
Abstract
Cell competition is an evolutionarily conserved quality control mechanism that eliminates less-fit cells to ensure optimal tissue integrity during development, homeostasis, and regeneration. Beyond these physiological roles, recent evidence implicates a role for cell competition in disease, particularly in cancer, where it can function by either suppressing or promoting malignant progression. In this review, we provide an overview of the different molecular mechanisms that drive cell competition and their impact on cancer development and progression. We will evaluate the current state-of-the-art in vitro experimental systems that can be employed to study these processes. Ranging from classical 2D co-culture systems to advanced organoid and organ-on-chip platforms, these model systems collectively enhance our understanding of the complex cellular interactions that underlie the competitive differences between cells. By integrating insights from diverse model systems, we highlight how cell competition shapes tumor dynamics and discuss how this knowledge could inspire novel therapeutic strategies to prevent or control tumor growth.
Similar content being viewed by others
Introduction
Throughout life, cells naturally accumulate both non-genetic and (epi)genetic alterations due to either intrinsic errors or extrinsic insults such as inflammation, irradiation, chemical exposure, or other environmental exposures [1, 2]. Since these alterations can have a significant impact on cell function, multicellular organisms rely on quality control mechanisms that are essential for proper embryonic development, tissue repair, and the maintenance of tissue homeostasis. One of the key processes to ensure that only the fittest cells contribute to tissue structure and function is competition, through which less fit or damaged cells are selectively eliminated and replaced by their neighbors (Fig. 1A).

A Cell competition: loser cells, with reduced fitness, are eliminated and replaced by adjacent wild-type cells. B Super competition: mutant cells with an acquired fitness advantage actively outcompete and replace wild-type cells.
The concept of cell competition was first established by Morata & Ripoll in Drosophila melanogaster cells [3]. They demonstrated that cells with heterozygous mutations in certain genes encoding ribosomal proteins (“Minute mutations”) were actively eliminated in the presence of wild-type cells. Whereas fully mutant flies developed normally, albeit somewhat slower than wild-type flies, in mosaic tissue, these less-fit mutant cells were effectively outcompeted.
Since its discovery, cell competition has been recognized as an evolutionarily conserved mechanism across multiple species, including mammals [4]. Beyond its essential role in embryonic development and in adult tissues, it has increasingly gained attention in the context of disease, particularly in cancer, where it can influence both the initiation and progression of the disease.
Intriguingly, cell competition in cancer is inherently paradoxical as it can safeguard tissue integrity by eliminating and replacing aberrant cells but also fuel tumorigenesis by enabling the expansion of oncogenic clones. Which of these opposing outcomes prevails is largely determined by the complex interplay between intrinsic cellular changes and extrinsic influences from the local microenvironment. Whereas most somatic mutations are phenotypically neutral, a subset may affect cancer driver genes that can confer a competitive advantage, allowing mutant cells to expand clonally within tissues. In this context, it is useful to distinguish between passive competition, where mutant cells gradually outcompete their neighbors through intrinsic advantages such as increased proliferation or survival, and so-called “supercompetition,” where mutant cells actively eliminate neighboring wild-type cells through non-cell-autonomous mechanisms (Fig. 1B) [5, 6]. This mechanism has been reported for various mutations in oncogenes and tumor suppressor genes, including Myc, JAK-STAT, Notch, YAP/TAZ, Ras, APC, and p53 [5,6,7,8,9,10,11,12,13,14]. Importantly, the outcome of cell competition is highly dependent on the relative fitness differences between neighboring cells. For example, cells with Kras-driven supercompetitor phenotype in the intestine [15, 16] may be efficiently removed from the pancreas [17, 18], suggesting that the context in which cell competition takes place may contribute to the distinct evolutionary trajectories as observed in different cancer types [19].
Much of our current understanding of cell competition has derived from in vivo animal studies, particularly in using fly and murine models. While these approaches have been invaluable, they come with limitations in scalability and mechanistic dissection. Recent advances in in vitro model systems, including two-dimensional (2D) and three-dimensional (3D) cultures, epithelioids, and organ-on-chip technologies, have opened new avenues to investigate cell competition under more controllable conditions. These systems enable detailed exploration of molecular pathways, biomechanical forces, and environmental factors that shape competitive interactions, and hold promise for bridging the gap between mechanistic insights and potential clinical application.
In this review, we will first describe some of the fundamental principles of cell competition across life stages (development, homeostasis, regeneration, and aging) and then focus on the role of cell competition during cancer development. We provide a comprehensive overview of the molecular mechanisms governing cell competition, focusing on contact-dependent, soluble factor-mediated, and mechanical modes of competition. We will discuss recent developments in the experimental systems used to model cell competition in vitro, highlighting how these platforms are contributing to our knowledge of the competitive dynamics in physiological and pathological contexts and how these approaches may inspire novel therapeutic strategies in cancer.
Physiological roles of cell competition
Embryonic development
Rapid cell proliferation during embryogenesis can generate replication and signaling errors, and it has been reported that somatic mutations and chromosome segregation errors frequently occur during normal human and mouse embryogenesis [20,21,22]. During this process, cell competition plays a crucial role in the removal of less-fit cells harboring these errors to ensure the formation of the fittest embryo possible [23]. For example, it was shown that already in forming epiblasts during preimplantation, extensive cell competition takes place, driven by naturally occurring relative expression differences in markers for cellular fitness, such as TEAD/YAP, MYC, or TP53. As a result of this selection, embryonic cells expressing relatively low levels of MYC are effectively eliminated through apoptosis, without disrupting embryonic development [9, 10]. Recent work has shown that this type of cell competition is the main reason that human pluripotent stem cells (hPSCs) are able to maintain diploid populations over generations despite mitotic errors that can lead to aneuploidy. Although aneuploid hPSC cells can still proliferate, these cells typically exhibit lower MYC or elevated TP53 compared to their diploid neighbors and will therefore often be eliminated during the competition within mosaic populations [24]. These findings indicate the importance of cell competition during early development, by maintaining a strong selective pressure, only the fittest and most capable cells will contribute to the formation of the organism.
Tissue homeostasis and regeneration
Adult stem cells have a central role in maintaining tissue homeostasis, growth, and regeneration of the different organs. To ensure the maintenance of the fittest pool of stem cells, cell competition has a key function in these processes as well. Whereas during embryonic development, less-fit cells are predominantly eliminated through apoptosis, in adult tissues, induction of differentiation is another important mechanism to outcompete these cells [9, 25,26,27,28]. A well-studied example of a specific form of cell competition during normal development and homeostasis is the competition between intestinal stem cells (ISCs) within the stem cell compartment of the small bowel, the crypts. The epithelial monolayer of the intestine completely renews approximately every 5–7 days [29, 30]. This rapid turnover is driven by ISCs located at the base of the crypt. Their number is limited, typically ranging from 5 to 16 cells depending on the definition and species, to ensure tissue homeostasis by maintaining a critical balance between proliferative and differentiated daughter cells within the tissue [31]. As their functionality is fully dependent on the signals produced by the niche cells at the crypt bottom, these equipotent ISCs continuously compete for limited space and signals within this niche, leading to stochastic loss and replacement of cells (neutral drift) and resulting in random clonal dominance [32,33,34]. Importantly, when an ISC acquires an oncogenic mutation conferring a competitive advantage, i.e., APC or KRAS mutations, these neutral stochastic dynamics might shift towards biased drift, increasing the probability of expansion of the mutant clone and potentially initiation of colorectal cancer (CRC) [15, 35]. Another example of differentiation-based competition in adult tissue homeostasis is found in the hematopoietic system, where p53-low stem cells have an advantage in the competition for space in the hematopoietic stem cell niche in the bone marrow, thereby forcing stem cells with relatively higher levels of p53 into the circulation and to differentiate. This process ensures a selection for cells with the lowest level of stress or damage to maintain the stem cell population [36].
On the other hand, during regeneration, micro-environmental changes and cellular plasticity can lead to alterations in cellular fitness, thereby reshaping cell-competition dynamics to enable dedifferentiation and replenishment of the pool of stem cells. For instance, in the well-studied setting of the intestine, WNT-dependent expression of the ISC marker ASCL2 in differentiated cells or resting stem cells induces dedifferentiation and confers a competitive advantage to these cells, rendering it essential for intestinal regeneration [37]. Other niche-defining factors, such as the Notch pathway and YAP/TAZ signaling, have been implicated in stem cell plasticity, dedifferentiation, and tissue regeneration, collectively altering the competition dynamics during regeneration [38]. Together, the regulation of cell competition is critical for both proper functioning and repair of the adult tissue.
Cell competition during ageing
During the lifespan of an organism, the gradual accumulation of damaged cells drives ageing, ultimately leading to a progressive, time-dependent decline in physiological functions and increased vulnerability to disease (reviewed in [39, 40]). Importantly, cellular ageing occurs asynchronously, creating disparities in fitness among cells in the population [41]. In this context, cell competition is pivotal for the elimination of damaged or suboptimal cells, which is essential for maintaining tissue homeostasis and ensuring healthy aging (reviewed in [42]). Studies in flies have demonstrated the importance of cell competition in the context of ageing, where expression levels of the gene Azot can strongly affect cell competition and thereby impacting on tissue regeneration and aging. Inhibition of this factor reduced competition between cells and resulted in shortened lifespan [43]. On the other hand, cell competition can also drive loser cells into senescence, i.e., mediated by differences in P53 levels, and thereby contributing to aging [36]. Furthermore, stem cell dynamics change during the lifespan of an organism. For example, the expression of the hemidesmosome component collagen XVII (COL17A1) in epidermal stem cells (EpSCs) is responsive to genomic and oxidative stress, leading to differential COL17A1 expression. This variation in COL17A1 drives the competition in the pool of EpSCs to remove cells with reduced fitness. In young skin, EpSCs divide symmetrically and orchestrate skin homeostasis; however, when COL17A1 expression declines during aging, EpSCs will undergo asymmetric divisions, thereby reducing the efficiency of competitive selection. This impairment compromises stem cell maintenance and contributes to age-related phenomena like depigmentation and skin aging. Notably, induction of COL17A1 expression in basal keratinocytes has been shown to reduce these effects, indicating that interfering with cell competition might result in promising therapeutic strategies for wound repair and age-associated skin degeneration [44].
Cell competition in cancer
Within epithelial tissues, normal cells can detect and eliminate transformed neighbor cells through a tumor-suppressive mechanism known as epithelial defense against cancer (EDAC) [45]. Previous studies using both in vitro and in vivo models have shown that transformed cells carrying oncogenic mutations, such as RAS, SRC, or constitutively active YAP, are recognized by surrounding normal epithelial cells and actively eliminated through apical extrusion [46,47,48,49]. In Madin–Darby canine kidney (MDCK) cells, an interesting example of EDAC occurs as constitutively active mutant YAP overexpressing cells are extruded apically when surrounded by normal epithelial cells. In addition, when YAP-overexpressing cells are co-cultured with K-RasG12V or v-Src expressing cells, their role shifts from losers to winners, underscoring the critical influence of neighboring cell status in determining competitive outcomes [48].
Despite the tumor suppressive role of EDAC, why do tumors still arise so frequently, and how do transformed cells escape this mechanism? Several factors may compromise EDAC efficiency, including cell density, cell–cell adhesion, and the ratio of normal to transformed cells, environmental factors, including metabolic changes or high-fat diet, and the accumulation of oncogenic mutations [46, 49,50,51,52,53,54]. For instance, RasV12-transformed cells undergo apical extrusion or form dynamic basal protrusions when surrounded by normal epithelial cells; however, neither mechanism occurs when RasV12 cells are surrounded by other RasV12 cells. In addition, E-cadherin expression in neighboring normal cells plays a critical role in determining the fate of RasV12 cells [46]. Similarly, whereas p53-R273H mutant cells are effectively outcompeted within healthy epithelium, elimination of mutant p53 cells is strongly impaired within RasV12 mutant epithelia, indicating the relevance of cell competition during different stages of tumorigenesis [53].
As tissues age, cells continuously accumulate somatic mutations; however, the majority of these alterations are functionally neutral and do not impact cellular expansion or survival [55,56,57]. In contrast, mutations in cancer driver genes may provide selective advantages that alter the relative fitness of cells. This leads to the elimination of less-fit neighboring cells and expansion of driver mutant cells, thereby initiating cancer [15, 58, 59].
Activation of several key oncogenes and loss of tumor suppressor genes have been shown to drive tumor development by functioning as supercompetitors [5,6,7,8,9,10,11,12,13,14]. For example, in glioblastoma (GBM), Yes-associated protein (YAP) is heterogeneously expressed, and cells with higher YAP activity drive tumorigenesis and outcompete neighboring clones [60]. In vitro 2D cultures and 3D tumor spheroid models also demonstrate that cells with elevated YAP activity gain a competitive advantage, outcompeting neighboring YAP-low cells. This clonal dominance is driven by the selective elimination of the less-fit YAP-low population through apoptosis [60].
Mechanisms of cell competition
Depending on the context, different types of cell competition might occur. For example, when the cellular fitness of competing cells is considered equal, neutral competition will be the main source of selection. Here, equipotent cells can be stochastically lost and replaced by neighboring cells, resulting in the random dominance of certain clones within the niche [61]. In contrast, when cells have a fitness advantage, competition may be biased in favor of the mutant cells. When cells negatively impact the fate of their competitors, this interaction has been termed active competition, a specific form of biased competition. The relative fitness disparities that drive cell competition can induce interactions that influence cellular survival and proliferation. Various types of interactions between cells can be involved in this process, including contact-dependent interactions, such as mechanical or molecular recognition-based interactions, or contact-independent interactions, i.e., mediated by soluble factors. Importantly, in the context of cancer, all types of cell competition may ultimately result in an opportunity for transformed cells to achieve clonal dominance and initiate tumor growth.
Mechanical cell competition is driven by differences in mechanical forces between cells that influence cell survival and spatial positioning within a tissue. Mechanical forces play a crucial role in regulating the number of cells within homeostatic tissues [62]. For instance, in epithelial tissues, cells must maintain strong intercellular adhesion and structural integrity to preserve overall tissue architecture and the barrier function. Using various model organisms, including humans, dogs, and zebrafish, it has been shown that epithelial tissues can become overcrowded due to increased proliferation or migration. In response, live cell extrusion acts as a regulatory mechanism to maintain tissue homeostasis [62]. When cells are subjected to mechanical stress, physically “weaker” cells are often more sensitive to extrusion; consequently, those exerting stronger forces can outcompete their neighboring cells [63, 64]. This process was illustrated using the MDCK epithelial cell line, where knockdown of the Scribble (Scrib) gene results in alterations in E-cadherin mediated cell adhesion between knockdown and wild-type cells. Together with Scrib-KD-induced activation of p53, this makes them more sensitive to compaction compared to wild-type cells, and consequently, these cells are rapidly outcompeted by their wild-type neighbors upon crowding [63,64,65].
Along the same line, Moreno et al. demonstrated that tissue compaction can trigger cell elimination by downregulating the EGFR/ERK signaling pathway while simultaneously upregulating the pro-apoptotic protein Hid. Interestingly, their findings revealed that premalignant cells with overactivation of the Ras oncogene were able to outcompete wild-type cells by suppressing ERK signaling and induction of apoptosis in neighboring cells through mechanical compaction. This suggests that this process not only promotes the elimination of surrounding less-fit cells during homeostasis but also drives the expansion of Ras clones during cancer initiation [13]. Besides p53 and ERK signaling, several other mechanosensitive pathways have been identified, such as the Hippo YAP/TAZ pathway and JNK signaling, that might have similar effects on cell competition under mechanical stress [66,67,68].
Molecular recognition-based cell competition involves direct molecular communication through cell surface markers that ultimately leads to the active elimination of less-fit competitors. Competition based on this kind of interaction can function as a way to recognize and remove damaged or neoplastic cells from the tissue. For example, pancreatic cells expressing oncogenic Ras can be recognized by neighboring wild-type cells, leading to their elimination from the tissue architecture [46]. The cell-surface molecule EPHA2, involved in cell repulsion and segregation, plays a key role in this process, as it is overexpressed in KRAS mutant cells within pancreatic tissue. In vitro studies showed that when in contact with EPHA2-low wild-type cells, ectopic EPHA2 boundaries are effectively formed that promote the segregation and elimination of mutant cells by neighboring wild-type cells [17, 69, 70]. To study these interactions, normal and Ras mutant (Ras-G12V) MDCK cells were seeded in separate compartments of a culture insert with a fixed gap. After removing the insert, both cell types migrated to fill the gap. Upon collision, contact with normal cells caused Ras mutant cells to adopt a rounded, contractile morphology and to separate from the normal cell layer. Eventually, these RasV12 cells were eliminated from the cell layer through apical extrusion [69, 70]. These observations were confirmed using in vivo models for pancreatic cancer, where KRAS mutant cells were effectively outcompeted when present in low numbers. However, in the absence of EPHA2, KRAS clones were able to persist, fostering the development of pancreatic intraepithelial neoplastic lesions [17]. A similar tumor suppressive role for recognition-based cell competition has been observed for multiple other potential oncogenic markers, such as v-SRC, ERBB2, YAP, or activated β-catenin expression, in various tissues, including kidney, mammary, and skin [47, 48, 71, 72]. Intriguingly, also aberrant expression of immunological ligands on precancerous cells can serve as recognition points for epithelial cell-mediated extrusion of transformed cells. High expression of MHC class I is often observed on transformed cells and can provoke mechanical extrusion by normal epithelial cells via LILRB3 interaction, independent of the immune system [73]. Together, these findings indicate that cell competition is an important mechanism to maintain tissue homeostasis, functioning as an immune-like surveillance system to protect the epithelium against cancer.
Soluble signal-mediated cell competition does not require direct cell-to-cell contact; instead, secreted signaling molecules in the microenvironment can influence the fitness of neighboring cells and ultimately determine cell fate. For instance, in Drosophila wing disc epithelium, Mychi cells gain a competitive advantage over their Myclow neighbors by modulation of their surroundings, ultimately leading to the displacement of competitors. During this process, Mychi cells not only rewire their own metabolism to support rapid proliferation and survival by increased glutamate production, but also actively reshape the local microenvironment by enhanced uptake of secreted lactate, thereby modulating the metabolic state of adjacent “loser” cells. These metabolic changes driven by Myc are essential for Myc-overexpressing cells to act as supercompetitor cells in the cancer setting [74]. Another example of supercompetition via modulation of the microenvironment can be found during the initiation of adenomas in the intestine. Several studies showed that during adenoma formation, APC mutant stem cells, which display constitutively active Wnt signaling, actively reduce the competitive fitness of their wild-type neighbors by secretion of Wnt inhibitory factors, similar to what was found previously in developing Drosophila wings [75]. This principle was demonstrated by exposing wild-type intestinal organoids to conditioned medium derived from APC mutant organoids, which was shown to induce their differentiation and inhibit their clonogenicity via WNT antagonists such as Notum, again highlighting how paracrine signaling can reinforce competitive dynamics [7, 8]. On the other hand, similar mechanisms can help maintain tissue integrity by removing transformed cells. Signals generated by aberrant cells can activate processes in normal cells that will shift the balance in their advantage. For example, extracellular ATP production by (partially) extruding transformed cells can stimulate ROS production of the surrounding cells, thereby inducing polarized movement of these cells towards the transformed cell in epithelial layers, which will promote the extrusion even further [76]. However, these processes are all highly context-dependent and can be exploited by tumor cells under the right circumstances. Whereas apical extrusion of RAS mutants is highly effective for the removal of these cells from normal epithelium, in the context of Wnt-activated epithelium, i.e., in early stages of CRC initiation, RAS mutations have been reported to drive basal extrusion, mediated by extracellular matrix (ECM) modulation via increased MMP21 secretion, thereby promoting invasion and metastasis [77].
These findings emphasize the urgency to study cell competition in various contexts and backgrounds, as the outcome of the competition between cells is fully determined by relative differences. Here, we will discuss the status of in vitro modeling of cell competition, where these conditions can be studied at scale in a highly controllable fashion.
2D cell competition models
Among the earliest and most widely used systems to study cell behavior is the 2D monolayer culture, valued for its simple and cost-effective maintenance of cell culture, reproducibility, and a large amount of comparative literature [78, 79]. These models have played a central role in advancing our understanding of cell competition, particularly within epithelial contexts. The MDCK cell line has been widely used to study epithelial cell competition due to its capacity to form confluent and polarized epithelial monolayers in vitro, effectively modeling epithelial tissue architecture. Within this setup, interactions and competition between wild-type and/or genetically modified cells can be studied using mono- or cocultures. These cultures are well suitable for modeling processes such as cell crowding, extrusion, or specifically migration and wound closing. The fate and behavior of different cells or cell populations can relatively easily be followed using (fluorescence) microscopy. In these settings, less-fit cells were shown to be selectively eliminated when surrounded by more competitive neighbors, despite appearing viable in isolation [63, 64, 80]. In these models, it was discovered that competitive elimination often depends on physical factors, including sensitivity to compactness, which can trigger apoptosis in cells unable to withstand elevated mechanical pressure. While many of these 2D studies emphasize the role of mechanical forces and cell recognition in cell competition, other reports have demonstrated that competition can also occur in the absence of direct cell-cell contact. Within the 2D culture system, several options to demonstrate the role of soluble factors in cell competition are available. Conditioned medium transfer assays can be employed to investigate the effect of secreted factors of a certain cell type on another cell type. In an indirect co-culture assay, two cell types can be separated by a membrane filter that permits the passage of large proteins while preventing direct mixing of the cell populations. This type of experiment revealed, for instance, that competition between cells with different levels of dMyc is mediated by soluble factors produced during co-culture, rather than requiring direct cell-cell interaction [81].
Another application of 2D monolayer culture models can be to perform CRISPR screening to identify genes involved in cell competition. For that purpose, GFP competition assays were further improved to study CRISPR-mediated cell competition [82,83,84]. In this setup, cells were transduced with GFP-expressing gRNA vectors at low MOI, creating mixed populations of GFP⁺ and GFP− cells. Here, mutations that impair cellular fitness lead to the loss of GFP⁺ cells, as they are outcompeted by their GFP⁻ counterparts. For example, introducing indels at the PCNA or RPA3 loci severely compromises cell fitness. gRNAs targeting these genes were tested across different cancer cell lines, and it was found that depletion of up to 90-fold occurred after five passages [83, 84].
Together, 2D monolayer cultures provide a well-established and informative framework for dissecting both physical and biochemical mechanisms of cell competition, particularly those involving direct cell–cell interactions. Despite the insights gained from 2D culture models, these systems cannot entirely replicate the complex architecture and microenvironment of native tissues and tumors.
3D cell competition models
3D organoid models have been employed to address some of the limitations of 2D cultures and to more closely mimic in vivo conditions. These organoid models are usually cultured in extracellular matrices, such as Matrigel or collagen, and often display a more hierarchical distribution of cell types, and their structural organization does resemble the tissue of origin. For these reasons, 3D organoid models more accurately replicate the biological properties and natural microenvironment of tumor tissues, making them valuable tools for personalized medicine; however, they are generally more expensive, less reproducible, and often more challenging to interpret than traditional 2D cultures [79, 85].
In the context of 3D models, co-culturing organoids with different genetic backgrounds offers a powerful approach to study both contact-dependent and contact-independent interactions between healthy and mutant cells [86, 87]. Previous studies have developed co-culture systems using mouse or human organoids labeled with fluorescent markers to study cell competition in 3D [7, 87, 88]. These studies demonstrated that such co-cultures can be robustly analyzed through flow cytometry and microscopy-based approaches, yielding critical insights into cell competition. In addition to 3D organoid models, researchers have developed assembloids by combining brain organoids derived from different regions using hPSCs [89]. These advanced models enable studying complex cellular interactions between distinct brain regions in vitro and provide valuable insights into neurodevelopmental and neurodegenerative diseases [89, 90]. More recently, advanced 3D culture models known as epithelioids have been introduced. This system was developed by culturing mouse esophageal epithelial explants on permeable membrane inserts in medium supplemented with growth factors. When these cultures reach confluence, stratified layers of keratinocytes will form. At this stage, the so-called epithelioids can be maintained in medium with reduced levels of growth factors. These esophageal epithelioids exhibit long-term self-renewal for over a year, recapitulate the native 3D epithelial structure, and mimic in vivo cellular dynamics. Moreover, this model enables the investigation of mutation-driven cell competition and provides a robust platform for testing anti-cancer compounds that modulate competitive interactions [91]. For instance, using this model combined with fluorescent tracing, the competitive advantage of activating PI3K pathway mutations in the esophagus could be demonstrated. Furthermore, the model could be used as a platform to screen for regulators of cellular fitness and test compounds that could potentially interfere with cell competition [92].
Whereas organoid models have advantages compared to 2D monocultures, a major limitation of both these models is the lack of a fully representative tumor microenvironment (TME). This constraint limits their ability to capture the complex interactions between cancer cells and stromal, immune, and vascular components, which are critical to tumor evolution and therapeutic response.
Organ-on-a-chip cell competition models
To better simulate in vivo conditions, research has increasingly focused on organ-on-a-chip (OoC) models, which more accurately mimic the dynamic TME. OoC models represent an advanced in vitro cell culture platform that combines microfabrication technology with 3D cell culture. These systems enable real-time monitoring and manipulation of cellular behaviors, microenvironmental dynamics, and tissue-level responses under physiologically relevant conditions (reviewed in [93, 94]). By incorporating features such as physiological fluid flow (e.g., blood circulation and interstitial flow) and mechanical cues experienced in the body, OoC systems serve as powerful tools for studying drug responses and advancing personalized medicine. Therefore, OoC models may transform cell competition studies by providing a physiologically relevant platform that bridges the gap between traditional in vitro assays and in vivo models. For example, researchers successfully generated mini-colons capable of undergoing tumorigenesis ex vivo by combining advanced bioengineering techniques. In this model, optogenetically inducible healthy colon epithelial cells that can acquire a set of oncogenic mutations (APC, KRAS, TP53) after exposure to blue light were cultured within a microfluidic platform designed to support the development of long-lived miniature colon tissues. This system enables real-time tracking of tumor initiation at single-cell resolution and can be monitored for several weeks [95]. These models pave the way for spatiotemporal analysis of the dynamics of tumorigenesis with high resolution and are applicable for high-throughput screening for compounds to prevent tumor initiation.
Despite their many advantages, OoC models still face limitations in fully replicating the complexity of the TME. However, integrating co-culture systems that include stromal cells into OoC models may be the next step to overcome these limitations.
Mimicking in vivo complexity
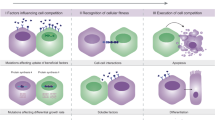
Given the limitations of the aforementioned culture systems, the use of complex in vitro co-culture models has been investigated (Fig. 2). To better recapitulate the complexity of the highly heterogeneous in vivo TME, co-culture systems have been developed, allowing different cell types to be studied together [81, 96]. To more accurately simulate the complexity of the TME, tumor cells have been co-cultured with cancer-associated fibroblasts (CAFs), immune cells, and other stromal components.

From basic 2D monolayer to advanced co-culture models. Created with BioRender.com.
The interactions between cancer cells and TME play a critical role in determining oncogenic fate. The TME comprises CAFs, different types of immune cells, endothelial cells, and the ECM, all of which engage in dynamic crosstalk with malignant cells. Among these components, CAFs are particularly abundant and have been shown to influence cancer progression through multiple mechanisms. These include directly promoting epithelial–mesenchymal transition to enhance cancer cell invasiveness and migration, as well as remodeling the ECM to facilitate tumor expansion and metastasis [97, 98]. Furthermore, we and others demonstrated that colon cancer cell clonogenicity in established cancer tissue is highly dependent on fibroblast signaling from the TME. These dynamics could be recapitulated in vitro using co-cultures of these cell types, but only when the culture medium was depleted of serum or growth factors [87, 99]. Culture models, including both tumor cells and fibroblasts under these conditions, can therefore have a large impact on cell competition within the system. Interestingly, Myc-mediated cell competition between stromal and cancer cells at the tumor border has been reported and might serve as a mechanism to promote tumor expansion and invasion into the surrounding tissue [100].
In addition to fibroblasts, immune cells represent another key cellular component of the TME that can profoundly influence tumor behavior. Co-culturing cancer cells with immune cells offers a powerful approach to study the complex interactions between tumors and the immune system [101]. A wide range of co-culture models has been developed to support these investigations, including direct co-cultures, the media transfer model, and transwell systems. These experimental systems help researchers dissect immune surveillance and evasion mechanisms and understand competitive interactions with tumor cells. Although the prevailing view is that immune cells play a secondary role in cell competition, which is thought to occur primarily among epithelial cells, some studies in animal models, such as fruit flies or mice, have demonstrated that immune cells can also play a primary, active role in this process [102, 103]. For instance, a study demonstrated that under certain circumstances, tumor-associated macrophages can outcompete MYC-overexpressing cancer cells, a process that could be manipulated using genetic or dietary interventions [103]. Although these mechanisms have been identified using in vivo models, the recent developments in immune-cancer co-culture systems provide an opportunity to explore these interactions further in vitro.
In addition to the co-culture models, machine learning can play a key role in studying cell competition by accurately identifying and classifying labeled cells, enabling detailed analysis of individual cell proliferation, migration, and elimination in response to local fitness differences [104, 105]. The combination of high-throughput live-cell imaging and machine learning provides a powerful platform for quantifying clonal dynamics in real time and uncovering mechanisms of cell competition [105]. Furthermore, integrating artificial intelligence (AI) into these systems can facilitate the analysis of large, complex datasets and enable real-time monitoring, which can be particularly valuable for testing the effects of various drugs and components on cell competition (reviewed in [106, 107]). Overall, co-culture systems offer enhanced physiological relevance to tumor biology by more closely replicating the hierarchical architecture and complex interactions of the TME.
Therapeutic perspectives
Cell competition plays a critical role in the initiation and progression of cancer, acting as a double-edged sword that can either be tumor-suppressive or tumor-promoting [108]. Manipulating competition by either reducing the competitive fitness of mutant cells or preventing elimination, or even boosting wild-type cells, could therefore help to prevent cancer initiation and progression [109]. A promising example of this principle was shown by Fernandez-Antoran et al., where p53 mutant cells could effectively be outcompeted by wild-type cells in the esophagus after radiation when first pre-treated with antioxidants. Whereas the competitive advantage of the mutant p53 cells lies in the high endogenous expression of antioxidants, which protects them from radiation damage, this advantage was abolished when the tissue was exposed to exogenous antioxidants first. Whereas initial findings were done using mouse models, elucidating the mechanism and evaluation of treatment strategies were performed using advanced in vitro cultures such as esophageal epithelium explants, 3D cultures, and co-culturing wild-type and mutant cells [59]. Furthermore, efforts have been made to identify compounds that selectively promote the elimination of (RasV12, or BRC-Abl) mutant cells in the presence of wild-type cells, using high-throughput screens on heterogeneous co-culture models including wild-type and mutant cells [110, 111].
However, translation to the clinic is challenging given the highly context-dependent effects of cell competition, which could be affected by, for instance, mutational load, location, and other factors. Nevertheless, as proof of principle, recent work of our lab demonstrated the possibility of reversing the competitive differences between mutant and wild-type cells. In the intestinal crypt, APC mutant stem cells secrete Wnt antagonists that suppress Wnt-signaling in wild-type stem cells, thereby increasing the competitive advantage of the mutant stem cells, which are unaffected by Wnt inhibition upstream of APC. In preclinical studies, it was shown that treatment with GSK3β (Glycogen Synthase Kinase-3) inhibitors, such as lithium chloride or CHIR, which act downstream of the Wnt antagonists produced by mutant cells, could boost the Wnt-pathway in wild-type cells. This abrogated the competitive advantage of Apc-mutant clones and was able to prevent adenoma formation in mice [7]. Whereas stimulation of the Wnt-pathway was able to restore the equilibrium between wild-type and mutant stem cells, further research suggested that additional low-dose treatment with inhibitors of BCL-XL, an anti-apoptotic protein upregulated in APC mutant stem cells, was sufficient to efficiently eliminate APC mutant cells from the intestinal crypt [7, 109]. These findings suggest that exploiting the mechanisms that determine competition between healthy and precancerous cells could provide promising new treatment strategies for high-risk individuals, such as Familial adenomatous polyposis (FAP) patients. Indeed, a phase II trial in FAP patients to investigate the effect of lithium on the spread of adenoma has recently been initiated [112]. These examples indicate the importance of improving our understanding of the mechanisms underlying cell competition, which requires proper experimental models mimicking various types of competition.
Conclusions and future perspectives
Cells constantly engage in dynamic interactions to establish relative fitness differences, which eventually will result in the elimination of less-fit neighbors via cell competition. This phenomenon serves as a safeguard, ensuring that only the fittest cells contribute to tissue architecture and homeostasis, but it can also be hijacked to promote the expansion of oncogenic clones. The dual role of cell competition highlights the importance of understanding the molecular and environmental factors that influence competitive fitness.
A wide array of experimental models has been developed to study cell competition that have contributed to our understanding of the different aspects of cell competition in vitro. Although these culture models have advanced significantly, they still fall short in fully replicating the spatiotemporal dynamics and microenvironmental complexity of native tissues. Bridging this gap will require integrating long-term tissue remodeling, vascularization, and diverse components of the TME to more accurately mimic in vivo conditions. Nevertheless, as in vitro systems increasingly recapitulate the structural and dynamic complexity of native tissues, they are becoming powerful platforms for identifying novel therapeutic targets linked to competitive cellular dynamics. Moreover, CRISPR-based and drug screening approaches enable manipulation of competing cell populations, facilitating detailed dissection of the molecular mechanisms underlying cell competition [111, 113, 114]. Finally, the integration of high-throughput imaging with AI might offer new opportunities to perform real-time tracking and high-throughput analysis of cellular behavior, offering a robust framework for uncovering new insights into dynamic cell interactions [105, 115]. Together, the application of these emerging techniques will greatly benefit from well-defined in vitro culture models, which have multiple advantages compared to animal models, including reduced costs and scalability.
References
Bjornsson HT, Daniele Fallin M, Feinberg AP. An integrated epigenetic and genetic approach to common human disease. Trends Genet. 2004;20:350–8.
Talens RP, Christensen K, Putter H, Willemsen G, Christiansen L, Kremer D, et al. Epigenetic variation during the adult lifespan: cross-sectional and longitudinal data on monozygotic twin pairs. Aging Cell. 2012;11:694–703.
Morata G, Ripoll P. Minutes: mutants of Drosophila autonomously affecting cell division rate. Dev Biol. 1975;42:211–21.
Oliver ER, Saunders TL, Tarlé SA, Glaser T. Ribosomal protein L24 defect in belly spot and tail (Bst), a mouse Minute. Development. 2004;131:3907–20.
de la Cova C, Abril M, Bellosta P, Gallant P, Johnston LA. Drosophila Myc regulates organ size by inducing cell competition. Cell. 2004;117:107–16.
Moreno E, Basler K. dMyc transforms cells into super-competitors. Cell. 2004;117:117–29.
van Neerven SM, de Groot NE, Nijman LE, Scicluna BP, van Driel MS, Lecca MC, et al. Apc-mutant cells act as supercompetitors in intestinal tumour initiation. Nature. 2021;594:436–41.
Flanagan DJ, Pentinmikko N, Luopajärvi K, Willis NJ, Gilroy K, Raven AP, et al. NOTUM from Apc-mutant cells biases clonal competition to initiate cancer. Nature. 2021;594:430–5.
Clavería C, Giovinazzo G, Sierra R, Torres M. Myc-driven endogenous cell competition in the early mammalian embryo. Nature. 2013;500:39–44.
Hashimoto M, Sasaki H. Epiblast formation by TEAD-YAP-dependent expression of pluripotency factors and competitive elimination of unspecified cells. Dev Cell. 2019;50:139–54.e5.
Alcolea MP, Jones PH. Cell competition: winning out by losing notch. Cell Cycle. 2015;14:9–17.
Rodrigues AB, Zoranovic T, Ayala-Camargo A, Grewal S, Reyes-Robles T, Krasny M, et al. Activated STAT regulates growth and induces competitive interactions independently of Myc, Yorkie, Wingless and ribosome biogenesis. Development. 2012;139:4051–61.
Moreno E, Valon L, Levillayer F, Levayer R. Competition for space induces cell elimination through compaction-driven ERK downregulation. Curr Biol. 2019;29:23–34.e8.
Murai K, Skrupskelyte G, Piedrafita G, Hall M, Kostiou V, Ong SH, et al. Epidermal tissue adapts to restrain progenitors carrying clonal p53 mutations. Cell Stem Cell. 2018;23:687–99.e8.
Vermeulen L, Morrissey E, Van Der Heijden M, Nicholson AM, Sottoriva A, Buczacki S, et al. Defining stem cell dynamics in models of intestinal tumor initiation. Science. 2013;342:995–8.
Yum MK, Han S, Fink J, Wu SHS, Dabrowska C, Trendafilova T, et al. Tracing oncogene-driven remodelling of the intestinal stem cell niche. Nature. 2021;594:442–7.
Hill W, Zaragkoulias A, Salvador-Barbero B, Parfitt GJ, Alatsatianos M, Padilha A, et al. EPHA2-dependent outcompetition of KRASG12D mutant cells by wild-type neighbors in the adult pancreas. Curr Biol. 2021;31:2550–60.e5.
Salvador-Barbero B, Alatsatianos M, Morton JP, Sansom OJ, Hogan C. KRASG12D cells override homeostatic cell elimination mechanisms in adult pancreas via Wnt5a and cell dormancy. Gastroenterology. 2025;169:983–99.e21.
Gerstung M, Jolly C, Leshchiner I, Dentro SC, Gonzalez S, Rosebrock D, et al. The evolutionary history of 2,658 cancers. Nature. 2020;578:122–8.
Currie CE, Ford E, Benham Whyte L, Taylor DM, Mihalas BP, Erent M, et al. The first mitotic division of human embryos is highly error prone. Nat Commun. 2022;13:1–13.
Mashiko D, Ikeda Z, Yao T, Tokoro M, Fukunaga N, Asada Y et al. Chromosome segregation error during early cleavage in mouse pre-implantation embryo does not necessarily cause developmental failure after blastocyst stage. Sci Rep. 2020;10:854.
Ju YS, Martincorena I, Gerstung M, Petljak M, Alexandrov LB, Rahbari R, et al. Somatic mutations reveal asymmetric cellular dynamics in the early human embryo. Nature. 2017;543:714–8.
Matsumoto K, Akieda Y, Haraoka Y, Hirono N, Sasaki H, Ishitani T. Foxo3-mediated physiological cell competition ensures robust tissue patterning throughout vertebrate development. Nat Commun. 2024;15:1–18.
Ya A, Deng C, Godek KM. Cell competition eliminates aneuploid human pluripotent stem cells. Stem Cell Rep. 2025;20:102506.
Sancho M, Di-Gregorio A, George N, Pozzi S, Sánchez JM, Pernaute B, et al. Competitive interactions eliminate unfit embryonic stem cells at the onset of differentiation. Dev Cell. 2013;26:19–30.
Kolahgar G, Suijkerbuijk SJE, Kucinski I, Poirier EZ, Mansour S, Simons BD, et al. Cell competition modifies adult stem cell and tissue population dynamics in a JAK-STAT-dependent manner. Dev Cell. 2015;34:297–309.
Rhiner C, Díaz B, Portela M, Poyatos JF, Fernández-Ruiz I, López-Gay JM, et al. Persistent competition among stem cells and their daughters in the Drosophila ovary germline niche. Development. 2009;136:995–1006.
Ellis SJ, Gomez NC, Levorse J, Mertz AF, Ge Y, Fuchs E. Distinct modes of cell competition shape mammalian tissue morphogenesis. Nature. 2019;569:497–502.
Barker N, Van Es JH, Kuipers J, Kujala P, Van Den Born M, Cozijnsen M, et al. Identification of stem cells in small intestine and colon by marker gene Lgr5. Nature. 2007;449:1003–7.
Barker N. Adult intestinal stem cells: critical drivers of epithelial homeostasis and regeneration. Nat Rev Mol Cell Biol. 2013;15:19–33.
Kozar S, Morrissey E, Nicholson AM, van der Heijden M, Zecchini HI, Kemp R, et al. Continuous clonal labeling reveals small numbers of functional stem cells in intestinal crypts and adenomas. Cell Stem Cell. 2013;13:626–33.
Ritsma L, Ellenbroek SIJ, Zomer A, Snippert HJ, De Sauvage FJ, Simons BD, et al. Intestinal crypt homeostasis revealed at single-stem-cell level by in vivo live imaging. Nature. 2014;507:362–5.
Lopez-Garcia C, Klein AM, Simons BD, Winton DJ. Intestinal stem cell replacement follows a pattern of neutral drift. Science. 2010;330:822–5.
Snippert HJ, van der Flier LG, Sato T, van Es JH, van den Born M, Kroon-Veenboer C, et al. Intestinal crypt homeostasis results from neutral competition between symmetrically dividing Lgr5 stem cells. Cell. 2010;143:134–44.
Snippert HJ, Schepers AG, Van Es JH, Simons BD, Clevers H. Biased competition between Lgr5 intestinal stem cells driven by oncogenic mutation induces clonal expansion. EMBO Rep. 2014;15:62–69.
Bondar T, Medzhitov R. p53-mediated hematopoietic stem and progenitor cell competition. Cell Stem Cell. 2010;6:309–22.
Murata K, Jadhav U, Madha S, van Es J, Dean J, Cavazza A, et al. Ascl2-dependent cell dedifferentiation drives regeneration of ablated intestinal stem cells. Cell Stem Cell. 2020;26:377–90.e6.
Hageman JH, Heinz MC, Kretzschmar K, van der Vaart J, Clevers H, Snippert HJG. Intestinal regeneration: regulation by the microenvironment. Dev Cell. 2020;54:435–46.
López-Otín C, Blasco MA, Partridge L, Serrano M, Kroemer G. The hallmarks of aging. Cell. 2013;153:1194–217.
Luo J, Mills K, le Cessie S, Noordam R, van Heemst D. Ageing, age-related diseases and oxidative stress: What to do next? Ageing Res Rev. 2020;57:100982.
Rando TA, Wyss-Coray T. Asynchronous, contagious and digital aging. Nat Aging. 2021;1:29–35.
Marques-Reis M, Moreno E. Role of cell competition in ageing. Dev Biol. 2021;476:79–87.
Merino MM, Rhiner C, Lopez-Gay JM, Buechel D, Hauert B, Moreno E. Elimination of unfit cells maintains tissue health and prolongs lifespan. Cell. 2015;160:461–76.
Liu N, Matsumura H, Kato T, Ichinose S, Takada A, Namiki T, et al. Stem cell competition orchestrates skin homeostasis and ageing. Nature. 2019;568:344–50.
Kajita M, Sugimura K, Ohoka A, Burden J, Suganuma H, Ikegawa M, et al. Filamin acts as a key regulator in epithelial defence against transformed cells. Nat Commun. 2014;5:1–13.
Hogan C, Dupré-Crochet S, Norman M, Kajita M, Zimmermann C, Pelling AE, et al. Characterization of the interface between normal and transformed epithelial cells. Nat Cell Biol. 2009;11:460–7.
Kajita M, Hogan C, Harris AR, Dupre-Crochet S, Itasaki N, Kawakami K, et al. Interaction with surrounding normal epithelial cells influences signalling pathways and behaviour of Src-transformed cells. J Cell Sci. 2010;123:171–80.
Chiba T, Ishihara E, Miyamura N, Narumi R, Kajita M, Fujita Y, et al. MDCK cells expressing constitutively active Yes-associated protein (YAP) undergo apical extrusion depending on neighboring cell status. Sci Rep. 2016;6:1–10.
Kon S, Ishibashi K, Katoh H, Kitamoto S, Shirai T, Tanaka S, et al. Cell competition with normal epithelial cells promotes apical extrusion of transformed cells through metabolic changes. Nat Cell Biol. 2017;19:530–41.
Pothapragada SP, Gupta P, Mukherjee S, Das T. Matrix mechanics regulates epithelial defence against cancer by tuning dynamic localization of filamin. Nat Commun. 2022;13:1–12.
Sato N, Yako Y, Maruyama T, Ishikawa S, Kuromiya K, Tokuoka SM, et al. The COX-2/PGE2 pathway suppresses apical elimination of RasV12-transformed cells from epithelia. Commun. Biol. 2020;3:1–11.
Sasaki A, Nagatake T, Egami R, Gu G, Takigawa I, Ikeda W, et al. Obesity suppresses cell-competition-mediated apical elimination of RasV12-transformed cells from epithelial tissues. Cell Rep. 2018;23:974–82.
Watanabe H, Ishibashi K, Mano H, Kitamoto S, Sato N, Hoshiba K, et al. Mutant p53-expressing cells undergo necroptosis via cell competition with the neighboring normal epithelial cells. Cell Rep. 2018;23:3721–9.
Kohashi K, Mori Y, Narumi R, Kozawa K, Kamasaki T, Ishikawa S, et al. Sequential oncogenic mutations influence cell competition. Curr Biol. 2021;31:3984–95.e5.
Martincorena I, Roshan A, Gerstung M, Ellis P, Van Loo P, McLaren S, et al. High burden and pervasive positive selection of somatic mutations in normal human skin. Science. 2015;348:880–6.
Lee-Six H, Olafsson S, Ellis P, Osborne RJ, Sanders MA, Moore L, et al. The landscape of somatic mutation in normal colorectal epithelial cells. Nature. 2019;574:532–7.
Martincorena I, Raine KM, Gerstung M, Dawson KJ, Haase K, Van Loo P, et al. Universal patterns of selection in cancer and somatic tissues. Cell. 2017;171:1029–41.e21.
Alcolea MP, Greulich P, Wabik A, Frede J, Simons BD, Jones PH. Differentiation imbalance in single oesophageal progenitor cells causes clonal immortalization and field change. Nat Cell Biol. 2014;16:612–9.
Fernandez-Antoran D, Piedrafita G, Murai K, Ong SH, Herms A, Frezza C, et al. Outcompeting p53-mutant cells in the normal esophagus by redox manipulation. Cell Stem Cell. 2019;25:329–41.e6.
Liu Z, Yee PP, Wei Y, Liu Z, Kawasawa YI, Li W. Differential YAP expression in glioma cells induces cell competition and promotes tumorigenesis. J Cell Sci. 2019;132:jcs225714.
Amoyel M, Simons BD, Bach EA. Neutral competition of stem cells is skewed by proliferative changes downstream of Hh and Hpo. EMBO J. 2014;33:2295–313.
Eisenhoffer GT, Loftus PD, Yoshigi M, Otsuna H, Chien C-B, Morcos PA, et al. Crowding induces live cell extrusion to maintain homeostatic cell numbers in epithelia. Nature. 2012;484:546–9.
Wagstaff L, Goschorska M, Kozyrska K, Duclos G, Kucinski I, Chessel A, et al. Mechanical cell competition kills cells via induction of lethal p53 levels. Nat Commun. 2016;7:1–14.
Norman M, Wisniewska KA, Lawrenson K, Garcia-Miranda P, Tada M, Kajita M, et al. Loss of Scribble causes cell competition in mammalian cells. J Cell Sci. 2012;125:59–66.
Qin Y, Capaldo C, Gumbiner BM, Macara IG. The mammalian Scribble polarity protein regulates epithelial cell adhesion and migration through E-cadherin. J Cell Biol. 2005;171:1061–71.
Pereira AM, Tudor C, Kanger JS, Subramaniam V, Martin-Blanco E. Integrin-dependent activation of the JNK signaling pathway by mechanical stress. PLoS ONE. 2011;6:e26182.
Levayer R, Dupont C, Moreno E. Tissue crowding induces caspase-dependent competition for space. Curr Biol. 2016;26:670–7.
Dupont S, Morsut L, Aragona M, Enzo E, Giulitti S, Cordenonsi M, et al. Role of YAP/TAZ in mechanotransduction. Nature. 2011;474:179–84.
Porazinski S, de Navascués J, Yako Y, Hill W, Jones MR, Maddison R, et al. EphA2 drives the segregation of ras-transformed epithelial cells from normal neighbors. Curr Biol. 2016;26:3220–9.
Hill W, Hogan C. Normal epithelial cells trigger EphA2-dependent RasV12 cell repulsion at the single cell level. Small GTPases. 2019;10:305–10.
Leung CT, Brugge JS. Outgrowth of single oncogene-expressing cells from suppressive epithelial environments. Nature. 2012;482:410–3.
Brown S, Pineda CM, Xin T, Boucher J, Suozzi KC, Park S, et al. Correction of aberrant growth preserves tissue homeostasis. Nature. 2017;548:334–7.
Ayukawa S, Kamoshita N, Nakayama J, Teramoto R, Pishesha N, Ohba K, et al. Epithelial cells remove precancerous cells by cell competition via MHC class I–LILRB3 interaction. Nat Immunol. 2021;22:1391–402.
Soares CC, Rizzo A, Maresma MF, Meier P. Autocrine glutamate signaling drives cell competition in Drosophila. Dev Cell. 2024;59:2974–89.e5.
Vincent J-P, Kolahgar G, Gagliardi M, Piddini E. Steep differences in wingless signaling trigger Myc-independent competitive cell interactions. Dev Cell. 2011;21:366–74.
Mori Y, Shiratsuchi N, Sato N, Chaya A, Tanimura N, Ishikawa S, et al. Extracellular ATP facilitates cell extrusion from epithelial layers mediated by cell competition or apoptosis. Curr Biol. 2022;32:2144–59.e5.
Nakai K, Lin H, Yamano S, Tanaka S, Kitamoto S, Saitoh H, et al. Wnt activation disturbs cell competition and causes diffuse invasion of transformed cells through NF-κB-MMP21 pathway. Nat Commun. 2023;14:1–17.
Urzì O, Gasparro R, Costanzo E, De Luca A, Giavaresi G, Fontana S, et al. Three-dimensional cell cultures: the bridge between in vitro and in vivo models. Int J Mol Sci. 2023;24:12046.
Kapałczyńska M, Kolenda T, Przybyła W, Zajączkowska M, Teresiak A, Filas V, et al. 2D and 3D cell cultures—a comparison of different types of cancer cell cultures. Arch Med Sci. 2016;14:910.
Ogawa M, Kawarazaki Y, Fujita Y, Naguro I, Ichijo H. FGF21 induced by the ASK1-p38 pathway promotes mechanical cell competition by attracting cells. Curr Biol. 2021;31:1048–57.e5.
Senoo-Matsuda N, Johnston LA. Soluble factors mediate competitive and cooperative interactions between cells expressing different levels of Drosophila Myc. Proc Natl Acad Sci USA. 2007;104:18543–8.
Shi J, Wang E, Milazzo JP, Wang Z, Kinney JB, Vakoc CR. Discovery of cancer drug targets by CRISPR-Cas9 screening of protein domains. Nat Biotechnol. 2015;33:661–7.
Girish V, Sheltzer JM. A CRISPR competition assay to identify cancer genetic dependencies. Bio Protoc. 2020;10:e3682.
Lin A, Giuliano CJ, Sayles NM, Sheltzer JM. CRISPR/Cas9 mutagenesis invalidates a putative cancer dependency targeted in on-going clinical trials. eLife. 2017;6:e24179.
Imamura Y, Mukohara T, Shimono Y, Funakoshi Y, Chayahara N, Toyoda M, et al. Comparison of 2D- and 3D-culture models as drug-testing platforms in breast cancer. Oncol Rep. 2015;33:1837–43.
van Neerven SM, Ramadan R, van Driel MS, Huels DJ, Vermeulen L. Intestinal organoid co-culture protocol to study cell competition. STAR Protoc. 2022;3:101050.
Baglamis S, Sheraton VM, Meijer D, Qian H, Hoebe RA, Lenos KJ, et al. Using picoliter droplet deposition to track clonal competition in adherent and organoid cancer cell cultures. Sci Rep. 2023;13:18832.
Krotenberg Garcia A, Fumagalli A, Le HQ, Jackstadt R, Lannagan TRM, Sansom OJ, et al. Active elimination of intestinal cells drives oncogenic growth in organoids. Cell Rep. 2021;36:109307.
Birey F, Andersen J, Makinson CD, Islam S, Wei W, Huber N, et al. Assembly of functionally integrated human forebrain spheroids. Nature. 2017;545:54–9.
Kim JL, Imaizumi K, Jurjuț O, Kelley KW, Wang D, Thete MV, et al. Human assembloid model of the ascending neural sensory pathway. Nature. 2025;642:143–53.
Herms A, Fernandez-Antoran D, Alcolea MP, Kalogeropoulou A, Banerjee U, Piedrafita G, et al. Self-sustaining long-term 3D epithelioid cultures reveal drivers of clonal expansion in esophageal epithelium. Nat Genet. 2024;56:2158–73.
Herms A, Colom B, Piedrafita G, Kalogeropoulou A, Banerjee U, King C, et al. Organismal metabolism regulates the expansion of oncogenic PIK3CA mutant clones in normal esophagus. Nat Genet. 2024;56:2144–57.
Tsai H-F, Trubelja A, Shen AQ, Bao G. Tumour-on-a-chip: microfluidic models of tumour morphology, growth and microenvironment. J R Soc Interface. 2017;14:20170137.
Seaman K, Sun Y, You L. Recent advances in cancer-on-a-chip tissue models to dissect the tumour microenvironment. Med-X. 2023;1:1–28.
Lorenzo-Martín LF, Hübscher T, Bowler AD, Broguiere N, Langer J, Tillard L, et al. Spatiotemporally resolved colorectal oncogenesis in mini-colons ex vivo. Nature. 2024;629:450–7.
De La Cova C, Senoo-Matsuda N, Ziosi M, Wu DC, Bellosta P, Quinzii CM, et al. Supercompetitor status of Drosophila Myc cells requires p53 as a fitness sensor to reprogram metabolism and promote viability. Cell Metab. 2014;19:470–83.
Kojima Y, Acar A, Eaton EN, Mellody KT, Scheel C, Ben-Porath I, et al. Autocrine TGF-β and stromal cell-derived factor-1 (SDF-1) signaling drives the evolution of tumor-promoting mammary stromal myofibroblasts. Proc Natl Acad Sci USA. 2010;107:20009–14.
Orimo A, Gupta PB, Sgroi DC, Arenzana-Seisdedos F, Delaunay T, Naeem R, et al. Stromal fibroblasts present in invasive human breast carcinomas promote tumor growth and angiogenesis through elevated SDF-1/CXCL12 secretion. Cell. 2005;121:335–48.
Lenos KJ, Miedema DM, Lodestijn SC, Nijman LE, van den Bosch T, Romero Ros X, et al. Stem cell functionality is microenvironmentally defined during tumour expansion and therapy response in colon cancer. Nat Cell Biol. 2018;20:1193–202.
Di Giacomo S, Sollazzo M, De Biase D, Ragazzi M, Bellosta P, Pession A, et al. Human cancer cells signal their competitive fitness through MYC activity. Sci Rep. 2017;7:1–12.
Cattaneo CM, Dijkstra KK, Fanchi LF, Kelderman S, Kaing S, van Rooij N, et al. Tumor organoid–T-cell coculture systems. Nat Protoc. 2019;15:15–39.
Zhu Y, Wunderlich Z, Lander AD. Epithelial cell competition is promoted by signaling from immune cells. Nat Commun. 2025;16:1–15.
Zhang X, Li S, Malik I, Do MH, Ji L, Chou C, et al. Reprogramming tumour-associated macrophages to outcompete cancer cells. Nature. 2023;619:616–23.
Bove A, Gradeci D, Fujita Y, Banerjee S, Charras G, Lowe AR. Local cellular neighborhood controls proliferation in cell competition. Mol Biol Cell. 2017;28:3215–28.
Gradeci D, Bove A, Charras G, Lowe AR, Banerjee S. Single-cell approaches to cell competition: High-throughput imaging, machine learning and simulations. Semin Cancer Biol. 2020;63:60–68.
Fetah KL, DiPardo BJ, Kongadzem EM, Tomlinson JS, Elzagheid A, Elmusrati M, et al. Cancer modeling-on-a-chip with future artificial intelligence integration. Small. 2019;15:1901985.
Shaw I, Ali YS, Nie C, Zhang K, Chen C, Xiao Y. Integrating artificial intelligence and microfluidics technology for psoriasis therapy: a comprehensive review for research and clinical applications. Adv Intell Syst. 2025;7:2400558.
Knapp K, Verchio V, Coburn-Flynn O, Li Y, Xiong Z, Morrison JC, et al. Exploring cell competition for the prevention and therapy of esophageal squamous cell carcinoma. Biochem Pharmacol. 2023;214:115639.
Zhang L, Atencia Taboada L, Baglamis S, de Kroon M, Elshout C, Ramesh P, et al. GSK-3 and BCL-XL inhibition mitigates the competitive advantage of APC-mutant colorectal cancer cells. Oncogenesis. 2025;14:1–8.
Yamauchi H, Matsumaru T, Morita T, Ishikawa S, Maenaka K, Takigawa I, et al. The cell competition-based high-throughput screening identifies small compounds that promote the elimination of RasV12-transformed cells from epithelia. Sci Rep. 2015;5:1–9.
Tadele DS, Robertson J, Crispin R, Herrera MC, Chlubnová M, Piechaczyk L, et al. A cell competition–based small molecule screen identifies a novel compound that induces dual c-Myc depletion and p53 activation. J Biol Chem. 2020;296:100179.
Linssen JDG, van Neerven SM, Aelvoet AS, Elbers CC, Vermeulen L, Dekker E. The CHAMP-study: the CHemopreventive effect of lithium in familial AdenoMatous Polyposis; study protocol of a phase II trial. BMC Gastroenterol. 2022;22:1–9.
Paraskevopoulos M, McGuigan AP. Application of CRISPR screens to investigate mammalian cell competition. Brief Funct Genom. 2021;20:135–47.
Meng X, Yao D, Imaizumi K, Chen X, Kelley KW, Reis N, et al. Assembloid CRISPR screens reveal impact of disease genes in human neurodevelopment. Nature. 2023;622:359–66.
Pylvänäinen JW, Gómez-de-Mariscal E, Henriques R, Jacquemet G. Live-cell imaging in the deep learning era. Curr Opin Cell Biol. 2023;85:102271.
Acknowledgements
SB is thankful for the scholarship provided by the Ministry of National Education of the Republic of Türkiye. This work was supported by the Oncode Institute, The New York Stem Cell Foundation, and grants from the European Research Council (ERC-CoG 101045612 - NIMICRY), Dutch Cancer Society grant (KWF 13435 / 2021-1), and ZonMw (Vici 09-15018-21-10029) awarded to LV.
Author information
Authors and Affiliations
Contributions
Conceptualization, LV, SB, KJL, PMK, SMvN; writing and original draft preparation, SB; review and editing, KJL, SMvN, LV, PMK; supervision, KJL, LV; funding acquisition, LV. All authors have read and agreed to the published version of the manuscript.
Corresponding author
Ethics declarations
Competing interests
This work was supported by a scholarship provided by the Ministry of National Education of the Republic of Türkiye (SB), by the Oncode Institute, The New York Stem Cell Foundation, and grants from the European Research Council (ERC-CoG 101045612 - NIMICRY), Dutch Cancer Society grant (KWF 13435/2021-1), and ZonMw (Vici 09-15018-21-10029) (LV). LV is presently an employee of Genentech Inc. All other authors have no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Baglamis, S., Krawczyk, P.M., Vermeulen, L. et al. In vitro models of cell competition: current approaches and future directions. Oncogene 45, 1287–1295 (2026). https://doi.org/10.1038/s41388-026-03751-7
Received:
Revised:
Accepted:
Published:
Version of record:
Issue date:
DOI: https://doi.org/10.1038/s41388-026-03751-7
