
Abstract
Atrial fibrillation (AF), the most prevalent sustained cardiac arrhythmia, is closely linked to disturbed intracellular Ca2+ homeostasis. Visinin-like protein 1 (VILIP-1), newly identified in cardiomyocytes, has been implicated in modulating Ca2+ signaling, yet its role in AF remains undefined. In this study, we integrated bulk RNA sequencing, single-cell transcriptomics, and electrophysiological profiling from human AF patients and rodent AF models to identify VILIP-1 as a key mediator of Ca2+ dysregulation in AF. VILIP-1 was significantly upregulated in atrial tissues from AF patients and in pacing-induced rat AF models, with enhanced membrane localization in cardiomyocytes. Atrial cardiomyocyte-specific overexpression of VILIP-1 led to pathological Ca2+ leakage, promoting delayed afterdepolarizations (DADs) and action potential duration (APD) alternans, which fostered AF substrate formation and increased arrhythmia susceptibility. Mechanistically, VILIP-1 augmented the surface abundance of sodium-calcium exchanger 1 (NCX-1) via a myristoylation-dependent trafficking mechanism, thereby disrupting Ca2+ handling and initiating AF. Pharmacologically, repaglinide and desloratadine, two FDA-approved drugs that identified to target VILIP-1 or its myristoylation, attenuated AF susceptibility by reducing NCX-1 surface expression and restoring intracellular Ca2+ homeostasis. Collectively, our findings define VILIP-1 as a critical upstream modulator of atrial Ca2+ homeostasis and establish it as a promising therapeutic target for AF, with efficacy validated in human and rodent models.
Similar content being viewed by others


Introduction
Atrial fibrillation (AF), the most common form of sustained cardiac arrhythmia, continues to rise globally, currently affecting more than 60 million individuals and placing a significant burden on healthcare systems both clinically and economically.1,2 Global burden-of-disease analyses indicate a steady increase in disability-adjusted life years attributable to AF.1 This trend is largely attributed to population aging, extended life expectancy, and persistent exposure to chronic yet modifiable risk factors, with improved detection further exacerbating the observed rise. Long-term follow-up studies of prospective cohorts highlight the ongoing challenge of mitigating the health consequences of AF.1 Collectively, these analyses underscore the need for research on AF beyond rhythm control, emphasizing the identification of upstream biological mechanisms that can be targeted therapeutically.
The pathophysiology of AF is tightly intertwined with the dysregulation of Ca2+ handling in atrial cardiomyocytes.3,4 For instance, aberrant Ca2+ leakage through ryanodine receptor type 2 (RyR2) can provoke spontaneous Ca2+ release events, subsequently activating the sodium-calcium exchanger (NCX), thereby depolarizing the membrane potential and initiating delayed afterdepolarizations (DADs).4,5,6 Furthermore, sustained Ca2+ overload combined with Na+ accumulation drives NCX reverse-mode activation, exacerbating Ca2+ influx and cytoplasmic Ca2+ accumulation, ultimately lowering the threshold for ectopic (triggered) activity and contributing to the initiation of AF.6,7,8,9
While the mechanistic link between abnormal Ca2+ handling and AF is well-established, the upstream molecular hierarchy that initiates this pathological cascade has not been fully explored, presenting a critical barrier to the development of targeted therapies. To address this, we employed an integrated multi-omics approach, combining single-cell RNA sequencing, bulk RNA sequencing, and functional Ca2+ dynamics analysis in atrial cardiomyocytes from human AF patients and rodent AF models. This approach allowed us to correlate molecular alterations with functional Ca2+ aberrancies and AF pathogenesis, ultimately leading to the identification of visinin-like protein 1 (VILIP-1) as a key candidate regulator of Ca2+ homeostasis in AF. VILIP-1 belongs to the neuronal Ca2+ sensors (NCSs), a diverse family of Ca2+-binding proteins that participate in Ca2+-dependent signaling and mediate various cellular responses across different tissues.10 In neurons, they increase synaptic plasticity and neurotransmitter release; in pancreatic cells, they promote insulin secretion; and in skin, they suppress the invasiveness of squamous cell carcinoma cells.10,11,12,13,14 Upon Ca2+ binding, VILIP-1 undergoes a conformational change, exposing its myristoyl group and facilitating translocation to the plasma membrane.15 This Ca2+-dependent translocation enhances VILIP-1’s interaction with target proteins, which increases their surface expression and activates specific signaling pathways. However, the role of VILIP-1 in the heart, particularly in atrial cardiomyocytes and AF, has remained underexplored.
In this study, we reveal a previously unprecedented association between VILIP-1 upregulation and AF in both human patients and animal models. Our findings demonstrate that VILIP-1 regulates Ca2+ homeostasis in atrial cardiomyocytes by modulating the surface expression of NCX-1 in a myristoylation-dependent manner. Furthermore, we identified a clinically approved pharmacological agent with potential VILIP-1-targeting activity capable of restoring Ca2+ homeostasis in atrial cardiomyocytes of AF patients and effectively reducing AF susceptibility in preclinical animal models. These findings suggest that targeting VILIP-1 represents a promising therapeutic strategy for AF intervention.
Results
Multi-omics analysis identifies VILIP-1 as a novel Ca2+ signaling regulator in AF
To identify novel AF-associated genes, we conducted an integrated multi-omics study combining bulk RNA sequencing (RNA-seq), full-length transcriptome profiling in single atrial cardiomyocytes and electrophysiological data (Fig. 1a). Specifically, we obtained 252 atrial cardiomyocytes from pacing-induced AF rats and control rats, recorded their Ca2+ dynamics, and integrated these data with single-cell expression profiles generated from the same cells via the Smart-seq3 protocol. Significant increases in AF susceptibility and Ca2+ release events were observed in AF rat models (Supplementary Fig. 1), with correlation analysis identifying 1500 genes associated with abnormal Ca2+ release events ( | Pearson’s (r) correlation coefficient | > 0.2, P < 0.01, Supplementary Table S1; blue circle in Fig. 1e).

Multi-omics analysis reveals that Vsnl1 is associated with Ca2+ homeostasis dysregulation in atrial fibrillation (AF). a Schematic workflow depicting the integration of bulk RNA sequencing (RNA-seq) and full-length single-cell RNA sequencing (scRNA-seq) data from human AF patients and rat AF models, alongside electrophysiological recordings. b UMAP analysis illustrating the transcriptional clustering of atrial cardiomyocytes, highlighting the distinct cellular subpopulations associated with AF pathophysiology. c Heatmap of differentially expressed genes in atrial cardiomyocytes during AF progression, derived from pseudotime trajectory analysis, demonstrating dynamic shifts in gene expression patterns over disease progression. The pseudotime trajectory is mapped onto the UMAP projection to visualize cellular state transitions. d Volcano plot displaying differentially expressed genes (DEGs) between AF and sinus rhythm (SR) patient samples, with Gene Ontology (GO) analysis indicating a significant enrichment of Ca2+ signaling pathway-related genes in AF. e Venn diagram illustrating the intersection of DEGs from human AF samples (P.adj < 0.05), genes whose expression levels correlated with abnormal Ca2+ dynamics ( | Pearson’s (r) correlation coefficient | > 0.2, P < 0.01), Cluster 3 marker genes (P.adj < 0.05), and genes from the pseudotime C4 module (q_value < 0.05 and morans_I > 0.25), identifying Vsnl1 (encoding VILIP-1) and Kcnip2 as a key AF-associated genes
UMAP analysis of the single-cell data from atrial cardiomyocytes revealed three distinct cell clusters. Among those clusters, Cluster 3 exhibited an expression pattern of typical AF-related genes, indicating its central role in AF pathogenesis (Supplementary Fig. 2). GO enrichment analysis also showed that Cluster 3 was significantly enriched in genes related to Ca2+ homeostasis, Ca2+ transport, and heart contraction (Fig. 1b). Most cells in Cluster 3 originated from AF rats, reinforcing the idea that this cluster plays a central role in AF pathogenesis. Additionally, pseudotime analysis uncovered a progressive transition from healthy to AF-associated states (Fig. 1c). Atrial cardiomyocytes from pacing-induced AF rats mainly occupied terminal positions (C4 module) compared to those from the control group. This terminal C4 module showed increased expression of key AF-related pathways, including Ca2+ signaling, oxidative phosphorylation, and energy metabolism (Fig. 1c). This convergence of key AF pathomechanisms establishes the C4 module as pivotal in AF progression.
To substantiate the relevance of our findings in rat AF models to clinical cases, we collected left atria (LA) samples from AF patients and sinus rhythm (SR) patients, ensuring that there were no significant age or sex differences between the two groups (Supplementary Table S2). Differential expression analysis identified 188 differentially expressed genes from 6 AF patients and 7 SR patients, with a significant enrichment in Ca2+ signaling pathways (Fig. 1d), suggesting a critical role of Ca2+ dysregulation in AF. Through an integrated analysis of differentially expressed genes (DEGs) from human patients, Ca2+-dynamics correlated genes, Cluster 3 marker genes and genes in the pseudotime C4 module, we identified that the Vsnl1 (encoding VILIP-1) and Kcnip2 (encoding KChIP2) are strongly correlated with AF and its pathological Ca2+ release events (Fig. 1e). Considering that VILIP-1 has never been reported in AF pathogenesis, whereas KChIP2 has been documented as a multimodal regulator of cardiac ionic currents and linked to AF,16,17,18 we propose VILIP-1 as a novel therapeutic target that warrants further investigation.
Validation of VILIP-1 upregulation in patients and animal models with AF
To validate the multi-omics findings, we conducted cross-species validation of VILIP-1 dysregulation in AF vs. SR cohorts. Through transcriptomic analysis of atrial tissues from patients with AF and rat AF models, we found that among all VILIP family members, VILIP-1 was selectively upregulated and exhibited the highest expression in atrial tissues (Supplementary Fig. 3). In patients, VILIP-1 was elevated 1.74-fold in atria from AF patients versus SR controls (Fig. 2a). Immunofluorescence staining showed that in human AF samples, VILIP-1 exhibited stronger membrane localization in AF atria (yellow triangles in the zoomed image, Fig. 2b). In rat AF models, Western blot analysis revealed a 2.01-fold upregulation of VILIP-1 in atria versus SR controls (Fig. 2c). Similarly, immunofluorescence staining further revealed increased VILIP-1 expression, particularly at the membrane of AF rat atrial cardiomyocytes (yellow triangles in the zoomed image highlight localized enrichment of surface VILIP-1; Fig. 2d). These results confirm that VILIP-1 is upregulated in both AF human patients and rat AF models, highlighting its potential involvement in AF pathogenesis.

Upregulation of VILIP-1 in patients and rat models with atrial fibrillation (AF). a Capillary western immunoassay confirming elevated VILIP-1 expression in human AF left atria compared to sinus rhythm (SR) controls. Left: representative immunoassay bands. Right: quantification of VILIP-1 protein levels normalized to total protein expression using the Total Protein Detection Module (DM-TP01, ProteinSimple, USA). *P < 0.05, Student’s t test, n = 6–12 per group. b Immunofluorescence staining of human left atria demonstrating increased VILIP-1 expression and membrane localization in AF patients compared to SR controls. c Western blot analysis of VILIP-1 expression in left atria from pacing-induced AF rat models and SR controls. Left: representative western blot images. Right: quantitative assessment of VILIP-1 protein levels, normalized to β-ACTIN expression. *P < 0.05, Student’s t test, n = 4 per group. d Immunofluorescence staining illustrating VILIP-1 localization in atrial cardiomyocytes from rat AF models, with increased membrane accumulation in rat AF samples
The role of VILIP-1 in governing AF susceptibility
To establish the causal role of VILIP-1 in AF pathophysiology, we first generated mice with atrial cardiomyocyte-specific VILIP-1 overexpression (VILIP-1-OE) via adeno-associated viral 9 (AAV9) under the control of an atrial cardiomyocyte-specific ANP promoter (Supplementary Fig. 4a). Western blot analysis confirmed the efficiency of VILIP-1 overexpression via AAV9 in the atria at the protein level (Supplementary Fig. 4b). On the other hand, the expression of VILIP-1 in ventricular tissues showed no significant alteration (Supplementary Fig. 4c). Baseline surface ECG analysis showed comparable parameters between VILIP-1-OE and control mice (Ctrl), including heart rate, PR interval, RR interval, and corrected QT interval (QTc), with no significant differences observed (Supplementary Fig. 5). However, programmed burst pacing-induced AF in 55.6% of VILIP-1-OE mice versus 0% of Ctrl mice (P = 0.0337), with significantly prolonged AF duration, establishing atrial VILIP-1 overexpression is sufficient to increased AF susceptibility (Fig. 3a). In addition, atrial effective refractory periods were measured using a programmed stimulation protocol, and no significant alterations were observed between Ctrl and VILIP-1-OE mice (Supplementary Fig. 6).

Atrial cardiomyocyte-specific overexpression of VILIP-1 promotes atrial fibrillation (AF) by increasing delayed afterdepolarizations (DADs), action potential duration (APD) alternans, and Ca2+ leakage. a Electrophysiological recordings of surface electrocardiogram (ECG) and intracardiac electrograms. Left: representative traces from Ctrl and VILIP-1-OE mice, indicating sinus rhythm (SR) in Ctrl and AF episodes in VILIP-1-OE mice following burst pacing. Right: incidence and duration of pacing-induced AF. *P < 0.05, Mann-Whitney test or Fisher’s exact test, n = 7–9 per group. b APD alternans analysis in isolated Langendorff-perfused hearts. Left: representative action potential traces of Ctrl and VILIP-1-OE mice. Right: quantification of APD alternans incidence. *P < 0.05, Fisher’s exact test, n = 11–12 per group. c Delayed afterdepolarizations (DADs) recordings in atrial cardiomyocytes isolated from Ctrl and VILIP-1-OE mice. Left: representative traces from Ctrl and VILIP-1-OE mice. Right: incidence of DADs. *P < 0.05, Fisher’s exact test, n = 19–21 per group. d Confocal Ca2+ imaging in isolated atrial cardiomyocytes. Left: representative Ca2+ wave recordings in Ctrl and VILIP-1-OE cells. Right: quantification of the frequency of spontaneous Ca2+ waves, indicating that VILIP-1-OE increased Ca2+ leakage in atrial cardiomyocytes. *P < 0.05, Mann-Whitney test, n = 16-19 per group. e Representative line-scan images of caffeine (10 mM)-induced Ca2+ transients in atrial cardiomyocytes from Ctrl and VILIP-1-OE mice. Quantification of sarcoplasmic reticulum Ca2+ content, measured by peak caffeine-induced Ca2+ release amplitude, showing significantly reduced sarcoplasmic reticulum Ca2+ stores in the VILIP-1-OE group (*P < 0.05, Student’s t test, n = 7 cells per group)
VILIP-1 overexpression promotes AF susceptibility by creating proarrhythmic substrates
AF susceptibility is associated with multiple pathological abnormalities in the atria, including structural remodeling, electrical remodeling, and Ca2+ homeostasis disruption.19,20 To determine whether atrial cardiomyocyte-specific VILIP-1 overexpression promotes AF susceptibility through structural remodeling, we comprehensively assessed cardiac function and atrial structure in VILIP-1-OE and Ctrl mice. As shown in Supplementary Figs. 7 and 8, echocardiographic analysis revealed no significant differences between the VILIP-1-OE and Ctrl mice in standard parameters of systolic function (e.g., EF, FS), ventricular dimensions (LVDs, LVDd), diastolic function (e.g., E/A ratio), as well as left atrial strain and area. Additionally, Sirius Red staining (Supplementary Fig. 9) demonstrated comparable collagen volume fractions in the atria and ventricles of VILIP-1-OE and Ctrl mice. These findings indicate that VILIP-1 overexpression increases AF susceptibility independently of structural remodeling.
Subsequently, we assessed electrical remodeling using optical mapping and patch-clamp techniques. While the mean action potential duration (APD) and conduction velocity (CV) remained unchanged, optical mapping revealed that the atria of VILIP-1-OE mice exhibited a significantly higher incidence of APD alternans, greater spatial dispersion of APD, increased CV variability, and more ectopic activity (Fig. 3b and Supplementary Fig. 10). Complementary patch-clamp data further demonstrated a higher incidence of delayed afterdepolarizations (DADs) in atrial cardiomyocytes from VILIP-1-OE mice (Fig. 3c). These results collectively suggest that the increased electrical heterogeneity and enhanced triggered activity underlie the increased AF susceptibility observed in VILIP-1-OE mice.
Considering the close association between APD alternans and DADs with disrupted Ca2+ homeostasis, characterized by abnormal intracellular Ca2+ elevations and enhanced sarcoplasmic reticulum Ca2+ release,21,22,23 we further evaluated intracellular Ca2+ handling in atrial cardiomyocytes obtained from Ctrl and VILIP-1-OE mice. Confocal line-scan imaging revealed an increased frequency of spontaneous Ca2+ waves and significantly reduced sarcoplasmic reticulum Ca2+ content in VILIP-1-OE atrial cardiomyocytes compared to the Ctrl group (Fig. 3d, e). These results indicate severe pathological Ca2+ leakage in atrial cardiomyocytes, further contributing to the formation of a proarrhythmic substrate.
Briefly, these findings establish that VILIP-1 overexpression promotes AF susceptibility by inducing proarrhythmic substrates characterized by Ca2+ leakage-driven electrical remodeling (e.g. APD alternans and DADs). Importantly, these effects occur independently of structural remodeling, highlighting Ca2+ homeostasis disruption as a central mechanism in VILIP-1-mediated AF susceptibility.
Identification of NCX-1 as a key downstream target of VILIP-1
To elucidate the molecular mechanisms by which VILIP-1 regulates Ca2+ homeostasis and increases AF susceptibility, we began by conducting immunoprecipitation-mass spectrometry (IP-MS) to identify downstream mediators in the atria of both mice and rats (Fig. 4a, left panel). This approach identified the potential VILIP-1 interactors (yellow and green circles in Fig. 4c). To further refine these findings, we utilized a VILIP-1-miniTurbo fusion construct for proximity labeling (PL) in neonatal rat cardiomyocytes (NRCMs), which exploits VILIP-1’s Ca2+-responsive activation to biotinylate proximal proteins.15,24,25 Under controlled Ca2+ concentration conditions, we performed PL and streptavidin bead enrichment for mass spectrometry analysis across four different Ca2+ concentrations (Fig. 4a, right panel). Statistical analysis revealed 115 proteins exhibiting Ca2+-dependent biotinylation patterns (proteins in Cluster 1, Fig. 4b). Integrating IP-MS and Ca2+-dependent PL interactome profiling identified 4 candidate VILIP-1 interactors (Fig. 4c).

Sodium-calcium exchanger (NCX-1) is identified as a key downstream target for VILIP-1. a Schematic workflow illustrating immunoprecipitation-mass spectrometry (left panel) and proximity labeling experiments using VILIP-1-V5-miniTurbo under varying Ca2+ concentrations (right panel) to identify VILIP-1 interactors. b MaSigPro (R package) identified four dynamic VILIP-1-proximity labeling patterns in response to increased Ca2+ concentrations. Proteins in Cluster 1 were identified as VILIP-1’s candidate interactors based on their progressive enrichment in VILIP-1-V5-miniTurbo-mediated biotinylation profiles with increasing Ca2+ concentrations. c Venn diagram illustrating the overlap between interactors identified by co-immunoprecipitation and proximity labeling, confirming high-confidence candidates. d Western blot analysis of biotinylated proteins from VILIP-1-V5-miniTurbo proximity labeling, showing Ca2+-dependent enrichment of NCX-1. e Confocal microscopy images confirming the localization of biotinylated VILIP-1-interacting proteins (within a 10 nm radius), NCX-1, in neonatal rat cardiomyocytes (NRCMs) expressing VILIP-1-V5-miniTurbo. Cells were treated with 500 μM biotin for 13 min at 37 °C, and biotinylated proteins were visualized using Neutravidin–Alexa Fluor 488 staining. f Representative immunoblots showing co-immunoprecipitation of VILIP-1 and NCX-1 in rat atrial lysate, demonstrating their interaction. g Western blot analysis of in vitro GST pull-down experiments from VILIP-1-GST and HA-NCX-1, confirming the direct interaction between VILIP-1 and NCX-1. h Confocal microscopy images displaying colocalization of VILIP-1 and NCX-1 in rat atrial cardiomyocytes
Among these candidates, NCX-1 emerged as the leading candidate effector of VILIP-1 due to its direct involvement in regulating intracellular Ca2+ homeostasis. Western blot analysis of the VILIP-1-miniTurbo proximity labeling experiment confirmed a Ca2+-dependent increase in biotinylated NCX-1 levels (Fig. 4d). Confocal microscopy further demonstrated strong colocalization between proteins biotinylated by VILIP-1-miniTurbo (visualized by Neutravidin) and NCX-1, supporting NCX-1 as a key downstream effector of VILIP-1 (Fig. 4e). Co-immunoprecipitation and GST pull-down experiments confirmed a direct interaction between VILIP-1 and NCX-1 (Fig. 4f, g), and their strong surface colocalization was further validated in adult rat atrial cardiomyocytes (Fig. 4h).
Molecular docking analysis and co-immunoprecipitation experiments identified Lys63 of VILIP-1 as a key residue mediating its interaction with NCX-1, supported by its predicted high contribution to binding and experimentally reduced interaction upon the K63A mutation (Supplementary Fig. 11a and b). Molecular dynamics simulations further revealed a destabilized binding geometry following the K63A mutation (Supplementary Fig. 11c) and identified Glu554 and Thr602 of NCX-1 as critical interaction residues frequently interacting with Lys63 of VILIP-1, ranking among the top ten contributors to binding energy at the NCX-1–VILIP-1 interface (Supplementary Fig. 11d and e). Experimentally, mutations of these residues (E554A and T602A) significantly impaired NCX-1 binding to VILIP-1 (Supplementary Fig. 11f). Collectively, these findings establish NCX-1 as a direct downstream effector of VILIP-1 that regulates atrial Ca2+ homeostasis, providing mechanistic insight into how VILIP-1 overexpression disrupts Ca2+ signaling and promotes AF susceptibility.
VILIP-1 overexpression enhances NCX-1 surface membrane expression
Considering that the proper functioning of NCX-1 in both electrophysiological processes and Ca2+ homeostasis maintenance has been shown to critically depend on its membrane localization,6,26 the effect of VILIP-1 overexpression on the surface membrane expression of NCX-1 was further evaluated by using the Jess automated protein analysis system. The results revealed a marked increase in NCX-1 expression at the cell surface, with no compensatory changes in total NCX-1 protein levels in the atria of VILIP-1-OE mice (Fig. 5a, b). In addition, we further isolated atrial cardiomyocytes from VILIP-1-OE mice, and the patch-clamp recordings demonstrated a significant increase in NCX-1 current (INCX) density in VILIP-1-OE atrial cardiomyocytes compared to Ctrl (Fig. 5c), implying that VILIP-1-OE also dramatically enhanced the function of NCX-1 in atrial cardiomyocytes.

Atrial cardiomyocyte-specific overexpression of VILIP-1 increases atrial fibrillation (AF) susceptibility through enhancing the membrane expression and function of NCX-1. a Western blot analysis of VILIP-1 and NCX-1 protein levels in the atria of VILIP-1-OE and Ctrl mice. Left: representative immunoblot images. Right: quantification of protein levels normalized to GAPDH. *P < 0.05, Student’s t test, n = 3 per group. b Capillary western immunoassay results showed elevated NCX-1 and VILIP-1 protein levels in membrane fractions of atria from Ctrl and VILIP-1-OE mice. Quantification was performed using the Total Protein Detection Module (DM-TP01, ProteinSimple, USA). *P < 0.05, Student’s t test, n = 4 per group. c Whole-cell patch-clamp recordings from isolated atrial cardiomyocytes illustrating a significant increase in the INCX current density in VILIP-1-OE mice compared to Ctrl. *P < 0.05, Student’s t test, n = 14 per group. d Confocal Ca2+ imaging of atrial cardiomyocytes from VILIP-1-OE mice pretreated with vehicle or SEA0400. Left: representative Ca2+ wave recordings. Right: quantification of the frequency of spontaneous Ca2+ waves, showing reduced spontaneous Ca2+ waves upon SEA0400 treatment. *P < 0.05, Mann-Whitney test, n = 13-18 per group. e Electrophysiological recordings of surface ECG and intracardiac electrograms in VILIP-1-OE mice. Left: representative traces of pacing-induced AF episodes in VILIP-1-OE mice with or without NCX-1 inhibition using SEA0400. Right: quantification of AF incidence and duration, showing a significant reduction in AF susceptibility upon SEA0400 treatment. *P < 0.05, Mann-Whitney test or Fisher’s exact test, n = 7–8 per group
To determine whether increased INCX mediates the effects of VILIP-1 overexpression on Ca2+ homeostasis and AF susceptibility, we conducted a rescue experiment using SEA0400, a NCX-1 reverse-mode inhibitor.27,28 Treatment with SEA0400 (1 μM) notably reduced the spontaneous Ca2+ waves in VILIP-1-OE atrial cardiomyocytes, indicating that blocking INCX alleviates the pathological Ca2+ overload induced by VILIP-1 overexpression (Fig. 5d). Furthermore, in vivo administration of SEA0400 (1 mg/kg) effectively reduced AF susceptibility in VILIP-1-OE mice, as evidenced by a 58.93% decrease in the AF induction rate and a shortened AF duration (Fig. 5e). These findings establish NCX-1 hyperactivity as a key proarrhythmic mechanism in atrial VILIP-1 overexpression models, linking increased NCX-1 membrane expression to AF susceptibility.
Myristoylation is essential for VILIP-1 to regulate the surface membrane expression of NCX-1
Normally, VILIP-1, like the other VILIP family members, requires myristoylation for membrane localization and Ca2+-dependent signal transduction.15,29 Thus, to investigate the contribution of VILIP-1 myristoylation to AF pathogenesis, we pharmacologically inhibited this modification using desloratadine which was previously found to inhibit the myristoylation of VILIPs.30 In vitro studies showed that desloratadine treatment significantly reduced surface NCX-1 in AF rat models (Fig. 6a). Subsequently, we isolated rat atrial cardiomyocytes from AF rat models to record the INCX current and Ca2+ dynamics, and the results showed that desloratadine dramatically reversed the elevated INCX current density and spontaneous Ca2+ waves (Fig. 6b and c). Corresponding electrophysiological analyses also revealed that desloratadine decreased AF susceptibility in rat AF models (Fig. 6d). These results establish myristoylation as an essential post-translational modification driving VILIP-1’s regulation of NCX-1 and arrhythmogenic activity in AF, highlighting its therapeutic relevance.

Inhibition of VILIP-1 myristoylation rescues atrial fibrillation (AF) susceptibility in rat AF models. a Capillary western immunoassay analysis and quantification of NCX-1 expression in atrial membrane fractions from AF rat models treated with vehicle or Desloratadine. Quantification was performed using the Total Protein Detection Module (DM-TP01, ProteinSimple, USA). *P < 0.05, Student’s t test, n = 4 per group. b Whole-cell patch-clamp recordings demonstrate that AF rats treated with vehicle exhibit significantly increased INCX current density compared to the control, while desloratadine treatment restores the INCX current density to control levels. *P < 0.05, one-way ANOVA, n = 14–18 per group. c Confocal Ca2+ imaging of atrial cardiomyocytes from rat AF models pretreated with vehicle or desloratadine. Left: representative Ca2+ wave recordings. Right: quantification of the frequency of spontaneous Ca2+ waves, showing reduced Ca2+ waves upon desloratadine treatment. *P < 0.05, Mann-Whitney test, n = 17 per group. d Electrophysiological recordings of surface ECG and intracardiac electrograms in pacing-induced AF rats. Left: representative traces showing AF episodes in vehicle-pretreated versus 5 mg/kg desloratadine-pretreated rats. Right: quantification of AF incidence and duration, demonstrating a significant reduction in AF susceptibility following desloratadine administration. *P < 0.05, Mann-Whitney test or Fisher’s exact test, n = 6–8 per group
Clinical drug repaglinide targets VILIP-1 to reduce AF susceptibility
Finally, we evaluated a clinically approved agent, repaglinide, which has been reported to inhibit the regulatory function of VILIP’s visinin-like domain, as a therapeutic strategy for AF.31 Molecular docking predicted a robust interaction between repaglinide and VILIP-1 (Fig. 7a), and thermal shift assays (TSA) demonstrated that repaglinide enhanced the thermal stability of VILIP-1, indicating high-affinity binding to VILIP-1 (Fig. 7b). Furthermore, surface plasmon resonance (SPR) assays confirmed that repaglinide binds to recombinant VILIP-1 in a dose-dependent manner (Kd = 7.52 µM, Fig. 7c). In our rat AF models, repaglinide treatment (1.5 mg/kg/day) reduced NCX-1 membrane expression, as confirmed by capillary western immunoassay (Fig. 7d) and immunofluorescence (Fig. 7e). In isolated rat atrial cardiomyocytes, repaglinide treatment effectively decreased the INCX density (Supplementary Fig. 12) and attenuated Ca2+ leakage (Fig. 7f). Additionally, the in vivo and in vitro AF inducibility were decreased (Fig. 7g and Supplementary Fig. 13). In line with these findings in the rat AF model, repaglinide treatment also diminished the membrane expression of NCX-1 in the human atria (Fig. 7h). Ca2+ imaging of atrial cardiomyocytes from AF patients showed that repaglinide mitigated intracellular Ca2+ overload, resulting in decreased spontaneous Ca2+ waves (Fig. 7i). These data additionally support that inhibition of VILIP-1 can stabilize Ca2+ homeostasis and ameliorate proarrhythmic imbalance, thereby reducing AF susceptibility.

VILIP-1 inhibition alleviates Ca2+ leakage and prevents atrial fibrillation (AF) inducibility in both human patients and rat AF models. a Molecular docking simulation demonstrating the interaction between repaglinide and VILIP-1 (docking score = -318.2), supporting its potential as a VILIP-1 inhibitor. b Thermal shift assay (TSA) analysis showing VILIP-1 protein stabilization following 10 μM repaglinide treatment at varying temperatures. c Binding affinities of repaglinide to VILIP-1 measured by surface plasmon resonance (SPR) assays. d Capillary western immunoassay demonstrating a reduction in NCX-1 expression in membrane fractions of atria from pacing-induced AF rat models treated with repaglinide. Quantification was performed using the Total Protein Detection Module (DM-TP01, ProteinSimple, USA). *P < 0.05, Mann-Whitney test, n = 4 per group. e Immunofluorescence staining of VILIP-1 and NCX-1 in atria from AF rat models pretreated with vehicle or repaglinide, showing reduced NCX-1 surface expression upon repaglinide treatment. f Confocal Ca2+ imaging of atrial cardiomyocytes from AF rat models pretreated with vehicle or repaglinide. Top: representative Ca2+ wave recordings. Bottom: quantification of the frequency of spontaneous Ca2+ waves, demonstrating a significant reduction in spontaneous Ca2+ waves following repaglinide treatment. *P < 0.05, Mann-Whitney test, n = 15–16 per group. g Electrophysiological recordings of surface ECG and intracardiac electrograms in pacing-induced AF rats pretreated with vehicle or repaglinide. Top: representative traces showing a decrease in pacing-induced AF episodes following repaglinide administration. Bottom: quantification of AF incidence and duration. *P < 0.05, Mann-Whitney test or Fisher’s exact test, n = 6 – 7 per group. h Capillary western immunoassay analysis of NCX-1 expression in membrane fractions of human atria, showing decreased NCX-1 surface expression following repaglinide treatment. Quantification was performed using the Total Protein Detection Module (DM-TP01, ProteinSimple, USA). *P < 0.05, Mann-Whitney test, n = 6-8 per group. i Confocal Ca2+ imaging of atrial cardiomyocytes from human AF patients pretreated with vehicle or 10 μM repaglinide. Left: representative Ca2+ wave recordings. Right: quantification of the frequency of spontaneous Ca2+ waves, demonstrating a significant reduction in spontaneous Ca2+ waves following repaglinide treatment. *P < 0.05, Mann-Whitney test, n = 17–19 per group
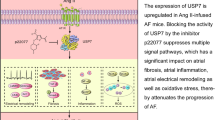
In summary, our study delineates how upregulated VILIP-1 disrupts Ca2+ homeostasis and drives AF pathogenesis through myristoylation-dependent potentiation of NCX-1 activity (Fig. 8). A therapeutic strategy targeting VILIP-1 exemplified by repaglinide, represents a promising strategy to restore Ca2+ homeostasis in atrial cardiomyocytes and treat AF.

Upregulated VILIP-1 promotes atrial fibrillation (AF) through a myristoylation-dependent pathway. In pathological conditions, upregulated VILIP-1 binds Ca2+, triggering myristoylation-dependent membrane anchoring. At the membrane, VILIP-1 directly increases the surface membrane expression of NCX-1 and enhances its function. The sustained Ca2+ overload combined with intracellular Na+ accumulation drives NCX-1 reverse-mode activation, exacerbating both Ca2+ influx and cytoplasmic Ca2+ accumulation. The resulting Ca2+ overload reinforces VILIP-1 membrane targeting by promoting its myristoylation, creating a self-perpetuating cycle driving NCX-1 hyperactivity and AF progression. The schematic illustration was created in Adobe Illustrator 2024
Discussion
In this study, we uncovered a novel association between VILIP-1 and AF according to data from patients and animal models with AF. We found that upregulated VILIP-1 is a crucial factor for the onset of AF and demonstrated that VILIP-1 regulates intracellular Ca2+ homeostasis by modulating the surface expression of NCX-1 through a myristoylation-dependent mechanism. This disruption in Ca2+ homeostasis leads to pathological Ca2+ leakage and electrical remodeling of atrial cardiomyocytes (DADs and APD alternans), ultimately triggering AF onset. Importantly, our study identifies two clinically available drugs targeting VILIP-1 (repaglinide) and its myristoylation (desloratadine), which can effectively restore Ca2+ homeostasis and AF susceptibility in both human and rodent AF models. These data suggest that VILIP-1 could serve as a promising therapeutic target for the prevention and treatment of AF.
NCSs are a diverse family of proteins essential for regulating Ca2+ homeostasis and signaling in various cell types. In neurons, these proteins facilitate synaptic plasticity and neurotransmitter release; in skeletal muscle, they regulate contraction and relaxation; in pancreatic cells, they modulate insulin secretion; and in skin, they suppress the invasiveness of squamous cell carcinoma cells.10,11,12,13,14 However, the roles of NCS in cardiac tissue, especially in atrial cardiomyocytes and AF, remain poorly characterized. In this study, we identified VILIP-1 as a novel contributor to atrial electrical remodeling and arrhythmogenesis through its abnormal upregulation and consequent dysfunction of NCX-1. Our findings demonstrate a primary role for VILIP-1 in regulating Ca2+ homeostasis via NCX-1, with potential secondary effects involving other key Ca2+-handling proteins, such as RyR2, L-type Ca2+ channels, and phospholamban. Further studies investigating these downstream interactions are warranted to fully elucidate the mechanisms underlying Ca2+ dysregulation in AF. Notably, several studies have reported that a shortened APD can enhance the inducibility of DADs,32,33,34 whereas others indicate that increased DAD susceptibility can also occur independently of APD shortening or even in the presence of APD prolongation, provided Ca2+ homeostasis is impaired.35,36,37,38,39 Clearly, the precise mechanisms governing intracellular Ca2+ dynamics in AF pathophysiology require additional investigation.
Our findings significantly strengthen NCX-1 as a critical player in AF. The expression, transport, and function of NCX-1 have long been known to be closely linked to cardiac arrhythmias, particularly AF.6 Dysregulation of NCX-1 has been associated with the development of DADs and abnormal APD alternans, conduction heterogeneity and facilitation of ectopic activity, both of which are implicated in the initiation and maintenance of AF.6,37,40 However, previous efforts to directly target NCX-1 for AF treatment have yielded limited success, with some inhibitors even leading to severe arrhythmias or sudden death. Here, we propose a novel Ca2+-VILIP-1-NCX-1-Ca2+ regulatory loop that plays an essential role in maintaining Ca2+ homeostasis in atrial cardiomyocytes. This feedback loop is disrupted in both human and rodent AF models, with a marked upregulation of VILIP-1 expression. Importantly, our data indicate that targeting upstream molecular signaling such as VILIP-1 may be more effective in preventing and treating AF than directly inhibiting NCX-1, positioning VILIP-1 as a critical intervention point.
The limitations of this study include the following: 1) Inherent cross-species differences: Pathophysiological disparities between humans and rodents limit the direct extrapolation of findings from rodent AF models to clinical practice. Nevertheless, despite variations in AF subtypes across species, dysregulated Ca2+ homeostasis remains a conserved mechanism driving AF pathogenesis. In this study, VILIP-1 upregulation plays a pivotal role in this core pathway by enhancing the surface expression of NCX-1, which leads to abnormal Ca2+ handling and increased arrhythmogenic susceptibility. Our functional experiments provide strong evidence supporting a causal role of VILIP-1 in promoting AF in the models used. However, further investigation is needed to confirm this causality across different AF phenotypes and clinical settings. 2) Lack of advanced preclinical models: Considering the known limitations of rodent models in accurately recapitulating human AF pathophysiology, future validation studies of therapeutic targets should incorporate large-animal models that more closely approximate human cardiac electrophysiology. 3) Limited specificity of currently available pharmacological inhibitors: While repaglinide and desloratadine were identified as potential VILIP-1 inhibitors, their target specificity remains suboptimal due to known off-target effects. This limitation undermines their potential as reliable therapeutic agents for VILIP-1 inhibition. Although our experimental results indicate that these agents effectively reduce VILIP-1-induced NCX-1 membrane expression, suppress INCX currents, attenuate Ca2+ overload, and decrease AF vulnerability in experimental models, the lack of specificity highlights a significant challenge for their clinical application. Therefore, these findings underscore the critical need for the development of novel, highly specific VILIP-1 inhibitors that can be more effectively translated into therapeutic strategies for AF.
In conclusion, our study establishes VILIP-1 as a crucial regulator of Ca2+ homeostasis in atrial cardiomyocytes, with its dysregulation serving as a key contributor to AF pathogenesis. Mechanistically, upregulated VILIP-1 enhances the surface membrane expression of NCX-1 through a myristoylation-dependent mechanism, which leads to intracellular Ca2+ leakage, subsequently driving atrial electrical remodeling, including increased APD alternans, DADs, spatial dispersion of APD, and conduction heterogeneity. These alterations promote triggered activity and create a reentrant substrate, facilitating the initiation and maintenance of AF. Targeting VILIP-1 offers a promising therapeutic strategy for AF by restoring Ca2+ homeostasis and reducing arrhythmogenic substrates.
Materials and methods
Patients and human samples
Left atria samples were collected from patients undergoing heart transplantation at Wuhan Union Hospital, Wuhan, China, who were diagnosed with either SR or AF. Informed consent was provided by all participants. This research complied with the ethical principles of the Declaration of Helsinki and received authorization from the Ethics Committee of Wuhan Union Hospital (approval number: 0858-01).
Animals
This study complies with the NIH Guide for the Care and Use of Laboratory Animals. All animal experiments received approval from the Animal Care and Use Committee of Tongji University School of Medicine (approval number: TJBB00921701). Animals in the study were randomly assigned to the different experimental groups, with the treatment allocation being blinded. All animals treated were included in the analyses. C57BL/6 mice and Sprague‒Dawley rats were purchased from Shanghai Sippr-BK Laboratory Animal Co., Ltd (China) and subsequently housed under standard specific pathogen-free conditions for the duration of the study.
Rodent AF model
The rodent AF model was set up as previously outlined, with modifications to suit our experimental requirements.41,42 The pacemaker from Ensense Biomedical Technologies (China) was connected to bipolar leads implanted in the right atrial appendage under 1% isoflurane anesthesia via mask ventilation. After a one-week post-operative recovery period, the animals were subjected to two weeks of rapid atrial pacing delivered to the right atrial appendage via the implanted pacemaker. The sham-operated control group underwent the same surgical procedures, but without delivering electrical stimulation.
Isolation of adult atrial cardiomyocytes
From adult male rats and mice, atrial cardiomyocytes were isolated acutely. In brief, animals were put under anesthesia with intraperitoneal injection of sodium pentobarbital solution, followed by a thoracotomy to expose the heart for further procedures. Following the incision of the descending aorta, the heart was quickly flushed with 15 mL of cold EDTA-Tyrode’s solution (this EDTA-Tyrode’s solution was the standard Tyrode’s solution formulation supplemented with 5 mM EDTA) injected into the ventricles. The heart underwent careful dissection and moved to a dish filled with cold EDTA-Tyrode’s solution to clear away surrounding connective and adipose tissues. Subsequently, the atrial tissues fragments were minced and rinsed twice in a low-Ca2+ solution with the following concentrations (mM): 140 NaCl, 5.4 KCl, 1.2 KH2PO4, 0.2 CaCl2, 18.5 glucose, 50 taurine, 1.0% BSA and its pH was set to 6.9 using NaOH. Next, the fragments underwent digestion for 15 min in a low-Ca2+ solution containing collagenase type II (0.8 mg/mL; Worthington, USA) while being gently agitated. The digestion was halted by transferring the supernatant to a Kraftbruhe solution with an addition of 1.0% BSA. A 100-μm filter was used to filter the cell suspensions, which were then centrifuged at 100× g for 1-2 min to collect cells. After removing the supernatant, the atrial cardiomyocytes in pellet form were resuspended in 4 mL of modified Kraftbruhe solution based on the yield. In electrophysiological experiments, acutely isolated cardiomyocytes underwent gradual recalcification by adding 1 M Ca2+ solution at 30-min intervals, until the Ca2+ concentration reached the standard extracellular level for recording.
The isolation of human atrial cardiomyocytes followed a previously described procedure, incorporating modifications.43 Atrial tissue was placed in ice-cold, Ca2+-free Tyrode’s solution supplemented with 30 mM BDM. The tissue was minced into smaller pieces and then washed three times, with the Ca2+-free Tyrode’s solution. Next, the tissue fragments were digested for 15 min in Ca2+-free Tyrode’s solution containing collagenase type I and protease type XXIV. This was followed by further digestion in Tyrode’s solution containing collagenase type I and 0.2 mM Ca2+, continuing until individual cardiomyocytes were liberated. Following centrifugation at 100 × g, the cardiomyocyte pellet was resuspended and maintained in Ca2+-free Tyrode’s solution supplemented with 0.5 mM Ca2+ (without BDM) and used in subsequent experiments.
Ca2+ imaging
In brief, isolated atrial cardiomyocytes were loaded in 1 mL Tyrode’s solution with 1 μM Cal-520 AM (AAT bioquest, USA; λex/λem = 488/500–540 nm) and 0.04% Pluronic F-127 (Sigma, USA) for 5 min. Ca2+ confocal imaging was performed with a STELLARIS 8 confocal system with a 63× objective lens. The kinetics of cytosolic Ca2+ dynamics (including sparks, waves and transients) were analyzed with the LAS X software and ImageJ 2.0 software (NIH, USA).
Single-cell RNA sequencing in isolated atrial cardiomyocytes
Single-cell RNA sequencing was performed according to a modified Smart-seq3 protocol.44 In brief, following Ca2+ imaging, individual atrial cardiomyocytes were manually picked and transferred into 0.5 μl lysis buffer under the microscope. Each single-cell lysate was then distributed into an individual well of 0.2 mL PCR 8-strip tubes pre-loaded with 2.5 μL of Smart-seq3 lysis buffer containing 0.5 U/μl RNase inhibitor (Takara, Japan), 0.1% Triton X-100, 0.5 mM dNTPs each (Thermo Fisher Scientific, USA), 0.5 µM Smart-seq3 OligodT30VN primer (5’-biotin-ACGAGCATCAGCAGCATACGAT30VN-3’) and 5% poly-ethylene glycol 8000 (Sigma-Aldrich, USA). The concentrations of dNTPs, OligodT30VN primer and poly-ethylene glycol were calculated for a total volume of 4 μl following the addition of 1 μl of reverse transcription mix as described by the Smart-seq3.44 Immediately after the transfer, the contents of each 8-strip tube were collected by centrifugation, cDNA libraries generation was performed according to established Smart-seq3 protocols published in protocols.io at https://doi.org/10.17504/protocols.io.bcq4ivyw (version 3), with 16 cycles of PCR amplification and 100 pg of amplified cDNA for Nextera tagmentation (FC-131-1096, Illumina, USA). After amplification, the tagmented libraries were pooled, cleaned with beads, and quantified with a Qubit fluorometer (Thermo Fisher Scientific, USA). Pooled libraries were sequenced with paired end 2 × 150 bp reads on an AVITI sequencer (Element Biosciences, USA). Single-cell analysis was performed using the R package (Seurat v4.4). Following standard preprocessing and clustering, normalized expression data from the relevant clusters were exported and subsequently analyzed in Monocle3 (v1.3.7) to infer pseudotemporal ordering and trajectory dynamics, largely following the developer’s recommended workflow.
Bulk RNA sequencing
All sequencing procedures, including library preparation and sequencing, were performed by Shanghai Genefund Biotechnology Co., Ltd. (Shanghai, China). In brief, human atrial tissues were processed for bulk RNA sequencing. Total RNA was extracted, and its integrity was verified using an Agilent Bioanalyzer. Sequencing libraries were then constructed from qualified RNA using a strand-specific RNA Kit (Illumina, USA), following the standard workflow provided by the manufacturer for Illumina platform sequencing. Sequencing libraries were constructed through poly-A selection, fragmentation, cDNA synthesis, and adapter ligation. The libraries were then quantified and pooled for sequencing. For read alignment, the raw sequencing data were mapped to the GRCh38.104 human reference genome using STAR aligner (v2.7.3). Following alignment, gene-level expression counts were quantified from the BAM files based on the GENCODE release 38 annotations.
Echocardiography
Cardiac function and left atrial size were assessed using the Visual Sonics Vevo 2100/Vevo F2 ultrasound system in mice and rats under anesthesia maintained with 1% isoflurane, while preserving spontaneous breathing throughout the procedure. Once a stable plane was secured, 2D B-mode imaging was utilized to obtain long-axis views of the heart, which subsequently served as a guide for positioning the M-mode cursor to acquire reproducible dimensional measurements. The LVEF was estimated from the M-mode ultrasound recordings using the Teichholz correction formula, based on measurements of the left ventricular internal diameter at end-diastole and end-systole. LA strain was measured and calculated using vendor-specific speckle-tracking software (Vevo Strain within Vevo Lab), as previously outlined.45
Optical mapping
High-resolution optical mapping was conducted following established methods.46 Briefly, adult mice or rats were anesthetized and subjected to thoracotomy. After opening the chest, surrounding connective and adipose tissues were carefully dissected away to fully expose the heart. The heart was then rapidly excised by cutting the great vessels, and immediately transferred to ice-cold Tyrode’s solution. Following establishment of Langendorff perfusion, the heart was continuously supplied with warm (35–37 °C), oxygenated Tyrode’s solution to allow for equilibration and recovery. To inhibit contractile motion, the myosin II inhibitor blebbistatin (MCE, USA) was included in the perfusate at 10 μM, thereby preventing motion artifacts. Once hemodynamic stability was achieved, the heart was loaded with Di-4-ANEPPS (10 μM, AAT Bioquest, USA), followed by a washout period. Image acquisition was performed utilizing a MiCAM ULTIMA high-speed camera (BrainVision, Japan). The camera frame rate was set to 1000 frames/second for all recordings. Data were collected and analyzed with the help of BV Workbench software, developed by SciMedia (USA). For each recording, the activation time was defined as the point corresponding to the maximum rate of fluorescence change (dF/dt).
Western blot analysis
For the isolation of membrane proteins, cells and tissues were processed using the Minute Plasma Membrane Protein Isolation Kit (Invent Biotechnologies, USA) following the instructions provided by the manufacturer. In parallel, for total protein analysis, samples were homogenized on ice using RIPA lysis buffer (Beyotime Biotechnology, China) supplemented with a protease inhibitor cocktail from Roche (USA). Using a BCA kit from Beyotime Biotechnology (China), protein concentrations were assessed, and equal amounts of protein were separated by electrophoresis on a 10% SDS‒PAGE gel before being transferred onto PVDF membranes from Millipore (USA). After blocking with QuickBlock Blocking Buffer (Beyotime Biotechnology, China), the membranes were incubated overnight at 4 °C with primary antibodies, including NCX-1 (Proteintech, USA), VILIP-1 (Proteintech, USA), β-ACTIN (Santa Cruz, USA) and GAPDH (Proteintech, USA), all diluted in the same blocking buffer. Following incubation with primary antibodies, a conjugated secondary antibody (Invitrogen, USA) was used to incubate the PVDF membranes according to antibody manufacturer’s recommendations. Immunoreactive bands were visualized by a gel imaging system (ChemiDoc Touch Gel Imaging System, Bio-Rad Laboratories, USA).
Capillary-based immunoassay
Capillary-based immunoassay was performed with a Jess automated protein analysis system (ProteinSimple, CA). According to the manufacturer’s recommendations, primary antibodies were diluted to detect the following proteins: NCX-1 (1:20, Proteintech, USA) and VILIP-1 (1:50, Proteintech, USA). The HRP-conjugated secondary antibodies were employed to detect the antibody targets. The total protein expression was determined using the Total Protein Detection Module (DM-TP01, ProteinSimple, USA). Digital images underwent analysis with Compass for SW software (V6.1.0, Protein Simple). The protein expression levels were determined according to the software’s automated analysis protocol, and the resulting quantitative values were compiled for statistical analysis.
RNA synthesis by in vitro transcription
The mRNA of VILIP, V5-VILIP-1-miniTurbo, and V5-miniTurbo were generated by in vitro transcription via the GSPure®T7 Thermostable and High Yield RNA Synthesis Kit (Geneseed Biotech. Co., Ltd, China) following the instructions provided by the manufacturer. The standard procedure begins with a linear DNA template that includes the T7 promoter. This is particularly important for co-transcriptional capping using the synthetic cap analog EasyCapTM GAG m7G(5’)ppp(5’)(2’OMeA)pG (Jiangsu Synthgene Biotechnology Co., Ltd, China), which is used at a 4:1 ratio relative to guanosine triphosphate. The transcripts were purified by RNA Clean Beads (Vazyme, China). To verify the presence and integrity of the 3’ Poly(A) tail, an aliquot of the purified RNA was treated with RNase T1 (Supplementary Fig. 14). The quality of the RNA was analyzed on a 2% agarose gel, and the concentration was determined by using a Nanodrop (NanoDrop Technologies, USA).
Proximity labeling and mass spectrometry
We performed proximity-dependent biotinylation assays as previously described.24,25 Briefly, the mRNA of V5-VILIP-1-miniTurbo and V5-miniTurbo were transfected into neonatal rat cardiomyocytes (NRCMs) by jetMESSENGER (Polyplus, France). To minimize endogenous biotin interference, cultures were maintained in dialyzed FBS-containing medium. After 16 h post-transfection, the NRCMs were treated with biotin (500 μM) for 15 min. After biotin treatment, the cells were lysed, and the biotinylated proteins were enriched using streptavidin beads. The enriched proteins were then subjected to mass spectrometry analysis. Identification and quantification of the targeted proteins was accomplished via a DIA mass spectrometry approach. The raw mass spectrometry data were processed with MaxQuant software (version 2.0.3.0). Identification of VILIP-1 target proteins were performed according to previously described methods with modification.24,25,47 Proteomic statistical analyses were performed using the R (v4.4) with the maSigPro and DEP packages.
Immunofluorescence
For immunofluorescence analysis, the atrial samples were first fixed by immersion in 4% paraformaldehyde for 24 h at 4 °C, followed by routine dehydration and embedded in paraffin. Consecutive sections (6 μm thick) were cut, cleared in xylene to remove paraffin and then gradually rehydrated through a descending ethanol series. To expose antigenic epitopes, the sections underwent heat-induced antigen retrieval in citric acid repair solution (Beyotime Biotechnology, China). After cooling and washing steps, these sections were blocked using QuickBlock Blocking Buffer (Beyotime Biotechnology, China) for a minimum of 1 h at room temperature. Next, an overnight incubation at 4 °C was performed with primary antibodies against NCX-1 (Invitrogen, USA), VILIP-1 (ABclonal, China) and ANP (Santa Cruz, USA). After washing thrice with PBST (0.1% Tween-20 in PBS), the sections were incubated for 1 h at room temperature in the dark with the appropriate fluorophore-conjugated secondary antibody (Invitrogen, USA) diluted in PBST. Following a final wash, coverslips were mounted with ProLong Glass Antifade Mountant containing DAPI (Invitrogen, USA) to preserve fluorescence and visualize nuclei. Confocal imaging was then performed on a Leica STELLARIS 8 system (Leica, Germany).
Atrial cardiomyocytes, for immunofluorescence analysis, were first fixed with fresh 4% paraformaldehyde (13 min) at room temperature. This was followed by two 5-min washes with PBS. Subsequently, the fixed cells were permeabilized with 0.1% Triton X-100-containing PBST for 10 min, blocked with QuickBlock Blocking Buffer (Beyotime Biotechnology, China) for a minimum of 1 h, and incubated with primary antibodies overnight at 4 °C. After the overnight incubation, the cells underwent three washes (5 min for per wash) with PBST. They were subsequently probed with the relevant fluorophore-conjugated secondary antibody (Invitrogen, USA) for 1 h. Following three additional PBST washes, confocal imaging was performed on a Leica STELLARIS 8 system (Leica, Germany) to capture high-resolution optical sections of the stained cells.
AAV generation and injection
The AAV9-mANF Promoter-VILIP-1-IRES-mCherry-pA adeno-associated virus and the control AAV9-mANF Promoter-mCherry-pA adeno-associated virus were custom-designed and synthesized by Taitool Biotech (China) following standard protocols. The virus was then administered via tail vein injection to adult male mice (6–8 weeks old) at a dose of 4 × 1011 viral particles/mouse. All relevant in vivo and in vitro experiments started at least 4 weeks after the virus injection.
Isolation of NRCMs and transfection
Primary NRCMs were isolated from 1- to 3-day-old neonatal rats. Briefly, the heart was excised, minced into small pieces (~1–3 mm2) and subjected to enzymatic digestion. The enzymatic digestion was performed at 37 °C in Ca2+-free PBS (Gibco, USA) containing an enzyme cocktail of 0.125 mg/mL trypsin (Gibco, USA), 0.1 mg/mL collagenase type IV (Sigma-Aldrich, USA) and 10 μg/mL DNase II (Sigma-Aldrich, USA). The tissue fragments were subjected to continuous stirring, with the supernatant being collected at 5-min intervals into PBS containing 10% FBS to neutralize protease activity. The digestion cycle was performed sequentially approximately 8–10 times until complete tissue dissociation. Upon completion, the pooled supernatants were centrifuged at 800 × g for 10 min. The resulting cell pellet was resuspended in DMEM (Gibco, USA) supplemented with 10% FBS and 100 μmol/L 5-bromo-2’-deoxyuridine (Sigma-Aldrich, USA). The cell suspension was then filtered through a 100-μm cell strainer (BD, USA) and subjected to differential attachment for 2 h to remove fibroblasts. The non-adherent NRCMs were collected and plated onto gelatin-coated dishes at a suitable density to facilitate adhesion. After 24 h of culture, the medium was replaced with a low-serum maintenance medium. For transfection of overexpression plasmids into NRCMs, Lipofectamine 3000 Reagent (Invitrogen, USA) was used following the protocol recommended by the manufacturer.
Thermal shift assay (TSA)
TSA experiments were performed as described in a previous study.48 Briefly, atrial tissues from rats were homogenized in RIPA lysis buffer with added protease and phosphatase inhibitors (Roche, USA), followed by a centrifugation step. The collected supernatant was equally divided for treatment with either repaglinide (10 μM) or solvent control. After a 1 h incubation, each reaction mixture was distributed into multiple aliquots. Each aliquot was then heated at a different designated temperature (ranging from 37 °C to 81 °C in 4 °C increments) for 3 min to assess protein thermal stability. Thereafter, all heated lysates were spun at 13,000 × g for 20 min under refrigeration and the resultant soluble fractions were collected and analyzed by immunoblotting against VILIP-1.
Immunoprecipitation
Immunoprecipitation assays were conducted according to an established protocol.49 In brief, cells or tissues were lysed using a total protein extraction kit (Abbkine, China). The lysates were spun at 13,000 × g for 15 min under refrigeration (4 °C) to clear out the cellular debris. For the immunoprecipitation, we added 1–2 µg of the target-specific antibody to the cleared supernatants and incubated the mixture overnight at 4 °C with gentle rotation to allow for immune complex formation. Immunocomplexes were then captured by adding 40 µL of pre-washed Protein A/G magnetic beads (Invitrogen, USA) and incubating for an additional 2 h at 4 °C under constant rotation. After the beads were washed four times with cold lysis buffer, the bound immunocomplexes were eluted from the beads by boiling in 4× loading buffer. For detection, the eluted samples were fractionated by electrophoresis on a 10% SDS-PAGE gel under denaturing conditions. Subsequently, the separated proteins were electrophoretically transferred to PVDF membranes and probed with specific antibodies for immunoblotting.
Pull-down assay
GST pull-down assays were conducted following the protocol of the Pierce GST Protein Interaction Pull-Down Kit (Invitrogen, USA). In brief, whole-cell lysates from transfected 293 cells were incubated with glutathione sepharose beads bound to either bacterially expressed and purified GST (control) or VILIP-1-GST fusion proteins. After washing steps, the proteins bound to the beads were eluted and then denatured by boiling in 4× loading buffer. The denatured proteins were separated by a 10% SDS-PAGE gel, transferred to PVDF membranes, and subsequently probed with indicated specific antibodies for Western blot analysis. Specifically, interactions were detected using rabbit anti-HA antibody (Proteintech, USA) and rabbit anti-GST antibody (Proteintech, USA) for detection.
Surface ECG recording
Surface ECG signals were continuously recorded from anesthetized animals. The recordings were performed using a Powerlab system (ADInstruments, USA) equipped with a BioAmp (ADInstruments, USA). Subcutaneous needle electrodes were placed to acquire a standard limb lead II configuration. Following signal acquisition, the ECG data were analyzed and quantified subsequently with ECG analysis module in LabChart8 (ADInstruments, USA).
Patch-clamp electrophysiology
To record membrane potentials and ionic currents, atrial cardiomyocytes were studied in the whole-cell patch-clamp configuration according to established protocols.50,51 Recordings were made using borosilicate glass electrodes with resistances of 2–5 MΩ when filled with the pipette solution. To record action potentials (APs), the pipette was filled with a pipette solution containing (in mM: K-glutamate 130; MgCl2 1; NaCl 5; KCl 15; Mg ATP 5; CaCl2 1; EGTA 5; HEPES 10; the pH was set to 7.20 with KOH). The external bath solution was the Tyrode’s solution at room temperature. To record delayed afterdepolarizations (DADs), the APs were stimulated with 900 pA 5 ms pulses at various pacing frequencies (1, 2, 4 and 5 Hz) for 10 sec, and then the pulse stimulation was stopped to record DADs. DADs were identified as depolarizations with an amplitude >2 mV relative to the diastolic membrane potential.
The Na+/Ca2+ exchange current (INCX) was elicited using a ramp voltage-clamp protocol.52 INCX was measured with the internal pipette solution containing (in mM: NaCl 15; EGTA 42; CaCl2 29; MgCl2 13; aspartic acid 42; tetraethylammonium chloride (TEA-Cl, a potassium channel blocker) 20; HEPES 5; and Na2ATP 5; pH adjusted to 7.2 with tetraethylammonium hydroxide. The external bath solution contained (in mM: NaCl 140; MgCl2 2; HEPES 5; glucose 10; CaCl2 1.8; Ouabain 0.02; BaCl2 1; CsCl 2 and verapamil 0.001; the pH was set to 7.4 with NaOH). The INCX blocker SEA0400 (1 µmol/L) was added to the extracellular bath solution at the end of the experiment to obtain the INCX current. The data were recorded using an Axopatch 700B amplifier (Axon Instruments, USA).
AF induction
The method for AF induction in mice and rats followed an established protocol.37 Briefly, animals were placed under 1% isoflurane anesthesia via mask ventilation, with body temperature maintained at 37 °C. A 1.1 F (mice) or 1.6 F (rats) octapolar electrophysiologic catheter was positioned in the right atrium via jugular vein access. To assess AF susceptibility, programmed electrical stimulation was delivered via an external stimulator (Multi-Channel Systems, Germany). Stimulation consisted of 5-sec burst pacing. The initial cycle length was 40 ms and was progressively reduced in 2-ms increments in subsequent bursts until a cycle length of 20 ms was reached. Following a 5-min stabilization period, the burst series was repeated 3 times. An AF episode was characterized by the immediate appearance of rapid, disorganized atrial electrograms associated with an irregular ventricular rhythm lasting for at least 1 s.
Molecular dynamics (MD) simulations and docking
The MD simulations of protein-protein complexes were conducted using Desmond software (Schrödinger, USA). Before simulation initiation, each complex system was solvated in an SPC cubic water box with periodic boundary conditions, ensuring at least 10 Å of space between the solute and box boundaries, using the System Builder module to form the complex-solvent system. The appropriate ions (Na+ or Cl-) ions were added to achieve electrical neutrality. The simulation protocol proceeded through the Molecular Dynamics module. The final 100 ns NPT simulation generated 10,000 trajectory frames (1 frame/10 ps). The simulation parameters were set at 300 K and 1.01325 bar during equilibration, with energy recorded every 1.0 ps and atomic coordinates sampled every 20 ps. Three-dimensional conformations were extracted and visualized via PyMOL.
For molecular docking, active drug components from PubChem and target proteins from the PDB database were obtained. Proteins were preprocessed in PyMOL. AutoDock Tools were used to add hydrogens and assign charges to the receptor and ligand, generating PDBQT files. HEX performed global blind docking for ligand-receptor pairs, evaluating the stability of the complexes based on binding affinity.
Statistical analysis
For the comparison of two groups, statistical significance was determined using unpaired two-tailed Student’s t-tests or nonparametric tests, as appropriate. For comparisons across more than two groups, one-way analysis of variance (ANOVA) was applied. Associations between categorical variables were examined using Fisher’s exact test. Data are presented as the mean ± SEM, and the threshold for statistical significance was set at P < 0.05. All analyses were conducted using GraphPad Prism 8.
Data availability
The raw sequence data generated in this study have been deposited in the China National GeneBank DataBase (https://db.cngb.org/cnsa/) of CNGBdb under the accession number CNP0008659.
References
Elliott, A. D., Middeldorp, M. E., Van Gelder, I. C., Albert, C. M. & Sanders, P. Epidemiology and modifiable risk factors for atrial fibrillation. Nat. Rev. Cardiol. 20, 404–417 (2023).
Lu, Y. et al. Non-traditional risk factors for atrial fibrillation: epidemiology, mechanisms, and strategies. Eur. Heart J. 00, 1–21 (2024).
Nattel, S. & Harada, M. Atrial remodeling and atrial fibrillation: recent advances and translational perspectives. J. Am. Coll. Cardiol. 63, 2335–2345 (2014).
Dridi, H. et al. Intracellular calcium leak in heart failure and atrial fibrillation: a unifying mechanism and therapeutic target. Nat. Rev. Cardiol. 17, 732–747 (2020).
Li, N. et al. Ryanodine receptor-mediated calcium leak drives progressive development of an atrial fibrillation substrate in a transgenic mouse model. Circulation 129, 1276–1285 (2014).
Voigt, N. et al. Enhanced sarcoplasmic reticulum Ca2+ leak and increased Na+-Ca2+ exchanger function underlie delayed afterdepolarizations in patients with chronic atrial fibrillation. Circulation 125, 2059–2070 (2012).
Bögeholz, N. et al. Suppression of Early and Late Afterdepolarizations by Heterozygous Knockout of the Na+/Ca2+ Exchanger in a Murine Model. Circ. Arrhythm Electrophysiol. 8, 1210–1218 (2015).
Schotten, U. et al. Blockade of atrial-specific K+-currents increases atrial but not ventricular contractility by enhancing reverse mode Na+/Ca2+-exchange. Cardiovasc. Res. 73, 37–47 (2007).
Sossalla, S. et al. Altered Na+ currents in atrial fibrillation effects of ranolazine on arrhythmias and contractility in human atrial myocardium. J. Am. Coll. Cardiol. 55, 2330–2342 (2010).
Burgoyne, R. D. Neuronal calcium sensor proteins: generating diversity in neuronal Ca2+ signalling. Nat. Rev. Neurosci. 8, 182–193 (2007).
McCue, H. V., Haynes, L. P. & Burgoyne, R. D. The diversity of calcium sensor proteins in the regulation of neuronal function. Cold Spring Harb. Perspect. Biol. 2, a004085 (2010).
Dai, F. F. et al. The neuronal Ca2+ sensor protein visinin-like protein-1 is expressed in pancreatic islets and regulates insulin secretion. J. Biol. Chem. 281, 21942–21953 (2006).
Gromada, J. et al. Neuronal calcium sensor-1 potentiates glucose-dependent exocytosis in pancreatic beta cells through activation of phosphatidylinositol 4-kinase beta. Proc. Natl. Acad. Sci. USA 102, 10303–10308 (2005).
Mahloogi, H. et al. Overexpression of the calcium sensor visinin-like protein-1 leads to a cAMP-mediated decrease of in vivo and in vitro growth and invasiveness of squamous cell carcinoma cells. Cancer Res 63, 4997–5004 (2003).
Li, C., Pan, W., Braunewell, K. H. & Ames, J. B. Structural analysis of Mg2+ and Ca2+ binding, myristoylation, and dimerization of the neuronal calcium sensor and visinin-like protein 1 (VILIP-1). J. Biol. Chem. 286, 6354–6366 (2011).
Lipovsky, C. E. et al. Chamber-specific transcriptional responses in atrial fibrillation. JCI insight 5, e135319 (2020).
Thomsen, M. B. et al. Accessory subunit KChIP2 modulates the cardiac L-type calcium current. Circ. Res. 104, 1382–1389 (2009).
Panama, B. K. et al. Nuclear factor kappaB downregulates the transient outward potassium current Ito,f through control of KChIP2 expression. Circ. Res. 108, 537–543 (2011).
Goyal, P. et al. Post-operative atrial fibrillation and risk of heart failure hospitalization. Eur. Heart J. 43, 2971–2980 (2022).
Nattel, S., Heijman, J., Zhou, L. & Dobrev, D. Molecular basis of atrial fibrillation pathophysiology and therapy: a translational perspective. Circ. Res. 127, 51–72 (2020).
Kanaporis, G. & Blatter, L. A. The mechanisms of calcium cycling and action potential dynamics in cardiac alternans. Circ. Res. 116, 846–856 (2015).
Andrade, J., Khairy, P., Dobrev, D. & Nattel, S. The clinical profile and pathophysiology of atrial fibrillation: relationships among clinical features, epidemiology, and mechanisms. Circ. Res. 114, 1453–1468 (2014).
Morciano, G. et al. Calcium dysregulation in heart diseases: targeting calcium channels to achieve a correct calcium homeostasis. Pharmacol. Res. 177, 106119 (2022).
Cho, K. F. et al. Proximity labeling in mammalian cells with TurboID and split-TurboID. Nat. Protoc. 15, 3971–3999 (2020).
Branon, T. C. et al. Efficient proximity labeling in living cells and organisms with TurboID. Nat. Biotechnol. 36, 880–887 (2018).
Wan, X. et al. New experimental evidence for mechanism of arrhythmogenic membrane potential alternans based on balance of electrogenic INCX/ICa currents. Heart Rhythm 9, 1698–1705 (2012).
Primessnig, U. et al. Novel pathomechanisms of cardiomyocyte dysfunction in a model of heart failure with preserved ejection fraction. Eur. J. Heart Fail. 18, 987–997 (2016).
Dong, Y. et al. Structural insight into the allosteric inhibition of human sodium-calcium exchanger NCX1 by XIP and SEA0400. EMBO J. 43, 14–31 (2024).
Spilker, C., Dresbach, T. & Braunewell, K.-H. Reversible translocation and activity-dependent localization of the calcium-myristoyl switch protein VILIP-1 to different membrane compartments in living hippocampal neurons. J. Neurosci. 22, 7331–7339 (2002).
Tan, X.-P. et al. Blockade of NMT1 enzymatic activity inhibits N-myristoylation of VILIP3 protein and suppresses liver cancer progression. Signal Transduct. Target. Ther. 8, 14 (2023).
Okada, M. et al. Neuronal calcium sensor proteins are direct targets of the insulinotropic agent repaglinide. Biochem J 375, 87–97 (2003).
Cao, Z. Z. et al. Barbaloin inhibits ventricular arrhythmias in rabbits by modulating voltage-gated ion channels. Acta Pharmacol. Sin. 39, 357–370 (2018).
Walker, M. A., Gurev, V., Rice, J. J., Greenstein, J. L. & Winslow, R. L. Estimating the probabilities of rare arrhythmic events in multiscale computational models of cardiac cells and tissue. PLoS Comput. Biol. 13, e1005783 (2017).
Fang, L. H. et al. Calcium-mediated DAD in membrane potentials and triggered activity in atrial myocytes of ETV1f/fMyHCCre/+ mice. J. Cell Mol. Med. 28, e70005 (2024).
King, J. H. et al. Atrial arrhythmia, triggering events and conduction abnormalities in isolated murine RyR2-P2328S hearts. Acta Physiol. 207, 308–323 (2013).
Roy, A. et al. Early atrial remodeling drives arrhythmia in fabry disease. Circ. Arrhythm. Electrophysiol. 18, e013352 (2025).
Wang, X. et al. Downregulation of FKBP5 promotes atrial arrhythmogenesis. Circ. Res. 133, e1–e16 (2023).
Bögeholz, N. et al. Triggered activity in atrial myocytes is influenced by Na+/Ca2+ exchanger activity in genetically altered mice. J Mol Cell Cardiol 101, 106–115 (2016).
Bers, D. M., Pogwizd, S. M. & Schlotthauer, K. Upregulated Na+/Ca2+ exchange is involved in both contractile dysfunction and arrhythmogenesis in heart failure. Basic Res Cardiol 97, I36–I42 (2002).
Fischer, T. H. et al. Late INa increases diastolic SR-Ca2+-leak in atrial myocardium by activating PKA and CaMKII. Cardiovasc. Res. 107, 184–196 (2015).
Yao, C. et al. Enhanced cardiomyocyte NLRP3 inflammasome signaling promotes atrial fibrillation. Circulation 138, 2227–2242 (2018).
Mulla, W. et al. Rapid atrial pacing promotes atrial fibrillation substrate in unanesthetized instrumented rats. Front. Physiol. 10, 1218 (2019).
Trum, M. et al. Empagliflozin inhibits increased Na influx in atrial cardiomyocytes of patients with HFpEF. Cardiovasc. Res. 120, 999–1010 (2024).
Hagemann-Jensen, M. et al. Single-cell RNA counting at allele and isoform resolution using Smart-seq3. Nat. Biotechnol. 38, 708–714 (2020).
Zhang, M. J. et al. Atrial myopathy quantified by speckle-tracking echocardiography in mice. Circ. Cardiovasc. Imaging 16, e015735 (2023).
Tsai, W.-C. et al. Complex arrhythmia syndrome in a knock-in mouse model carrier of the N98S Calm1 mutation. Circulation 142, 1937–1955 (2020).
Zhong, X. et al. A proximity proteomics pipeline with improved reproducibility and throughput. Mol. Syst. Biol. 20, 952–971 (2024).
Jafari, R. et al. The cellular thermal shift assay for evaluating drug target interactions in cells. Nat Protoc 9, 2100–2122 (2014).
Zhao, H. et al. The Cep63 paralogue Deup1 enables massive de novo centriole biogenesis for vertebrate multiciliogenesis. Nat Cell Biol 15, 1434–1444 (2013).
Wang, Y. et al. CIB2 is a novel endogenous repressor of atrial remodeling. Circulation 147, 1758–1776 (2023).
Li, D. et al. Spexin diminishes atrial fibrillation vulnerability by acting on galanin Receptor 2. Circulation 150, 111–127 (2024).
Ren, A.-J. et al. ZBTB20 regulates SERCA2a activity and myocardial contractility through phospholamban. Circ. Res. 134, 252–265 (2024).
Acknowledgements
The research leading to these results received funding through grants from the National Natural Science Foundation of China (82088101, 82222008, 82070338, 82470330, 82500397 and 82400385), the Key Research Center Construction Project of Shanghai (2022ZZ01008), the Noncommunicable Chronic Diseases–National Science and Technology Major Project (2024ZD0521503), National Key Clinical Specialty, the Shuguang Program of Shanghai Education Development Foundation, the Shanghai Tongji University Education Development Foundation and the China Postdoctoral Science Foundation under Grant Number 2025T180588. Prof. Y.-H.C. is a fellow at the Collaborative Innovation Center for Cardiovascular Disease Translational Medicine, Nanjing Medical University.
Author information
Authors and Affiliations
Contributions
The authors contributed as follows: K.X., G.W., D.L., B.S., and Z.C. designed and conducted the experiments. Q.Z., X.Z., Y.D., X.Z., Y.Y., and H.X. carried out the experimental procedures and analyzed the resulting data. Y.L., D.L., L.W., and B.Z. analyzed the resulting data and revised the manuscript. N.D. and D.X. reviewed and revised the manuscript. Y.-H.C. conceived and designed the study, supervised and funded the projects, and wrote the manuscript. All authors reviewed and approved the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Xiong, K., Wang, G., Li, D. et al. Visinin-like protein 1 disrupts calcium homeostasis and promotes atrial fibrillation in human and rodent models. Sig Transduct Target Ther 11, 105 (2026). https://doi.org/10.1038/s41392-026-02615-6
Received:
Revised:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41392-026-02615-6
