
Abstract
Plasma proteomic biomarkers hold significant promise for enhancing clinical assessments and improving early detection of psychosis conversion; however, concerns about their reproducibility and generalizability persist. Previous studies, largely conducted in Caucasian cohorts, have identified proteomic biomarkers predictive of psychosis conversion in individuals at ultra-high risk (UHR) of psychosis. In this study, we acquired plasma proteomics data from an Asian UHR cohort, the Longitudinal Youth at Risk Study (LYRIKS). We established a robust machine learning framework, through which we developed and evaluated prediction models for psychosis conversion. We showed that proteomic signatures previously identified in a predominantly Caucasian UHR cohort generalized to the LYRIKS cohort (best AUC = 0.81). Furthermore, we developed three prediction models using the LYRIKS dataset that demonstrated superior performance (best AUC = 0.96). Through these models, we identified novel proteomic signatures with high predictive performance. Despite the differences in individual protein composition between Asian- and Caucasian-derived signatures, functional convergence was observed across key pathways and protein families, namely the complement and coagulation cascade, apolipoproteins, inter-alpha-trypsin inhibitor heavy chain proteins, and serine protease inhibitors. Although current literature is divided on the utility of blood plasma biomarkers in psychiatric diagnosis, our study supports their use by demonstrating cross-population generalizability of Caucasian-derived signatures and deriving signatures with high predictive value from an Asian cohort that converge functionally.
Similar content being viewed by others


Introduction
Mental illnesses, including schizophrenia, major depressive disorder and generalized anxiety disorder, rank among the leading global health challenges, significantly contributing to morbidity and economic burden [1]. Current clinical diagnoses of these conditions primarily rely on interviews and behavioural assessments, which are inherently subjective and prone to variability [2]. The integration of objective molecular biomarkers into psychiatric evaluation holds promise for enhancing diagnostic precision, enabling early detection, improving prognostication, and optimizing resource allocation through risk stratification.
Previous studies have highlighted the potential of blood proteomic biomarkers in predicting transition to psychosis. For instance, Mongan et al. [3] developed machine learning (ML) models incorporating blood proteomic biomarkers that achieved remarkable predictive performance with an area under the receiver operating characteristic curve (AUC) of 0.95. Despite these findings, the use of blood proteomic biomarkers for prognosis and diagnosis of mental illnesses remains contentious due to concerns regarding reproducibility and generalizability [4]. This inconsistency underscores the need for further investigation into the reliability of blood proteomic biomarkers for prediction of psychosis conversion in ultra-high risk (UHR) individuals, who are at elevated risk of developing psychosis as well as other mental illnesses such as depression. Moreover, most studies have been conducted in predominantly Caucasian cohorts, and the generalizability of their reported signatures to Asian populations has not been examined.
In this study, we acquired plasma proteomics data from an Asian cohort of individuals at UHR for psychosis – the Longitudinal Youth at Risk Study (LYRIKS) – using the Orbitrap Astral mass spectrometer. From these data, we established a robust ML framework through which we developed and evaluated prediction models for psychosis conversion. Through these models, we addressed three key questions:
-
1.
To what extent do previously published signatures developed in predominantly Caucasian cohorts [3] generalize to an Asian population?
-
2.
What is the performance of prediction models developed on the LYRIKS dataset, and which proteins constitute novel signatures derived from these models?
-
3.
Which biological pathways and protein families are shared between previously published and LYRIKS signatures, and what insights do they provide into the molecular mechanisms underlying psychosis development?
Methods
Participants and study design
The Longitudinal Youth at Risk Study (LYRIKS) was a two-year prospective observational study conducted in Singapore investigating the social, clinical, and biological risk factors associated with youths at ultra-high risk of developing psychosis [5]. Youth participants aged between 14 to 29 were recruited between 2009 to 2012 from outpatient clinics at the Institute of Mental Health, community mental health agencies, educational institutions, or through self-referral [6]. Exclusion criteria were predefined in the study protocol and included individuals with a history of psychosis, active substance use, intellectual disability, use of antipsychotics or mood stabilizers, or medical conditions associated with psychosis. Recruitment and assessments were conducted by trained research psychologists. This study received ethics approval from the National Healthcare Group Domain Specific Review Board (Approval Number: 2009/00167). Informed consent was obtained from all participants, or from a legally authorized representative for minors under the age of 21. All methods were performed in accordance with relevant institutional guidelines and regulations.
UHR positive participants were ascertained on the Comprehensive Assessment of At-Risk Mental State (CAARMS) instrument [7]. These participants were further evaluated for comorbid psychiatric disorders through the Structured Clinical Interview for DSM-IV Axis I disorders (SCID-I) [8]. Controls were UHR negative participants with no psychiatric disorder and no family history of psychosis. Follow-up clinical assessments were conducted for all participants at six-month intervals over the two-year study period, during which psychometric assessments were administered and blood samples were collected at each visit.
In this study, we investigated plasma proteomics data acquired via mass spectrometry from 135 participants in the LYRIKS cohort. Sixty-five out of the 135 participants were UHR positive. Of these, 13 developed their first episode of psychosis (FEP) within the study period and are termed as converters (Fig. 1A). Among the 52 remaining UHR positive participants (non-converters), 30 remitted to become UHR negative over two years.

A: Stratification of participants from the LYRIKS cohort with available proteomic data. Ultra-high risk (UHR) positive participants are divided into converters (blue) who developed their first episode of psychosis (FEP) within the two-year study period and non-converters (orange). Machine learning models were trained to predict transition to psychosis among UHR positive participants. B: Clinical follow-up assessments were conducted for all participants at six-month intervals over the two-year study period, with blood samples collected at each visit. For converters, blood samples were collected from baseline up to the point of FEP, at which time a final blood sample is collected. All available blood samples from converters were retrieved for proteomic analysis. For non-converters and healthy controls, only blood samples from baseline and at yearly intervals were retrieved for proteomic analysis. Syringe and computer icons denote blood collection and proteomic analysis, respectively.
Blood samples were collected at baseline and at six-month intervals for all study participants into standard EDTA tubes (BD Vacutainer®), centrifuged at 3000 rpm for 10 min for plasma collection and stored at −80 oC. Sample collection and processing was performed on-site and plasma stored rapidly to minimize ex-vivo proteolytic activity, in accordance with established plasma proteomics protocols [9]. Protease inhibitors were not used during plasma collection to avoid the potential introduction of technical variability and additional cost. This is consistent with plasma proteomics practices adopted in other large-scale cohort studies [3, 10]. For converters, blood samples were collected from baseline up to the point of transition to psychosis, at which time a final blood sample is collected. All available blood samples from converters were sent for proteomic analysis. For non-converters and healthy controls, only blood samples at baseline, 12 months and 24 months were retrieved for proteomic analysis (Fig. 1B).
Sample preparation and mass spectra acquisition and analysis
Plasma samples were depleted of high-abundance proteins using the ThermoFisher Scientific™ High-Select™ Top14 Abundant Protein Depletion Resin, which removes the top 14 abundant proteins that constitutes approximately 95% of all plasma proteins (Supplementary Table 1). Subsequently, proteolytic digestion using trypsin (Promega, V5280) was performed. Both steps were done to improve protein detection. Protein concentration and quality were assessed using the Bradford Assay and SDS-PAGE, respectively. Subsequently, 200 ng of each sample was injected into a Vanquish Neo Ultra-High Performance Liquid Chromatography system coupled to a ThermoFisher Scientific™ Orbitrap™ Astral™ mass spectrometer. Mass spectra were acquired using the Orbitrap Astral mass spectrometer in Data-Independent Acquisition (DIA) mode and analysed using DIA-NN [11], with peptide identification and quantification performed against the UniProtKB/Swiss-Prot Homo sapiens database (release dated 2024-07-26).
Data processing
Each sample was normalized according to standard procedure as follows. Total protein intensity was first computed by summing all reported protein intensities within a sample. The maximum total intensity among all samples was used to compute a scaling factor for each sample, defined as the ratio of the maximum total intensity to the total intensity of the given sample. All protein intensities in a sample were then scaled by this factor. To facilitate compatibility with parametric statistical analyses, normalized protein intensities were log2-transformed.
Exploratory analysis revealed significant batch effects associated with sample extraction dates (Supplementary Figure 1). To account for this during missing value imputation, samples were split by batch and proteins with more than 50% missing values in any batch were excluded, retaining a total of 605 of 1,757 proteins for subsequent analysis. Of these, 340 proteins contained missing values ranging from 0.2 to 46.5% per protein, representing 7.1% of total measurements. Subsequently, missing values were imputed for each batch independently using the k-nearest neighbours (k-NN) algorithm (k = 5) implemented in the impute (version 1.72.3) package [12] in R (version 4.2.1) [13]. The choice of imputation method and parameter was guided by systematic evaluation of imputation accuracy (Supplementary Figure 2). Imputed data for all batches were then combined and corrected for batch effects using ComBat [14] from the sva (version 3.46.0) package [15] (Supplementary Figure 3).
Machine learning modeling and evaluation
Five psychosis conversion prediction models were trained and evaluated on the LYRIKS dataset (Supplementary Table 2). Two models were trained on published biomarkers from the European Network of National Schizophrenia Networks Studying Gene-Environment Interactions (EU-GEI) study [16], while the remaining three models were developed from the LYRIKS dataset.
Psychosis conversion biomarkers were identified based on model coefficients or feature selection statistics from the LYRIKS models. Elastic net logistic regression was used as the primary prediction algorithm, with default hyperparameters fixed a priori (C = 1, L1 ratio = 0.5) across models. When support vector machines (SVMs) were employed, default hyperparameters were likewise used (linear kernel, L2 penalty, C = 1). In models where the same ML algorithm was employed for both feature selection and prediction, identical hyperparameter settings were used at both stages. To ensure that models were not biased toward the majority class, samples were weighted inversely proportional to their class frequencies using the standard class weighting implementation in scikit-learn. Hyperparameter optimization was not performed, as the additional data partitioning required would have decreased the effective training sample size and increased the risk of overfitting. All models were implemented using the scikit-learn (version 1.2.2) library [17] in Python (version 3.11) [18], and performance was assessed using the area under the receiver operating characteristic curve (AUC).
The five prediction models are described below; each is named according to its originating study, with the feature selection method indicated in parentheses.
EU-GEI (ANCOVA) model
Thirty-five proteins were identified as differentially expressed between baseline samples of converters and non-converters in the EU-GEI dataset using analysis of covariance (ANCOVA) with multiple testing correction (Supplementary Table 3). Of these, 33 proteins were quantified in the LYRIKS dataset and were subsequently validated using elastic net logistic regression (L1 ratio = 0.5) via stratified 4-fold cross validation (Fig. 2A).

A: Framework used for training and evaluating the EU-GEI (ANCOVA), EU-GEI (SVM) and LYRIKS (ANCOVA) prediction models. Benjamini-Hochberg adjusted ANCOVA P-values were consolidated across four cross-validation splits to identify proteomic biomarkers predictive of psychosis conversion in the LYRIKS (ANCOVA) model. B: Framework used for training and evaluating the LYRIKS (elastic net) and LYRIKS (SVM) model. For the LYRIKS (elastic net) model, elastic net coefficients from inner splits were consolidated across outer splits to identify biomarkers predictive of psychosis conversion.
EU-GEI (SVM) model
This model was based on a previously published signature comprising 10 proteins predictive of psychosis conversion in the EU-GEI dataset (Supplementary Table 4). These proteins were selected based on the coefficients of a SVM model trained on 162 quantified proteins. We validated these 10 proteins using the same experimental and validation framework as described for the EU-GEI (ANCOVA) model.
LYRIKS (SVM) model
This model was developed largely based on the protocol of Mongan et al. [3], albeit with a more robust evaluation framework. Instead of evaluating model performance on a single hold-out test set, we performed nested cross validation with four outer folds and three inner folds (Fig. 2B). Within each outer split, feature selection was performed using coefficients of SVM models fitted on the inner splits (Supplementary Table 5). A separate SVM model was independently trained on the selected proteins for final prediction.
LYRIKS (elastic net) model
This model was trained and evaluated via nested cross validation with four outer folds and three inner folds (Fig. 2B). Within each outer split, an elastic net logistic regression model (L1 ratio = 0.5) was fitted to all quantified proteins in each of the three inner splits. Feature were selected based on coefficients from the three fitted models, and a final elastic net logistic regression model was trained on the selected proteins. Psychosis conversion biomarkers were identified based on coefficients from all 12 inner models (see Supplementary Table 5 for details). These biomarkers are termed collectively as the LYRIKS (elastic net) signature (Supplementary Table 6).
LYRIKS (ANCOVA) model
This model was trained and evaluated via stratified 4-fold cross validation (Fig. 2A). We opted for four folds due to the small number of converters (n = 13). Within each split, ANCOVA was performed independently on each protein with class labels as the group variable and age and gender as covariates. Resulting P-values were corrected for multiple testing using the Benjamini-Hochberg (BH) procedure, and proteins with BH-adjusted ANCOVA P-values < 0.05 were selected as features for training an elastic net logistic regression model. Psychosis conversion biomarkers were identified by averaging BH-adjusted ANCOVA P-values across all splits and retaining proteins with mean values below 0.05. These biomarkers are collectively termed as the LYRIKS (ANCOVA) signature (Supplementary Table 7).
Negative control analysis
To assess the robustness of the predictive performance estimates and safeguard against optimistic performance bias, we conducted a negative control analysis in which protein signatures were randomly sampled [19] and used to train prediction models using an identical training and evaluation framework to the LYRIKS (ANCOVA) model. A total of 1,000 random signatures, each comprising 15 proteins (matching the size of the LYRIKS (ANCOVA) signature), were sampled from the set of 605 proteins found in the LYRIKS dataset. An elastic net logistic regression model was trained on each random signature and evaluated via 4-fold cross-validation to determine the null distribution of average AUC values. A P-value for each model is calculated as 1 – F(x) where F is the cumulative distribution function of a normal distribution parameterized by the mean and standard deviation (SD) of the null distribution of average AUC values [20].
Functional enrichment analysis
Functional enrichment analysis was conducted using the hypergeometric test (with BH-correction) in the STRING web tool (https://string-db.org/) [21], using the 605 proteins quantified in the LYRIKS dataset as the background proteome. Specifically, analysis was performed on included pathways such as the Kyoto Encyclopedia of Genes and Genomes (KEGG) pathways [22], and STRING network clusters, which are smaller in size. The STRING clusters are obtained from hierarchically clustering the full STRING network.
Statistical analysis
Sample size estimation was performed to ensure sufficient precision to achieve a 95% confidence interval for the AUC with a width less than 0.15, assuming a true AUC of 0.75 and a 2-year incidence risk of psychosis of 15% [23]. A minimum sample size of 317 (47 converters) was required. While recruitment fell short of the requirement due to practical constraints of recruiting high-risk cohorts and the low rate of psychosis transition, all available participants were included to maximize the reliability of the estimates. Sample size was calculated using the powerROC web tool (https://fcgrolleau.github.io/powerROC/) [24].
Assumptions for parametric tests were evaluated qualitatively through visual inspection of data distributions using Q-Q plots and histograms. Parametric tests were applied to variables that met normality assumptions. Statistical analyses were conducted using two-sided t-tests for continuous variables and the chi-squared tests for categorical variables in R (version 4.2.1) [13]. For prediction modeling and differential expression analysis, ANCOVA and the BH procedure were performed using the statsmodels (version 0.14.0) library [25], while paired t-tests were performed using the scipy (version 1.10.1) library [26] in Python (version 3.11.3).
Results
LYRIKS proteomics dataset
We profiled 402 blood plasma samples from 135 participants over the two-year study period at six-month intervals (Fig. 1). Table 1 presents the sociodemographic and clinical characteristics of UHR converters and non-converters. Overall, 18.6% of UHR participants transited to psychosis. No statistically significant differences based on age, body mass index (BMI), gender, ethnicity, smoking status, antidepressants/anxiolytics use, and psychiatric comorbidities were observed. A total of 1,757 proteins were detected, of which 605 proteins were present in more than 50% of samples in all batches and were retained for all subsequent analyses (Fig. 3A).

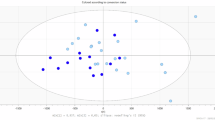
A: Number of proteins identified, quantified, and selected via ANCOVA in the LYRIKS and EU-GEI datasets. Quantification criteria: >50% of samples per batch (LYRIKS) and >80% of samples (EU-GEI). Bracketed numbers indicate number of EU-GEI proteins also detected in LYRIKS. B: Mean area under the receiver operating characteristic curve (AUC) for EU-GEI signatures evaluated on LYRIKS via 4-fold cross-validation. Error bars show standard deviation (SD) across folds. C: PCA of baseline LYRIKS samples using 33 ANCOVA-identified biomarkers from EU-GEI show distinct distributions between converters and non-converters. Concentric ellipses represent two and four SD intervals around each group centroid.
EU-GEI signatures generalize moderately well to the LYRIKS dataset
We first examined whether EU-GEI proteomic signatures could predict transition to psychosis in the LYRIKS dataset. In the EU-GEI dataset, 35 out of 166 proteins were identified as differentially expressed using ANCOVA (p < 0.05). We refer to these proteins collectively as the EU-GEI (ANCOVA) signature (Supplementary Table 3). Of the 35 proteins in the EU-GEI (ANCOVA) signature, 33 (94.3%) were successfully quantified in our dataset. Predictive modeling using these 33 proteins resulted in a mean AUC of 0.81 (Fig. 3B), suggesting that the EU-GEI (ANCOVA) signature generalizes moderately well to the LYRIKS dataset. This result was further supported by principal components analysis (PCA), which showed that converters and non-converters are linearly separable on the EU-GEI (ANCOVA) signature (Fig. 3C).
We next evaluated the condensed 10-protein EU-GEI (SVM) signature (Supplementary Table 4), which achieved a mean AUC of 0.75 (Fig. 3B), reflecting a modest but expected decrease in performance given the reduction in signature size from 33 to 10 proteins. Altogether, these results indicate that both EU-GEI signatures demonstrate acceptable performance on the LYRIKS dataset.
LYRIKS signatures outperform EU-GEI signatures
To investigate the potential of a proteomic signature derived from an Asian cohort, we developed three prediction models using the LYRIKS dataset. The LYRIKS (ANCOVA) model showed the highest performance among the three models, achieving a mean AUC of 0.96 (Fig. 4A). Through the model, 15 proteins with an average BH-adjusted P-value below 0.05 across all cross-validation splits were incorporated into a novel LYRIKS (ANCOVA) signature (Table 2 and Supplementary Table 7). In the second model, we employed a modeling framework similar to that of Mongan et al. [3] and identified a set of 10 proteins through the coefficients of fitted SVM models from all inner cross-validation splits. This alternative approach also yielded good performance, achieving a mean AUC of 0.90. The LYRIKS (elastic net) model likewise performed well with a mean AUC of 0.95 (Fig. 4A). The top 10 proteins were selected based on the magnitude of model coefficients from all inner cross-validation splits and are collectively termed as the LYRIKS (elastic net) signature (Table 2 and Supplementary Table 6). All three models derived from the Asian LYRIKS cohort outperformed models built on EU-GEI signatures, which achieved lower mean AUCs of 0.75 and 0.81. Performance estimates of the LYRIKS (elastic net), LYRIKS (ANCOVA) and LYRIKS (SVM) models were stable across cross-validation splits, exhibiting percentage coefficients of variation for AUC of 4.9% (SD = 0.047), 4.6% (SD = 0.044), and 9.4% (SD = 0.085), respectively. Notably, variability in model performance across splits for all LYRIKS models were consistently lower than the variability in performance of random signature models (Fig. 4A).

A: Mean area under the receiver operating characteristic curve (AUC) from cross-validation on the LYRIKS dataset. LYRIKS (random) refers to the null distribution of predictive performance estimates based on the mean AUCs of 1,000 iterations of models trained on randomly selected 15-protein signatures. Error bars indicate standard deviation (SD). B: PCA of baseline LYRIKS samples (converters vs. non-converters) using the 10-protein LYRIKS (elastic net) signature. Concentric ellipses represent two and four SD intervals around each group centroid.
LYRIKS signatures greatly outperform random signatures
As a negative control, 1,000 random 15-protein signatures were sampled from the 605 quantified proteins in the LYRIKS dataset, and elastic net logistic regression models were trained on these signatures to estimate chance-level performance. All three LYRIKS models significantly outperformed the negative control, which exhibited a mean AUC of 0.66 (SD = 0.123). The LYRIKS (elastic net), LYRIKS (ANCOVA), and LYRIKS (SVM) models, achieved P-values of 0.010, 0.014, and 0.024, respectively. These results demonstrate that LYRIKS models can robustly identify biomarkers associated with psychosis conversion.
LYRIKS and EU-GEI signatures are compositionally distinct but share similar pathways and protein families
We examined overlaps between the LYRIKS (ANCOVA), LYRIKS (elastic net) and EU-GEI (ANCOVA) signatures to assess their concordance (Supplementary Figure 4). A sizeable overlap (Jaccard coefficient = 25%) was observed between the LYRIKS (ANCOVA) and LYRIKS (elastic net) signatures, with five shared proteins out of the 20 proteins present in both signatures. However, of these five overlapping proteins (IGKV1-5, APOB, RPSA, ZNF607 and SERPIND1), only SERPIND1 was present in all three signatures. Additionally, there was limited overlap of five proteins (Jaccard coefficient = 10%) between biomarkers from either of the LYRIKS signatures and the EU-GEI (ANCOVA) signature.
Despite modest overlap in protein composition, several protein families were consistently represented across all three signatures. These included the inter-alpha-trypsin inhibitor heavy chain (ITIH), apolipoprotein, complement, coagulation factor families, as well as the serine protease inhibitors (SERPINs) superfamily. Most overlapping proteins belonged to either the ITIH or SERPIN protein families. Notably, we observed consistent downregulation of proteins across the ITIH family, viz. ITIH1, ITIH2, ITIH3 and ITIH4 (Supplementary Figure 5), although only ITIH1 and ITIH3 were identified in either the LYRIKS or EU-GEI signatures.
Functional enrichment analysis of the 15 biomarkers comprising the LYRIKS (ANCOVA) signature did not reveal significantly enriched KEGG pathways. However, among STRING network clusters, the “complement and coagulation cascades, and protein-lipid complex” cluster was significantly enriched (FDR = 0.0362).
Discussion
Our study substantiates previous findings that plasma proteins are predictive of psychosis conversion in UHR individuals [3, 27,28,29,30,31,32,33]. Importantly, we demonstrated the generalizability of EU-GEI plasma proteomic signatures on our cohort of Asian UHR individuals and developed prediction models using the LYRIKS proteomics dataset which demonstrated superior performance. Our observation that key sociodemographic and clinical covariates such as age, BMI, gender, ethnicity, comorbidities, antidepressants/anxiolytics use and smoking status were not significantly associated with psychosis conversion (Table 1) motivated our decision to restrict the prediction models to proteomic features. Taken together, these findings highlight the potential of using plasma biomarkers for early detection of psychosis conversion.
Importance of population diversity in mental health research
The stronger performance of LYRIKS prediction models underscores the need to include diverse populations in mental health research, which has been largely focused on Caucasian cohorts. Our study provides one of the largest plasma proteomics datasets of an Asian UHR cohort, expanding the ethnic scope of mental health research and demonstrating that while Caucasian-derived signatures demonstrate acceptable discriminatory performance in an Asian cohort, population-specific models may yield more accurate predictions.
Functional convergence of Asian- and Caucasian-derived signatures
We identified novel proteomic signatures through the LYRIKS prediction models. Although there was limited overlap in individual proteins between LYRIKS and EU-GEI signatures, we observed functional convergence across key pathways and protein families. First, many constituent biomarkers in both the LYRIKS and EU-GEI signatures were immune-related and mapped to the complement and coagulation cascade pathway (KEGG: hsa04610), supporting recent evidence implicating the dysregulation of these pathways in schizophrenia and psychosis [34]. In addition, previous studies have reported upregulation of the coagulation and complement cascades and an enhanced inflammatory state before FEP and other mental disorders [3, 27, 28]. Second, we observed recurrent representation of proteins from the ITIH, SERPIN and apolipoprotein families, which is consistent with previous genome-wide association studies associating single nucleotide polymorphisms in ITIH proteins with schizophrenia, bipolar disorder and depression in Caucasian [35,36,37], Japanese [38] and Han Chinese [39, 40] populations.
Dysregulation of lipid-associated proteins in mental health disorders
Functional enrichment analysis of the LYRIKS (ANCOVA) signature revealed significant enrichment of the “complement and coagulation cascades, and protein-lipid complex” STRING network cluster. Specifically, the enzyme LCAT from the cluster was downregulated in baseline samples of converters (Table 2 and Supplementary Table 7); LCAT is essential for the maturation of nascent glial-derived lipoproteins and has been shown to influence APOE and APOA1 levels in CSF [41]. In addition, we observed downregulation of APOB in baseline samples of converters, which corroborates previous findings showing negative association of APOB with cognitive impairment [42] and negative symptoms [43] in Han Chinese patients with schizophrenia. Similarly, the EU-GEI (ANCOVA) signature also contains other apolipoproteins such as APOE, consistent with earlier reports of elevated APOE and reduced APOB, APOD, APOF, APOM in schizophrenia patients. The consistent representation of apolipoproteins in both LYRIKS and EU-GEI signatures support previous literature that associate differential levels of lipoproteins and lipids with various mental health disorders. There is increasing evidence that brain lipids, particularly those involved in membrane forming and signaling, play an important role in the etiologies of mental health disorders [44, 45]. This underscores the value of lipidomics and multiomics approaches in the prognosis, diagnosis, and monitoring of mental health disorders.
Strengths and limitations
To the best of our knowledge, this study presents the largest LC-MS proteomics dataset profiling an Asian UHR cohort to date. Proteins were quantified using the Orbitrap Astral mass spectrometer, ensuring deep proteome coverage, thus enabling the detection of previously reported signatures. Importantly, this study makes a crucial contribution towards expanding mental health research into Asian populations, which addresses concerns regarding the generalizability of findings derived from predominantly Caucasian datasets and ensures the broader applicability of proteomic biomarkers in psychiatry.
We acknowledge several technical limitations and outline the measures taken to address them. First, the number of converters was small (n = 13) relative to non-converters (n = 52), resulting in class imbalance. This was mitigated through class weighting during model training. Related concerns regarding overfitting, model instability and biased performance estimates were addressed through careful design choices, including nested k-fold cross-validation, negative control analysis and constraints on model complexity. Notably, hyperparameters were fixed across prediction models rather than optimized. This decision was made because hyperparameter optimization would have required an additional layer of data partitioning, further reducing the effective training sample size and increasing the risk of overfitting, which could lead to optimistically biased performance estimates. This rationale is supported by recent work demonstrating that hyperparameter optimization in small-sample settings can lead to severe model miscalibration, as well as by evidence that hyperparameter optimization yields only marginal performance gains over default hyperparameter settings [46, 47]. Hyperparameters were therefore fixed to prioritize model stability and consistency. Nevertheless, performance estimates should be interpreted with caution and validated in external cohorts.
Second, analyses were restricted to proteins present in at least 50% of samples for all batches to ensure reliability. Although this is consistent with standard proteomic analysis, filtering proteins with high missingness reduces proteome coverage and may have excluded low-abundance proteins of potential biological relevance from analysis. In future work, we aim to incorporate robust imputation and network algorithms that will allow us to investigate low-abundance proteins and further extend our findings. Finally, although LYRIKS and EU-GEI signatures show functional convergence, external validation in independent cohorts across different ethnicities remains essential to confirm their generalizability and clinical utility.
Conclusion
We present the first single site plasma proteomics study of an Asian UHR cohort for psychosis using the Orbitrap Astral mass spectrometer. By combining deep coverage plasma proteomics with robust machine learning frameworks, we demonstrate that proteomic signatures derived from a predominantly Caucasian sample generalize moderately well to an Asian dataset but are outperformed by prediction models developed from the Asian dataset itself. We derive proteomic signatures from the LYRIKS prediction models, which differ in protein composition from the Caucasian-derived signatures. However, the Asian- and Caucasian-derived signatures consistently included members of the same protein families with shared functions. This convergence corroborates existing literature associating the dysregulation of the complement and coagulation cascade, as well as specific members from the ITIH and apolipoprotein families, with various mental health disorders.
Data availability
The LYRIKS dataset contains sensitive patient information and is not publicly available to protect participant privacy. Requests for access to data can be directed to the corresponding authors and will be considered on the criteria of confidential access and institutional requirements.
Code availability
All codes are available at: https://github.com/dblux/astral-lyriks.
References
Wu Y, Wang L, Tao M, Cao H, Yuan H, Ye M, et al. Changing trends in the global burden of mental disorders from 1990 to 2019 and predicted levels in 25 years. Epidemiol Psychiatr Sci. 2023;32:e63.
Andreassen OA, Hindley GFL, Frei O, Smeland OB. New insights from the last decade of research in psychiatric genetics: discoveries, challenges and clinical implications. World Psychiatry. 2023;22:4–24.
Mongan D, Föcking M, Healy C, Susai SR, Heurich M, Wynne K, et al. Development of proteomic prediction models for transition to psychotic disorder in the clinical high-risk state and psychotic experiences in adolescence. JAMA Psychiatry. 2021;78:77.
Byrne JF, Healy C, Föcking M, Susai SR, Mongan D, Wynne K, et al. Proteomic biomarkers for the prediction of transition to psychosis in individuals at clinical high risk: a multi-cohort model development study. Schizophrenia Bulletin. 2024;50:579–88.
Lee J, Rekhi G, Mitter N, Bong YL, Kraus MS, Lam M, et al. The Longitudinal Youth at Risk Study (LYRIKS) — An Asian UHR perspective. Schizophrenia Research. 2013;151:279–83.
Mitter N, Nah GQR, Bong YL, Lee J, Chong S. Longitudinal Youth-At-Risk Study (LYRIKS): outreach strategies based on a community-engaged framework. Early Intervention Psych. 2014;8:298–303.
Yung AR, Yung AR, Pan Yuen H, Mcgorry PD, Phillips LJ, Kelly D, et al. Mapping the onset of psychosis: the comprehensive assessment of at-risk mental states. Aust N Z J Psychiatry. 2005;39:964–71.
First MB, editor. Structured clinical interview for DSM-IV axis I disorders: SCID-I; administration booklet; [compatible with both DSM-IV and DSM-IV-TR]. Clinician version, 9. [print.]. Arlington, Va: American Psychiatric Publ; 2008. 84 p.
Tuck MK, Chan DW, Chia D, Godwin AK, Grizzle WE, Krueger KE, et al. Standard operating procedures for serum and plasma collection: early detection research network consensus statement Standard Operating Procedure Integration Working Group. J Proteome Res. 2009;8:113–7.
English JA, Lopez LM, O’Gorman A, Föcking M, Hryniewiecka M, Scaife C, et al. Blood-Based protein changes in childhood are associated with increased risk for later psychotic disorder: evidence from a nested case–control study of the ALSPAC longitudinal birth cohort. Schizophrenia Bulletin. 2018;44:297–306.
Demichev V, Messner CB, Vernardis SI, Lilley KS, Ralser M. DIA-NN: neural networks and interference correction enable deep proteome coverage in high throughput. Nat Methods. 2020;17:41–4.
Trevor Hastie RT impute [Internet]. Bioconductor; 2017 [cited 2025 June 16]. Available from: https://bioconductor.org/packages/impute
R Core Team. R: A Language and Environment for Statistical Computing [Internet]. Vienna, Austria: R Foundation for Statistical Computing; 2022. Available from: https://www.R-project.org/
Johnson WE, Li C, Rabinovic A. Adjusting batch effects in microarray expression data using empirical Bayes methods. Biostatistics. 2007;8:118–27.
Jeffrey T Leek <Jtleek@Gmail. Com> WEJE. sva [Internet]. Bioconductor; 2017 [cited 2025 June 16]. Available from: https://bioconductor.org/packages/sva
Jongsma HE, Gayer-Anderson C, Lasalvia A, Quattrone D, Mulè A, Szöke A, et al. Treated incidence of psychotic disorders in the multinational EU-GEI study. JAMA Psychiatry. 2018;75:36.
Pedregosa F, Varoquaux G, Gramfort A, Michel V, Thirion B, Grisel O, et al. Scikit-learn: Machine learning in python. Journal of Machine Learning Research. 2011;12:2825–30.
Van Rossum G, Drake FL Python 3 Reference Manual. Scotts Valley, CA: CreateSpace; 2009.
Goh WWB, Wong L. Why breast cancer signatures are no better than random signatures explained. Drug Discovery Today. 2018;23:1818–23.
Ho SY, Phua K, Wong L, Bin Goh WW. Extensions of the external validation for checking learned model interpretability and generalizability. Patterns. 2020;1:100129.
Szklarczyk D, Kirsch R, Koutrouli M, Nastou K, Mehryary F, Hachilif R, et al. The STRING database in 2023: protein–protein association networks and functional enrichment analyses for any sequenced genome of interest. Nucleic Acids Research. 2023;51:D638–46.
Kanehisa M, Furumichi M, Sato Y, Matsuura Y, Ishiguro-Watanabe M. KEGG: biological systems database as a model of the real world. Nucleic Acids Research. 2025;53:D672–7.
Silvey S, Liu J. Sample size requirements for popular classification algorithms in tabular clinical data: empirical study. J Med Internet Res. 2024;26:e60231.
Grolleau F, Tibshirani R, Chen JH. powerROC: An Interactive web tool for sample size calculation in Assessing models’ discriminative abilities. AMIA Jt Summits Transl Sci Proc. 2025;2025:196–204.
Seabold S, Perktold J statsmodels: Econometric and statistical modeling with python. In: 9th Python in Science Conference. 2010.
Virtanen P, Gommers R, Oliphant TE, Haberland M, Reddy T, Cournapeau D, et al. SciPy 1.0: fundamental algorithms for scientific computing in Python. Nat Methods. 2020;17:261–72.
Perkins DO, Jeffries CD, Addington J, Bearden CE, Cadenhead KS, Cannon TD, et al. Towards a psychosis risk blood diagnostic for persons experiencing high-risk symptoms: preliminary results from the NAPLS project. Schizophrenia Bulletin. 2015;41:419–28.
Chan MK, Krebs MO, Cox D, Guest PC, Yolken RH, Rahmoune H, et al. Development of a blood-based molecular biomarker test for identification of schizophrenia before disease onset. Transl Psychiatry. 2015;5:e601–e601.
Levin Y, Wang L, Schwarz E, Koethe D, Leweke FM, Bahn S. Global proteomic profiling reveals altered proteomic signature in schizophrenia serum. Mol Psychiatry. 2010;15:1088–100.
Li Y, Zhou K, Zhang Z, Sun L, Yang J, Zhang M, et al. Label-free quantitative proteomic analysis reveals dysfunction of complement pathway in peripheral blood of schizophrenia patients: evidence for the immune hypothesis of schizophrenia. Mol BioSyst. 2012;8:2664.
Sabherwal S, English JA, Föcking M, Cagney G, Cotter DR. Blood biomarker discovery in drug-free schizophrenia: the contribution of proteomics and multiplex immunoassays. Expert Review of Proteomics. 2016;13:1141–55.
Rodrigues-Amorim D, Rivera-Baltanás T, del Vallejo-Curto MC, Rodriguez-Jamardo C, de las Heras E, Barreiro-Villar C, et al. Proteomics in schizophrenia: a gateway to discover potential biomarkers of psychoneuroimmune pathways. Front Psychiatry. 2019;10:885.
Woo JJ, Pouget JG, Zai CC, Kennedy JL. The complement system in schizophrenia: where are we now and what’s next?. Mol Psychiatry. 2020;25:114–30.
Kopczynska M, Zelek W, Touchard S, Gaughran F, Di Forti M, Mondelli V, et al. Complement system biomarkers in first episode psychosis. Schizophrenia Research. 2019;204:16–22.
Hamshere ML, Walters JTR, Smith R, Richards AL, Green E, Grozeva D, et al. Genome-wide significant associations in schizophrenia to ITIH3/4, CACNA1C and SDCCAG8, and extensive replication of associations reported by the Schizophrenia PGC. Mol Psychiatry. 2013;18:708–12.
The Schizophrenia Psychiatric Genome-Wide Association Study (GWAS) Consortium. Genome-wide association study identifies five new schizophrenia loci. Nat Genet. 2011;43:969–76.
Psychiatric GWAS Consortium Bipolar Disorder Working Group, Sklar P, Ripke S, Scott LJ, Andreassen OA, Cichon S, et al. Large-scale genome-wide association analysis of bipolar disorder identifies a new susceptibility locus near ODZ4. Nat Genet. 2011;43:977–83.
Miyake Y, Tanaka K, Arakawa M. ITIH3 and ITIH4 polymorphisms and depressive symptoms during pregnancy in Japan: the Kyushu Okinawa Maternal and Child Health Study. J Neural Transm. 2018;125:1503–9.
He K, Wang Q, Chen J, Li T, Li Z, Li W, et al. ITIH family genes confer risk to schizophrenia and major depressive disorder in the Han Chinese population. Progress in Neuro-Psychopharmacology and Biological Psychiatry. 2014;51:34–8.
Li K, Li Y, Wang J, Huo Y, Huang D, Li S, et al. A functional missense variant in ITIH3 affects protein expression and neurodevelopment and confers schizophrenia risk in the Han Chinese population. Journal of Genetics and Genomics. 2020;47:233–48.
Clay MA, Pyle DH, Rye KA, Barter PJ. Formation of spherical, reconstituted high density lipoproteins containing both apolipoproteins A-I and A-II is mediated by lecithin:Cholesterol acyltransferase. Journal of Biological Chemistry. 2000;275:9019–25.
Rao W, Zhang Y, Li K, Zhang XY. Association between cognitive impairment and apolipoprotein A1 or apolipoprotein B levels is regulated by apolipoprotein E variant rs429358 in patients with chronic schizophrenia. Aging. 2021;13:16353–66.
Rao W, Meng X, Li K, Zhang Y, Zhang XY. Association between clinical symptoms and apolipoprotein A1 or apolipoprotein B levels is regulated by apolipoprotein E variant rs429358 in patients with chronic schizophrenia. Ann Gen Psychiatry. 2021;20:56.
Schneider M, Levant B, Reichel M, Gulbins E, Kornhuber J, Müller CP. Lipids in psychiatric disorders and preventive medicine. Neuroscience & Biobehavioral Reviews. 2017;76:336–62.
Couttas TA, Jieu B, Rohleder C, Leweke FM. Current state of fluid lipid biomarkers for personalized diagnostics and therapeutics in schizophrenia spectrum disorders and related psychoses: a narrative review. Front Psychiatry. 2022;13:885904.
Dunias ZS, Van Calster B, Timmerman D, Boulesteix A, Van Smeden M. A comparison of hyperparameter tuning procedures for clinical prediction models: A simulation study. Statistics in Medicine. 2024;43:1119–34.
Wong J, Manderson T, Abrahamowicz M, Buckeridge DL, Tamblyn R. Can hyperparameter tuning improve the performance of a super learner?: A case study. Epidemiology. 2019;30:521–31.
Acknowledgements
This research was supported by the National Medical Research Council (NMRC), Ministry of Health, Singapore, under its Population Health Research Grant scheme, Project No. PHRGOC24jul-0026. The LYRIKS study was supported by the National Research Foundation Singapore under the NMRC Translational and Clinical Research Flagship Programme (grant number NMRC/TCR/003/2008). Jimmy Lee acknowledges funding support from NMRC (grant number MOH-000013-00, MOH-000839-00, MOH-001414-00). W.W.B. Goh acknowledges support from a Ministry of Education, Singapore Tier 1 grant (Grant No. RS08/21).
Author information
Authors and Affiliations
Contributions
W.X. Chan conducted the machine learning modeling, performed data analysis, and wrote the manuscript. J.J. Wong contributed to data analysis and manuscript writing. Z.X. Yang and J.Y. Yee performed the experiments and assisted in data analysis and interpretation. Justin Lee generated the Orbitrap Astral dataset and provided practical advice. Jimmy Lee designed the LYRIKS cohort and collected patient blood samples. W.W.B. Goh conceptualized the project and wrote the manuscript. Jimmy Lee and W.W.B. Goh co-supervised and acquired funding for the project.
Corresponding authors
Ethics declarations
Competing interests
Jimmy Lee has received honoraria and served as a consultant or advisory board member from Otsuka, Janssen, Lundbeck, Sumitomo Pharmaceuticals, Boehringer Ingelheim, ThoughtFull World Pte. Ltd. and Singapore Deep-Tech Alliance. The remaining authors declare no competing interests, financial or otherwise.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Chan, W.X., Wong, J.J., Yang, Z. et al. Blood plasma proteomic biomarkers for forecasting transition to psychosis in an Asian cohort. Transl Psychiatry 16, 219 (2026). https://doi.org/10.1038/s41398-026-04004-7
Received:
Revised:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41398-026-04004-7
