting-state oscillatory brain dynamics in Alzheimer's disease. J Clin Neurophysiol. 2008;25:187–93. https://doi.org/10.1097/WNP.0b013e31817da184.
Larramona-Arcas R, González-Arias C, Perea G, Gutiérrez A, Vitorica J, García-Barrera T, et al. Sex-dependent calcium hyperactivity due to lysosomal-related dysfunction in astrocytes from APOE4 versus APOE3 gene-targeted replacement mice. Mol Neurodegener. 2020;15:35. https://doi.org/10.1186/s13024-020-00382-8.
Li G, Bien-Ly N, Andrews-Zwilling Y, Xu Q, Bernardo A, Ring K, et al. GABAergic interneuron dysfunction impairs hippocampal neurogenesis in adult apolipoprotein E4 knockin mice. Cell Stem Cell. 2009;5:634–45. https://doi.org/10.1016/j.stem.2009.10.015.
Scheinman SB, Tseng KY, Alford S, Tai LM. Higher neuronal facilitation and potentiation with APOE4 suppressed by angiotensin II. Mol Neurobiol. 2024;61:120–31. https://doi.org/10.1007/s12035-023-03556-9.
Jain A, Huang GZ, Woolley CS. Latent sex differences in molecular signaling that underlies excitatory synaptic potentiation in the hippocampus. J Neurosci. 2019;39:1552–65. https://doi.org/10.1523/JNEUROSCI.1897-18.2018.
Mosconi L, Nerattini M, Matthews DC, Jett S, Andy C, Williams S, et al. In vivo brain estrogen receptor density by neuroendocrine aging and relationships with cognition and symptomatology. Sci Rep. 2024;14:12680. https://doi.org/10.1038/s41598-024-62820-7.
Tabatadze N, Huang G, May RM, Jain A, Woolley CS. Sex differences in molecular signaling at inhibitory synapses in the hippocampus. J Neurosci. 2015;35:11252. https://doi.org/10.1523/JNEUROSCI.1067-15.2015.
Terstege DJ, Epp JR. Parvalbumin as a sex-specific target in Alzheimer’s disease research—a mini-review. Neurosci Biobehav Rev. 2023;153:105370. https://doi.org/10.1016/j.neubiorev.2023.105370.
Oliva M, Muñoz-Aguirre M, Kim-Hellmuth S, Wucher V, Gewirtz ADH, Cotter DJ, et al. The impact of sex on gene expression across human tissues. Science. 2020;369:eaba3066. https://doi.org/10.1126/science.aba3066.
Hafizi S, Rajji TK. Modifiable risk factors of dementia linked to excitation-inhibition imbalance. Ageing Res Rev. 2023;83:101804. https://doi.org/10.1016/j.arr.2022.101804.
Targa Dias Anastacio H, Matosin N, Ooi L. Neuronal hyperexcitability in Alzheimer’s disease: what are the drivers behind this aberrant phenotype?. Transl Psychiatry. 2022;12:1–14. https://doi.org/10.1038/s41398-022-02024-7.
Franco R, Vargas MR. Redox biology in neurological function, dysfunction, and aging. Antioxid Redox Signal. 2018;28:1583–6. https://doi.org/10.1089/ars.2018.7509.
Beltrán González AN, López Pazos MI, Calvo DJ. Reactive oxygen species in the regulation of the GABA-mediated Inhibitory neurotransmission. Neuroscience. 2020;439:137–45. https://doi.org/10.1016/j.neuroscience.2019.05.064.
Atlante A, Calissano P, Bobba A, Giannattasio S, Marra E, Passarella S. Glutamate neurotoxicity, oxidative stress and mitochondria. FEBS Lett. 2001;497:1–5. https://doi.org/10.1016/S0014-5793(01)02437-1.
de Prado Bert P, Mercader EMH, Pujol J, Sunyer J, Mortamais M. The effects of air pollution on the brain: a review of studies interfacing environmental epidemiology and neuroimaging. Curr Environ Health Rep. 2018;5:351–64. https://doi.org/10.1007/s40572-018-0209-9.
Nguyen TT, Ta QTH, Nguyen TKO, Nguyen TTD, Giau VV. Type 3 diabetes and its role implications in Alzheimer’s Disease. Int J Mol Sci. 2020;21:3165. https://doi.org/10.3390/ijms21093165.
Kciuk M, Kruczkowska W, Gałęziewska J, Wanke K, Kałuzińska-Kołat Ż, Aleksandrowicz M, et al. Alzheimer’s Disease as type 3 diabetes: understanding the link and implications. Int J Mol Sci. 2024;25:11955. https://doi.org/10.3390/ijms252211955.
Cai W, Zhang X, Batista TM, García-Martín R, Softic S, Wang G, et al. Peripheral insulin regulates a broad network of gene expression in hypothalamus, hippocampus, and nucleus accumbens. Diabetes. 2021;70:1857–73. https://doi.org/10.2337/db20-1119.
Arnold SE, Arvanitakis Z, Macauley-Rambach SL, Koenig AM, Wang HY, Ahima RS, et al. Brain insulin resistance in type 2 diabetes and Alzheimer disease: concepts and conundrums. Nat Rev Neurol. 2018;14:168–81. https://doi.org/10.1038/nrneurol.2017.185.
Petrou M, Pop-Busui R, Foerster BR, Edden RA, Callaghan BC, Harte SE, et al. Altered excitation-inhibition balance in the brain of patients with diabetic neuropathy. Acad Radiol. 2012;19:607–12. https://doi.org/10.1016/j.acra.2012.02.004.
Larsson M, Lietzau G, Nathanson D, Östenson CG, Mallard C, Johansson ME, et al. Diabetes negatively affects cortical and striatal GABAergic neurons: an effect that is partially counteracted by exendin-4. Biosci Rep. 2016;36:e00421. https://doi.org/10.1042/BSR20160437.
Lyoo IK, Yoon SJ, Musen G, Simonson DC, Weinger K, Bolo N, et al. Altered prefrontal glutamate–glutamine–γ-aminobutyric acid levels and relation to low cognitive performance and depressive symptoms in type 1 diabetes mellitus. Arch Gen Psychiatry. 2009;66:878–87. https://doi.org/10.1001/archgenpsychiatry.2009.86.
Chang MY, Boulden J, Katz JB, Wang L, Meyer TJ, Soler AP, et al. Bin1 ablation increases susceptibility to cancer during aging, particularly lung cancer. Cancer Res. 2007;67:7605–12. https://doi.org/10.1158/0008-5472.CAN-07-1100.
Voskobiynyk Y, Roth JR, Cochran JN, Rush T, Carullo NV, Mesina JS, et al. Alzheimer’s disease risk gene BIN1 induces Tau-dependent network hyperexcitability. Zoghbi HY, Fryer JD, Reed M, Kraemer B, eds. eLife. 2020;9:e57354. https://doi.org/10.7554/eLife.57354.
Devinsky O, Vezzani A, Najjar S, De Lanerolle NC, Rogawski MA. Glia and epilepsy: excitability and inflammation. Trends Neurosci. 2013;36:174–84. https://doi.org/10.1016/j.tins.2012.11.008.
Dubé C, Vezzani A, Behrens M, Bartfai T, Baram TZ. Interleukin-1β contributes to the generation of experimental febrile seizures. Ann Neurol. 2005;57:152–5. https://doi.org/10.1002/ana.20358.
Ravizza T, Lucas SM, Balosso S, Bernardino L, Ku G, Noé F, et al. Inactivation of caspase-1 in rodent brain: a novel anticonvulsive strategy. Epilepsia. 2006;47:1160–8. https://doi.org/10.1111/j.1528-1167.2006.00590.x.
Balosso S, Maroso M, Sanchez-Alavez M, Ravizza T, Frasca A, Bartfai T, et al. A novel non-transcriptional pathway mediates the proconvulsive effects of interleukin-1β. Brain. 2008;131:3256–65. https://doi.org/10.1093/brain/awn271.
Beattie EC, Stellwagen D, Morishita W, Bresnahan JC, Ha BK, Von Zastrow M, et al. Control of synaptic strength by glial TNFα. Science. 2002;295:2282–5. https://doi.org/10.1126/science.1067859.
Santello M, Bezzi P, Volterra A. TNFα controls glutamatergic gliotransmission in the hippocampal dentate gyrus. Neuron. 2011;69:988–1001. https://doi.org/10.1016/j.neuron.2011.02.003.
Frigerio F, Pasqualini G, Craparotta I, Marchini S, van Vliet EA, Foerch P, et al. n-3 Docosapentaenoic acid-derived protectin D1 promotes resolution of neuroinflammation and arrests epileptogenesis. Brain. 2018;141:3130–43. https://doi.org/10.1093/brain/awy247.
Lagarde S, Villeneuve N, Trébuchon A, Kaphan E, Lepine A, McGonigal A, et al. Anti–tumor necrosis factor alpha therapy (adalimumab) in Rasmussen’s encephalitis: an open pilot study. Epilepsia. 2016;57:956–66. https://doi.org/10.1111/epi.13387.
Takahashi K, Rochford CDP, Neumann H. Clearance of apoptotic neurons without inflammation by microglial triggering receptor expressed on myeloid cells-2. J Exp Med. 2005;201:647–57. https://doi.org/10.1084/jem.20041611.
Lenz KM, Nelson LH. Microglia and beyond: innate immune cells as regulators of brain development and behavioral function. Front Immunol. 2018;9. https://doi.org/10.3389/fimmu.2018.00698.
Umpierre AD, Wu LJ. How microglia sense and regulate neuronal activity. Glia. 2021;69:1637–53. https://doi.org/10.1002/glia.23961.
Wu W, Li Y, Wei Y, Bosco DB, Xie M, Zhao MG, et al. Microglial depletion aggravates the severity of acute and chronic seizures in mice. Brain Behav Immun. 2020;89:245–55. https://doi.org/10.1016/j.bbi.2020.06.028.
Akiyoshi R, Wake H, Kato D, Horiuchi H, Ono R, Ikegami A, et al. Microglia enhance synapse activity to promote local network synchronization. eNeuro. 2018;5. https://doi.org/10.1523/ENEURO.0088-18.2018.
Kofuji P, Newman EA. Potassium buffering in the central nervous system. Neuroscience. 2004;129:1043–54. https://doi.org/10.1016/j.neuroscience.2004.06.008.
Wang F, Qi X, Zhang J, Huang JH. Astrocytic modulation of potassium under seizures. Neural Regener Res. 2020;15:980. https://doi.org/10.4103/1673-5374.270295.
Alterations in Expression of Glutamatergic Transporters and Receptors in Sporadic Alzheimer’s Disease - C.P, Jacob, E, Koutsilieri, J, Bartl, E, Neuen-Jacob, T, Arzberger, N, et al. Grünblatt, 2007. Accessed July 26, 2025. https://journals-sagepub-com.ezproxy.library.wisc.edu/doi/10.3233/jad-2007-11113?url_ver=Z39.88-2003&rfr_id=ori:rid:crossref.org&rfr_dat=cr_pub%20%200pubmed.
Scott HA, Gebhardt FM, Mitrovic AD, Vandenberg RJ, Dodd PR. Glutamate transporter variants reduce glutamate uptake in Alzheimer’s disease. Neurobiol Aging. 2011;32:553.e1–553.e11. https://doi.org/10.1016/j.neurobiolaging.2010.03.008.
Li S, Mallory M, Alford M, Tanaka S, Masliah E. Glutamate transporter alterations in Alzheimer disease are possibly associated with abnormal APP expression. J Neuropathol Exp Neurol. 1997;56:901–11. https://doi.org/10.1097/00005072-199708000-00008.
Pirttimaki TM, Codadu NK, Awni A, Pratik P, Nagel DA, Hill EJ, et al. α7 Nicotinic receptor-mediated astrocytic gliotransmitter release: Aβ effects in a preclinical Alzheimer’s mouse model. PLOS ONE. 2013;8:81828. https://doi.org/10.1371/journal.pone.0081828.
Buffo A, Rolando C, Ceruti S. Astrocytes in the damaged brain: Molecular and cellular insights into their reactive response and healing potential. Biochem Pharmacol. 2010;79:77–89. https://doi.org/10.1016/j.bcp.2009.09.014.
Ortinski PI, Dong J, Mungenast A, Yue C, Takano H, Watson DJ, et al. Selective induction of astrocytic gliosis generates deficits in neuronal inhibition. Nat Neurosci. 2010;13:584–91. https://doi.org/10.1038/nn.2535.
Yamamoto K, Tanei ZI, Hashimoto T, Wakabayashi T, Okuno H, Naka Y, et al. Chronic optogenetic activation augments Aβ pathology in a mouse model of Alzheimer Disease. Cell Rep. 2015;11:859–65. https://doi.org/10.1016/j.celrep.2015.04.017.
Traikapi A, Konstantinou N. Gamma oscillations in Alzheimer’s Disease and their potential therapeutic role. Front Syst Neurosci. 2021;15. https://doi.org/10.3389/fnsys.2021.782399.
Majdi A, van Boekholdt L, Sadigh-Eteghad S, Mc Laughlin M. A systematic review and meta-analysis of transcranial direct-current stimulation effects on cognitive function in patients with Alzheimer’s disease. Mol Psychiatry. 2022;27:2000–9. https://doi.org/10.1038/s41380-022-01444-7.
De Paolis ML, Paoletti I, Zaccone C, Capone F, D'amelio M, Krashia P. Transcranial alternating current stimulation (tACS) at gamma frequency: an up-and-coming tool to modify the progression of Alzheimer’s Disease. Transl Neurodegener. 2024;13:33. https://doi.org/10.1186/s40035-024-00423-y.
Weiler M, Stieger KC, Long JM, Rapp PR. Transcranial magnetic stimulation in alzheimer’s disease: are we ready? eNeuro. 2020;7:ENEURO.0235-19.2019. https://doi.org/10.1523/ENEURO.0235-19.2019.
Picton B, Wong J, Lopez AM, Solomon SS, Andalib S, Brown NJ, et al. Deep brain stimulation as an emerging therapy for cognitive decline in Alzheimer Disease: systematic review of evidence and current targets. World Neurosurg. 2024;184:253–66. https://doi.org/10.1016/j.wneu.2023.12.083.
Kumar A, Maini K, Kadian R. Levetiracetam. In: StatPearls. StatPearls Publishing; Accessed June 23, 2025. http://www.ncbi.nlm.nih.gov/books/NBK499890/ 2025
Bakker A, Krauss GL, Albert MS, Speck CL, Jones LR, Stark CE, et al. Reduction of hippocampal hyperactivity improves cognition in amnestic mild cognitive impairment. Neuron. 2012;74:467–74. https://doi.org/10.1016/j.neuron.2012.03.023.
Bakker A, Albert MS, Krauss G, Speck CL, Gallagher M. Response of the medial temporal lobe network in amnestic mild cognitive impairment to therapeutic intervention assessed by fMRI and memory task performance. Neuroimage Clin. 2015;7:688–98. https://doi.org/10.1016/j.nicl.2015.02.009.
Mohs R, Bakker A, Rosenzweig-Lipson S, Rosenblum M, Barton RL, Albert MS, et al. The HOPE4MCI study: a randomized double-blind assessment of AGB101 for the treatment of MCI due to AD. Alzheimer's Dement. 2024;10:12446. https://doi.org/10.1002/trc2.12446.
Bakker A, Rani N, Mohs R, Gallagher M. The HOPE4MCI study: AGB101 treatment slows progression of entorhinal cortex atrophy in APOE ε4 non-carriers with mild cognitive impairment due to Alzheimer’s disease. Alzheimer’s Dement. 2024;10:70004. https://doi.org/10.1002/trc2.70004.
Cumbo E, Ligori LD. Levetiracetam, lamotrigine, and phenobarbital in patients with epileptic seizures and Alzheimer’s disease. Epilepsy Behav. 2010;17:461–6. https://doi.org/10.1016/j.yebeh.2010.01.015.
Sen A, Akinola M, Tai XY, Symmonds M, Davis Jones G, Mura S, et al. An Investigation of Levetiracetam in Alzheimer’s Disease (ILiAD): a double-blind, placebo-controlled, randomised crossover proof of concept study. Trials. 2021;22:508. https://doi.org/10.1186/s13063-021-05404-4.
Sen A, Toniolo S, Tai XY, Akinola M, Symmonds M, Mura S, et al. Safety, tolerability and efficacy outcomes of the Investigation of Levetiracetam in Alzheimer’s Disease (ILiAD) trial. Epilepsia Open. 2024;9:2353–64. https://doi.org/10.1002/epi4.13070.
Vossel K, Ranasinghe KG, Beagle AJ, La A, Ah Pook K, Castro M, et al. Effect of Levetiracetam on Cognition in Patients With Alzheimer Disease With and Without Epileptiform Activity. JAMA Neurol. 2021;78:1–10. https://doi.org/10.1001/jamaneurol.2021.3310.
Rao NR, Savas JN. Levetiracetam treatment normalizes levels of presynaptic endocytosis machinery and restores nonamyloidogenic APP processing in App knock-in mice. J Proteome Res. 2021;20:3580–9. https://doi.org/10.1021/acs.jproteome.1c00180.
Sanchez PE, Zhu L, Verret L, Vossel KA, Orr AG, Cirrito JR, et al. Levetiracetam suppresses neuronal network dysfunction and reverses synaptic and cognitive deficits in an Alzheimer’s disease model. Proc Natl Acad Sci. 2012;109:2895–903. https://doi.org/10.1073/pnas.1121081109.
Zheng XY, Zhang HC, Lv YD, Jin FY, Wu XJ, Zhu J, et al. Levetiracetam alleviates cognitive decline in Alzheimer’s disease animal model by ameliorating the dysfunction of the neuronal network. Front Aging Neurosci. 2022;14:888784. https://doi.org/10.3389/fnagi.2022.888784.
Thompson JC, Levis Rabi M, Novoa M, Nash KR, Joly-Amado A. Evaluating the efficacy of levetiracetam on non-cognitive symptoms and pathology in a Tau mouse model. Biomedicines. 2024;12:2891. https://doi.org/10.3390/biomedicines12122891.
Funding
R21AG087888-01.
Author information
Authors and Affiliations
Contributions
APB: conceptualization (lead), project administration (lead), writing – original draft preparation (lead), writing – review & editing (lead), project administration (lead); MID: conceptualization (supporting), writing – original draft preparation (supporting), writing – review & editing (lead); IF: conceptualization (supporting), writing – review & editing (lead); OL: conceptualization (supporting); LZ: conceptualization (supporting); BB: conceptualization (supporting); AL: conceptualization (lead), supervision, writing – review & editing (lead).
Corresponding author
Ethics declarations
Competing interests
The authors declare the following competing interests: AL is a cofounder of KeyWise and a scientific advisor to Buoy Health and OTSUKA US. All other authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Burns, A.P., Duran, M.I., Fortel, I. et al. Excitation–inhibition homeostasis in Alzheimer’s disease: a selective multiscale review of mechanisms, sex differences, and therapeutic opportunities. Neuropsychopharmacol. (2026). https://doi.org/10.1038/s41386-026-02334-0
Received:
Revised:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41386-026-02334-0
This article is cited by
-
Neuropsychiatric illness in the later years of life: summary and synthesis
Neuropsychopharmacology (2026)
