
Abstract
Capecitabine has been commonly used for the treatment of early-stage triple-negative breast cancer (TNBC) patients; however, the resistance limits its curative potential. Here, we perform multi-omics data analysis and immunohistochemical (IHC) staining of biological samples from patients in the CBCSG010 clinical trial who were randomized to receive adjuvant docetaxel-anthracycline-based chemotherapy with or without capecitabine. We find that patients with a better prognosis in the capecitabine group exhibited an immune-inflamed microenvironment and upregulation of interferon pathways. Moreover, we identify interferon-related TANK-binding kinase 1-binding protein 1 (TBKBP1) as the key gene involved in capecitabine resistance. We uncover that TBKBP1 promotes capecitabine resistance through impairment of activated immune cells infiltration in vivo. Mechanistically, TBKBP1 negatively regulates type I interferon pathway activated by capecitabine treatment, by promoting autophagy-mediated protein degradation of TANK binding kinase 1 (TBK1). In summary, our study implicates TBKBP1 in mediating capecitabine resistance and may serve as a potential therapeutic target for the treatment of TNBC.

Similar content being viewed by others


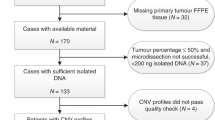
Introduction
Triple-negative breast cancer (TNBC) is a highly heterogeneous subtype that accounts for approximately 10–15% of all breast carcinomas [1]. Owing to the lack of clear therapeutic targets, anthracycline-taxane-based chemotherapy is still the standard treatment for TNBC patients [2]. Many clinical trials including CBCSG010, FinXX and SYSUCC-001, have demonstrated that adding capecitabine to standard adjuvant chemotherapy could significantly prolong the survival of TNBC patients [3,4,5]. Capecitabine, an oral prodrug of 5-fluorouracil (5-FU) that works by inhibiting the biosynthetic process of DNA or RNA, is preferentially delivered to the tumor site after metabolism by three enzymes [6,7,8]. However, not all patients could benefit from capecitabine, and some of the TNBC patients experience recurrence, which may be related to chemoresistance. Although various mechanisms have been associated with capecitabine resistance, such as thymidylate synthase (TP) polymorphism, the overexpression of ATP-binding cassette (ABC) transporter proteins ABCB1 or ABCG2, the aberrant expression of stemness related genes, metabolic reprogramming which leads to high oxidative phosphorylation, the upregulation of angiogenesis and extensive DNA repair potentiality [9,10,11,12,13,14,15,16,17], the mechanisms underlying the capecitabine resistance are still unclear.
TANK-binding kinase 1-binding protein 1 (TBKBP1), also called SINTBAD, is an adaptor protein that interacts with TANK-binding kinase 1 (TBK1) [18]. In previous studies, TBKBP1 was shown to play important roles in improving multiple sclerosis susceptibility, affecting tumorigenesis and tumor-mediated immunosuppression, regulating reactive oxygen species (ROS) production, mitochondrial function, and autophagy and mediating type I interferon expression and antiviral immune responses [19,20,21,22,23,24,25,26]. The association between TBKBP1 and capecitabine resistance has not been reported previously, and the underlying mechanisms need to be elucidated.
In this study, we found that among TNBC patients, long survival was associated with the upregulation of interferon pathway expression, and overexpression of TBKBP1 indicated capecitabine resistance. Mechanistically, TBKBP1 negatively regulates the expression of interferon-related genes via capecitabine-induced autophagy-mediated TBK1 protein degradation, thereby regulating the distribution of immune cells in the tumor microenvironment (TME). These findings suggest that TBKBP1 is an essential regulator of capecitabine sensitivity and may serve as a chemo-sensitizing therapeutic biomarker for the treatment of TNBC patients.
Materials and methods
Clinical samples
All human TNBC patient samples used in this study was retrospectively collected from CBCSG010 clinical trial (Clinicaltrials.gov registration: NCT01642771) [3]. In brief, among these 585 patients, 26 patients with whole-exome sequencing (WES) data, 36 patients with RNA-seq data, and 207 patients with formalin-fixed, paraffin-embedded (FFPE) specimens were included in our study for screening candidate genes for capecitabine resistance. Approval for the use of these samples was granted by independent ethics committees at each participating center, and each patient provided written informed consent for data and tissue use.
DNA sequencing and data analysis
Genomic DNA from tissue samples was prepared for WES. For each sample, 300 ng of DNA as quantified by Qubit, was fragmented on a Bioruptor Plus sonication system (Diagenode, Liege, Belgium). Sheared DNA underwent end repair, A-tailing, and adapter ligation using the Agilent SureSelectXT Library Prep Kit (Agilent Technologies, Santa Clara, CA, USA) according to the manufacturer’s protocol. Genome-wide copy number analysis was performed using the OncoScan CNV Assay Kit (Affymetrix, Santa Clara, CA, USA). Somatic mutation analysis and mutational signature extraction were conducted with the “maftools” and “deconstructSigs” packages [27, 28].
RNA sequencing and data analysis
Total RNA was extracted from RNAlater-preserved tumor specimens using the MiRNeasy Mini Kit (Qiagen, Hilden, Germany) and sequenced on the IIlumina HiSeq platform (Illumina Inc., San Diego, CA, USA). Transcript abundance was estimated as Fragments Per Kilobase of exon model per Million mapped fragments (FPKM) using the TopHat–Cufflinks pipeline aligned to the human reference genome (Hg19, GRCh37_snp_tran) [29]. Single sample gene set enrichment analysis (ssGSEA, “GSVA” function in R) was used to calculate the abundance of each cell subset in each sample [30]. GSEA was applied for pathway enrichment analysis using GSEA software (version 4.3.3) [31]. Differentially expressed genes (|log₂FC | > 0.5, p < 0.05) identified using the “limma” package were subjected to functional annotation through GO and KEGG analyses [32].
Cell lines and reagents
The human embryonic kidney cell line HEK293T, human TNBC cell lines Hs 578 T, MDA-MB-468, BT-549, SUM159, LM2-4175, HCC1806, and the mouse TNBC cell line 4T1 were obtained from American Type Culture Collection (ATCC). The mouse TNBC cell lines 4T07, TS/A, and AT3 were kindly provided by Y.Kang’s laboratory (Princeton University, USA). All the cell lines were cultured in high-glucose DMEM (Basal Media, #L110) supplemented with 10% fetal calf serum (FBS; ExCell Biol, #FSP500) and 1% penicillin-streptomycin (Basal Media, #L540KJ) at 37 °C in a 5% CO2 incubator. The cell lines were regularly confirmed to be negative for mycoplasma contamination with a MycoBlue Mycoplasma Detector Kit (Vazyme, #D101).
Immunohistochemical (IHC) staining
Immunohistochemical staining was performed on human FFPE specimens with the following antibodies: anti-TBKBP1 (Novus Biologicals, #NBP2-37989, 1:200), anti-CD4 (Cell Signaling Technology, #48272, 1:100), anti-CD8 (Cell Signaling Technology, #85336, 1:200), anti-CD86 (Cell Signaling Technology, #91882, 1:100), and anti-CD206 (Cell Signaling Technology, #24595, 1:200), and antibodies for mouse tumor FFPE specimens were the following: anti-CD4 (Cell Signaling Technology, #25229, 1:100), anti-CD8 (Cell Signaling Technology, #98941, 1:400), anti-CD86 (Cell Signaling Technology, #19589, 1:200), and anti-CD206 (Cell Signaling Technology, #24595, 1:200). The sections were stained with goat anti-rat IgG (Servicebio Cat# GB21302, 1:200) at room temperature for 1 h and counterstained with hematoxylin. The percentage of TBKBP1-positive tumor cells refered to the proportion of tumor cells with positive cytoplasmic TBKBP1 staining relative to the total tumor cells. Similarly, the evaluation of CD4/CD8/CD86/CD206-positive cells was based on the proportion of immune cells exhibiting positive staining for each marker among the total immune cells assessed. Every IHC-stained slide was assessed by two independent pathologists.
Short hairpin RNA (shRNA), small interfering RNA (siRNA), plasmid, and the transfection
The two shRNAs with the best knockdown efficiency for human TNBC cell lines (shTBKBP1#1:GACAGGGCGAGGAGCAGA;shTBKBP1#3:GACAGGGCGAGGAGCAGAG) and for mouse TNBC cell lines (shTbkbp1#2: CCTCAAAGTCTATGAGATCAA; shTbkbp1#5: GATGCTCTCATCAAACACATA) were cloned into the pLKO.1 vector with AgeI and EcoRI. The pCDH-TBKBP1/Tbkbp1-CMV-MCS-EF1-GFP-puro-3xFLAG was purchased from GeneChem, China. The shRNAs and pCDH-TBKBP1/Tbkbp1-CMV-MCS-EF1-GFP-puro-3xFLAG plasmids were co-transfected with the packing vectors (psPAX2 and pMD2.G) into HEK293T cells by using Lipofectamine 2000 (Invitrogen) according to the manufacturer’s protocol. The siRNA (siTbk1: UGACGGCGCAUAAGAUUUA) used in this study was generated by GenePharm, China. Cells infected with above lentiviruses were cultured for up to 48 h, and were screened with 2 μg/mL puromycin after 72 h from infection for at least 5 days.
RNA preparation and real-time quantitative reverse transcription (RT-qPCR)
FastPure Cell/Tissue Total RNA Isolation Kit V2 (Vazyme, #RC112) was used for the purification of total RNA from breast cancer cells following the manufacturer’s recommendations. We first employed HiScript III RT SuperMix for qPCR (+gDNA wiper) Kit (Vazyme, #R323) to synthesize cDNA and then used ChamQ SYBR qPCR Master Mix (Vazyme, #RQ311) to perform RT-qPCR on a QuantStudio 6 Flex Real-Time PCR system (Applied Biosystems). GAPDH was used for normalization. The sequences of the primers used for RT-qPCR in Supplementary Table 1.
Western blotting
We extracted cellular protein by using sodium dodecyl sulfate (SDS) lysis buffer (Beyotime, #P0013G), along with protease inhibitor cocktail (Yeasen, #20124ES, 100x) and phosphatase inhibitor cocktail (Yeasen, #20109ES, 100x), and quantified by using the BCA Protein Assay Kit (Solarbio, #PC0020). Proteins were separated by SDS-PAGE (polyacrylamide gel electrophoresis) and transferred onto a 0.22 μm polyvinylidene difluoride (PVDF) membrane (Merck Millipore). The membranes were blocked with 10% skim milk dissolved in PBST for 2 h at room temperature (RT), and were incubated with primary antibodies overnight at 4 °C, followed by an HRP-conjugated secondary antibodies for 1 h at RT. Finally, the membranes were detected by enhanced chemiluminescence (NCM Biotech, #P10200) and scanned with the Tanon 5200 Multi System. Antibodies: TBKBP1 (Cell Signaling Technology, #8605, 1:1000), α-tubulin (Proteintech, #66031-1-Ig, 1:20000), GAPDH (Proteintech, #60004-1-Ig, 1:10000), cGAS (Cell Signaling Technology, #31659, 1:1000), STING (Cell Signaling Technology, #13647, 1:1000), TBK1 (Cell Signaling Technology, #3504, 1:1000), IRF3 (Cell Signaling Technology, #4302, 1:1000), and IFNGR1 (Abcam, #ab280353, 1:1000). All results are derived from at least three independent biological replicates, and representative results were shown.
Cell growth and colony formation assay
For cell growth assay, 1 × 103 (4T1, 4T07, TS/A, AT3) and 3 × 103 (Hs 578 T, MDA-MB-468, BT-549, SUM159, LM2-4175, HCC1806) cells were preseeded per well in 96-well plates and then incubated with 10% Cell Counting Kit-8 (CCK-8) solution (Vazyme, #A311-02) at 37 °C for 2 h. Absorbance was measured at A450 nm. For the colony formation assay, 250 cells were seeded into 6-well plates in triplicate and treated with or without 5-FU for 10 days. Finally, 6-well plates were fixed and stained with 0.25% crystal violet staining solution.
IC50 assay
For the assessment of the effect of TBKBP1 expression level on breast cancer cell viability under 5-FU treatment, cells were plated in 96-well plates at a density of 2 × 103 cells per well and exposed to varying concentrations of 5-FU for 72 h. Then the cells were incubated with 10% CCK-8 (Vazyme, #A311-02) solution at 37 °C for 1 h. The absorbance value of each well was measured at A450 nm.
In vivo mouse model
The animal protocols were approved by the Animal Welfare Committee of Shanghai Medical College at Fudan University (Protocol number: IACUC-2023141). Female BALB/c mice at 4 weeks of age were used for in vivo mouse models. A total of 1.5 × 105 4T1 breast cancer cells (with or without Tbkbp1 knockdown) were injected subcutaneously into the mammary fat pad region. The mice were randomly divided into 6 groups: (1) injected with 4T1 shNC cells and treated with PBS, (2) injected with 4T1 shNC cells and treated with capecitabine (25 mg/kg, intragastric injection every two days), (3) injected with 4T1 shTbkbp1#2 cells and treated with PBS, (4) injected with 4T1 shTbkbp1#2 cells and treated with capecitabine (25 mg/kg, intragastric injection every two days), (5) injected with 4T1 shTbkbp1#5 cells and treated with PBS, (6) injected with 4T1 shTbkbp1#5 cells and treated with capecitabine (25 mg/kg, intragastric injection every two days). Tumor size was measured every two days using a caliper. Tumor volume in mm3 was calculated by the following formula: tumor volume = 0.5×L×W2, where L is the longest dimension and W is the perpendicular dimension. We randomly selected 3 tumors per group for RNA-sequencing, while one shTbkbp1#2 tumor treated with capecitabine with excessive necrosis failed quality control and was removed from analysis.
Flow cytometry analysis
For cell apoptosis assessment, Annexin V-PE/7-AAD Apoptosis Detection Kit (Vazyme, #A213) was used for mouse breast tumor cells following the manufacturers’ instructions. For measurement of mouse tumor immune microenvironment, single cell suspensions were prepared from freshly excised mouse tumors by mechanically dissociating with scissors in sterile PBS followed by digestion in combination of 20 mg/ml collagenase D (Roche, #COLLD-RO), 20 mg/ml DNase I (Roche, #11284932001), and 20 mg/ml collagenase I (Sigma, #SCR103) for 1 h at 37 °C with rotation. Samples were then passed through 70μm cell stainers, and were lysed with red blood cell lysis buffer (eBioscience, #00-4333-57) for 5 min at RT. Subsequently, single cell suspensions were washed in Cell Staining Buffer (BioLegend, #420201) and incubated with the indicated flow antibodies at 4 °C for 30 min. All the antibodies and reagents used for flow cytometry included Zombie-RED (ECD, #423110), CD45 (APC/Cy7, clone 30-F11, #103116), CD3e (PC5.5, clone 145-2C11, #100328), CD4 (AF700, clone RM4-5, #100536), CD8 (FITC, clone 53-6.7, #100706), F4/80 (PC5.5, clone BM8, #123127), CD11b (FITC, clone M1/70, #101206), CD86 (PE, clone GL-1, #105006), CD206 (APC, clone C068C2, #141719) and IFN-γ (PE/Cy7, XMG1.2, #505825) were from BioLegend. Stained cells were stimulated with Leukocyte Activation Cocktail (BD Biosciences, #550583) for 5 h at 37 °C and permeabilized with Fixation Buffer (BioLegend, #420801) and Intracellular Staining Permeabilization Wash Buffer (BioLegend, #421002) according to the manufacturers’ instructions. Then these cells were incubated with antibodies against IFN-γ (clone XMG1.2; BD Biosciences, Cat#563854) for 30 min at 4 °C for intracellular protein analysis. A CytoFLEX S flow cytometer (Beckman Coulter) and FlowJo software (Version10.8.1, TreeStar) were used for further analyses.
Inhibitor treatment assay
4T1 or 4T07 cells with or without Tbkbp1 knockdown were treated with 4μM 5-FU for 18 h. Then the cells were treated with the combination with proteasome inhibitor MG132 (MedChemExpress, #HY-13259, 10 μM), or autophagosome inhibitor 3-MA (Medchemexpress, #HY-19312, 5 mM), or BafA1 (Medchemexpress, # HY-100558, 200 nM) for 18 h, and the lysates were examined by Western blotting analysis.
Statistical analysis
We used R software (version 2024.04.2) and GraphPad (version 9.3.1) to analyze data. Two-way ANOVA was used to analyze the variance between two growth curves. One-way ANOVA and unpaired or paired Student’s t tests were used to compare data between two groups. Correlation coefficients were calculated using the Spearman test or Pearson test. The survival curves were generated by the Kaplan–Meier method and compared with the log-rank test. Two-sided P < 0.05 was considered statistically significant.
Results
High TBKBP1 expression is correlated with capecitabine resistance in TNBC
We employed our CBCSG010 TNBC dataset, which included 26 samples with WES data, 36 samples with transcriptome data and 207 FFPE specimens with IHC staining to explore the intrinsic molecular features underlying different prognoses and investigate the key genes associated with capecitabine resistance (Fig. 1A). First, as demonstrated in Fig. 1B, the most prominent cancer-related mutated gene observed in this cohort was TP53 (69% in tumors), followed by RYR2 (19%), TTN (15%), MYH14 (15%), and PIK3CA (15%), which were similar in frequency to previously published gene mutations in Chinese TNBC patients [33]. Due to the limited sample size, we were unable to identify the key mutated genes indicating capecitabine resistance. Utilizing previously established immune microenvironment classification systems for TNBC, we sought to investigate the correlation between capecitabine therapeutic efficacy and tumor immune microenvironment characteristics [34]. Patients with immune-inflamed tumors treated with capecitabine showed no recurrence (0/5, 0%) compared to a 66.7% recurrence rate (2/3) in patients with similar immune profiles in the control group (Fisher’s exact test p = 0.107) (Fig. 1C and Supplementary Fig. 1A). Although this difference did not reach statistical significance due to small sample size, the trend may suggest an association between capecitabine response and immune-activated tumor microenvironment characteristics. GSEA was performed in capecitabine group, to understand the potential immune pathways resulting in better outcomes. Compared with patients with a worse prognosis (with recurrence), interferon pathway was found upregulated in those with a better prognosis (without recurrence) (Fig. 1D). Interferon-stimulated gene (ISG) signature scores were calculated to explore the status of IFN signaling and higher ISG scores were observed in patients treated with capecitabine without recurrence (Fig. 1E) [35]. To further explore the key genes affecting capecitabine efficacy, we analyzed transcriptome data and paired clinical data of patients in the capecitabine group, and selected the top 30 genes by ranking them by hazard ratios (HR) (Supplementary Fig. 1B, C, and Supplementary Table 2). Venn analysis was performed to determine the intersection of the capecitabine response-related top 30 genes and interferon-related genes, and TBKBP1 was screened out as the key gene mediating capecitabine resistance (Fig. 1F) [24,25,26, 36]. We found that low TBKBP1 expression was correlated with high interferon-related gene expression in the capecitabine group (Supplementary Fig. 1D, E).

A A sketch map of this study. B The genomic landscape of patients in the CBCSG010 clinical trial. CAP, capecitabine. C Landscape of the microenvironment phenotypes of patients in the CBCSG010 clinical trial. D GSEA analysis of the upregulated pathways associated with the differentially expressed genes (DEGs) among patients with and without relapse in the capecitabine group. E Violin plots showing the associations between ISG signature scores and different prognostic outcomes between two treatment groups in the CBCSG010 clinical trial. ISG, interferon-stimulated gene. F A Venn diagram showing the interactions between the capecitabine response-related top30 genes and interferon-related genes. G Representative IHC images of TBKBP1 staining (upper). Boxplots showing the percentage of TBKBP1-positive cells in patients in two treatment groups with different relapse statuses (bottom). Data were compared via the Mann–Whitney U test. Scale bar, 20 μm.
To investigate the association between TBKBP1 expression and capecitabine treatment response, we subsequently performed IHC staining of TBKBP1 on FFPE samples from 207 TNBC patients from the CBCSG010 clinical trial. TBKBP1 expression was higher in tissues from patients with worse prognosis than in those from patients with better prognosis; nevertheless, there was no significant difference in TBKBP1 expression between patients in the control group regardless of the recurrence status, indicating that TBKBP1 is a capecitabine resistant gene (Fig. 1G and Supplementary Fig. 1F). Additionally, we found that TBKBP1 expression did not affect patient survival outcomes in the FUSCCTNBC cohort (Supplementary Fig. 1G) [33]. In summary, high TBKBP1 expression is correlated with capecitabine resistance in TNBC patients.
TBKBP1 promotes capecitabine resistance through regulating tumor immune microenvironment in vivo
To investigate the function of TBKBP1 in TNBC cellular behaviors, we first investigated the expression levels of TBKBP1 in 6 human TNBC cell lines by immunoblotting. The results demonstrated that BT-549 and SUM159 cell lines expressed relatively higher levels of TBKBP1 than the other cell lines did (Supplementary Fig. 2A). Based on the expression levels of TBKBP1, we next knocked down its expression via two independent shRNAs (shTbkbp1#1 and #3) and a negative control shRNA (shNC) in BT-549 and SUM159 cells (Supplementary Fig. 2B). Notably, TBKBP1 knockdown did not affect the proliferation ability of TNBC cells (Supplementary Fig. 2C). Similarly, we transfected Flag-tagged TBKBP1 into HCC1806 and LM2-4175 cells, respectively, and found that cell proliferation ability was not affected by the overexpression of TBKBP1 (Supplementary Fig. 2D, E). Additionally, we repeated the above experiments in mouse TNBC cell lines, and equally Tbkbp1 expression did not affect mouse TNBC cell proliferation (Supplementary Fig. 2F–J).
Capecitabine undergoes three metabolic steps in the liver and is ultimately converted to the effective component 5-FU [37]. However, the process requires the liver to initiate metabolism, which is relatively limited in cases of in vitro metabolism and sensitivity of capecitabine evaluation, therefore we employed 5-FU, the prodrug of capecitabine, to conduct subsequent experiments in vitro [38]. Capecitabine has been reported to induce tumor cell apoptosis with antitumor effects [39, 40]. Therefore, we next examined the effects of TBKBP1 expression level on 5-FU-induced tumor cell apoptosis. Tbkbp1 knockdown did not affect apoptosis rates under 5-FU treatment, nor its overexpression (Supplementary Fig. 3). Additionally, neither depletion nor overexpression of TBKBP1 affected the cytotoxic effect of 5-FU in vitro, whether in human or mouse cell lines (Supplementary Fig. 4). We concluded that TBKBP1 did not affect TNBC cell proliferation, apoptosis or 5-FU sensitivity in vitro.
To explore the effects of TBKBP1 on the anti-tumor effect of capecitabine in vivo, we implanted 4T1 shNC cells and 4T1 shTbkbp1 cells, respectively, into the mammary fat pads of BALB/c mice on Day 0, followed by intraperitoneal injection of PBS or intragastric administration of capecitabine every two days since Day 9 (Fig. 2A). When treated with capecitabine, Tbkbp1 knockdown resulted in a significant reduction in tumor growth and weight, indicating that Tbkbp1-depleted tumors were more sensitive to capecitabine treatment (Fig. 2B–D). 5-FU, the prodrug of capecitabine, reportedly increases the number of infiltrating natural killer (NK) cells to enhance cytotoxicity [41], promotes tumor antigen presentation by dendritic cells (DCs) to T cells [42, 43], improves IFN-γ production by tumor-specific CD8+ T cells and boosts T-cell-dependent antitumor responses [44]. Therefore, we speculated that TBKBP1 depletion may inhibit tumor growth by influencing the tumor microenvironment under capecitabine treatment, and we subsequently evaluated the immune responses via flow cytometry analysis (Supplementary Fig. 5A, B). When treated with capecitabine, the inhibition of Tbkbp1 significantly increased the proportions of CD4+ cells, CD8+ cells especially interferon-γ (IFNγ)-positive CD8+ T cells, and M1 macrophages (CD86+) and simultaneously decreased the proportion of M2 macrophages (CD206+) (Fig. 2E and Supplementary Fig. 5C). Consistent with the results of the flow cytometric analysis, IHC staining demonstrated that Tbkbp1 depletion effectively led to the recruitment of CD4+ cells, CD8+ cells, and CD86+ cells and reduced the number of CD206+ cells (Fig. 2F, G). These results suggest that TBKBP1 promotes capecitabine resistance especially through depleting IFNγ-positive CD8+ T cells in vivo.

A Schematic outline of Tbkbp1-knockdown in tumors induced by capecitabine: 4T1 mouse breast cancer cells stably expressing shNC or shTbkbp1 were subcutaneously injected into BALB/c mice. The mice were treated with capecitabine (25 mg/kg i.g.), or PBS every two days since Day 9. (n = 8 mice/group). B Representative images of the tumors illustrating the effect of TBKBP1-depletion in the PBS and CAP groups. (n = 8 mice/group). C Effects of Tbkbp1 knockdown on tumor growth in the PBS and CAP groups. The data were represented as the mean ± SEM. The two-way ANOVA was used to compare the data. D Tumor weights of the six groups. The data were represented as the mean ± SEM. Data were compared by using the Student’s t test. E Bar plots showing the percentages of CD3e+ cells among CD45+ cells, CD4+ cells among CD3e+CD45+ cells, CD8+ cells among CD3e+CD45+ cells, interferon-γ (IFNγ)-positive cells among CD8+ T cells, CD86+ cells among CD45+CD11b+F4/80+ cells, and CD206+ cells among CD45+CD11b+F4/80+ cells. Data were compared by using the Student’s t test. F Representative IHC images of CD4, CD8, CD86, and CD206 expression in shNC and shTbkbp1 tumors in the PBS and capecitabine group. Scale bar, 20 μm. G Boxplots showing the percentages of CD4-, CD8-, CD86-, and CD206-positive cells. The data were represented as the mean ± SEM. Data were compared by using the Student’s t test.
TBKBP1 negatively regulates type I interferon pathway under capecitabine treatment
To elucidate the mechanisms underlying TBKBP1-induced capecitabine resistance, we performed transcriptome sequencing of mouse tumors. Specifically, the RNA-seq data were generated from whole shNC or shTbkbp1 tumors (not sorted epithelial cell populations) harvested from BALB/c mice treated with either PBS or capecitabine. By analyzing the data, we found that in the capecitabine group, Tbkbp1 knockdown strongly upregulated immune-related pathways, including type I/II interferon production, interferon-mediated signaling pathway, T-cell proliferation, and antigen processing and presentation (Fig. 3A). GSEA and differential gene expression analysis also revealed the upregulation of interferon-alpha and interferon-gamma responses with Tbkbp1 knockdown (Fig. 3B, C). These results were consistent with the findings that the interferon-related responses were upregulated in patients without recurrence following capecitabine treatment, which confirmed that Tbkbp1 was the key gene mediating capecitabine resistance (Fig. 1F). Increased expression of interferon-related genes, including Tbk1, Irf3 (interferon regulatory factor 3) and Ifngr1 (interferon gamma receptor 1), was further validated at the mRNA and protein levels in 4T1 and 4T07 cells expressing Tbkbp1 shRNA vectors, whereas overexpressing Tbkbp1 in AT3 and TS/A cells significantly decreased the protein levels of Tbk1, Irf3 and Ifngr1 (Fig. 3D–G). Collectively, these results indicate that TBKBP1 negatively regulates type I interferon signaling pathway induced by capecitabine.

A Bar plots of GO and KEGG analysis results showing that Tbkbp1 knockdown in CAP-treated breast tumors upregulated several pathways and biological processes. B GSEA analysis revealed that Tbkbp1 knockdown promoted CAP-induced upregulation of genes involved in interferon-related pathways. C Heatmaps showing that Tbkbp1-depletion upregulated genes involved in IFN-α pathway (left) and IFN-γ pathway (right). D qRT-PCR results showing that knockdown of Tbkbp1 upregulated the mRNA levels of Tbk1, Irf3, and Ifngr1 in 4T1 cells (upper) and 4T07 cells (bottom) under 5-FU treatment. The data were represented as the mean ± SEM. Data were compared by using the Student’s t test. E qRT-PCR results showing that overexpression of Tbkbp1 downregulated the mRNA levels of Irf3 and Ifngr1 in AT3 cells (upper) and TS/A cells (bottom) under 5-FU treatment. The data were represented as the mean ± SEM. Data were compared by using the Student’s t-test. F Western blot analysis showing that Tbkbp1 knockdown increased the protein levels of Tbk1, Irf3, and Ifngr1 in 4T1 cells (left) and 4T07 cells (right) induced by 5-FU. Densitometric quantification of Western blots was performed using ImageJ software. G Western blot analysis revealed that Tbkbp1 overexpression decreased the protein levels of Tbk1, Irf3, and Ifngr1 in AT3 cells (left) and TS/A cells (right) induced by 5-FU. Densitometric quantification of Western blots was performed using ImageJ software.
Capecitabine activates the cGAS-STING pathway
Transcriptome sequencing was performed on tumors from PBS- and capecitabine-treated BALB/c mice. Differential gene expression analysis revealed that capecitabine upregulated cell cycle checkpoint pathway, DNA damage signaling pathway and immune-related pathways (Fig. 4A). Additionally, capecitabine markedly increased the expression of genes involved in DNA damage and the cytosolic DNA-sensing pathway (Fig. 4B). 5-FU was previously reported to produce micronuclei-like DNA in colon cancer cells, and many studies have already demonstrated that micronuclei-like DNA can activate the cGAS (cyclic GMP-AMP synthase) - STING (stimulator of interferon genes protein) pathway [45,46,47,48,49]. As expected, the transcription and protein levels of Cgas and Sting were greatly increased induced by 5-FU, regardless of whether Tbkbp1 was knocked down or overexpressed (Fig. 4C–F). Taken together, these results suggest that type I interferon production is induced by cGAS-STING pathway activation under capecitabine treatment.

A Bar plots of GSEA, GO, KEGG, and Reactome analysis results based on RNA-seq data of breast tumors from BALB/c mice treated with PBS, or CAP. B Heatmaps showing that CAP upregulated genes involved in DNA damage (upper) and cytosolic DNA-sensing pathway (bottom). C qRT-PCR results showing that 5-FU upregulated the mRNA levels of Cgas, and Sting in 4T1 cells (upper) or 4T07 cells (bottom) stably expressing shNC or shTbkbp1. The data were represented as the mean ± SEM. Data were compared by using the Student’s t test. D qRT-PCR results showing that 5-FU upregulated the mRNA levels of Cgas, and Sting in AT3 cells (left) or TS/A cells (right) stably expressing Vector or Flag-Tbkbp1. The data were represented as the mean ± SEM. Data were compared by using the Student’s t test. E Western blot analysis showing that 5-FU increased the protein levels of Cgas, and Sting in 4T1 cells (left) or 4T07 cells (right) stably expressing shNC or shTbkbp1. Densitometric quantification of Western blots was performed using ImageJ software. F Western blot analysis showing that 5-FU increased the levels of Cgas, and Sting protein in AT3 cells (left) or TS/A cells (right) stably expressing Vector or Flag-Tbkbp1. Densitometric quantification of Western blots was performed using ImageJ software.
TBKBP1 promotes the autophagy-mediated lysosomal degradation of TBK1 after capecitabine treatment
TBKBP1 is known as an adaptor of TBK1, and the above findings revealed that TBKBP1 negatively regulates interferon signaling, including TBK1 expression [18]. Notably, Tbk1 protein expression was inhibited by overexpression of Tbkbp1 under capecitabine treatment, while the Tbk1 mRNA level remained unchanged, indicating that TBKBP1 may participate in the degradation of TBK1 protein (Fig. 3E, G). The degradation of cellular proteins mainly involves the ubiquitin-proteasome, autophagosome, or lysosome pathway [50]. We next used AT3 and TS/A cells transfected with the FLAG-Tbkbp1 vector and then treated them with the proteasome inhibitor MG132, the autophagosome inhibitor 3-methyladenine (3-MA), or the lysosomal inhibitor bafilomycin A1 (Baf-A1) in combination with 5-FU for 18 h. 5-FU plus 3-MA strongly affected the viability of AT3 cells, and we found that the reduced expression of Tbk1 caused by Tbkbp1 overexpression was rescued by Baf-A1 and 3-MA (Fig. 5A, B). Since p62 acts as a well-established selective autophagy receptor that mediates the degradation of ubiquitinated cargo via the autophagy-lysosome pathway, we noticed that consistent with the changing state of p62 protein levels, Tbk1 expression was upregulated by using BafA1 in a time-dependent manner (Fig. 5C) [51]. Additionally, knocking down Tbk1 in Tbkbp1-depleted 4T1 and 4T07 cells, the expression of Tbk1, Irf3 and Ifngr1 decreased under 5-FU treatment (Fig. 5D). Taken together, these findings indicate that TBKBP1 inhibits the interferon pathway through promoting the autophagy-mediated lysosomal degradation of TBK1.

A Viability of AT3 cells stably overexpressing Tbkbp1 (AT3-Tbkbp1, left) and TS/A cells stably overexpressing Tbkbp1 (TS/A-Tbkbp1, right) treated with DMSO (negative control), 5-FU (4 μM), 5-FU (4 μM) with MG132 (10 μM), 5-FU (4 μM) with BafA1 (200 nM), or 5-FU (4 μM) with 3-MA (5 mM) for 18 h, respectively. The data were represented as the mean ± SEM. Data were compared by using the Student’s t test. B Western blot analysis showing the expression of Tbk1 in AT3-Tbkbp1 cells (left) and TS/A-Tbkbp1 cells (right) treated with DMSO, 5-FU, or 5-FU plus the indicated inhibitors. Densitometric quantification of Western blots was performed using ImageJ software. C AT3-Tbkbp1 cells (left) and TS/A-Tbkbp1 cells (right) were treated with or without 200 nM BafA1 in combination of 4 μM 5-FU for the indicated times, and then subjected to Western blot analysis with Tbk1 and p62 antibodies. Densitometric quantification of Western blots was performed using ImageJ software. D Western blot analysis showing the expression of Tbk1, Irf3, and Ifngr1 when Tbkbp1-knockdown 4T1 cells (left) and 4T07 cells (right) were co-transfected with siTbk1 under 5-FU treatment for 18 h. Densitometric quantification of Western blots was performed using ImageJ software.
Discussion
Chemoresistance is one of the major reasons for treatment failure in cancer patients. Capecitabine in combination with paclitaxel and anthracycline drugs, has been used as an adjuvant chemotherapy regimen for early-stage TNBC patients. A fraction of signaling genes, such as ARF6, CD16, TYMP, RB1, SHMT2, SPOCK1, and METTL3, have been identified as predictive biomarkers of capecitabine or 5-FU resistance; however, most of these studies lack clinical evidence [52,53,54,55,56,57]. Therefore, in this study, we identified a novel factor, TBKBP1, that drives capecitabine resistance in TNBC patients in the CBCSG010 clinical trial. TBKBP1 did not affect tumor cell proliferation, sensitivity to capecitabine or apoptosis in vitro, but in vivo, TBKBP1 mediated capecitabine resistance by influencing tumor microenvironment. Mechanistically, capecitabine activated the cGAS-STING pathway, and TBKBP1 promoted autophagy-mediated protein degradation of TBK1 and negatively regulated the interferon pathway under capecitabine treatment. These functions of TBKBP1 may be essential not only for understanding the underlying mechanisms of capecitabine resistance but also for identifying novel therapeutic targets.
Capecitabine is a cytotoxic agent that exerts anticancer effects by inhibiting DNA synthesis [38]. Our transcriptome data analysis revealed upregulation of cell cycle-related pathways and increased expression of genes involved in DNA damage and the cytosolic DNA-sensing pathway. cGAS is a cytosolic DNA sensor that recognizes microbial pathogens, leaks DNA from tumor cell nuclei, and interacts with double-stranded DNA (dsDNA) [58,59,60]. The activation of cGAS subsequently stimulates the adaptor protein STING, which leads to the downstream activation of IRF3 and the expression of type I IFN [61, 62]. Previous studies have demonstrated that the cGAS-STING pathway plays a vital role in immune responses to viral infections, and emerging evidence has suggested its role in antitumor immunity as well [63,64,65,66,67,68]. In addition to the upregulation of interferon-related genes, activation of the cGAS-STING pathway also mediates the secretion of senescence associated secretory phenotype (SASP), including pro-inflammatory cytokines, chemokines, and growth factors, which are involved in restricting tumorigenesis [46, 69].
There are three methods of interferon induction, including cellular DNA activated the cGAS-STING pathway, viral RNA, and pathogen-associated molecular patterns (PAMPs) [70, 71]. Our results revealed that the cGAS-STING pathway may be the main mechanism of interferon induction under capecitabine treatment. Following cGAS-STING activation, the interaction facilitates the recruitment of TBK1 and subsequent phosphorylation of IRF3 [72, 73]. Phosphorylated IRF3 binds to interferon-stimulated response elements (ISREs) and the promoters of the IFN-I genes, leading to the transcriptional induction of type I interferons [74].
We previously reported that capecitabine efficacy was correlated with immune infiltration, specifically “immune-hot” TNBC patients tended to have better survival when treated with the combination of capecitabine with anthracycline-taxane-based chemotherapy [75]. To identify the key genes mediating capecitabine resistance, we analyzed multi-omics data and collected biological samples. We selected TBKBP1, an immune-related gene, for further study. Previous studies demonstrated that TBKBP1 was dispensable for TBK1 activation and IFN-I induction by innate immune stimuli [20, 76, 77]. Nevertheless, our data suggested that under capecitabine treatment, TBKBP1 overexpression promoted the degradation of the TBK1 protein and subsequently inhibited interferon pathway activation. The cascade of immune-stimulatory factors from the cGAS-STING pathway, and interferon can recruit immune cells for tumor clearance [78]. The interaction of interferons and interferon receptors of immune cells in turn triggers immune cells [79]. It was previously reported that dendritic cells (DCs) activated by IFN-I-IFNAR (interferon alpha receptor) signaling may promote antitumor CD8+ T cell responses [80]. In addition, IFN-γ may enhance the function of tumor infiltrating immune cells such as CD4+T cells, CD8+ T cells, and macrophages, inhibit the function of regulatory T cells (Tregs) and regulate the function of stromal cells to induce antitumor immunity [81].
In summary, our studies revealed the predictive value of TBKBP1 in mediating capecitabine resistance and the elucidation of the underlying mechanisms may be of great importance for the exploration of new therapeutic targets or combination therapies, such as immunotherapy, to reverse capecitabine resistance. Limitations of our work need to be mentioned. First, due to the retrospective sample collection of the CBCSG010 clinical trial, we did not have enough samples for DNA and RNA sequencing. Second, the detailed mechanism by which TBKBP1 mediates the degradation of the TBK1 protein and the effect of interferon on the regulation of immune cells need to be investigated. Third, trials regarding capecitabine in combination with immunotherapy in TNBC patients warrant further exploration.
Data availability
The datasets generated in this study are available from the corresponding author on reasonable request.
References
Brown M, Tsodikov A, Bauer KR, Parise CA, Caggiano V. The role of human epidermal growth factor receptor 2 in the survival of women with estrogen and progesterone receptor-negative, invasive breast cancer: the California Cancer Registry, 1999–2004. Cancer. 2008;112:737–47.
Bianchini G, Balko JM, Mayer IA, Sanders ME, Gianni L. Triple-negative breast cancer: challenges and opportunities of a heterogeneous disease. Nat Rev Clin Oncol. 2016;13:674–90.
Li JJ, Yu KD, Pang D, Wang CQ, Jiang J, Yang S, et al. Adjuvant capecitabine with docetaxel and cyclophosphamide plus epirubicin for triple-negative breast cancer (CBCSG010): an open-label, randomized, multicenter, phase III trial. J Clin Oncol. 2020;38:1774–84.
Joensuu H, Kellokumpu-Lehtinen PL, Huovinen R, Jukkola-Vuorinen A, Tanner M, Kokko R, et al. Adjuvant capecitabine, docetaxel, cyclophosphamide, and epirubicin for early breast cancer: final analysis of the randomized FinXX trial. J Clin Oncol. 2012;30:11–8.
Wang X, Wang S, Huang H, Cai L, Zhao L, Peng R, et al. Effect of capecitabine maintenance therapy using lower dosage and higher frequency vs. observation on disease-free survival among patients with early-stage triple-negative breast cancer who had received standard treatment: the SYSUCC-001 randomized clinical trial. JAMA. 2021;325:50–58.
Longley DB, Harkin DP, Johnston PG. 5-Fluorouracil: mechanisms of action and clinical strategies. Nat Rev Cancer. 2003;3:330–8.
Venturini M. Rational development of capecitabine. Eur J Cancer. 2002;38:3–9.
Wagstaff AJ, Ibbotson T, Goa KL. Capecitabine: a review of its pharmacology and therapeutic efficacy in the management of advanced breast cancer. Drugs. 2003;63:217–36.
Sethy C, Kundu CN. 5-Fluorouracil (5-FU) resistance and the new strategy to enhance the sensitivity against cancer: implication of DNA repair inhibition. Biomed Pharmacother. 2021;137:111285.
Marin JJG, Macias RIR, Monte MJ, Herraez E, Peleteiro-Vigil A, de Blas BS, et al. Cellular mechanisms accounting for the refractoriness of colorectal carcinoma to pharmacological treatment. Cancers (Basel). 2020;12:2605.
Tecza K, Pamula-Pilat J, Lanuszewska J, Butkiewicz D, Grzybowska E. Pharmacogenetics of toxicity of 5-fluorouracil, doxorubicin and cyclophosphamide chemotherapy in breast cancer patients. Oncotarget. 2018;9:9114–36.
Bartmann C, Raman SRJ, Floter J, Schulze A, Bahlke K, Willingstorfer J, et al. Beta-hydroxybutyrate (3-OHB) can influence the energetic phenotype of breast cancer cells, but does not impact their proliferation and the response to chemotherapy or radiation. Cancer Metab. 2018;6:8.
Xie P, Yuan F, Huang M, Zhang W, Zhou H, Li X, et al. DCBLD2 affects the development of colorectal cancer via EMT and angiogenesis and modulates 5-FU drug resistance. Front Cell Dev Biol. 2021;9:669285.
Zheng X, Liu J, Li X, Tian R, Shang K, Dong X, et al. Angiogenesis is promoted by exosomal DPP4 derived from 5-fluorouracil-resistant colon cancer cells. Cancer Lett. 2021;497:190–201.
Yang Y, Ma L, Xu Y, Liu Y, Li W, Cai J, et al. Enalapril overcomes chemoresistance and potentiates antitumor efficacy of 5-FU in colorectal cancer by suppressing proliferation, angiogenesis, and NF-κB/STAT3-regulated proteins. Cell Death Dis. 2020;11:477.
Park JM, Huang SB, Tougeron D, Sinicrope FA. MSH3 mismatch repair protein regulates sensitivity to cytotoxic drugs and a histone deacetylase inhibitor in human colon carcinoma cells. PLoS One. 2013;8:e65369.
Mori R, Yoshida K, Tanahashi T, Yawata K, Kato J, Okumura N, et al. Decreased FANCJ caused by 5-FU contributes to the increased sensitivity to oxaliplatin in gastric cancer cells. Gastric Cancer. 2013;16:345–54.
Ryzhakov G, Randow F. SINTBAD, a novel component of innate antiviral immunity, shares a TBK1-binding domain with NAP1 and TANK. EMBO J. 2007;26:3180–90.
Sorosina M, Barizzone N, Clarelli F, Anand S, Lupoli S, Salvi E, et al. A multi-step genomic approach prioritized TBKBP1 gene as relevant for multiple sclerosis susceptibility. J Neurol. 2022;269:4510–22.
Zhu L, Li Y, Xie X, Zhou X, Gu M, Jie Z, et al. TBKBP1 and TBK1 form a growth factor signalling axis mediating immunosuppression and tumourigenesis. Nat Cell Biol. 2019;21:1604–14.
Zhu L, Xie X, Zhang L, Wang H, Jie Z, Zhou X, et al. TBK-binding protein 1 regulates IL-15-induced autophagy and NKT cell survival. Nat Commun. 2018;9:2812.
Murphy MP. How mitochondria produce reactive oxygen species. Biochem J. 2009;417:1–13.
Boyle KB, Ravenhill BJ, Randow F. CALCOCO2/NDP52 initiates selective autophagy through recruitment of ULK and TBK1 kinase complexes. Autophagy. 2019;15:1655–56.
Unterholzner L, Sumner RP, Baran M, Ren H, Mansur DS, Bourke NM, et al. Vaccinia virus protein C6 is a virulence factor that binds TBK-1 adaptor proteins and inhibits activation of IRF3 and IRF7. PLoS Pathog. 2011;7:e1002247.
Lei X, Dong X, Ma R, Wang W, Xiao X, Tian Z, et al. Activation and evasion of type I interferon responses by SARS-CoV-2. Nat Commun. 2020;11:3810.
Hamrashdi MA, Brady G. Regulation of IRF3 activation in human antiviral signaling pathways. Biochem Pharm. 2022;200:115026.
Mayakonda A, Lin DC, Assenov Y, Plass C, Koeffler HP. Maftools: efficient and comprehensive analysis of somatic variants in cancer. Genome Res. 2018;28:1747–56.
Rosenthal R, McGranahan N, Herrero J, Taylor BS, Swanton C. DeconstructSigs: delineating mutational processes in single tumors distinguishes DNA repair deficiencies and patterns of carcinoma evolution. Genome Biol. 2016;17:31.
Trapnell C, Roberts A, Goff L, Pertea G, Kim D, Kelley DR, et al. Differential gene and transcript expression analysis of RNA-seq experiments with TopHat and Cufflinks. Nat Protoc. 2012;7:562–78.
Hanzelmann S, Castelo R, Guinney J. GSVA: gene set variation analysis for microarray and RNA-seq data. BMC Bioinforma. 2013;14:7.
Suvramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gilette MA, et al. Gene set enrichment analysis: a knowledge-based approach for interpreting gegnome-wide expression profiles. Proc Natl Acad Sci USA. 2005;102:15545–50.
Ritchie ME, Phipson B, Wu D, Hu Y, Law CW, Shi W, et al. limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 2015;43:e47.
Jiang Y, Ma D, Suo C, Shi J, Xue M, Hu X, et al. Genomic and Transcriptomic Landscape of Triple-Negative Breast Cancers: Subtypes and Treatment Strategies. Cancer Cell. 2019;35:428–40.
Xiao Y, Ma D, Zhao S, Suo C, Shi J, Xue M, et al. Multi-omics profiling reveals distinct microenvironment characterization and suggests immune escape mechanisms of triple-negative breast cancer. Clin Cancer Res. 2019;25:5002–14.
Liu H, Golji J, Brodeur LK, Chung FS, Chen JT, deBeaumont RS, et al. Tumor-derived IFN triggers chronic pathway agnosim and sensitivity to ADAR loss. Nat Med. 2019;25:95–102.
Liberzon A, Birger C, Thorvaldsdottir H, Ghandi M, Mesirov JP, Tamayo P. The molecular signatures database (MSigDB) hallmark gene set collection. Cell Syst. 2015;1:417–25.
Judson IR, Beale PJ, Trigo JM, Aherne W, Crompton T, Jones D, et al. A human capecitabine excretion balance and pharmacokinetic study after administration of a single oral dose of 14C-labelled drug. Invest N Drugs. 1999;17:49–56.
McGavin JK, Goa KL. Capecitabine: a review of its use in the treatment of advanced or metastatic colorectal cancer. Drugs. 2001;61:2309–26.
Zhang S, Fan S, Wang Z, Hou W, Liu T, Yoshida S, et al. Capecitabine regulates HSP90AB1 expression and induces apoptosis via AKT/SMARCC1/AP-1/ROS axis in T cells. Oxid Med Cell Longev. 2022;2022:1012509.
Alzahrani SM, Doghaither HAA, AI-Ghafari AB, Pushparaj PN. 5-fluorouracil and capecitabine therapies for the treatment of colorectal cancer (Review). Onocl Rep. 2023;50:175.
Khallouf H, Marten A, Serba S, Teichgraber V, Buchler MW, Jager D, et al. 5-fluorouracil and interferon-α immunochemotherapy enhances immunogenicity of murine pancreatic cancer through upregulation of NKG2D ligands and MHC class I. J Immunother. 2012;35:245–53.
Buttiglieri S, Galetto A, Forno S, De Andrea M, Matera L. Influence of drug-induced apoptotic death on processing and presentation of tumor antigens by dendritic cells. Int J Cancer. 2003;106:516–20.
Galetto A, Buttiglieri S, Forno S, Moro F, Mussa A, Matera L. Drug- and cell-mediated antitumor cytotoxicities modulate cross-presentation of tumor antigens by myeloid dendritic cells. Anticancer Drugs. 2003;14:833–43.
Vincent J, Mignot G, Chalmin F, Ladoire S, Bruchard M, Chevriaux A, et al. 5-fluorouracil selectively kills tumor-associated myeloid-derived suppressor cells resulting in enhanced T cell-dependent antitumor immunity. Cancer Res. 2010;70:3052–61.
Tian J, Zhang D, Kurbatov V, Wang Q, Wang Y, Fang D, et al. 5-fluorouracil efficacy requires anti-tumor immunity triggered by cancer-cell-intrinsic STING. EMBO J. 2021;40:e106065.
Dou Z, Ghosh K, Vizioli MG, Zhu J, Sen P, Wangensteen KJ, et al. Cytoplasmic chromatin triggers inflammation in senescence and cancer. Nature. 2017;550:402–6.
Harding SM, Benci JL, Irianto J, Discher DE, Minn AJ, Greenberg RA. Mitotic progression following DNA damage enables pattern recognition within micronuclei. Nature. 2017;548:466–70.
Mackenzie KJ, Carroll P, Marin C, Murina O, Fluteau A, Simpson DJ, et al. cGAS surveillance of micronuclei links genome instability to innate immunity. Nature. 2017;548:461–5.
Gluck S, Guey B, Gulen MF, Wolter K, Kang T, Schmacke NA, et al. Innate immune sensing of cytosolic chromatin fragments through cGAS promotes senescence. Nat Cell Biol. 2017;19:1061–70.
Zhao L, Zhao J, Zhong K, Tong A, Jia D. Targeted protein degradation: mechanisms, strategies and application. Signal Transduct Target Ther. 2022;7:113.
Lee DH, Park JS, Lee YS, Han J, Lee D, Kwon SW, et al. SQSTM1/p62 activates NFE2L2/NRF2 via ULK1-mediated autophagic KEAP1 degradation and protects mouse liver from lipotoxicity. Autophagy. 2020;16:1949–73.
Geng D, Wu H. Abrogation of ARF6 in promoting erastin-induced ferroptosis and mitigating capecitabine resistance in gastric cancer cells. J Gastrointest Oncol. 2022;13:958–67.
Lu Y, Huang Y, Huang L, Xu Y, Wang Z, Li H, et al. CD16 expression on neutrophils predicts treatment efficacy of capecitabine in colorectal cancer patients. BMC Immunol. 2020;21:46.
Marangoni E, Laurent C, Coussy F, El-Botty R, Chateau-Joubert S, Servely J, et al. Capecitabine efficacy is correlated with TYMP and RB1 expression in PDX established from triple-negative breast cancers. Clin Cancer Res. 2018;24:2605–15.
Pranzini E, Pardella E, Muccillo L, Leo A, Nesi I, Santi A, et al. SHMT2-mediated mitochondrial serine metabolism drives 5-FU resistance by fueling nucleotide biosynthesis. Cell Rep. 2022;40:111233.
Qu Y, Liu X, Zhao S, Zhai X. SPOCK1 silencing decreases 5-FU resistance through PRRX1 in colorectal cancer. Pathol Res Pr. 2022;234:153895.
Li M, Xia M, Zhang Z, Tan Y, Li E, Guo Z, et al. METTL3 antagonizes 5-FU chemotherapy and confers drug resistance in colorectal carcinoma. Int J Oncol. 2022;61:106.
Sun L, Wu J, Du F, Chen X, Chen ZJ. Cyclic GMP-AMP synthase is a cytosolic DNA sensor that activates the type I interferon pathway. Science. 2013;339:786–91.
Wu X, Wu F, Wang X, Wang L, Siedow JN, Zhang W, et al. Molecular evolutionary and structural analysis of the cytosolic DNA sensor cGAS and STING. Nucleic Acids Res. 2014;42:8243–57.
Civril F, Deimling T, de Oliveira Mann CC, Ablasser A, Moldt M, Witte G, et al. Structural mechanism of cytosolic DNA sensing by cGAS. Nature. 2013;498:332–7.
Ishikawa H, Ma Z, Barber GN. STING regulates intracellular DNA-mediated, type I interferon-dependent innate immunity. Nature. 2009;461:788–92.
Chen Q, Sun L, Chen ZJ. Regulation and function of the cGAS-STING pathway of cytosolic DNA sensing. Nat Immunol. 2016;17:1142–9.
Stetson DB, Medzhitov R. Recognition of cytosolic DNA activates an IRF3-dependent innate immune response. Immunity. 2006;24:93–103.
Hartlova A, Erttmann SF, Raffi FA, Schmalz AM, Resch U, Anugula S, et al. DNA damage primes the type I interferon system via the cytosolic DNA sensor STING to promote antimicrobial innate immunity. Immunity. 2015;42:332–43.
Lam AR, Bert NL, Ho SS, Shen YJ, Tang LF, Xiong GM, et al. RAE1 ligands for the NKG2D receptor are regulated by STING-dependent DNA sensor pathways in lymphoma. Cancer Res. 2014;74:2193–203.
Ohkuri T, Ghosh A, Kosaka A, Zhu J, Ikeura M, David M, et al. STING contributes to antiglioma immunity via triggering type I IFN signals in the tumor microenvironment. Cancer Immunol Res. 2014;2:1199–208.
Woo SR, Fuertes MB, Corrales L, Spranger S, Furdyna MJ, Leung MYK, et al. STING-dependent cytosolic DNA sensing mediates innate immune recognition of immunogenic tumors. Immunity. 2014;41:830–42.
Zhu Q, Man SM, Gurung P, Liu Z, Vogel P, Lamkanfi M, et al. Cutting edge: STING mediates protection against colorectal tumorigenesis by governing the magnitude of intestinal inflammation. J Immunol. 2014;193:4779–82.
Coppe JP, Patil CK, Rodier F, Sun Y, Munoz DP, Goldstein J, et al. Senescence-associated secretory phenotypes reveal cell-nonautonomous functions of oncogenic RAS and the p53 tumor suppressor. PLoS Biol. 2008;6:2853–68.
Ivashkiv LB, Donlin LT. Regulation of type I interferon responses. Nat Rev Immunol. 2014;14:36–49.
Bourden M, Manet C, Montagutelli X. Host genetic susceptibility to viral infections: the role of type I interferon induction. Genes Immun. 2020;21:365–79.
Ishikawa H, Barber GN. STING is an endoplasmic reticulum adaptor that facilitates innate immune signalling. Nature. 2008;455:674–8.
Zhong B, Yang Y, Li S, Wang Y, Li Y, Diao F, et al. The adaptor protein MITA links virus-sensing receptors to IRF3 transcription factor activation. Immunity. 2008;29:538–50.
Garcia-Sastre A, Biron CA. Type 1 interferons and the virus-host relationship: a lesson in détente. Science. 2006;312:879–82.
Wu W, Yang Y, Yang W, Pang D, Liu Y, Sheng Y, et al. Tailoring escalation adjuvant therapy for early-stage triple-negative breast cancer in the CBCSG010 clinical trial biomarker analysis. J Natl Compr Canc Netw. 2024;22:528–36.
Kawagoe T, Takeuchi O, Takabatake Y, Kato H, Isaka Y, Tsujimura T, et al. TANK is a negative regulator of Toll-like receptor signaling and is critical for the prevention of autoimmune nephritis. Nat Immunol. 2009;10:965–72.
Fukasaka M, Ori D, Kawagoe T, Uematsu S, Maruyama K, Okazaki T, et al. Critical role of AZI2 in GM-CSF- induced dendritic cell differentiation. J Immunol. 2013;190:5702–11.
Kwon J, Bakhoum SF. The cytosolic DNA-sensing cGAS-STING pathway in cancer. Cancer Discov. 2020;10:26–39.
Lukhele S, Rabbo DA, Guo M, Shen J, Elsaesser HJ, Quevedo R, et al. The transcription factor IRF2 drives interferon-mediated CD8+ T cells exhaustion to restrict anti-tumor immunity. Immunity. 2022;55:2369–85.
Duong E, Fessenden TB, Lutz E, Dinter T, Yim L, Blatt S, et al. Type I interferon activates MHC class I-dressed CD11b+ conventional dendritic cells to promote protective anti-tumor CD8+ T cell immunity. Immunity. 2022;55:308–23.
Ivashkiv LB. IFNγ: signalling, epigenetics and roles in immunity, metabolism, disease and cancer immunotherapy. Nat Rev Immunol. 2018;18:545–58.
Acknowledgements
We greatly appreciate all the patients enrolled in the CBCSG010 clinical trial who contribute their specimens for our analysis. This work was supported by grant from the National Natural Science Foundation of China (82172576).
Author information
Authors and Affiliations
Contributions
W-YW: Conceptualization, data curation, formal analysis, validation, investigation, methodology, writing-original draft, writing-review and editing. Y-SY: Conceptualization, validation, investigation, methodology, writing-review, and editing. LA: Resources, methodology. Y-FX: Resources, methodology. G-HD: Conceptualization, supervision, writing-review, and editing. Z-MS: Conceptualization, supervision, resources, funding acquisition, writing-review, and editing. J-JL: Conceptualization, supervision, resources, funding acquisition, project administration. writing-review and editing.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Ethics approval and consent to participate
All methods were performed in accordance with the relevant guidelines and regulations. Approval for the use of specimens from the CBCSG010 clinical trial was granted by independent ethics committees at each participating center. The designed animal experiments were approved by the Animal Welfare Committee of Shanghai Medical College at Fudan University (Protocol number: IACUC-2023141).
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Wu, WY., Yang, YS., Andriani, L. et al. TBKBP1 induces capecitabine resistance through negative regulation of type I interferon pathway in triple-negative breast cancer. Oncogene 45, 703–714 (2026). https://doi.org/10.1038/s41388-025-03598-4
Received:
Revised:
Accepted:
Published:
Version of record:
Issue date:
DOI: https://doi.org/10.1038/s41388-025-03598-4
