
Abstract
Although there are several genome-wide association studies available which highlight genetic variants associated with Alzheimer’s disease (AD), often the X chromosome is excluded from the analysis. We conducted an X-chromosome-wide association study (XWAS) in three independent studies with a pathologically confirmed phenotype (total 1970 cases and 1113 controls). The XWAS was performed in males and females separately, and these results were then meta-analysed. Four suggestively associated genes were identified which may be of potential interest for further study in AD, these are DDX53 (rs12006935, OR = 0.52, p = 6.9e-05), IL1RAPL1 (rs6628450, OR = 0.36, p = 4.2e-05; rs137983810, OR = 0.52, p = 0.0003), TBX22 (rs5913102, OR = 0.74, p = 0.0003) and SH3BGRL (rs186553004, OR = 0.35, p = 0.0005; rs113157993, OR = 0.52, p = 0.0003), which replicate across at least two studies. The SNP rs5913102 in TBX22 achieves chromosome-wide significance in meta-analysed data. DDX53 shows highest expression in astrocytes, IL1RAPL1 is most highly expressed in oligodendrocytes and neurons and SH3BGRL is most highly expressed in microglia. We have also identified SNPs in the NXF5 gene at chromosome-wide significance in females (rs5944989, OR = 0.62, p = 1.1e-05) but not in males (p = 0.83). The discovery of relevant AD associated genes on the X chromosome may identify AD risk differences and similarities based on sex and lead to the development of sex-stratified therapeutics.
Similar content being viewed by others
Introduction
Alzheimer’s Disease (AD) is the most common form of dementia accounting for between 60 and 80% of dementia cases [1]. The prevalence of AD in women is higher than that in men [1, 2]. This may be because women tend to live longer than men [3, 4]. However, some studies have suggested that women over 80 years may be more likely to have AD than men of the same age [5]. The effect of sex has a varying impact on AD over the course of the disease [6]. The duration of ovarian hormone exposure protects against dementia [7], i.e., shorter oestrogen exposure in women was associated with higher dementia risk in the UK Biobank [8]. A number of genetic loci have sex-specific effects on AD [6], for example, the risk of AD associated with a given APOE genotype changes with sex [9]. The presentation of disease can differ by the sex of the individual with AD, highlighting the impact of sex on disease heterogeneity. Men and women demonstrate different cognitive and psychiatric symptoms. After diagnosis of either MCI or AD, women show faster cognitive decline. For those with mild cognitive impairment (MCI), brain atrophy is faster in women compared to men [10]. Females have been shown to have a higher prevalence and severity of neuropsychiatric symptoms associated with AD, and males have been shown to have more severe apathy [11].
Although there are several genome-wide association studies (GWAS) available which highlight genetic variants associated with AD, often the X chromosome is excluded from the analysis. The X chromosome is often excluded because of analytical challenges caused by unique features such as transcriptional silencing of one allele in females, hemizygosity in males and recombination patterns [12]. A PubMed search of chromosome X specific association analyses and AD from 2000 to present day identified only three relevant manuscripts. The first is a method to estimate risk on the X-chromosome but does not use X chromosome data directly [13]. The second investigated the number of single nucleotide variants on the X chromosome and compared this number between AD cases and controls, highlighting two genes UBE2NL and ATXN3L [14]. But only one study found have performed a chromosome X-wide association study (XWAS) for AD [15], but no associations reached genome-wide significance. A recent study investigating X chromosome gene expression found that several genes (including GRIA3, GPRASP2, and GRIPAP1) were associated with slower cognitive decline in women but not men. In contrast, X chromosome gene expression, like UBL4A, which encodes a protein folding factor and sorts proteins to the proteasome or to the endoplasmic reticulum, is associated with neuropathological tau burden in men but not women [16].
The aim of this study is to highlight single nucleotide polymorphisms (SNPs) and the most proximal genes to XWAS identified SNPs which are associated with AD risk on the X chromosome. To investigate potential sex differences in AD highlighted by genetics, we performed an XWAS (a genome-wide like association study focused on the X-chromosome) of AD risk by meta-analysing results from sex-stratified analyses. We conducted this in three collections of samples; (i) KRONOS/Tgen data which contains 994 AD cases and 572 controls, (ii) Brains for Dementia Research (BDR) data which contains 356 AD cases and 164 controls, and (iii) Religious Orders Study/Memory and Aging Project (ROSMAP) + Mount Sinai Brain Bank (MSBB) + Mayo Clinic Brain Bank (MAYO) data which contains 702 AD cases and 486 controls. The AD diagnosis in these data was pathologically confirmed. We used these studies to determine AD associated SNPs which replicate in at least two studies. We then meta-analysed the results with the view to increase power. In addition, the XWAS results for each study were used to perform a gene-based analysis to better identify associated genes with AD risk accounting for multiple, independent associations in the gene.
Materials and methods
Data description
The KRONOS/Tgen dataset is obtained from 21 National Alzheimer’s Coordinating Center (NACC) brain banks and from the Miami Brain Bank as previously described [17,18,19,20]. The criteria for inclusion were: self-defined ethnicity of European descent, neuropathologically confirmed AD or no neuropathology present, and age of death greater than 65. Neuropathological diagnosis was defined by board-certified neuropathologists according to the standard NACC protocols [21]. Samples derived from subjects with a clinical history of stroke, cerebrovascular disease, Lewy body dementia, or comorbidity with any other known neurological disease were excluded. AD or control neuropathology was confirmed by plaque and tangle assessment with 45% of the entire series undergoing Braak staging [22]. The cohort consists of 912 AD cases and 454 controls. Samples were de-identified and the study met human studies institutional review board and HIPPA regulations. This work is declared not human-subjects research and is IRB exempt under regulation 45 CFR 46. This data was imputed using the Michigan Imputation Server [23] using the TOPMed panel [24], and SNPs with an INFO score less than 0.7 were removed.
The Brains for Dementia Research (BDR, brainsfordementiaresearch.org.uk) data [25, 26] is a longitudinal cohort of dementia samples and controls. Currently there are approximately 1200 DNA samples from brain tissue or blood and this is expected to increase to 3200 samples with genetic information. BDR is a world-class brain tissue resource supported by the Alzheimer’s Society and Alzheimer’s Research UK establishing a network of brain banks in England and Wales. In addition, other data is collected related to cognition, general health, and lifestyle every 1–5 years. BDR data was imputed on the Michigan Imputation Server using Minimac4 pipeline and the TOPMed reference panel [24] is available through the Dementias Platform UK (DPUK, https://portal.dementiasplatform.uk). The genotyped cohort includes 354 cases confirmed with AD as the primary dementia (age at onset >65 years) and 163 cognitively normal controls without additional neuropathology; all diagnoses were neuropathologically confirmed [15].
We harmonised the Religious Orders Study/Memory and Aging Project [27] (ROSMAP), Mount Sinai Brain Bank (MSBB) and Mayo Clinic Brain Bank (MAYO) whole-genome sequenced data into one cohort which we then analysed together. Datasets were downloaded from the AMP-AD portal via the Synapse platform and https://www.radc.rush.edu. ROSMAP is a longitudinal study investigating AD and ageing [28, 29], MSBB has gene expression, genetic variant, neuropathological and proteomic data for brain specimens and MAYO is a cohort containing genetic, neuropathological, biochemistry and cell biology data. Quality control analysis was carried out in the combined data, as described in [30]. AD cases are defined using a subject’s clinical definition for AD and Braak score of 5 or 6 and controls are defined as those without a clinical AD diagnosis and Braak score less than or equal to 4. This sample contains 1188 individuals: 702 AD cases and 486 controls.
The demographic data for all cohorts is seen in Table 1. The age in cases and controls are comparable between KRONOS/Tgen, BDR and ROSMAP/MAYO/MSBB.
Quality control
In all data sets, the chromosome X data was QC’ed in males and females separately. The amount of missingness in individuals and SNPs were checked, but no missingness was found. SNPs out of Hardy-Weinberg Equilibrium (p < 1e-6), tested in females [31], were removed and SNPs with minor allele frequency (MAF) < 1% were also removed. In KRONOS/Tgen 225,873 and 227,323 SNPs, in BDR 228,716 and 230,052 SNPs and in ROSMAP/MAYO/MSBB 167,018 and 169,794 SNPs were retained, in males and females respectively. SNPs in all datasets are genome build 38.
Chromosome X-wide Association Studies (XWAS)
The XWAS was carried out in males and females separately in Plink v1.9 [32, 33] using option --xchr-model 2. The models were adjusted for age and principal components (PCs); 5 PCs were used for KRONOS/Tgen and 10 PCs were used in BDR and ROSMAP/MAYO/MSBB. The number of PCs necessary for adjustment was determined from visual inspection of PC plots.
The XWAS from males and females were meta-analysed together using GWAMA [34] which also reports differentiation and heterogeneity between results in males and females. These results were represented using a Manhattan plot using the manhattan() function in R [35]. An FDR multiple testing correction was applied to identify significantly associated SNPs.
Meta-analysis of AD XWAS
The XWAS were meta-analysed together using METAL [36], results in males and females were meta-analysed separately and then GWAMA was used to join results in males and females. These results were represented using a Manhattan plot using the manhattan() function in R [35]. An FDR multiple testing correction was applied to identify significantly associated SNPs.
Gene-based analysis
A gene-based analysis of the XWAS was carried out in MAGMA v.1.08 [37], SNPs were assigned to genes based on gene locations from the NCBI site using a window of 35 kb upstream and 10 kb downstream and the original data was used to estimate linkage disequilibrium (LD) between SNPs. The mean-chi2 approach was used, which averages the effect of SNPs in the gene. The KRONOS/Tgen and BDR data annotates to 800 genes and ROSMAP/MAYO/MSBB annotates to 665 genes. An FDR multiple testing correction was applied to identify significantly associated genes.
Using expression data to gain insights into genes of interest
To gain insights into how these chromosome X putative risk genes may contribute to AD, we searched a series of publicly open datasets, including our own, containing expression data for these genes from bulk and single-cell RNA-seq datasets from human and mouse [20, 38,39,40,41,42,43,44,45,46,47,48,49]. We have also examined RNA samples of AD mouse models [50,51,52]. STRING database (string-db.org) was used to assess Protein-Protein Interaction Networks for the identified putative genes. Ingenuity analysis (digitalinsights.qiagen.com) was performed with candidate genes across all mammalian species for tissues and cell types curated in Ingenuity.
Results
XWAS results
The XWAS results in the KRONOS/Tgen data, with males and females meta-analysed together are presented in Supplementary Fig. 1. There are no SNPs with association above the chromosome-wide significant threshold, but there are several peaks which reach suggestive significance (p < 1.1e-03). The GWAMA software provides a p value for a sex differentiated effect. Of the top SNPs presented in Supplementary Table 1, rs5910591 has a differential effect between males and females (p = 3.2e-05), the effect is driven by females (OR = 0.48, p = 2.4e-05, Ref/Alt Allele=G/A, MAF = 0.20), and there is no significant effect in males (0.82, p = 0.10, Ref/Alt Allele= G/A, MAF = 0.21).
The Manhattan plot of the BDR data with males and females combined is seen in Supplementary Fig. 2. Similarly, to the KRONOS/Tgen data, no SNPs reach chromosome-wide significance but a number reach suggestive significance (1.1e-03). The top SNPs from these peaks can be seen in Supplementary Table 2. The top SNPs from BDR are different to those in KRONOS/Tgen but rs186553004 and rs5913102 both replicate (OR = 0.39, p = 0.031, Ref/Alt Allele=G/C, MAF = 0.02; OR = 0.69, p = 0.009, Ref/Alt Allele=C/T, MAF = 0.18 respectively).
The ROSMAP/MAYO/MSBB XWAS results in males and females combined is seen in Supplementary Fig. 3. There are several suggestive SNPs (1.4e-03) but none are chromosome-wide significant, the top SNPs from these peaks are seen in Supplementary Table 3. The ROSMAP/MAYO/MSBB XWAS identifies another set of SNPs associated with AD, however, the SH3BGRL gene is identified by both ROSMAP/MAYO/MSBB and KRONOS/Tgen and IL1RAPL1 is highlighted by both ROSMAP/MAYO/MSBB and BDR. In addition to these replicated genes, SNPs rs5913102 and rs12006935 identified previously are replicated in ROSMAP (OR = 0.78, p = 0.008, Ref/Alt Allele=C/T, MAF = 0.19; OR = 0.79, p = 0.028, Ref/Alt Allele=A/C, MAF = 0.23 respectively).
Meta-analysis
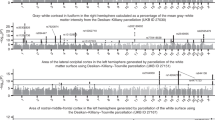
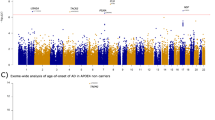
Since there was apparent overlap between results, we meta-analysed all three cohorts together. The meta-analysis produced results for 264,793 SNPs. The results can be seen in Fig. 1, two peaks can be observed which are chromosome-wide significant based on an FDR correction of chromosome X SNPs. Table 2 shows all SNPs which were highlighted by a single cohort which also replicate in the meta-analysis; the effect sizes and p values in each single cohort and also from the meta-analysis are presented. The Manhattan plots in males and females separately are seen in Supplementary Figs. 4 and 5 respectively. There is a peak in females which reaches chromosome-wide significance based on an FDR correction which is not seen in males, these SNPs map to gene NXF5.

Each dot represents a SNP, the x-axis is the SNPs base position and the y-axis is the p-value (–log10(p)), the red line shows the chromosome X wide significance threshold and the blue line shows the suggestive significance threshold.
The results across all cohorts are summarised in Fig. 2, this highlights that evidence is strongest for 4 genes; DDX53, IL1RAPL1, TBX22 and SH3BGRL, which show replication in at least two independent cohorts. So, although individual SNP association p values are only suggestive, we can be more confident in these findings as they replicate across multiple studies. In addition, in the meta-analysed data SNP rs5913102 in TBX22 reaches chromosome-wide significance based on an FDR correction. The LocusZoom [53] plots for these four genes in the meta-analysed (KRONOS/Tgen + BDR + ROSMAP/MAYO/MSBB) data is seen in Supplementary Fig. 6.

The four panels represent each of the four genes (grey) highlighted in this study, the relevant identified SNPs (blue/green) for each genes, and the effect sizes and p-values in the different cohorts (red/orange/yellow) and the meta-analysis of all cohorts (pink).
SNPs in Table 2 were uploaded to RegulomeDB [54], SNPs rs5913102 and rs12848641 had rank “1f” indicating that these SNPs are likely to be located in a functional region and affect transcription factor binding. All other SNPs had rank > 4 suggesting minimal binding evidence. We also computed a combined annotation dependent depletion (CADD) score [55] for each of these variants which integrates several diverse annotations, but no SNPs had a score > 20 suggesting that variants may not be functional.
We have also found SNPs significantly associated with AD in the genes which have been identified in [16], which also used ROSMAP but gene expression data. We replicated signals in three genes in the meta-analysis of all three cohorts, namely GRIA3 (rs6649016, OR = 1.15, p = 0.0028, Ref/Alt Allele=A/G), GRIPAP1 (rs5906732, OR = 1.53, p = 0.028, Ref/Alt Allele=T/C) and UBL4A (rs45463798, OR = 1.16, p = 0.05, Ref/Alt Allele=A/G).
The SNPs presented in Table 2 have effect sizes in the same direction in males and females, see Supplementary Table 4. For the SNPs replicating across multiple studies we also investigated the impact of APOE status on these associations, by adjusting for both number of APOEe4 alleles and the interaction between the SNP and number of APOE e4 alleles, the p value for this model is presented in Supplementary Table 4. In general, the p-value adjusted for APOE status has only changed slightly, with the largest change for the SH3BGRL gene (p = 5.2e-5, padj = 0.014). We have identified SNPs in NXF5 gene to be chromosome-wide significant in females but not in males (rs5944989 MAF = 0.60 and 0.58, OR = 0.62 and 0.98, p = 1.1e-05 and 0.830, in females and males, respectively, Ref/Alt Allele=A/G), see Table 3.
When meta-analysing males and females using GWAMA, a sex heterogeneity p-value is computed; the top SNPs with the smallest p-values for heterogeneity are also seen in Table 3. These SNPs show an opposite effect direction in males and females.
Gene-based analysis
The gene-based analysis in the KRONOS/Tgen and ROSMAP/MAYO/MSBB data does not provide any genes which surpass the gene-wide threshold (based upon the total number of chromosome X genes analysed). In the KRONOS/Tgen data, the third most significant gene is SLC25A5 (p = 0.0048, pfdr = 0.74) which was also highlighted as a proximal gene to a SNP identified in the XWAS. In the BDR data, there were two genes which reached gene-wide significance, these were BGN and HAUS7 (pfdr = 0.004 for both genes). HAUS7 was also identified as chromosome-wide significant from the XWAS in the meta-analysed data. A gene-based analysis from the meta-analysis also did not produce any gene-wide significant results.
Using expression data to gain insights into genes of interest
We investigated the meta-analysis XWAS significant genes (Table 2) for their relevance to neurodegeneration by searching several public datasets containing expression data from bulk and single-cell RNA-seq datasets from human and mouse samples to assess conservation of gene responses between species. We saw DDX53 was expressed at low levels in several cell types across the human brain but showed highest expression in astrocytes (Supplementary Fig. 7A). In some human AD studies DDX53 showed increased expression in AD compared to age-matched controls (Supplementary Fig. 7B). IL1RAPL1 was expressed at highest levels in human oligodendrocytes and neurons (Supplementary Fig. 8A) and showed decreased expression in mouse models of AD compared to age-matched wild-type mice (Supplementary Fig. 8C). SH3BGRL was expressed at the highest levels in human microglia (Supplementary Fig. 9A), and showed decreased expression in AD compared to age-matched controls in one data set (Supplementary Fig. 9B), and increased expression in another (Supplementary Fig. 9C). In mouse models of AD, SH3BGRL showed increased expression compared to age-matched controls (Supplementary Fig. 9D, E). Data was not available for TBX22, and so was likely lowly expressed in the datasets examined. We saw that these genes of interest could be linked to known familial neurodegenerative disease genes (APP and HTT), and AD risk genes (APOE and PLCG2) from prior experimental studies using the Ingenuity database (Fig. 3).

Input genes are denoted by gene symbols encircled with grey filled nodes, while solid lines signify direct interactions, such as protein-protein interactions or phosphorylation events.
BTK is an additional gene which was highlighted in only one study but show interesting expression results. BTK was expressed at the highest levels in human microglia (Supplementary Fig. 10A). BTK showed increased expression in AD compared to age-matched controls (Supplementary Fig. 10B), and increased expression in mouse models of AD (Supplementary Fig. 10E). In the STRING database, the BTK-based network includes PLCG2, SYK, TLR4, TGFB1 and TREM2 genes which belong to microglial pathways important in AD (Supplementary Fig. 11). Thus, the new chromosome X genes we have identified are likely to contribute to AD by modifying existing pathways that are known to control AD risk.
Discussion
This study, which uses a pathologically confirmed diagnosis of AD, identifies four potential genes, DDX53, IL1RAPL1, TBX22 and SH3BGRL, associated with AD, which replicate across at least two of the sub-studies.
TBX22 has previously been shown to be associated with cleft lip and cleft palate [56]. The SNP in TBX22 has a RegulomeDB [54] rank of “1f” indicating the SNPs likelihood of being in a functional region and affects transcription factor binding.
SH3BGRL has been linked to Parkinson’s disease, where higher expression is shown in cases compared to controls [57] and is highly expressed in breast cancers [58]. In addition, a proteome analysis in AD has identified increased SH3 protein in the brain [59].
DDX53 is an intronless gene which is linked to Autism Spectrum Disorder (ASD), although the DDX53 mutations were shown to have no effect on synaptic transmission [60].
IL1RAPL1 is a synaptic adhesion molecule located at the postsynaptic membrane, it regulates dendrite formation and impacts activity of IL-1β on dendrite morphology [61]. Literature suggests that there are other genes in the IL1 family that have some relevance to neurodegenerative or brain disorders. IL1RAP is highly expressed in the brain [62] and SNPs located in IL1RAP were found to be associated with longitudinal change in brain amyloid [63]. There were also associations found between the most significant SNP in IL1RAP and progression from MCI to AD, cognitive decline, temporal cortex atrophy and microglial activity [63]. rs1921622, a variant in IL1RL1 has been shown to lower the risk effects of APOE-ε4 in female AD patients by lowering soluble ST2 [64].
Several genes were identified as being associated with sex-differentiated effects. NXF5 was found to be chromosome-wide significant in females but not in males. NXF5 has been linked to intellectual disability [65] and is known to be involved in brain development [66]. SNPs in SPIN4, LOC105373237, ZC3H12B and LOC105373347 genes were identified as the most significant sex heterogeneous SNPs in the XWAS meta-analysis. The SPIN4 gene inhibits cell proliferation, binds specific histone modifications and negatively regulates body growth [67]. ZC3H12B has been identified as being associated with AD using a Bayesian genome-wide transcriptome-wide association study [68].
Three genes identified in a previous study [16] were replicated from the meta-analysis XWAS of all three cohorts; these genes were GRIA3, GRIPAP1 and UBL4A. As well as being associated with slower cognitive decline in women but not men, GRIA3 is known to be involved in memory and learning and is highly correlated to HLA-DRB5, which is associated with AD [69].
Two additional gene-wide significant genes were identified in the BDR data in the gene-based analysis; these genes are BGN and HAUS7. BGN has been associated with amyloid metabolism in AD [70], inflammatory state in obesity and type 2 diabetes [71] and is known to be a central gene in a network in the brain in response to fructose consumption [72]. HAUS7 is necessary for cytokinesis and regulates mitotic spindle and centrosome integrity (https://www.ncbi.nlm.nih.gov/). SNPs in HAUS7 also reach chromosome-wide significance after FDR multiple testing correction in the meta-analysis of all three cohorts.
BTK was found to be strongly connected to PLCG2 [73]. PLCG2 activation leads to the B cell receptor (BCR) signalling and BTK is in the BCR signalling complex. In the protein network, Toll Like Receptor 4 (TLR4) connects BGN to the BTK network [73, 74] including the PLCG2 pathway and epigenetic silencing of the immunosuppressive response. PLCG2 is a well validated AD risk gene [75, 76]. The BTK-BGN network includes strong microglial genes (PLCG2, SYK, TLR4, TGFB1) and TREM2 links strongly to these [76].
DDX53 shows highest expression in astrocytes, IL1RAPL1 is most highly expressed in oligodendrocytes and neurons and SH3BGRL is most highly expressed in microglia. Collectively, the expression data suggests that the putative chromosome X risk genes act through different cell types and pathways to modulate risk for AD, with some genes increasing risk and some being protective.
The strength of this study is the utilisation of available chromosome X data in the KRONOS/Tgen, BDR and ROSMAP/MAYO/MSBB data in relation to AD, which until now has been understudied. It also uses three independent cohorts with a pathologically confirmed phenotype to investigate potential replication and increase power by meta-analysing independent XWAS together.
The limitation of this study is the relatively small sample size of the available studies; however, we have attempted to improve power by meta-analysing these cohorts together. Despite the small sample sizes, we report consistent results across studies which reinforce our findings.
In conclusion, this study has highlighted several potential target genes on chromosome X associated with AD risk which may be relevant for further study, with the end goal of identifying differences in AD progression between males and females and potentially developing sex-stratified therapeutics.
Data availability
The XWAS summary statistics for the meta-analysis of KRONOS/Tgen and BDR is available at the DRI GitHub repository (https://github.com/UKDRI/XWAS_AD_summary_stats). The Manhattan plot for this meta-analysis is presented in Supplementary Fig. 12. Mouseac, this paper and Matarin et al. [51]: www.mouseac.org WGCNA (Langfelder and Horvath, 2008): https://horvath.genetics.ucla.edu/html/CoexpressionNetwork/Rpackages/WGCNA/index.html (accessed September 2018) Braineac (Ramasamy et al., 2014): www.braineac.org (accessed September 2018) 1,000 genomes (Genomes Project et al., 2015): www.1000genomes.org and http://www.internationalgenome.org (accessed September 2018) MAGMA de Leeuw et al. [37]: www.ctg.cncr.nl/software/magma (accessed May 2019) Coloc, version 3.1, (Giambartolomei et al., 2014): https://github.com/chr1swallace/coloc (accessed September 2018) ROS/MAP (Bennett et al. [29]; Bennett et al. [29]; De Jager et al., 2018): https://www.synapse.org/#!Synapse:syn3219045 (accessed September 2018) i-CisTarget (Imrichova et al., 2015): https://gbiomed.kuleuven.be/apps/lcb/i-cisTarget (accessed September 2018) GTEx V6 gene expression (Consortium GT, 2015): https://gtexportal.org/home (accessed September 2018) Coexp (Botia et al., 2017): https://github.com/juanbot/CoExpNets (accessed September 2018) Myeloid landscape datasets (Friedman et al., 2018): http://research-pub.gene.com/BrainMyeloidLandscape/# (accessed June 2019).
References
Alzheimer’s Association. 2023 Alzheimer’s disease facts and figures. Alzheimer’s Dement. 2023. https://www.alz.org/alzheimers-dementia/factsfigures#:~:text=The%20lifetime%20risk%20for%20Alzheimer's,valued%20at%20nearly%20%24350%20billion.
Hebert LE, Weuve J, Scherr PA, Evans DA. Alzheimer disease in the United States (2010-2050) estimated using the 2010 census. Neurology. 2013;80:1778–83.
Mielke MM. Sex and gender differences in Alzheimer’s disease dementia. Psychiatr Times. 2018;35:14–7.
Plassman BL, Langa KM, Fisher GG, Heeringa SG, Weir DR, Ofstedal MB, et al. Prevalence of dementia in the United States: the aging, demographics, and memory study. Neuroepidemiology. 2007;29:125–32.
Roberts RO, Knopman DS, Mielke MM, Cha RH, Pankratz VS, Christianson TJ, et al. Higher risk of progression to dementia in mild cognitive impairment cases who revert to normal. Neurology. 2014;82:317–25.
Guo L, Zhong MB, Zhang L, Zhang B, Cai D. Sex differences in Alzheimer’s disease: insights from the multiomics landscape. Biol Psychiatry. 2022;91:61–71.
Gilsanz P, Lee C, Corrada MM, Kawas CH, Quesenberry CP, Whitmer RA. Reproductive period and risk of dementia in a diverse cohort of health care members. Neurology. 2019;92:e2005–e14.
Gong J, Harris K, Peters SAE, Woodward M. Reproductive factors and the risk of incident dementia: a cohort study of UK Biobank participants. PLoS Med. 2022;19:e1003955.
Farrer LA, Cupples LA, Haines JL, Hyman B, Kukull WA, Mayeux R, et al. Effects of age, sex, and ethnicity on the association between apolipoprotein E genotype and Alzheimer disease: a meta-analysis. JAMA. 1997;278:1349–56.
Ferretti MT, Iulita MF, Cavedo E, Chiesa PA, Schumacher Dimech A, Santuccione Chadha A, et al. Sex differences in Alzheimer disease - the gateway to precision medicine. Nat Rev Neurol. 2018;14:457–69.
Eikelboom WS, Pan M, Ossenkoppele R, Coesmans M, Gatchel JR, Ismail Z, et al. Sex differences in neuropsychiatric symptoms in Alzheimer’s disease dementia: a meta-analysis. Alzheimers Res Ther. 2022;14:48.
Gorlov IP, Amos CI. Why does the X chromosome lag behind autosomes in GWAS findings? PLoS Genet. 2023;19:e1010472.
Armon C, Wolfson S, Margalit R, Avraham L, Bugen Y, Cohen A, et al. Estimating the X chromosome-mediated risk for developing Alzheimer’s disease. J Neurol. 2022;269:2479–85.
Gomez-Ramos A, Podlesniy P, Soriano E, Avila J. Distinct X-chromosome SNVs from some sporadic AD samples. Sci Rep. 2015;5:18012.
Young J, Gallagher E, Koska K, Guetta-Baranes T, Morgan K, Thomas A, et al. Genome-wide association findings from the brains for dementia research cohort. Neurobiol Aging. 2021;107:159–67.
Davis EJ, Solsberg CW, White CC, Miñones-Moyano E, Sirota M, Chibnik L, et al. Sex-specific association of the X chromosome with cognitive change and tau pathology in aging and Alzheimer Disease. JAMA Neurol. 2021;78:1249–54.
Corneveaux JJ, Myers AJ, Allen AN, Pruzin JJ, Ramirez M, Engel A, et al. Association of CR1, CLU and PICALM with Alzheimer’s disease in a cohort of clinically characterized and neuropathologically verified individuals. Hum. Mol. Genet. 2010;19:3295–301.
Myers AJ, Gibbs JR, Webster JA, Rohrer K, Zhao A, Marlowe L, et al. A survey of genetic human cortical gene expression. Nat. Genet. 2007;39:1494–9.
Petyuk, Chang VA, Ramirez-Restrepo R, Beckmann M, Henrion MYR ND, Piehowski PD, et al. The human brainome: network analysis identifies HSPA2 as a novel Alzheimer disease target. Brain. 2018;141:2721–39.
Webster JA, Gibbs JR, Clarke J, Ray M, Zhang W, Holmans P, et al. Genetic control of human brain transcript expression in Alzheimer disease. Am J Hum Genet. 2009;84:445–58.
Beekly DL, Ramos EM, van Belle G, Deitrich W, Clark AD, Jacka ME, et al. The National Alzheimer’s Coordinating Center (NACC) Database: an Alzheimer disease database. Alzheimer Dis Assoc Disord. 2004;18:270–7.
Braak H, Braak E. Staging of alzheimer’s disease-related neurofibrillary changes. Neurobiol Aging. 1995;16:271–8.
Das S, Forer L, Schonherr S, Sidore C, Locke AE, Kwong A, et al. Next-generation genotype imputation service and methods. Nat Genet. 2016;48:1284–7.
Taliun D, Harris DN, Kessler MD, Carlson J, Szpiech ZA, Torres R, et al. Sequencing of 53,831 diverse genomes from the NHLBI TOPMed Program. Nature. 2021;590:290–9.
Francis PT, Costello H, Hayes GM. Brains for dementia research: evolution in a longitudinal brain donation cohort to maximize current and future value. J Alzheimers Dis. 2018;66:1635–44.
Francis PT, Hayes GM, Costello H, Whitfield DR. Brains for dementia research: the importance of cohorts in brain banking. Neurosci Bull. 2019;35:289–94.
Bennett DA, Buchman AS, Boyle PA, Barnes LL, Wilson RS, Schneider JA. Religious orders study and rush memory and aging project. J Alzheimer’s Dis. 2018;64:S161–S89.
Bennett D, Schneider J, Arvanitakis Z, Wilson R. Overview and findings from the religious orders study. Curr Alzheimer Res. 2011;9:628–45.
Bennett DA, Schneider JA, Buchman AS, Barnes LL, Boyle PA, Wilson RS. Overview and findings from the rush memory and aging project. Curr Alzheimer Res. 2012;9:646–63.
Leonenko G, Baker E, Stevenson-Hoare J, Sierksma A, Fiers M, Williams J, et al. Identifying individuals with high risk of Alzheimer’s disease using polygenic risk scores. Nat Commun. 2021;12:4506.
Konig IR, Loley C, Erdmann J, Ziegler A. How to include chromosome X in your genome-wide association study. Genet Epidemiol. 2014;38:97–103.
Purcell S, Neale B, Todd-Brown K, Thomas L, Ferreira MA, Bender D, et al. PLINK: a tool set for whole-genome association and population-based linkage analyses. Am J Hum Genet. 2007;81:559–75.
Chang CC, Chow CC, Tellier LC, Vattikuti S, Purcell SM, Lee JJ. Second-generation PLINK: rising to the challenge of larger and richer datasets. Gigascience. 2015;4:7.
Mägi R, Morris AP. GWAMA: software for genome-wide association meta-analysis. BMC Bioinform. 2010;11:288.
R Development Core Team. R: A language and environment for statistical computing. R Foundation for Statistical Computing. 2008.
Willer CJ, Li Y, Abecasis GR. METAL: fast and efficient meta-analysis of genomewide association scans. Bioinformatics. 2010;26:2190–1.
de Leeuw CA, Mooij JM, Heskes T, Posthuma D. MAGMA: generalized gene-set analysis of GWAS data. PLoS Comput Biol. 2015;11:e1004219.
Zhang B, Gaiteri C, Bodea LG, Wang Z, McElwee J, Podtelezhnikov AA, et al. Integrated systems approach identifies genetic nodes and networks in late-onset Alzheimer’s disease. Cell. 2013;153:707–20.
Berchtold NC, Cribbs DH, Coleman PD, Rogers J, Head E, Kim R, et al. Gene expression changes in the course of normal brain aging are sexually dimorphic. Proc Natl Acad Sci USA. 2008;105:15605–10.
Srinivasan K, Friedman BA, Etxeberria A, Huntley MA, van der Brug MP, Foreman O, et al. Alzheimer’s patient microglia exhibit enhanced aging and unique transcriptional activation. Cell Rep. 2020;31:107843.
Olah M, Patrick E, Villani AC, Xu J, White CC, Ryan KJ, et al. A transcriptomic atlas of aged human microglia. Nat Commun. 2018;9:539.
Zhang Y, Chen K, Sloan SA, Bennett ML, Scholze AR, O’Keeffe S, et al. An RNA-sequencing transcriptome and splicing database of glia, neurons, and vascular cells of the cerebral cortex. J Neurosci. 2014;34:11929–47.
Zhang Y, Sloan SA, Clarke LE, Caneda C, Plaza CA, Blumenthal PD, et al. Purification and characterization of progenitor and mature human astrocytes reveals transcriptional and functional differences with mouse. Neuron. 2016;89:37–53.
Morabito S, Miyoshi E, Michael N, Shahin S, Martini AC, Head E, et al. Single-nucleus chromatin accessibility and transcriptomic characterization of Alzheimer’s disease. Nat Genet. 2021;53:1143–55.
Sala Frigerio C, Wolfs L, Fattorelli N, Thrupp N, Voytyuk I, Schmidt I, et al. The major risk factors for Alzheimer’s disease: age, sex, and genes modulate the microglia response to Abeta plaques. Cell Rep. 2019;27:1293–306.e6.
Lake BB, Chen S, Sos BC, Fan J, Kaeser GE, Yung YC, et al. Integrative single-cell analysis of transcriptional and epigenetic states in the human adult brain. Nat Biotechnol. 2018;36:70–80.
Swarup V, Chang TS, Duong DM, Dammer EB, Dai J, Lah JJ, et al. Identification of conserved proteomic networks in neurodegenerative dementia. Cell Rep. 2020;31:107807.
Wang Y, Cella M, Mallinson K, Ulrich JD, Young KL, Robinette ML, et al. TREM2 lipid sensing sustains the microglial response in an Alzheimer’s disease model. Cell. 2015;160:1061–71.
Ulland TK, Song WM, Huang SC, Ulrich JD, Sergushichev A, Beatty WL, et al. TREM2 maintains microglial metabolic fitness in Alzheimer’s disease. Cell. 2017;170:649–63.e13.
Orre M, Kamphuis W, Osborn LM, Jansen AHP, Kooijman L, Bossers K, et al. Isolation of glia from Alzheimer’s mice reveals inflammation and dysfunction. Neurobiol Aging. 2014;35:2746–60.
Matarin M, Salih DA, Yasvoina M, Cummings DM, Guelfi S, Liu W, et al. A genome-wide gene-expression analysis and database in transgenic mice during development of amyloid or tau pathology. Cell Rep. 2015;10:633–44.
Salih DA, Bayram S, Guelfi S, Reynolds RH, Shoai M, Ryten M, et al. Genetic variability in response to amyloid beta deposition influences Alzheimer’s disease risk. Brain Commun. 2019;1:fcz022.
Boughton AP, Welch RP, Flickinger M, VandeHaar P, Taliun D, Abecasis GR, et al. LocusZoom.js: interactive and embeddable visualization of genetic association study results. Bioinformatics. 2021;37:3017–8.
Boyle AP, Hong EL, Hariharan M, Cheng Y, Schaub MA, Kasowski M, et al. Annotation of functional variation in personal genomes using RegulomeDB. Genome Res. 2012;22:1790–7.
Kircher M, Witten DM, Jain P, O’Roak BJ, Cooper GM, Shendure J. A general framework for estimating the relative pathogenicity of human genetic variants. Nat Genet. 2014;46:310–5.
Marcano AC, Doudney K, Braybrook C, Squires R, Patton MA, Lees MM, et al. TBX22 mutations are a frequent cause of cleft palate. J Med Genet. 2004;41:68–74.
Werner CJ, Heyny-von Haussen R, Mall G, Wolf S. Proteome analysis of human substantia nigra in Parkinson’s disease. Proteome Sci. 2008;6:8.
Li H, Zhang M, Wei Y, Haider F, Lin Y, Guan W, et al. SH3BGRL confers innate drug resistance in breast cancer by stabilizing HER2 activation on cell membrane. J Exp Clin Cancer Res. 2020;39:81.
Shiozaki A, Tsuji T, Kohno R, Kawamata J, Uemura K, Teraoka H, et al. Proteome analysis of brain proteins in Alzheimer’s disease: subproteomics following sequentially extracted protein preparation. J Alzheimer’s Dis. 2004;6:257–68.
Faheem M, Deneault E, Alexandrova R, Rodrigues DC, Pellecchia G, Shum C, et al. Disruption of DDX53 coding sequence has limited impact on iPSC-derived human NGN2 neurons. BMC Med Genom. 2023;16:5.
Montani C, Gritti L, Beretta S, Verpelli C, Sala C. The synaptic and neuronal functions of the X-linked intellectual disability protein Interleukin-1 Receptor Accessory Protein Like 1 (IL1RAPL1). Dev Neurobiol. 2019;79:85–95.
Kang HJ, Kawasawa YI, Cheng F, Zhu Y, Xu X, Li M, et al. Spatio-temporal transcriptome of the human brain. Nature. 2011;478:483–9.
Ramanan VK, Risacher SL, Nho K, Kim S, Shen L, McDonald BC, et al. GWAS of longitudinal amyloid accumulation on 18F-florbetapir PET in Alzheimer’s disease implicates microglial activation gene IL1RAP. Brain. 2015;138:3076–88.
Jiang Y, Zhou X, Wong HY, Ouyang L, Ip FCF, Chau VMN, et al. An IL1RL1 genetic variant lowers soluble ST2 levels and the risk effects of APOE-epsilon4 in female patients with Alzheimer’s disease. Nat Aging. 2022;2:616–34.
Callaerts-Vegh Z, Ahmed T, Vermaercke B, Marynen P, Balschun D, Froyen G, et al. Nxf7 deficiency impairs social exploration and spatio-cognitive abilities as well as hippocampal synaptic plasticity in mice. Front Behav Neurosci. 2015;9:179.
Jun L, Frints S, Duhamel H, Herold A, Abad-Rodrigues J, Dotti C, et al. NXF5, a novel member of the nuclear RNA export factor family, is lost in a male patient with a syndromic form of mental retardation. Curr Biol. 2001;11:1381–91.
Lui JC, Wagner J, Zhou E, Dong L, Barnes KM, Jee YH, et al. Loss-of-function variant in SPIN4 causes an X-linked overgrowth syndrome. JCI Insight. 2023;8:e167074.
Luningham JM, Chen J, Tang S, De Jager PL, Bennett DA, Buchman AS, et al. Bayesian genome-wide TWAS method to leverage both cis- and trans-eQTL information through summary statistics. Am J Hum Genet. 2020;107:714–26.
Bodily PM, Fujimoto MS, Page JT, Clement MJ, Ebbert MT, Ridge PG, et al. A novel approach for multi-SNP GWAS and its application in Alzheimer’s disease. BMC Bioinform. 2016;17:268.
Lam V, Takechi R, Pallebage-Gamarallage MM, Galloway S, Mamo JC. Colocalisation of plasma derived apo B lipoproteins with cerebral proteoglycans in a transgenic-amyloid model of Alzheimer’s disease. Neurosci Lett. 2011;492:160–4.
Babelova A, Moreth K, Tsalastra-Greul W, Zeng-Brouwers J, Eickelberg O, Young MF, et al. Biglycan, a danger signal that activates the NLRP3 inflammasome via toll-like and P2X receptors. J Biol Chem. 2009;284:24035–48.
Ying Z, Byun HR, Meng Q, Noble E, Zhang G, Yang X, et al. Biglycan gene connects metabolic dysfunction with brain disorder. Biochim Biophys Acta Mol Basis Dis. 2018;1864:3679–87.
Li K, Ran B, Wang Y, Liu L, Li W. PLCgamma2 impacts microglia-related effectors revealing variants and pathways important in Alzheimer’s disease. Front Cell Dev Biol. 2022;10:999061.
Hu L, Zang MD, Wang HX, Li JF, Su LP, Yan M, et al. Biglycan stimulates VEGF expression in endothelial cells by activating the TLR signaling pathway. Mol Oncol. 2016;10:1473–84.
Sims R, van der Lee SJ, Naj AC, Bellenguez C, Badarinarayan N, Jakobsdottir J, et al. Rare coding variants in PLCG2, ABI3, and TREM2 implicate microglial-mediated innate immunity in Alzheimer’s disease. Nat Genet. 2017;49:1373.
Magno L, Bunney TD, Mead E, Svensson F, Bictash MN. TREM2/PLCgamma2 signalling in immune cells: function, structural insight, and potential therapeutic modulation. Mol Neurodegener. 2021;16:22.
Acknowledgements
This work was supported by the Dementia Research Institute [UKDRI supported by the Medical Research Council (UKDRI-3003), Alzheimer’s Research UK, and Alzheimer’s Society], Welsh Government, Joint Programming for Neurodegeneration (MRC: MR/T04604X/1), Dementia Platforms UK (MRC: MR/L023784/2) and MRC Centre for Neuropsychiatric Genetics and Genomics (MR/L010305/1). DAS and JH are also supported by the Dolby Foundation, and by the National Institute for Health Research University College London Hospitals Biomedical Research Centre. UY is funded by the YLSY Programme of Turkish Ministry of Education. For the Tgen/Kronos series many data and biomaterials were collected from several National Institute on Aging (NIA) and National Alzheimer’s Coordinating Center (NACC, grant #U01 AG016976) funded sites. Amanda J. Myers, PhD (University of Miami, Department of Cell Biology) and John A. Hardy, PhD (Reta Lila Weston Institute, University College London) collected and prepared the series. A.J.M. is supported by grant # AG069008 from the National Institute on Aging. Marcelle Morrison-Bogorad, PhD., Tony Phelps, PhD and Walter Kukull PhD are thanked for helping to co-ordinate this collection. The directors, pathologist and technicians involved include: National Institute on Aging: Ruth Seemann, John Hopkins Alzheimer’s Disease Research Center (NIA grant # AG05146): Juan C. Troncoso, MD, Dr. Olga Pletnikova, University of California, Los Angeles (NIA grant # P50 AG16570):Harry Vinters, MD, Justine Pomakian, The Kathleen Price Bryan Brain Bank, Duke University Medical Center (NIA grant #AG05128, NINDS grant # NS39764, NIMH MH60451 also funded by Glaxo Smith Kline): Christine Hulette, MD, Director, John F. Ervin, Stanford University: Dikran Horoupian, MD, Ahmad Salehi, MD, PhD, New York Brain Bank, Taub Institute, Columbia University (NYBB): Jean Paul Vonsattel, MD, Katerina Mancevska, Massachusetts Alzheimer’s Disease Research Center (P50 AG005134): E. Tessa Hedley-Whyte, MD, MP Frosch, MD, Karlotta Fitch, University of Michigan (NIH grant P50-AG08671): Dr. Roger Albin, Lisa Bain, Eszter Gombosi, University of Kentucky (NIH #AG05144): William Markesbery, MD, Sonya Anderson, Mayo Clinic, Jacksonville: Dennis W. Dickson, MD, Natalie Thomas, University Southern California: Caroll A. Miller, MD, Jenny Tang, M.S., Dimitri Diaz, Washington University, St Louis Alzheimer’s Disease Research Center (NIH #P50AG05681): Dan McKeel, MD, John C. Morris, MD, Eugene Johnson, Jr., PhD, Virginia Buckles, PhD, Deborah Carter, University of Washington, Seattle (NIH #P50 AG05136):Thomas Montine, MD, PhD, Aimee Schantz, MEd., University of Pennsylvania School of Medicine, Alzheimer’s Disease Research Center: John Q Trojanowski, MD, Virginia M Lee, MD, Vivianna Van Deerlin, MD, Terry Schuck, Boston University Alzheimer’s Disease Research Center (NIH grant P30-AG13846): Ann C. McKee, Carol Kubilus Sun Health Research Institute Brain Donation Program of Sun City, Arizona (NIA #P30 AG19610; Arizona Alzheimer’s Disease Core Center, Arizona Department of Health Services, contract 211002, Arizona Alzheimer’s Research Center; Arizona Biomedical Research Commission, contracts 4001, 0011, 05-901 and 1001 to the Arizona Parkinson’s Disease Consortium; Michael J. Fox Foundation for Parkinson’s Research): Joseph Rogers, PhD, Thomas G. Beach, MD, PhD, Lucia I. Sue Emory University: Bruce H. Wainer, MD, PhD, Marla Gearing, PhD, University of Texas, Southwestern Medical School: Charles L. White, III, M.D., Roger Rosenberg, Marilyn Howell, Joan Reisch, University of California, Davis: William Ellis, MD, Mary Ann Jarvis, Rush University Medical Center, Rush Alzheimer’s Disease Center (NIH #AG10161): David A. Bennett, M.D. Julie A. Schneider, MD, MS, Karen Skish, MS, PA (ASCP)MT, Wayne T Longman, University of Miami/NPF Brain Endowment Bank: Deborah C. Mash, MD, Margaret J Basile, Mitsuko Tanaka Oregon Health & Science University: Randy Wotljer, PhD Additional tissues include samples from the following sites: Newcastle Brain Tissue Resource (funding via the Medical Research Council, local NHS trusts and Newcastle University): C.M. Morris, MD, Ian G McKeith, Robert H Perry MRC London Brain Bank for Neurodegenerative Diseases (funding via the Medical Research Council): Simon Lovestone, Md PhD, Safa Al-Sarraj. MD, Claire Troakes, South West Dementia Brain Bank (funding via numerous sources including the Higher Education Funding Council for England (HEFCE), Alzheimer’s Research Trust (ART), BRACE as well as North Bristol NHS Trust Research and Innovation Department and DeNDRoN): Seth Love, MD, Patrick Kehoe, PhD, Laura Palmer, The Netherlands Brain Bank (funding via numerous sources including Stichting MS Research, Brain Net Europe, Hersenstichting Nederland Breinbrekend Werk, International Parkinson Fonds, Internationale Stiching Alzheimer Onderzoek): Inge Huitinga, MD, Marleen Rademaker, Michiel Kooreman, Institut de Neuropatologia, Servei Anatomia Patologica, Universitat de Barcelona: Isidre Ferrer Abizanda, MD, PhD, Susana Casas Boluda. We would like to gratefully acknowledge all donors and their families for the tissue provided for this study. Human post-mortem tissue was obtained from the South West Dementia Brain Bank, London Neurodegenerative Diseases Brain Bank, Manchester Brain Bank, Newcastle Brain Tissue Resource and Oxford Brain Bank, members of the Brains for Dementia Research (BDR) Network. The BDR is jointly funded by Alzheimer’s Research UK and the Alzheimer’s Society in association with the Medical Research Council. We also wish to acknowledge the neuropathologists at each centre and BDR Brain Bank staff for the collection and classification of the samples. Brains for Dementia Research has ethics approval from London – City and East NRES committee 08/H0704/128 + 5 and has deemed all approved requests for tissue to have been approved by the committee. BDR is supported by funding provided by an ARUK project grant, entitled ‘Enabling high-throughput genomic approaches in Alzheimer’s disease’ (ARUK-PG2014-2) awarded to KM, and an ARUK extension grant entitled ‘NeuroChip analysis of the entire Brains for Dementia Research (BDR) resource of 2000 samples’, (ARUK-EXT2017A-1) awarded to KM and KJB. The results published here are in whole or in part based on data obtained from the AD Knowledge Portal (https://adknowledgeportal.org). ROSMAP data were provided by the Rush Alzheimer’s Disease Center, Rush University Medical Center, Chicago. Data collection was supported through funding by NIA grants P30AG10161 (ROS), R01AG15819 (ROSMAP; genomics and RNAseq), R01AG17917 (MAP), R01AG30146, R01AG36042 (5hC methylation, ATACseq), RC2AG036547 (H3K9Ac), R01AG36836 (RNAseq), R01AG48015 (monocyte RNAseq) RF1AG57473 (single nucleus RNAseq), U01AG32984 (genomic and whole exome sequencing), U01AG46152 (ROSMAP AMP-AD, targeted proteomics), U01AG46161(TMT proteomics), U01AG61356 (whole genome sequencing, targeted proteomics, ROSMAP AMP-AD), the Illinois Department of Public Health (ROSMAP), and the Translational Genomics Research Institute (genomic). Additional phenotypic data can be requested at www.radc.rush.edu. MSBB data were generated from postmortem brain tissue collected through the Mount Sinai VA Medical Center Brain Bank and were provided by Dr. Eric Schadt from Mount Sinai School of Medicine. The Mayo RNAseq study data was led by Dr. Nilüfer Ertekin-Taner, Mayo Clinic, Jacksonville, FL as part of the multi-PI U01 AG046139 (MPIs Golde, Ertekin-Taner, Younkin, Price). Samples were provided from the following sources: The Mayo Clinic Brain Bank. Data collection was supported through funding by NIA grants P50 AG016574, R01 AG032990, U01 AG046139, R01 AG018023, U01 AG006576, U01 AG006786, R01 AG025711, R01 AG017216, R01 AG003949, NINDS grant R01 NS080820, CurePSP Foundation, and support from Mayo Foundation. Study data includes samples collected through the Sun Health Research Institute Brain and Body Donation Program of Sun City, Arizona. The Brain and Body Donation Program is supported by the National Institute of Neurological Disorders and Stroke (U24 NS072026 National Brain and Tissue Resource for Parkinsons Disease and Related Disorders), the National Institute on Aging (P30 AG19610 Arizona Alzheimers Disease Core Center), the Arizona Department of Health Services (contract 211002, Arizona Alzheimers Research Center), the Arizona Biomedical Research Commission (contracts 4001, 0011, 05-901 and 1001 to the Arizona Parkinson’s Disease Consortium) and the Michael J. Fox Foundation for Parkinsons Research.
Author information
Authors and Affiliations
Contributions
ES ran all analyses and wrote the initial manuscript draft, GL contributed to data QC and revising manuscript, UY and DS investigated expression data for identified genes, assisted with biological interpretation and revised the manuscript, EB assisted with biological interpretation and revised the manuscript, AM and JH contributed the KRONOS/Tgen data and revised the manuscript, KM and KB contributed the BDR data and revised the manuscript, and VEP designed the study, provided guidance for analyses and wrote and revised the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Ethics approval and consent to participate
All methods were performed in accordance with the relevant guidelines and regulations. The KRONOS/Tgen study is declared not human-subjects research and is IRB exempt under regulation 45 CFR 46. The Brains for Dementia Research (BDR) datahas ethics approval from London – City and East NRES committee 08/H0704/128 + 5 and has deemed all approved requests for tissue to have been approved by the committee. For ROSMAP the parent studies and sub-studies were all approved by an Institutional Review Board of Rush University Medical Center. All participants signed an informed consent and Anatomical Gift Act; in addition, they signed a repository consent allowing their data to be shared. Data documentation and sharing documents can be obtained at www.radc.rush.edu.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Simmonds, E., Leonenko, G., Yaman, U. et al. Chromosome X-wide association study in case control studies of pathologically confirmed Alzheimer’s disease in a European population. Transl Psychiatry 14, 358 (2024). https://doi.org/10.1038/s41398-024-03058-9
Received:
Revised:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41398-024-03058-9
