
Abstract
Parkinson’s disease (PD) involves α-synuclein (αSyn) oligomerization and aggregation, processes facilitated by glycosphingolipids. Defective glycosphingolipid transport and degradation—especially via the lipid-degrading enzyme glucocerebrosidase 1 (GCase, gene GBA1)—aggravate PD and increase dementia risk. Ambroxol is a mucolytic drug and has emerged as a promising add-on therapy for PD since it acts as a chaperone for misfolded GCase, thereby increases the likelihood that mutated and misfolded GCase eludes ER-associated degradation (ERAD) and is transported to its destination, the lysosome. In this study we investigated whether and how ambroxol provided therapeutic benefits for PD irrespective of the GBA1 mutation status. Pink1−/−/SNCAA53T double mutant PD mice were administered ambroxol either via the drinking water (120–150 mg·kg−1·d−1) or via food pellets (75–100 mg·kg−1·d−1) for approximately 6 months. During the treatments mice were observed in IntelliCages; and in motor, sensory and cognitive functions tests. After mice were euthanized, tissues were dissected for protein, lipidomic and metabolomic analyses. We showed that high-dose long-term ambroxol was well tolerated and led to mild behavioral and metabolic improvements but had adverse effects on brain sulfatides, lysosomal functions and mitochondrial cardiolipins. Notably, brain levels of glucosylceramides (GlcCer 16:0) were normalized, while sulfatides (SHexCer) further increased. Western blots revealed a modest reduction of αSyn and phosphorylated αSyn (P-Ser129). IntelliCage assessments showed increased exploratory activity with ambroxol, suggesting reduced bradykinesia, though sensory and motor functions remained unchanged. Lipidomic profiles of mitochondria showed accumulation of HexCer and triglycerides in PD mitochondria, regardless of treatment, while ambroxol led to an additional decline of cardiolipins including the most abundant tetralinoleoyl cardiolipins. In HT22 hippocampal neurons preloaded with αSyn pre-formed fibrils, ambroxol accumulated within lysosomes, increased lysosomal mass and sphingolipid content and promoted lysosomal enzyme release. Collectively, these results suggest that ambroxol confers transient behavioral benefits and modestly reduces αSyn pathology, albeit with potential drawbacks. In addition, its lysosomal accumulation may further disrupt sphingolipid metabolism and impair mitochondrial compensatory mechanisms. Ambroxol-induced lysosomal exocytosis may transiently relieve αSyn burden, but further interventions would be required to ensure αSyn clearance from the brain.
Similar content being viewed by others


Introduction
Parkinson’s disease (PD) is characterized by the progressive loss of midbrain dopaminergic neurons [1], manifesting in characteristic PD motor symptoms including tremors, muscle rigidity, bradykinesia and postural instability [2, 3]. PD also leads to non-motor symptoms, including chronic pain, REM sleep disorder, autonomic, olfactory and cognitive dysfunctions, in part preceding the motor disease [1, 4, 5]. The predominant morphological feature of PD consists in oligomeric alpha synuclein (αSyn, SNCA gene) deposits in Lewy bodies [6,7,8], which are cause and consequence of dysfunctions of the proteasome [9, 10] and of autophagolysosomal pathways of protein and organelle degradation [11, 12], which lead, among others to insufficient mitochondrial segregation and renewal [13, 14]. The removal of oligomeric αSyn deposits depends on the autophagolysosomal pathway [15,16,17]. If degradation fails, αSyn-laden lysosomes can be expelled, thereby momentarily unburdening affected neurons but allowing further spreading of αSyn [18,19,20].
It has been recognized recently that Lewy-bodies not only consist of αSyn-aggregates but rather αSyn-sphingolipid conglomerates, and glucosyl-sphingolipids precipitate the formation of αSyn oligomers [21, 22] and increase their toxicity [23,24,25]. The accumulation of αSyn in lysosomes leads to a malfunction of lysosomal enzymes, in particular the lipid-degrading enzyme glucocerebrosidase 1 (GCase, gene GBA1) [26, 27], which catalyzes the metabolism of glucosylceramides (GlcCer) to ceramides (Cer) and D-glucose. GlcCer are components of membrane barriers and are involved in the maintenance of organelle curvature, but overloading interferes with the lysosomal membrane charge, thereby leading to leakage [28, 29]. Consequently, GBA1 mutations and GCase malfunctioning aggravate deficits in αSyn degradation. Some heterozygous GBA1 mutations have been recognized as the cause of GBA1-associated PD, which is often rapidly progressive and has a high risk of PD dementia complex [26, 30,31,32,33]. In addition, we and others have shown that low GCase activity and accumulation of GlcCer also occur in PD patients without known causative mutations of GBA1 [26, 34, 35]. High levels of GlcCer were associated with high pain ratings and sensory loss in PD patients and PD mice [34, 36]. The discovery of GBA1 as a critical PD-risk gene has encouraged research in GBA1 refolding drugs [37] such as ambroxol or enzyme replacements as PD therapeutics.
Ambroxol is a mucolytic drug and has emerged as a promising add-on therapy for PD because ambroxol acts as a chaperone for misfolded GCase, and thereby increases the likelihood that mutated and misfolded GCase eludes ER-associated degradation (ERAD) and is transported to its destination, the lysosome [38,39,40,41,42]. It has been shown that ambroxol binds to and mobilizes segregated mutant GCase from the endoplasmic reticulum, induces a conformational change and facilitates the transport to the lysosome, which is carried out by the integral membrane protein LIMP2 (Scarb2 = gene name) [43]. The most frequent PD-associated E326K mutation does not alter the structural conformation of the catalytic site but makes the dimeric complex of GBA1 unstable, especially at lysosomal pH, and interferes with the LIMP2-interaction and hence transport [44, 45]. In the acidic environments of the lysosome, ambroxol detaches from GCase, which allows GCase to resume its catalytic activity [46]. In initial clinical studies, ambroxol met the primary required endpoints of bioavailability, brain penetrance and absence of relevant adverse effects [42]. Efficacy studies are still ongoing [47, 48]. At high doses, ambroxol was found to improve the outcome in transgenic Gaucher mice [38], and in human Gaucher patients [49,50,51]. PD-associated GBA1 mutations are heterozygous and mostly do not lead to relevant misfolding or a serious drop of GCase activity [52]. Still, even without GBA1 mutations, ceramides and glucosylceramides are associated with PD pathology [53] and it is therefore clinically important if and how ambroxol might provide a therapeutic benefit for PD irrespective of the GBA1 mutation status.
Therefore, we used a previously described Pink1−/−SNCAA53T double mutant mouse model of PD [54] to study the efficacy of ambroxol treatment on sensory, motor and cognitive functions and on plasma and brain lipidomic and metabolomic patterns. In addition, we used HT22 hippocampal neurons and αSyn pre-formed fibrils as an in vitro model to study effects of ambroxol on sphingolipid biology and lysosomal functions.
Materials and methods
Double mutant Pink1 −/− SNCA A53T Parkinson-model mice
Homozygous Pink1−/− plus SNCAA53T double mutant mice were generated as described in [36, 54] by crossing Pink1−/− mice (MGI:3850370) with PrPmtA mice expressing human mutant SNCA A53T (mtA) under the prion promoter (PrP) (MGI:3723258) and then interbreeding the littermates. Mice are available from Jackson lab as cryopreserved sperm (FVB;129-Pink1tm1Aub X Tg(Prnp-SNCA*A53T)AAub/J; Strain #:017678) Double mutant mice are referred to as Pink1−/−SNCAA53T or in short, Pink1SNCA.
We used Pink1−/−SNCAA53T double mutant mice because unlike other PD models, they develop spontaneous PD-like motor symptoms with an onset at >15 months of age and a frequency of about 20-30 percent [54] and they combine the complex PD pathophysiology of mitochondrial and autophagolysosomal dysfunctions [55,56,57,58]. Further, in contrast to single mutant mice they develop sensory premotor deficits reminiscent of the human premotor phase that manifests with a sensory loss and pain [59] and is associated in humans with increased plasma GlcCer species [34]. Sensory deficits in Pink1−/−SNCAA53T double mutant mice are first detectable at around 6 months of age and are progressive [59]. At 1 year of age, PD-like pathology is well developed (mitochondrial morphology [59], respiratory dysfunction [59], synuclein aggregates [54], ceramide accumulation [59]) but still without overt serious motor deficits and suffering of the animals. Overall, Pink1−/−SNCAA53T double mutant mice show many features of human PD.
Mice had free access to water and food and were kept in climate-controlled rooms with a 12 h light-dark cycle. The behavioral studies were approved by the local Ethics Committee for animal research (Darmstadt, Germany) under FK1026, FK1059, and FK1096. The maintenance of the breeding colony of Pink1−/−SNCAA53T and observation up to onset of PD symptoms were approved under FK1032 (12.02.205-11.02.2020) and FK1131 (17.02.2020-31.12.2023). The studies adhered to the guidelines of the Society of Laboratory Animals (GV-SOLAS) and were in line with the European and German regulations for animal research and the ARRIVE guideline. Mice were observed for approximately 12 months and were euthanized to obtain tissue and plasma samples for lipid analyses before onset of overt clinical symptoms which occur in about 20 percent of mice > 18 months [36, 54].
Ambroxol treatment
Pink1−/−SNCAA53T mice were treated with high doses of ambroxol orally, either via the drinking water (cohort 1, n = 10 per group) or via food pellets (cohorts 2 and 3; n = 6–7 per group). Treatments were continuous for approximately 6 months. Ambroxol has a high > 90% oral bioavailability, distributes to the brain and increases GCase activity in the brain in a GBA1 mutant mouse model [38]. For the drinking water cohort, the daily dose for a mouse with a body weight of 25 grams and drinking of ̴4 ml per day was ~120–150 mg·kg−1·d−1. Because of a mildly bitter taste, controls received water with slightly bitter taste. For ambroxol food pellets, 100 ml 1:2 diluted Mucosolvan® liquid (15 mg/ml) was sprayed onto 1 kg pellets, which were then dried. Depending on the food consumption (about 4 grams per day) and the body weight (25–30 g) the mice received 75–100 mg·kg−1·d−1. The doses were based on a previous study where 4 mM ambroxol drinking water for 12 days increased brain GCase activity in a mouse Gaucher model [38]. Assuming a daily drinking volume of 4 ml and mouse body weight of 25 g, this would be 240 mg·kg−1·d−1. Because our treatment period was much longer we used about half of this dose to avoid toxicity such as phospholipidosis, which is a putative side effect of cationic amphiphilic drugs including ambroxol [60,61,62]. In comparison, Gaucher patients received up to 27 mg·kg−1·d−1 [63], and patients with PD-associated dementia shall receive 15–20 mg·kg−1·d−1 for 6 months (NCT05778617; status recruiting).
Administration via drinking water or food pellets was chosen because it allows for stress-free continuous long-term oral drug treatment with low daily fluctuations of the concentrations, which align with the circadian rhythms of activity and drinking/feeding habits of the mice. It is therefore compatible with complex behavioral studies including IntelliCage observations.
General health and weight were monitored 1x per month. During treatments mice were observed in IntelliCages, Thermal Gradient Ring, Hotplate and Rotarod tests to assess motor, sensory and cognitive functions with and without ambroxol treatment. Finally, mice were euthanized and tissues were dissected for protein, lipidomic and metabolomic analyses.
Mice tissue collection: brain and plasma
Mice were euthanized with slowly rising carbon dioxide and blood withdrawal by cardiac puncture, whereby blood was collected into K3+ EDTA tubes, centrifugated at 1300 × g for 5 min, and plasma was transferred to a fresh tube and snap frozen on dry ice or in liquid nitrogen. The brain was dissected for lipidomic and proteomic analyses. Cerebellum and olfactory bulb were removed, and the brain was cut sagittally. Left and right halves were weighed with precision scales and snap frozen on dry ice. In some experiments, the spinal cord, dorsal root ganglia, prefrontal cortex and hippocampus were separately dissected. Samples were stored at −80 °C until analysis.
Behavior
Behavioural analyses were done with unbiased video-based Thermal Gradient Ring, Hotplate, Rotarod tests or IntelliCage. Mice were habituated to rooms and test environments before baseline measurements.
Assessment of thermal sensitivity (hotplate and thermal gradient ring (TGR))
The sensitivity to painful heat stimuli was assessed as paw withdrawal latency with a Hot Plate at 52 °C according to standard procedures [36, 64].
A thermal gradient ring (TGR) was used to assess the temperature preferences and the exploration of the ring platform that consists of a circular running ring platform that allows free choice of the comfort zone [36, 64]. The dimensions of inner and outer ring diameters are 45 cm and 57 cm. The inner walls consist of plexiglass and the outer walls of aluminum. Both are 12 cm high and build a 6 cm wide circular running arena. The aluminum surface provides a temperature gradient that is controlled with two Peltier elements and constantly measured with infrared cameras. The arena is divided into mirror-image semicircles of 12 temperature zones, so that duplicate readouts are provided for each zone. During measurements, the running track is illuminated, and the mouse behavior is videotaped with a regular CCD camera, mounted above the mid-point of the ring. The time spent in zones and temperature preferences are analyzed with the TGR ANY-Maze video tracking software (Stoelting).
Assessment of motor function (rotarod tests)
Motor coordination and running performance were assessed with the accelerating Rotarod test (cohort 1: 16–32 rpm, ramp 3 rpm/min, cutoff 5 min; cohort 2 and 3: 10–40 rpm, ramp 4 rpm/min, cutoff 5 min; Ugo Basile). Mice were trained in short training runs for adaptation before the baseline measurements. The running time from three test trials was averaged. The cut-off time was 300 s.
IntelliCage set up and tasks
The IntelliCage (TSE, Berlin Germany) consists of four operant corners, each with two water bottles, sensors, LEDs, and doors that control the access to the water bottles. The system fits into a large cage (20 cm× 55 cm× 38 cm, Tecniplast, 2000P) and allows housing of 16 mice per cage. Four triangular red houses are placed in the center to serve as sleeping quarters and as stands to reach the food. The floor is covered with standard bedding. Mice are tagged with radiofrequency identification (RFID)-transponders, which are read with an RFID antenna integrated in the corner entrances. The corners give access to two holes with water bottles, which can be opened and closed by automated doors. Mice must make nosepokes (NP) i.e. peak through a light barrier to open the doors for water access. The IntelliCage is controlled by IntelliCage Plus software, which executes pre-programmed experimental tasks and schedules. The numbers and duration of corner visits, NP, and licks are recorded continuously without the need for handling of the mice during the recording times. Learning and memory can be supported by LEDs.
IntelliCage tasks address several different aspects of cognition as well as circadian rhythms and social interactions and were run sequentially. The tasks followed previously established protocols [65,66,67]. The IntelliCage experiments were done in female mice to avoid fighting. Up to 16 mice were housed per cage (n = 8 per group of Sv129FVB-WT versus Pink1−/−SNCAA53T double mutant; n = 10 per group of Pink1−/−SNCAA53T with Ambroxol or vehicle). The first experiment of WT versus double mutant PD mice was done in 2015, the Ambroxol experiment in 2020. The schedules of the tasks were comparable, and details are presented in Supplementary Table 1A, B. Mice were adapted to the cages for 1–2 weeks with free access to every corner, with all doors open, and water and food ad libitum. This free adaptation (FA) was followed by “nosepoke adaptation” (NP) for 1–2 weeks in which the doors were closed. The first NP of the visit opened the door for 5 s, or 2 s in special tasks. To drink more, a mouse must leave the corner and start a new visit. In the place preference learning (PPL) task mice learned to prefer a specific corner in which an NP opened the door to get water access. Each 4 mice were assigned to one corner. After conditioning to the corner for 1–2 weeks, the rewarding corner was switched to the opposite side (PPL reversal learning (PPLrev) for another 1–2 weeks. In an opposite switching protocol (Osw), the correct corner switches every 12 or 24 h. In a social competition experiment, all mice of a cage were assigned to one correct corner.
Protein extraction, Western blot and isolation of mitochondria
Protein extracts were prepared from brain, hippocampus and spinal cord in RIPA lysis buffer (Sigma) containing a protease inhibitor cocktail (Roche). The protein amount was quantified using Bradford reagent.
Proteins (30 µg/lane) were separated on a Mini-PROTEAN® TGX Protein Gel (4-15%, Bio-Rad) and transferred to nitrocellulose membranes (Amersham) by electro-blotting (Bio-Rad). Blots were fixated in 0.4% PFA in 1× PBS for 30 min at room temperature, then blocked in 5% milk powder in 1× PBS or Odyssey blocking buffer in 1× PBS/Tween 20 for 90 min at room temperature. The primary and secondary antibodies were either diluted in Odyssey blocking buffer in 1× PBS/Tween 20 or 3% BSA in PBS depending on the target protein. Incubation with primary antibodies (Supplementary Table 2) was done at 4 °C overnight with overhead rotation. The first incubation was for phospho-S129 SNCA, followed by SNCA and finally the loading control, beta actin. Secondary antibodies conjugated with IRDye 680 or 800 (1:1000; LI-COR Biosciences, Bad Homburg, Germany) were used for detection (1–2 h at RT). Blots were sequentially developed and analyzed on the Odyssey Infrared Imaging System (RRID:SCR_022510, LI-COR Biosciences), superimposed and quantified using ImageStudio Light (RRID:SCR_013715, LICOR Biosciences).
Mitochondria were isolated from mouse brain or heart using discontinuous Percoll density gradient centrifugation (Qproteome mitochondria, Qiagen) according to the manufacturer’s instructions. The tissue was homogenized using a Precellys 24-Dual tissue homogenizer coupled with a Cryolys cooling module with 10 zirconium dioxide grinding balls (3×20 s at 6500 ×g with 60 s breaks), operated at <6 °C. Mitochondrial yield was assessed using a bicinchoninic acid assay of mitochondrial protein. Mitochondria were processed for untargeted lipidomic and cardiolipin analyses.
Primary DRG neuron culture
To assess mitochondrial respiration, primary neurons of dorsal root ganglia (DRG) of each nine mice per genotype were prepared by dissecting DRGs of adult mice into Hank’s balanced salt solution (HBSS, Merck), followed by digestion with 5 mg/ml collagenase A (Millipore) and 1 mg/ml dispase II (Roche Diagnostics, Germany) before treatment with DNase (Sigma, 250 U per sample). Triturated cells were centrifuged through a 15% fat-free bovine serum albumin (BSA) solution. Neurons were seeded in poly-L-lysine coated Seahorse miniplates. Cells were cultured in serum-free Neurobasal medium containing 1× B27 supplement, 1× Pen/Strep, 200 ng/ml nerve growth factor and 2 mM L-glutamine at 37 °C and 5% CO2 and 95% humidity.
Seahorse analysis of respiration in primary neurons
Mitochondrial respiration was analyzed with the Mito Stress Test (Agilent Technologies) on an XFp Seahorse analyzer. It is a microplate-based live cell assay for monitoring the oxygen consumption rates (OCR) and extracellular acidification rates (ECAR) in living cells. Neurons were prepared as described above and seeded in XFp miniplates in complete medium. Twenty four hours after seeding, the culture medium was switched to Seahorse XF medium supplemented with 1 mM pyruvate, 2 mM glutamine and 10 mM glucose, and neurons were allowed to equilibrate in a non CO2-inbubator. Cultures were then transferred to the XFp analyzer and subjected to the mitochondrial stress test protocol. Basal respiration was measured under full nutrient supply. Subsequently, oligomycin (1.5 µM) was added to assess ATP generation, proton leakage and extracellular acidification. Oligomycin blocks ATP synthase, manifesting in a drop of OCR and raise of ECAR. In the next step, FCCP (carbonyl cyanide-4-(trifluoromethoxy)-phenylhydrazone; 0.5 µM and 1.0 µM) was injected to assess maximum oxygen consumption. FCCP causes a leak of H+ across the mitochondrial membrane leading to a collapse of the membrane potential. Finally, residual non-mitochondrial respiration was determined after inhibition of complex I with rotenone (0.5 µM) and complex-III with antimycin A (0.5 µM). Finally, cultures were imaged for evaluation of equal density. Oxygen consumption rate (OCR) and extracellular acidification rate (ECAR) were analyzed using the Seahorse XF Wave® software.
HT22 cell culture, αSyn-PFF loading, and immunofluorescence analysis
HT22 mouse immortalized hippocampal neurons (RRID:CVCL_0321) were grown in High Glucose Dulbecco’s modified Eagle’s medium (DMEM, Gibco) containing 10% fetal bovine serum (FBS, Gibco), 2 mM L-glutamine (Gibco) and 1× penicillin/streptomycin (Sigma) at 37 °C and in 5% CO2 atmosphere in a humidified cell culture incubator.
For the stimulation of HT22 neurons with pre-formed active αSyn fibrils (αSyn-PFF) (A53T Mutant Alpha Synuclein Active Protein, MyBioSource), sub-confluent cells were trypsinized, pelleted, and resuspended in full medium containing αSyn-PFF and seeded in T75 flasks (Greiner) to achieve a final concentration of 2 µg/ml PFF or 6 µg/ml PFF and cell density of 0.4 × 106 cells/flask. Control cells were plated in full medium without PFF. Treatments (ambroxol (40 µM), pimozide (5 µM), ambroxol (40 µM) plus pimozide (5 µM) (Abx & PIM) or the vehicle (DMSO) were added by partial medium exchange. The target treatment concentrations were to be non-toxic (no cell death over 72 h) based on IC50 values in the WST-1 assay.
For lipid analyses cells were incubated for 72 h and harvested by trypsinization. Cells were washed 3-times with 1× PBS, resuspended in 1 ml 1× PBS, counted and adjusted to 250,000 cells per sample, and finally pelleted. The supernatant was collected for LDH activity assay, Cathepsin D activity assay and αSyn ELISA and pellets were stored at −80 °C until analysis. For lipid analysis, 8 replicates were generated for each treatment.
For live imaging of αSyn-PFF uptake under the influence of the respective treatments, HT22 cells (5000 cells per well) were seeded in 8-well poly-L-lysine coated (0.01%) cover glass bottom culture slides in full medium in the presence of αSyn-PFF labeled with a fluorophore (Alexa Fluor 555 Microscale Protein Labeling Kit, Invitrogen) for 48 h. Then ambroxol (40 µM), pimozide (5 µM), Abx & PIM or vehicle (DMSO) were added by medium exchange followed by incubation for 24 h. Live culture images were obtained on a Leica Stellaris 8 fluorescent confocal microscope (RRID:SCR_024660). To visualize the plasma membrane, cultures were incubated 15 min before imaging with wheat germ agglutinin-Alexa Fluor 488 reagent (WGA, Invitrogen). Hoechst 33342 was used as nuclear stain (Thermo Scientific).
For live imaging of lysosomes under the influence of ambroxol or pimozide treatment, HT22 cells were seeded and treated as described above. To visualize the lysosomes the Lyso-ID®Green detection kit (Enzo Life Sciences) was used. Hoechst 33342 was used as nuclear stain (included in the kit). Live culture images were obtained on a Leica Stellaris 8 fluorescent confocal microscope (RRID:SCR_024660).
For quantification, images were converted to binary images using auto-threshold settings in FIJI ImageJ and subsequent analysis using the particle counter plugin. The αSyn-PFF-AF555 area or the Lyso-ID-Green immunofluorescent area was divided by the Hoechst immunofluorescent area representing the nuclei to normalize the results on the number of cells in the respective image. The experiments were done 3-times, and from each 4–6 images were analyzed per condition. Data were submitted to one-way ANOVA and subsequent post hoc Tukey adjusted t-tests. ****P < 0.0001.
WST-1 assay
The WST-1 cell proliferation reagent (Roche) was used to measure cell viability or cell proliferation of HT22 cells treated with different concentrations of ambroxol or pimozide. It was used to determine the IC50 of the drugs. HT22 cells were cultured for 24 h in a 96-well plate (3500 cells/well) in full DMEM medium. Ambroxol or pimozide was added in culture medium to reach final concentrations of ambroxol 0-800 µM and pimozide 0–20 µM, and cells were incubated for another 24–48 h. The medium was then replaced with 100 μl of fresh medium (per well), and 10 µl WST-1 solution was added to each well. After 90 min in the incubator, the absorbance was read on a multimode microplate reader (SpectraMax i3X, RRID:SCR_026346) at 450 nm. The reference wavelength was set to 620 nm. The absorbance was normalized to the respective reference wavelength and the average absorbance of blanks (WST-1 without cells) was subtracted from the sample absorbances.
Lactate dehydrogenase (LDH) and cathepsin D activity assay
LDH activity was measured in cell culture supernatants using the LDH activity assay kit (Sigma). The supernatant (5 µl) and the NADH standards (0–12.5 nmol/well) were pipetted into the wells of a 96-well plate. The master reaction mix consisting of assay buffer and substrate mix was added to the wells. The absorbance was measured on a microplate reader at 450 nm every 5 min until the value of the most active sample exceeded the value of the highest standard. The absorbance was corrected with the blank, and LDH activity was calculated as described in the manufacturer’s protocol.
Cathepsin D activity was measured in culture supernatants using a fluorescent cathepsin D activity assay kit (Abcam). 25 µl of the supernatant was pipetted into a 96-well plate. The reaction mix consisting of assay buffer and substrate was added to the wells and after a 90 min incubation at 37 °C, the fluorescence intensity was measured at Ex/Em = 328/460 nm. The cathepsin D activity is expressed by relative units (RFU) compared to the DMSO control sample.
Untargeted lipidomic and metabolomic analyses
Mouse blood was collected in 500 µl K3 EDTA tubes, centrifuged at 1300 ×g or 1500 ×g and plasma was transferred into 1.5 ml tubes and stored at −80 °C until further use. Mouse tissue samples were homogenized by adding ethanol:water (1:3, v/v, tissue concentration 0.02 µg/ml) using a Precellys 24-Dual tissue homogenizer coupled with a Cryolys cooling module (both Bertin Technologies, Montigny-le-Bretonneux, France) with 10 zirconium dioxide grinding balls (3*20 s at 6500 ×g with 60 s breaks), operated at <6 °C. Subsequently, 20 µl of the homogenate containing 1 mg of tissue was extracted using a liquid-liquid-extraction method. Cells were collected by trypsinization, washed, pelleted, resuspended in 100 µl counted and adjusted to yield 1.5 × 106 cells (experiment-1) or 2.5 × 105 cells (experiment-2) per pellet. Finally, the cells were pelleted at high speed and supernatant removed with 100 µl and subsequently 20 µl pipet. The “dry” pellet was frozen at −80 °C until lipid/metabolite extraction. For mouse plasma 10 µl was extracted.
Lipidomic analysis and metabolomic analysis were conducted by applying the same procedure as previously described [68]. Further protocol details for extraction and analyses of different matrices (cells, tissue, plasma, mitochondria) are described in the supplementary methods (Excel file).
Briefly, a methyl-tert-butyl-ether (MTBE) and methanol-based liquid-liquid extraction was used to allow for the simultaneous analysis of polar metabolites and lipids from the same sample. For analysis of cell pellets, some experiments also included the targeted analyses of sphingoid bases and ceramides from the same samples.
For chromatographic separation of lipids, a Zorbax RRHD Eclipse Plus C8 1.8 µm 50 × 2.1 mm ID column (Agilent, Waldbronn, Germany) with a pre-column of the same type was used. The mobile phases were (A) 0.1% formic acid and 10 mM ammonium formate and (B) 0.1% formic acid in acetonitrile:isopropanol (2:3, v/v). For metabolomic analysis, polar metabolites were separated on a SeQuant ZIC-HILIC, 3.5 µm, 100 mm × 2.1 mm I.D. column coupled to a guard column with the same chemistry (both Merck, Darmstadt, Germany) and a KrudKatcher inline filter (Phenomenex, Aschaffenburg, Germany). Using 0.1% formic acid in water (solvent A) and 0.1% formic acid in acetonitrile (solvent B), binary gradient elution was performed with a run time of 25 min.
Analyses of lipids and polar metabolites were performed on an Orbitrap Exploris 480 with a Vanquish Horizon UHPLC system (both Thermo Fisher Scientific, Dreieich, Germany). Data was acquired using Thermo Scientific XCalibur v4.4 (RRID:SCR_014593) and relative quantification was performed in Thermo Scientific TraceFinder 5.1 (RRID:SCR_023045). Full scan spectra were acquired from 180-1500 m/z (lipidomics), 70–700 m/z (metabolomics positive ion mode) or 59-590 m/z (metabolomics negative ion mode) at 120,000 mass resolution each for 0.6 s, and data dependent MS/MS spectra at 15,000 mass resolution in between.
For all lipid analyses, the area under the curve (AUC) was used for quantification. AUCs of lipid species were divided by the AUC of the respective internal standard, referred to as AUC/IS. AUCs were transformed to square root of the AUC (sqrt AUC or sqrt AUC/IS) for data analysis. For multivariate analyses sqrt AUCs were scaled to have a common mean and variance of 1 (autoscaling in MetaboAnalyst). Mitochondrial lipidomic data were adjusted to the mitochondrial mass based on protein and total lipid content. Relative quantification of metabolites was performed based on peak areas in extracted ion chromatograms, normalized by median-based probabilistic quotient normalization (PQN).
Semitargeted cardiolipin analysis
Based on the same extraction protocol, software and instrumentation as used for the lipidomics analysis, cardiolipins were analyzed using an adapted chromatography from Seneviratne et al. [69] by injecting the identical mitochondrial lipid extract used for lipidomics. The chromatographic separation was done with a Zorbax RRHD Eclipse Plus C8 1.8 µm 50 × 2.1 mm ID column (Agilent, Waldbronn, Germany) and an 8 min binary gradient. Mobile phases were (A) Acetonitrile:water (6:4) with 0.1% formic acid and 10 mM ammonium formate, and (B) Acetonitrile:isopropanol (1:9) with 0.1% formic acid and 10 mM ammonium formate. Full Scan spectra were acquired from 200 to 2000 m/z at 240,000 mass resolution. MS/MS spectra were acquired from representative samples using AcquireX iterative scan at 120,000 mass resolution for MS1 and 15,000 mass resolution for MS2. The AUC was used for quantification, was divided by 1000 to reduce numbers and then transformed to sqrt AUC for analysis. Analytical details can be found in the supplementary Excel file.
Targeted lipid analyses
Targeted sphingolipid analyses were done in plasma, cell and tissue extracts essentially as described previously [34] using liquid chromatography-electrospray ionization-tandem mass spectrometry (LC-ESI-MS/MS), according to procedures described in detail in [70]. Endocannabinoids were analyzed in mouse tissue with LC-ESI-MS/MS as described in [70, 71]. Further details are described in the supplementary methods (Excel file). Tissue samples for analysis of endocannabinoids were prepared in the same manner as for untargeted lipidomics. Tissues for analysis of sphingolipids were homogenized accordingly or by using a ball mill mixer (MM400, Retsch, Haan, Germany) with 4 zirconium oxide grinding balls (25 Hz for 2.5 min). Subsequently, lipids were extracted from the homogenates (sphingolipids: whole tissue piece extracted (DRG & sciatic nerve), 100 µl of 0.01 mg/µl (brain), endocannabinoids: 40 µl of 0.025 mg/µl sciatic nerve, 20 µl of 0.1 mg/µl, additionally samples were measured after a 1:25 (sciatic nerve) or 1:100 (brain) dilution with ethanol:water (1:3, v/v)) using a liquid-liquid-extraction method after spiking with the internal standards. For plasma samples, we used 10 μl for lipid extraction. Sphingolipids were separated using an Agilent 1260 Infinity II UHPLC System (RRID:SCR_019365) equipped with a Zorbax C18 Eclipse Plus UHPLC column (50 Å~ 2.1 mm, 1.8 μm, Agilent, Waldbronn, Germany). Endocannabinoids were separated using an Agilent 1290 Infinity II UHPLC system equipped with an Acquity UPLC BEH C18 UPLC column (100 Å~ 2.1 mm, 1.7 μm, Waters, Eschborn, Germany). For mass spectrometry, we used a hybrid triple quadrupole-ion trap mass spectrometer QTRAP 6500+ (Sciex, Darmstadt, Germany), equipped with a Turbo-V-source operating in positive ESI mode.
Quality control samples of three different concentration levels (low, middle, high) were run as initial and final samples of each run. For all analytes, the concentrations of the calibration standards, quality controls and samples were evaluated by Analyst®TF Software (RRID:SCR_015785) v.1.7.1 and MultiQuant Software 3.02 (Sciex) using the internal standard method (isotope-dilution mass spectrometry). Calibration curves were calculated by linear or quadratic regression with 1/x weighting or 1/x2 weighting. Variations in accuracy of the calibration standards were less than 15% over the range of calibration, except for the lower limit of quantification (LLOQ), where a variation in accuracy of 20% was accepted. Lipid concentrations in tissue are expressed as ng/ml (or pg/ml) of plasma, or pg/2.5 × 105 cells. For multivariate statistical analyses, lipid data were scaled to have a common mean and standard deviation of 1 (referred to as “autoscaling” in MetaboAnalyst).
Proteomics
Tissue sampling and protein extraction
Each seven Pink1−/−SNCAA53T and wildtype Sv129-FVB mice were euthanized by slowly rising carbon dioxide. The blood was removed and collected in K3-EDTA tubes by cardiac puncture. The brain was rapidly excised, and the prefrontal cortex was snap frozen and stored in liquid nitrogen pending protein extraction. Tissue, frozen and stored in liquid nitrogen, was directly transferred in a glass potter containing 2 ml of 20% TCA and homogenized with 10 strokes at 1300 rpm. Proteins were precipitated by centrifugation followed by two washing steps with 10% TCA and 5% TCA, respectively. Pellets were resolved in 6 M guanidine hydrochloride (GdmCl), 100 mM Tris/HCl, pH 8.5, 10 mM TCEP and incubated at 95 °C for 5 min. Reduced thiols were alkylated with 40 mM chloroacetamide and samples were diluted with 25 mM Tris/HCl, pH 8.5, 10% acetonitrile to obtain a final GdmCl concentration of 0.6 M. 100 µg of protein was digested with 1 µg trypsin (sequencing grade, Promega) overnight at 37 °C under gentle agitation [72]. Digestion was stopped by adding trifluoroacetic acid to a final concentration of 0.5%. Peptides were loaded on multi-stop-and-go tips (StageTip) containing six C18-disks. Purification and elution of peptides was performed as described [73]. Peptides were eluted in wells of microtiter plates and peptides were dried and resolved in 1% acetonitrile, 0.1% formic acid.
Proteomics: label free mass spectrometry
For proteome analysis, the LC/MS was performed on a Thermo Scientific™ Q Exactive Plus equipped with an ultra-high-performance liquid chromatography unit (Thermo Scientific Easy-nLC) and a Nanospray Flex Ion-Source (Thermo Scientific). Peptides were loaded on a C18 reversed-phase pre-column (Thermo Scientific) followed by separation on in-house packed picotip emitter tips (diameter 100 µm, 15 cm long from New Objectives) with 2.4 µm Reprosil C18 resin (Dr. Maisch GmbH). The gradient was from mobile phase A (4% acetonitrile, 0.1% formic acid) to 30% mobile phase B (80% acetonitrile, 0.1% formic acid) for 60 min followed by a second gradient to 60% B for 30 min at a flow rate of 300 nl/min.
MS data were recorded by data dependent acquisition Top10 method selecting the most abundant precursor ions in positive ion mode for HCD fragmentation. The full MS scan range was 350–2000 m/z with resolution of 70,000 and an automatic gain control (AGC) value of 3E6 total ion counts. The maximal ion injection time was 160 ms. Higher charged ions (2 + ) were selected for MS/MS scans with a resolution of 17,500, an isolation window of 1.6 m/z and an automatic gain control value set to E5 ions. The maximal ion injection time was 80 ms. Ions were excluded if they occurred within a time-window of 60 s following a fragmentation event. Full scan data were acquired in profile mode and fragments in centroid mode by Xcalibur software.
Data processing protocol
Xcalibur raw files were analyzed using the proteomics software Max Quant (1.6.1.0) [74]. The enzyme specificity was set to Trypsin, missed cleavages were limited to 2. Acetylation of N-terminus ( + 42.01) and oxidation of methionine ( + 15.99) were set as variable modifications, and carbamidomethylation ( + 57.02) on cysteines was set as fixed modification.
The reference mouse proteome from Uniprot (reviewed 17 K entries) was used to identify peptides and proteins. The False discovery rate (FDR) was set to 1%. Label free quantification values were obtained from at least one identified peptide. Identifications from the reverse decoy database, identified by site only and known contaminants were excluded.
Data deposited to PRIDE; Project accession: PXD039172.
Username: reviewer_pxd039172@ebi.ac.uk; Password: OoFBNmAy.
Data analysis and statistics
Group data are presented as mean ± SD, mean ± sem for behavioral data, or median ± IQR as specified in the respective figure legends. Data were analyzed with SPSS 29 (RRID:SCR_016479) and GraphPad Prism 9 or 10 (RRID:SCR_002798), Origin Pro 2024 (RRID:SCR_014212), and MetaboAnalyst 5.0 (RRID:SCR_016723) (https://www.metaboanalyst.ca) [75]. Bioinformatic analysis of “omic” data (proteome, lipidomic, metabolomic) is explained in the respective paragraphs. Area under the curve (AUC) data of lipidomic and metabolomic analyses were transformed to square root values to adjust skewed distributions. For testing the null-hypothesis that groups were identical, two groups were compared with 2-sided, unpaired Student’s t-tests. Lipidomic and metabolomic data were submitted to 2-way analysis of variance (ANOVA) using e.g., the factors “feature” (e.g. lipid, metabolite) and ‘group’ (e.g. PD with/without ambroxol versus wildtype). In case of significant differences, groups were mutually compared using post hoc t-tests according to Šidák or false discovery rate (FDR). The meaning of asterisks in figures is explained in legends. Partial Least Square discrimination analyses (PLS-DA) and Random Forest supervised learning algorithms were used to assess the prediction of group membership and classification according to their importance. For multivariate analyses, data were normalized to have a common average and variance of 1 (Z-scores = (x-x̄)/SD). Volcano plots were used to assess fold differences of proteins or lipids versus the negative logarithm (Log10) of the t-test P value according to standard procedures. Hierarchical clustering according to Euclidean distance metrics and Ward method were used to visualize lipidomic and metabolomic results as heatmaps with dendrograms. The selection of top 100 or top 50 regulated metabolites was based on ANOVA P-values.
Results
Proteomics showed increased αSyn and reduced Fgf1 and Scarb2/LIMP2 in PD mice
Proteomic and Western blot studies confirmed increased αSyn levels in cortex or brain tissue of Pink1−/−SNCAA53T versus wildtype Sv129-FVB mice (Fig. 1, Supplementary Figure S1). Brain tissue samples consisted of cortex, subcortex and midbrain without olfactory bulb and cerebellum. Western blot analyses of the hippocampus showed an increase of the pathogenic phosphorylated S129 αSyn but not of total αSyn (Supplementary Fig. S1). Proteome studies revealed a downregulation of fibroblast growth factor 1 (Fgf1), which is known to be involved in the development and/or progression of PD. Further strongly reduced proteins included ubiquitin ligase Ube2j, GABA receptor Gabrb3, and G-protein receptor kinase Grk6, whereas TBC1 Domain Family Member 22 A was increased (Fig. 1a, b). Grk6 is required for dopamine receptor functions and contributes to phosphorylation of αSyn [76, 77]. It has been suggested that accumulation of αSyn in lysosomes leads to a disruption of lysosomal biology and lysosomal enzyme abundance and activity. The proteome analyses, however, did not detect a loss of GBA1 or other lysosomal enzymes, which are involved in sphingolipid metabolism. But the expression of the GBA1 transporter LIMP2 (Scarb2) was strongly reduced. The results suggested that increased mutant αSyn and Pink1 deficiency in our mouse model did not lead to a secondary deficiency of GBA1 at the protein level but might affect its transport.

a Volcano plots of protein expression as assessed by proteomic analysis of the prefrontal cortex from adult Pink1−/−SNCAA53T versus wildtype Sv129-FVB mice. The x-axis shows the Log2(Fold change) of label free intensities. The y-axis shows the negative logarithm of the t-test P-value. The data are from n = 9 mice per group. Upregulated proteins are shown in red, downregulated in blue. b Candidate proteins were selected according to the FDR adjusted P-values and are sorted according to abundance. Functional descriptions of candidate proteins are shown in a Supplementary Excel file. c Western blot analysis of aSyn and phosphorylated Ser129 aSyn in the brain of adult Pink1−/−SNCAA53T mice treated with ambroxol (n = 7) or vehicle (n = 6) versus wildtype Sv129-FVB mice (n = 10). Treatment started at 6 months of age and tissue was obtained at 12 months of age. Details about mouse groups are in Supplementary Tables. Raw intensities from individual blots were range normalized, and the ratio versus loading control β-actin is shown as box/scatter plots. Each scatter is one mouse. The box is the interquartile range, the line is the median and whiskers show minimum to maximum. Data were submitted to 2-way ANOVA and subsequent posthoc t-tests. The asterisks show Šidák adjusted P values. *<0.05, ***<0.001.
Western blot analyses of the brain of ambroxol-treated mice (ambroxol-chow) showed a statistical therapeutic lowering of elevated phosphorylated S129 αSyn or of total αSyn in comparison with placebo treated PD mice (Fig. 1c). However, the effect was variable and required statistical analyses of range-normalized Western blots from 6-9 mice per group (WT, PD-placebo, PD-ambroxol) to become significant. The difference was not obvious on single blots.
Proteomic data:
Data deposited to PRIDE; Project accession: PXD039172.
Candidate regulated proteins and descriptions in Supplementary Excel file.
IntelliCage studies showed a mild improvement of exploratory behavior with ambroxol
In the first part we compared Pink1−/−SNCAA53T versus wildtype Sv129-FVB mice, and in the second part Pink1−/−SNCAA53T mice with/without ambroxol treatment. To investigate the therapeutic effects of ambroxol, three cohorts of PD mice were treated with ambroxol versus placebo, 1× as drinking water (n = 10/10), 2× with the food pellets (n = 7/6 and n = 6/6). The treatment started at about 6 months of age and continued until the end of the experiment. During the time of the observation and at the end at 13–16 months of age the mice had no PD-like motor dysfunctions but had general minor age-related health problems such as inflammation of the eyes and loss of whiskers in both treatment groups. With an average drinking and feeding amount of 3–4 ml and 3–4 g per day, respectively, the doses were approximately 120–150 mg·kg−1·d−1 (drinking water) and 75–100 mg·kg−1·d−1 (pellets). In comparison, Gaucher patients received up to 27 mg·kg−1·d−1 [63] and patients with PD-associated dementia shall receive 15–20 mg·kg−1·d−1 (NCT05778617). Gaucher mice were treated with 100-300 mg/kg/d in previous studies [38]. We confirmed that ambroxol was orally bioavailable and reached the CNS in mice at the doses used in the present study. The results are presented with the metabolomic results below.
Ambroxol was well tolerated and had no obvious negative effects at the chosen dosages which are at a maximum for continuous long-term administration via food or drinking water. As a cationic amphiphilic lysomotropic drug, ambroxol might cause phospholipidosis owing to a disruption of lysosomal phospholipases [60, 61] which would manifest as renal water and electrolyte loss and compensatory high intake of water. Throughout our studies, ambroxol treatment had no effect on drinking volume or licking in IntelliCages. We infer that ambroxol had no apparent effect on renal function. Body weight time courses were also equal in both groups (Supplementary Fig. S2).
To assess if ambroxol prevented or reduced a cognitive decline or other behavioral indices of PD-like dementia and slowness, we first studied Pink1−/−SNCAA53T versus wildtype SV129-FVB mice in long-term observations in IntelliCages (Fig. 2), which allows the evaluation of multiple cognitive dimensions. Most parameters were surprisingly equal. The analysis of 12 h intervals shows a disruption of circadian rhythms in Pink1−/−SNCAA53T mice (Fig. 2a), which was induced by a short period of day-pattern-restricted drinking and was not resolved up until the end of the observation. The frequency of corner visits with licks was mildly increased (LVisits/h, Fig. 2a), and the licks were strongly increased throughout the tasks irrespective of the door opening times (Fig. 2b). Visits without licks did not differ between groups (Fig. 2c, d) and Pink1−/−SNCAA53T mice had no deficit of place preference learning.

a Time course of corner visits with licks per hour (LVisits/h) during different tasks in IntelliCages. b Time course of Licks per visit with licks (Licks/LVisits). c Time course of exploratory visits with nosepokes but without licks (NPVisits/h). d Time course of erroneous visits without nosepokes and without licks (SVisits/h, s=sine). SVisits have no purpose. The tasks are shown in the header and described at the bottom of the graph. Further details about the tasks and IntelliCage abbreviations are shown in Supplementary Tables. The data show means ± sem of 8 female mice per group. The IC experiment started at 41 weeks of age and lasted until 64 weeks. The fluctuation of the behaviour reveal nighttime and daytime differences (12 h Bins) and show the circadian rhythm. Data were compared with 2-way ANOVA for the factors “time” × “genotype” and posthoc comparison for “genotype” with adjustment of alpha according to Šidák. Pink1−/−SNCAA53T mice made more LVisits/h and Licks/LVisits than wildtype mice throughout most of the tasks.
With the knowledge of these behavioral features, we then set up an IntelliCage experiment to compare Pink1−/−SNCAA53T mice with and without ambroxol treatment. Ambroxol was delivered through drinking water (Fig. 3). Control mice received placebo-water with slightly bitter taste resembling ambroxol-water. The tasks were similar to the WT vs PD experiment, but day patterns were not disrupted and hence, circadian rhythms were perfect in both groups through all tasks. Like in the WT vs PD experiment, licks were increased in PD mice (Licks/LVisits) with and without ambroxol (Fig. 3b). The dotted line shows the average of wildtype mice. The results also show a higher rate of visits without licks (Fig. 3c, d) in ambroxol treated mice during adaptation tasks. Visits without licks but with nosepokes (NPVisits, Fig. 3c) can be interpreted as exploratory behavior, which is a characteristic feature of young mice, whereas visits without nosepokes and without licks (SVisits, Fig. 3d) are without purpose and are often increased in mice with dementia [66, 78]. Differences between treatment groups were fading during learning tasks because the need to find the correct corner also forced the vehicle treated group to increase their exploratory visits so that they caught up with the ambroxol group.

a Time course of corner visits with licks per hour (LVisits/h) during different tasks in IntelliCages. b Time course of Licks per visit with licks (Licks/LVisits). c Time course of exploratory visits with nosepokes but without licks (NPVisits/h). d Time course of erroneous visits without nosepokes and without licks (SVisits/h, s=sine). SVisits have no purpose. The tasks are shown in the header and described at the bottom of the graph. Further details about the tasks and IntelliCage abbreviations are shown in Supplementary Tables. The data show means ± sem of 8 female mice per group. The IC experiment started at 41 weeks of age and lasted until 64 weeks. The fluctuation of the behaviour reveals nighttime and daytime differences (12 h Bins) and shows the circadian rhythm. Data were compared with 2-way ANOVA for the factors “time” × “genotype” and posthoc comparison for “treatment” with adjustment of alpha according to Šidák. Ambroxol treated Pink1−/−SNCAA53T mice made more exploratory NPVisits/h during adaptation tasks (i.e. tasks without need to learn). LVisits/h and SVisits/h were also mildly increased in ambroxol treated mice, mostly during adaptation tasks. Data were compared with 2-way ANOVA for the factors “time” × “genotype” and posthoc comparison for “treatment” with adjustment of alpha according to Šidák.
To further assess the therapeutic anti-slowness / pro-exploratory effect of ambroxol we compared the nosepokes per visit (NP/Visit), which is a very stable feature and indicator of exploratory behavior [79]. Wildtype mice make on average about 3-5 nosepokes per visit (Fig. 4). The NP/Visit ratio was lower in Pink1−/−SNCAA53T mice throughout different tasks (Fig. 4a), and ambroxol treated Pink1−/−SNCAA53T mice had a higher ratio than vehicle treated mice but still lower than wildtype mice (Fig. 4b). The result suggested a therapeutic benefit with ambroxol, which is further revealed in the scatter plots (Fig. 4b) showing NP/Visit ratios throughout different tasks in individual mice. Despite the interindividual variability, one can see that the majority of the vehicle treated mice was more seriously affected by the disease than ambroxol treated mice.

a Time course of NP/Visit ratios during different IntelliCage tasks. For direct comparison, tasks were selected which matched between part-1 (comparison of genotypes) and part-2 (comparison of ambroxol vs vehicle treatments) of the IntelliCage studies. The NP/Visit ratio is an age-dependent characteristic feature of mouse lines and disease models. The wildtype midlife average is about 3.5 (3–4) NPs per visit, depicted at dotted line. b Nested analysis of NP/Visit ratios in Pink1−/−SNCAA53T versus Sv129-FVB wildtype mice and in Pink1−/−SNCAA53T mice treated with ambroxol versus vehicle. For this analysis NP/visit ratios at each time point are stacked in sub-columns, presented as scatters for each mouse. The nested t-tests compares treatments (or genotypes) and individual mice. The within subject variation was higher in part-2 of the study suggesting that the ambroxol versus vehicle cohort of Pink1−/−SNCAA53T mice was more seriously affected than the cohort comparing PD versus WT mice (part-1).
Ambroxol did not prevent or reduce the sensory or motor decline in PD mice
Our previous clinical quantitative sensory studies in PD patients had suggested that high GlcCer are associated with the extent of sensory loss and pain. Therefore, we studied sensory and motor functions with and without ambroxol in Pink1−/−SNCAA53T mice to assess if ambroxol was able to prevent the sensory decline which often precedes the motor stage and probably is a good prognostic indicator. We used unbiased Thermal Gradient Ring (TGR) preference behavior (Fig. 5) and standard paw withdrawal latency based nociceptive tests (Fig. 6).

a Times spent in temperature zones on a Thermal Gradient Ring (TGR) in Pink1−/−SNCAA53T versus Sv129-FVB wildtype mice and in Pink1−/−SNCAA53T mice treated with ambroxol versus vehicle with a temperature gradient of 15–40 °C. The groups comprised n = 9–10 female mice, which were 43-46 weeks old for part-1 comparing genotypes and 62.5–65.4 weeks for part-2 comparing ambroxol versus vehicle. The data show means ± sem. The test is divided into four time quarters, Q1-Q4 shown in Supplementary Fig. 3. b Distances travelled during the 30 min habituation and the 60 min TGR test. Mice could freely move in the ring gateway and were tracked with TGR camera and software. The box is the interquartile range, the line the median, and whiskers show minimum to maximum. The scatters represent the mice. Data were compared with 2-way ANOVA and subsequent posthoc t-tests using an adjustment of alpha according to Šidák. Pink1−/−SNCAA53T mice travelled a longer distance than wildtype controls. *P < 0.05. c–e Box-Scatter plots group comparisons of (c) travel distance in the test period, (d) body rotations leading to inversion of travel direction (clockwise-anticlockwise), (e) preference temperature based on time spent in zones. The violin plots show pooled data of wildtype controls at about 1 year of age. The boxes show the interquartile range, the line is the median, whiskers show minimum to maximum. The scatters show individual mice. The data were compared with one-way ANOVA and subsequent posthoc analysis versus wildtype using an adjustment of alpha according to Dunnett. Asterisks show significant differences versus wildtype. *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001. There was no difference between ambroxol versus vehicle.

a Time courses of Hotplate withdrawal latencies in Pink1−/−SNCAA53T versus Sv129-FVB wildtype mice and in Pink1−/−SNCAA53T mice treated with ambroxol versus vehicle. Ambroxol was administered via the drinking water (120–150 mg·kg−1·d−1) or via food pellets (75–100 mg·kg−1·d−1). The boxes show the interquartile range, the line is the median, whiskers show minimum to maximum. The scatters show individual mice. The data were compared with two-way ANOVA and subsequent posthoc analysis according to Šidák comparing genotypes or treatments. There was no difference between ambroxol versus vehicle except for the last time point at 63-68 weeks of age in the drinking water cohort. *P < 0.05, **P < 0.01. b Rotarod running behaviour. For each age, mice performed three tests on consecutive test days. The data were compared with 2-way ANOVAs for the factors “time” × “treatment”. The posthoc analysis according to Šidák did not show significant differences between treatment groups, but ANOVA revealed that overall running times were somewhat longer in mice treated with ambroxol drinking water.
Thermal preference behavior showed that Pink1−/−SNCAA53T mice were more indifferent to the floor temperature as compared to wildtype mice (WT vs PD experiment), which is shown by a flattening of the zone preference curves (Fig. 5a, left), and was reproducible in the second experiment comparing ambroxol vs vehicle (Fig. 5a, right) but was not affected by ambroxol. Preferences per quarter are shown in Supplementary Fig. S3. During the test and habituation Pink1−/−SNCAA53T mice were more active than wildtype controls as revealed by longer travel distances (Fig. 5b, left). This overactivity was not evident in the first quarter but in subsequent quarters when wildtype mice settle down. The second cohort (ambroxol versus vehicle) showed even stronger restlessness (Fig. 5b, right), which is also shown in the summary graphs of motor behavior (Fig. 5c, d) but was not influenced by ambroxol. Ambroxol also had no influence on the overall temperature preference (Fig. 5e). Hence, the previously observed reproducible loss of thermal perception in PD mice was not prevented by ambroxol.
In standard longitudinal Hotplate experiments we could confirm the previously observed decline of heat pain sensitivity in Pink1−/−SNCAA53T mice compared to wildtype mice [36] (Fig. 6a, left). Ambroxol drinking water appeared to reduce the decline >50 weeks of age, but the mild therapeutic advantage was not reproducible with ambroxol food pellets. Ambroxol had also no effect on the Rotarod running time (Fig. 6b).
Ambroxol did not reduce elevated brain ceramides or glucosylceramides
In previous studies we [34] and others [80] have observed that plasma concentrations of ceramides and glucosylceramides or glucosylsphingosines are increased in patients with idiopathic PD without known GBA1 mutations, and ceramides were increased in the brain and peripheral nervous tissue of PD mice in our previous studies [36, 81]. Therefore, we now analysed if ambroxol treatment was able to prevent or reduce derangements of ceramides and other sphingolipids at either site (CNS and PNS) or systemically in plasma.
Targeted (Fig. 7) and untargeted (Fig. 8) lipidomic studies revealed increased ceramides in the brain of double mutant Pink1−/−SNCAA53T mice compared with wildtype Sv129-FVB control mice. Importantly, Pink1−/−SNCAA53T mice had no clinical symptoms of PD motor disease at the time of tissue sampling at approximately 12-13 months of age. Targeted sphingolipid analyses of brain tissue revealed that treatment with ambroxol (drinking water) reduced elevated GlcCer 16:0 in the brain to the wildtype level (Fig. 7a) but had no effect on raised GlcCer in the DRGs and sciatic nerve (Fig. 7b). Ambroxol treatment also did not prevent the loss of anandamide in DRGs and sciatic nerve that occurs upon aging and particularly in PD mice (Supplementary Fig. S4). The restoration of normal GlcCer 16:0 levels achieved by ambroxol-drinking water was not reproducible with ambroxol food pellets, which provided a slightly lower daily dosage of ambroxol but still very high compared with human doses for Gaucher disease. Metabolomic studies showed a high peak of ambroxol in the ambroxol group (no peak in the vehicle group) confirming that ambroxol in food pellets was bioavailable as expected and provided stable plasma concentrations (Figs. 7d, 9). Unexpectedly, ambroxol via drinking water or food pellets increased glucosylceramides in the periphery in plasma (Fig. 7e), which were not affected by the PD genotype per se (Supplementary Fig. S5). The result suggested that ambroxol triggers the extracellular release of glucosylceramides possibly via lysosomal exocytosis. As a cationic amphiphilic drug (CAD), ambroxol itself accumulates in lysosomes owing to its amphiphilic nature and high pKa. The resulting raise of the lysosomal pH and binding to hydrophobic luminal parts of membrane phospholipids leads to an impairment of lysosomal phospholipid metabolism via lysosomal phospholipase, and the resulting accumulation of phospholipids triggers exocytosis [82].

a Ceramides (Cer) and hexosylceramides (GlcCer and LacCer) in brain tissue (cortex, subcortex, midbrain) in Sv129-FVB wildtype mice (n = 10 at 59–69 weeks) and in Pink1−/−SNCAA53T mice treated with ambroxol via drinking water (n = 10) or vehicle (n = 9) (age 65–68 weeks). Quantitative analyses were obtained by targeted LC-MS/MS analyses. b Glucosylceramides in brain tissue (cortex, subcortex, midbrain), dorsal root ganglia (DRG) and sciatic nerve in the mice shown in a. Endocannabinoids at these sites are shown in Supplementary Fig. S4. c Ceramides and hexosylceramides in brain tissue in Sv129-FVB wildtype mice (n = 6 at 46 wks, n = 4 at 58 weeks) and in Pink1−/−SNCAA53T mice treated with ambroxol food pellets (n = 7) or vehicle (n = 6) (age 58 weeks). d Confirmation of systemic ambroxol bioavailability by plasma analysis from mice shown in c. e Plasma ceramides and hexosylceramides in mice treated with ambroxol drinking water (left) or ambroxol food pellets (right) versus the respective control groups in mice presented in a and c. The comparison of Pink1−/−SNCAA53T mice versus wildtype mice is shown in Supplementary Fig. S5. The boxes show the interquartile range, the line is the median, whiskers show minimum to maximum. The scatters show individual mice. The data were compared with two-way ANOVA and subsequent posthoc analysis according to Šidák comparing genotypes or treatments. Asterisks show adjusted P-values, *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001.

a, b Volcano plots showing lipid species in the brain (cortex, subcortex, midbrain) in Pink1−/−SNCAA53T mice versus Sv129FVB wildtype control mice (n = 10, age 46 or 58 weeks). Pink1−/−SNCAA53T mice were treated with ambroxol food pellets (a; n = 7, age 58 weeks) or vehicle (b; n = 6, age 58 weeks). Lipids were analysed by untargeted UHPLC-MS/MS lipidomic screen. The X-axis shows the log2(Fold change), the Y-axis -Log10 of the t-test P-value. Lipids that were beyond threshold in plot a PD-ambroxol versus wildtype) are in pink or colour coded according to the lipid class. The respective lipids maintain their colour in plot b and plot d to reveal the change of their position. Thresholds are indicated as dotted lines. c Comparison of summed sulfatides (SHexCer) in wildtype and Pink1−/−SNCAA53T mice with/without ambroxol food pellets. Mice as in a. Data are compared with one-way ANOVA and posthoc Šidák, **P < 0.01. d Volcano plots showing lipid species in the brain in direct comparison of ambroxol treated versus vehicle treated Pink1−/−SNCAA53T mice. Mice and lipid colours as in a. e Hierarchical clustering of mice according to their brain lipid profiles. The tree shows Pearson distance metrics, Ward method. Clusters do not differentiate ambroxol versus vehicle treated mice. Abbreviations: CER ceramides, CE cholesterol ester, DG diglycerides, FA fatty acids, HexCer,hexosylceramides, GlcCer glucosylceramides, LacCer lactosylceramides, LPC lysophosphatidylcholines, LPE lysophosphatidylethanolamines, LPG lysophosphatidylglycerols, LPI lysophosphatidylinositols, PC phosphatidylcholines, PE phosphatidylethanolamines, PG phosphatidylglycerols, PI phosphatidylinositols, SM sphingomyelins, ST sterols, TG triglycerides, UbiQ ubiquitin, –O ether bound lipids.

a Plasma lipids were analysed by untargeted UHPLC-MS/MS lipidomic screen in Pink1−/−SNCAA53T mice versus Sv129FVB wildtype control mice. Mice and treatments are as described in Fig. 8. For comparison of lipidomic patterns in plasma, AUC/IS values of individual species of a lipid class were summed, and the summed AUC/IS were normalized to have a common mean and standard deviation of 1 (Z-score). The median of Sv129-FVB wildtype mice was used as reference. The lipid classes are sorted from “increased in PD” (left) to “reduced in PD” (right). Data were submitted to 2-way ANOVA and subsequent posthoc analysis using an FDR adjustment of alpha. Asterisks indicate discoveries. Cholesteryl esters (CE) were significantly different between ambroxol treated and placebo treated PD mice. b Polar metabolites in mouse plasma was analysed from the same mice as in a using a UHPLC-MS/MS metabolomic screen. The AUCs of polar metabolites were square root transformed and subsequently normalized to have a common mean and standard deviation of 1 and are presented as Z-scores. The metabolites are sorted from “increased in PD” (left) to “reduced in PD” (right). Candidate metabolites which differed between ambroxol treated and placebo treated Pink1−/−SNCAA53T mice are highlighted in pink and are selected and plotted in c. c Candidate polar metabolites in plasma which differed between ambroxol treated and placebo treated Pink1−/−SNCAA53T mice. The box shows the interquartile range, the line is the median, the whiskers show minimum to maximum, the scatters show the mice. Data were submitted to 2-way ANOVA and subsequent posthoc analysis using an FDR adjustment of alpha. Asterisks indicate discoveries. *P < 0.05.
Ambroxol aggravated the rise of sulfatides in the PD mouse brain
Untargeted lipidomic analyses of brain tissue (Fig. 8) showed that ceramides, hexosylceramides (GlcCer or GalCer) and sulfatides (SHexCer) were increased in Pink1−/−SNCAA53T brains. Lipidomic analyses further revealed increased diacylglycerols (DG) and lysophosphatidylethanolamines (LPE) in Pink1−/−SNCAA53T brains. High brain DGs are considered as indicators of CNS disease [83] because they are normally low. LPEs are contained in Lewy bodies and likely precipitate αSyn aggregation, depending on C-chain length and saturation. Comparison of the Volcano plots of Ambroxol-treated Pink1−/−SNCAA53T versus wildtype mice (Fig. 8a) and vehicle-treated Pink1−/−SNCAA53T versus wildtype mice (Fig. 8b), showed almost identical patterns, and no significant differences between ambroxol versus vehicle on direct comparison (Fig. 8c), but a summed analysis of sulfatide species (SHexCer) revealed that these sphingolipids were significantly stronger increased in ambroxol-treated PD mice than in vehicle-treated PD mice.
Source data are available at https://www.ebi.ac.uk/biostudies/studies/S-BSST1946?key=1b15a58b-643a-4357-a489-f1081592dad4.
Ambroxol restored some deranged plasma metabolites to wildtype level
To follow up on the unexpected rise of plasma ceramides with ambroxol we subjected plasma to further untargeted lipidomic and metabolomic analyses (Fig. 9). For comparison of lipids, species per class were summed and the sums normalized to the median of wildtype (Fig. 9a). There were substantial differences in several lipid classes between wildtype and Pink1−/−SNCAA53T mice regardless of ambroxol treatment. Vehicle and ambroxol treated mice were mostly similar except for sterols (ST) and sterol (cholesterol)-ester (CE) which were low in PD mice but almost restored in ambroxol-treated PD mice. A similar trend for ambroxol-mediated restoration of deranged metabolites was observed in metabolomic studies of polar metabolites (Fig. 9b, c). Specifically, ambroxol restored plasma levels of biogenic amine metabolites (N-acetyl-dopamine, kynurenine) and normalized some amino acids or their metabolites. Overall, the profile of polar metabolites of ambroxol treated PD mice was closer to wildtype mice (Fig. 9b).
Source data are available at https://www.ebi.ac.uk/biostudies/studies/S-BSST1946?key=1b15a58b-643a-4357-a489-f1081592dad4.
Mitochondria accumulated triglycerides and HexCer regardless of ambroxol
Glucosylceramides are components of organelle membranes and are enriched at inter-organelle contact sites including mitochondria-to-lysosome contacts. Lysosomal build-up of glycosphingolipids disrupts these contacts and consequently, mitophagy [84, 85]. Therefore, we studied if and how PD-associated alterations of sphingolipids impacted on mitochondria. Previously, transmission electron microscopy had shown pathologic swollen mitochondria in neurons of old Pink1−/−SNCAA53T mice [36], and we confirmed now via Seahorse experiments that the FCCP-stimulated maximal respiratory capacity of primary neurons of Pink1−/−SNCAA53T mice was impaired (Supplementary Fig. S6a). For the present study of mitochondrial lipid composition, we used the mouse heart as a defined source of highly energetic mitochondria (Fig. 10). Triglycerides (TG) and HexCer were increased in PD-mitochondria (Fig. 10a–f, Supplementary Fig. S6), regardless of ambroxol treatment. TGs were previously found to accumulate in mitochondria in models of myopathy, axonal injury or MPTP-induced PD [86,87,88]. Mitochondrial lipid overload suggests chronic metabolic dysregulation in PD mitochondria—possibly due to impaired β-oxidation or lipid droplet-mitochondria crosstalk.

a Mitochondrial lipids were analysed by untargeted UHPLC-MS/MS lipidomic screen in heart mitochondria of Pink1−/−SNCAA53T (PD) versus Sv129FVB wildtype control mice (WT) (experiment-1), and in Pink1−/−SNCAA53T mice treated with vehicle (PD-Vehicle) or ambroxol-pellets (PD-Abx) (experiment-2). To allow for combined analyses AUC/IS values of experiment-2 were scaled to experiment-1 based on the median of the PD mice (untreated, vehicle treated). For comparison of lipidomic patterns, AUC/IS values of individual species were summed per class. Lipid classes are sorted from high-to-low and the counts of individual species are shown in the right Y-axis. Data were submitted to 2-way ANOVA and subsequent posthoc analysis using an adjustment of alpha according to Dunnett versus WT. Asterisks indicate significant differences, *P < 0.05, **P < 0.01. The relevant classes are highlighted in orange letters and shown in more detail below. HexCer species in Supplementary Fig. S6. Each scatter is one mouse, the line is the average, and whiskers show the standard deviation. b Mitochondrial triglycerides, diglycerides and fatty acids. c Ceramides, hexosylceramides (HexCer (glucosylceramides and galactosylceramides), Hex2Cer (lactosylceramides) and sulfatides (SHexCer). Analyses as in (a). d, e Cardiolipins sorted according to C-chain length and saturation. The insert shows the sum of CL species. The most abundant tetralineoyl-CL (CL 72 species) and total CL were reduced in mitochondria from ambroxol treated PD mice. CL 0_XX are monolyso-cardiolipins (MLCL). Statistics as in a, Welch-ANOVA for total CL and subsequent posthoc analysis according to Dunnett T3. *P < 0.05. Multivariate PLS-DA analysis of CL is shown in Supplementary Fig. S6b, c. Distributions of CL species show a left-shift of mitochondrial CL in ambroxol treated mice. PD and PD-vehicle were pooled. f Long chain triglyceride species. Similar differences were observed for TGs with 48-54 chain length. For clarity, only the long-chain TGs are shown. *P < 0.05.
Treatment with ambroxol was associated with reduced mitochondrial cardiolipins
While mitochondria of untreated or vehicle treated PD mice showed an increase of some cardiolipin species including the most abundant tetralineoyl cardiolipin alongside the increase of TG, mitochondria of ambroxol-treated mice showed a reduction of cardiolipin species mostly below the WT level (Fig. 10a, d, Supplementary Fig. S6b, S6c), and additionally increased diglycerides (DG). The increase in cardiolipins in non-treated PD mice might be a mitochondrial attempt to stabilize respiratory complexes [89] under lipid stress or under the specific PD-model-associated stress caused by Pink1 deficiency and mutant SNCA. The fact that TG and HexCer remain elevated despite ambroxol treatment implies that ambroxol may not alleviate upstream lipid dysregulation or HexCer metabolism. A reduction of cardiolipins below wildtype level suggests that the accumulation of ambroxol in lysosomes (please see below) impacts inter-organelle lipid exchange.
Ambroxol increased sphingolipids in HT22 cells and triggers lysosomal exocytosis
The analyses of plasma sphingolipids suggested that ambroxol may increase the extracellular release of lipids which are degraded in lysosomes, in particular sphingolipids. To further study these effects, we used HT22 mouse hippocampal neurons with/without loading of preformed αSyn fibrils. It was confirmed via confocal live imaging microscopy that HT22 cells ingested αSyn PFF, thus creating PD-like HT22. For lipidomic studies, pimozide was used as a positive control because it is like ambroxol a cationic amphiphilic drug (CAD) and is known to lead to lysosomal accumulation of phospholipids and sphingolipids and eventually, depending on the concentration, to lysosomal membrane leakage [90].
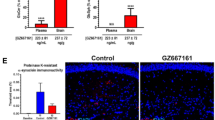
The IC50 for ambroxol in HT22 cells was about 100 µM as assessed in WST viability assays (Fig. 10a). Subsequent experiments were carried out with 40 µM ambroxol which did not impair viability, but at 40 µM, ambroxol caused a significant increase of lactate dehydrogenase (LDH) activity in the culture supernatant (Fig. 11b) pointing to the extracellular release of lysosomal content. Autophagolysosomes mostly harbour LDHB activity [91]. In support, ambroxol treatment also increased cathepsin D activity in cell culture supernatants (Fig. 11c), which was independent of the presence of αSyn-fibrils. CTSD is a lysosomal enzyme. Its activity in the extracellular medium is highly suggestive of lysosomal exocytosis, which is one mechanism how cells expel excess αSyn [20], at least temporarily. At concentrations which were non-toxic according to viability tests, ambroxol increased the lysosomal mass and caused lysosomal trapping of acidic fluorescent dye. The morphology of lysosomes was visualized microscopically using the fluorescent lysosomal tracer Lyso-ID (Supplementary Fig. 7). The effect of ambroxol was stronger than that of pimozide and comparable with effects of chloroquine (positive control in this assay).

a Analysis of IC50 concentrations of ambroxol in HT22 mouse hippocampal neurons as assessed by WST viability assays. The WST absorbance (range scaled) is plotted versus Log2 ambroxol concentrations, and IC50 values are calculated with a standard sigmoidal Emax model. b Lactate dehydrogenase (LDH) activity was measured in the culture supernatant of HT22 cells treated with 40 µM ambroxol versus vehicle with/without preloading of the cells with αSyn preformed fibrils (PFF, 2 µg/ml). Each scatter shows one culture. The experiment was done twice. c In analogy to B, the plots show cathepsin D (CTSD) activity in HT22 cell culture supernatants. d Lipidomic analysis of HT22 cells treated with ambroxol (Abx), pimozide (PIM) or the combination (A + P) with/without preloading of the cells with αSyn PFF at 2 µg/ml. Control cells were treated with the respective volume of the DMSO (vehicle) or were left untreated (naïve HT22) or received high dose PFF at 6 µg/ml. Each group consisted in 8 replicates. Sample and group IDs are given at the bottom of the heatmap. The heatmap shows top 100 regulated lipids with hierarchical clustering of lipid species (rows) and samples (columns) using Euclidean distance metrics and the Ward method. The top 100 lipids were selected by ANOVA P-value. For depiction as heatmap, lipid AUC/IS values were square root transformed and auto-scaled to have a common mean and variance of 1 (z-scores). Multivariate analyses are shown in Supplementary Fig. S6. Abbreviations: CER ceramides, CE cholesterol ester, DG diglycerides, FA fatty acids, HexCer hexosylceramides, LPC lysophosphatidylcholines, LPE lysophosphatidylethanolamines, LPG lysophosphatidylglycerols, LPI lysophosphatidyl-inositols, PC phosphatidylcholines, PE phosphatidylethanolamines, PG phosphatidylglycerols, PI phosphatidylinositols, SM sphingomyelins, ST sterols, TG triglycerides, –O ether bound lipids.
For lipidomic studies, αSyn-PFF was added in excess to the culture medium in the presence or absence of the treatments ambroxol (40 µM), pimozide (5 µM), the combination Abx+PIM (40 + 5), or the vehicle DMSO, or cells were left untreated (naïve HT22). Owing to high pKa, both ambroxol and pimozide are trapped in lysosomes, and raise lysosomal pH albeit not as much as strong alkaline drugs such as chloroquine. Ambroxol treatment of HT22 cells without/with ingestion of αSyn-PFF led to a significant increase of sphingolipids (Cer, HexCer, SM) and of cholesteryl ester (CE), which are metabolized in lysosomes (Fig. 10d, Supplementary Fig. S8). At the chosen concentrations, ambroxol caused a stronger accumulation of sphingolipids than pimozide. In addition, ambroxol treatment reduced several phosphatidylcholine species (PC), while these PC species were increased with pimozide (Fig. 11d). The lipidomic patterns under ambroxol and pimozide agree well with expectation of CAD-mediated inhibition of lysosomal phospholipase (Pla2g15) [61, 62, 92], and acidic sphingomyelinase [93], albeit with differences in the preferential accumulation of SM and PC species. PCs are connected with GlcCer via the phospholipid translocase ATP10 (ATP10A, 10B and 10D), which flips PC in exchange with GlcCer in the plasma membrane, and contributes thereby to membrane dynamics [94,95,96]. It was recently suggested that ATP10 defects rather than malfunctioning of GBA1 account for elevated plasma ceramides in PD [53]. The results suggest that lysosomal accumulation of ambroxol disrupts lysosomal lipid metabolisms and triggers lysosomal exocytosis. Theoretically, this could be a route of relief for a neuron with a high αSyn-fibril load.
HT22 lipidomic and metabolomic data and metadata are available at https://www.ebi.ac.uk/biostudies with the accession number S-BSST1897.
https://www.ebi.ac.uk/biostudies/studies/S-BSST1897?key=d2a20a65-a88f-44a4-9654-672a72f8a9ac.
Discussion
We show that high-dose continuous “preventive” ambroxol treatment in Pink1−/−SNCAA53T mice was well tolerated and provided a modest therapeutic benefit consisting in a statistic reduction of elevated αSyn and phosphorylated Ser129 αSyn and of GlcCer16:0 in the brain, the latter (GlcCer) only in the drinking water cohort. At the behavioral level, ambroxol treated PD mice showed higher exploratory activity in IntelliCages in adaptation tasks. The behavior suggests a beneficial therapeutic reduction of bradykinesia. However, ambroxol had no effect on the sensory decline, which is in humans a good indicator for the progression of the disease [97, 98]. Ambroxol also had no effect on elevated GlcCer or loss of anandamide in the PNS, and ambroxol did not prevent the PD-associated accumulation of triglycerides and HexCer in mitochondria, which is an indicator of mitochondrial stress and dysfunction [86, 99]. In fact, in the ambroxol treated mice, this TG accumulation was associated with a reduction of cardiolipin species in mitochondria below wildtype level, which did not occur without ambroxol treatment suggesting that mitochondrial lipid homeostasis was more seriously impaired in the ambroxol treatment group. Furthermore, ambroxol caused a further increase of sulfatides in the brain, suggesting that ambroxol may cause a further impairment of lysosomal sphingolipid metabolism in the brain. Our in vitro experiments in HT22 neuronal cells also show that ambroxol may perturb lysosomal metabolic functions. Lysosome morphology and trapping of the acidic Lyso-ID dye in the lysosomes under ambroxol treatment resembled effects of the alkaline drug [100], chloroquine which is a well-known lysosome perturbation agent. Like chloroquine and pimozide, which was used as a positive control in our in vitro studies, ambroxol is a cationic amphiphilic drug that is trapped in lysosomes, raises the intraluminal pH, binds to the luminal hydrophobic site of membrane phospholipids and interferes thereby with lysosomal phospholipase (LPLA2, gene Pla2g15) [92] and acidic sphingolipid metabolizing enzymes (e.g. ASM, gene Smpd1) [82]. As a consequence of these lysosomal effects, ambroxol treatment of HT22 cells resulted in an accumulation of phosphatidylcholine species, sphingolipids and cholesteryl ester, which are metabolized through lysosomal pathways, strongly suggesting that ambroxol inhibited both LPLA2 and ASM, similar to other cationic amphiphilic drugs [61, 101]. Interestingly, though, ambroxol did not kill the cells at concentrations that were sufficient to accumulate sphingolipids but instead increased the extracellular release of lysosomal enzymes which was detected as extracellular cathepsin D and LDHB activity.
The results suggest that ambroxol interferes with lysosomal phospholipid and sphingolipid metabolism and triggers lysosomal exocytosis, which depends on lysosomal calcium dynamics [102, 103] and pH [104]. Lysosomal exocytosis may be triggered by sphingolipid or glycerophospholipid overload [105], the latter resulting from a defective hydrolysis of bis(monoacylglycero)phosphate (BMP), which is a key substrate of LPLA2/Pla2g15 [62] and crucial component of lysosomal membranes in control of endolysosomal traffic [106]. It has been suggested before in cell experiments that ambroxol leads to lysosomal exocytosis [82, 107], whereas its activity as pharmacologic chaperone was rather low [108]. Theoretically, expelling lysosomes or their content could be a route of relief for a neuron with a high burden of lysosomal αSyn-fibrils, if such fibrils or oligomers were secreted along with lysosomal enzymes. Indeed, it has been shown that αSyn can be expelled via exocytosis or lysosomal exocytosis [20, 109]. It has to be considered that lysosomal exocytosis creates a hostile microenvironment and contributes to αSyn spreading if extracellular fibrils cannot be phagocytosed and degraded by microglia or astrocytes [110] or exported out of the brain, which would require capture and carrier through the blood brain barrier.
The disruption of lysosomal lipid metabolism in cells exposed to ambroxol could possibly explain why mitochondrial lipids were more deranged in the ambroxol treatment group than in the controls and why long-term therapeutic benefits in vivo were modest. In particular, the sensory decline was not prevented, and the improvement of exploratory behavior was only partial and temporary. The increase of overall activity also extended to “useless” (erroneous) corner visits without licks or nosepokes (SVisits), which is rather suggestive of pathologic overactivity. Nonetheless, the behavioral studies suggested that ambroxol-treated mice had a modest advantage over vehicle treated mice. Interestingly, beneficial effects of ambroxol were also manifested in a systemic restoration of some polar metabolites to wildtype levels. Since GBA1-dependent lipid metabolism has been the focus of ambroxol’s effect in PD research such peripheral metabolic effects on polar metabolites have not yet been reported and require confirmation in human studies.
From our experiments, we infer that ambroxol leads to a temporary relief of αSyn-fibril overloaded neurons but expelling of αSyn-fibrils does not eliminate αSyn and may result in further spreading or perineuronal inflammation, particularly if expelled together with glucosylsphingolipids, which precipitate αSyn oligomerization in acidic environments in vitro [111]. αSyn uses several means of cell-to-cell prion-like spreading [7, 112, 113] including transsynaptic transfer from neuron to neuron, travelling through tunnelling nanotubes [114,115,116], or spreading via extracellular vesicles [117, 118]. Glycosphingolipids are enriched together with αSyn in Lewy bodies [22] and per se may form ribbon band amyloid-like structures [21]. They are also components of exosomal membranes [119] and may assist in packaging of αSyn into exosomes [120]. Although mechanistically complex, lipidomic research suggests that an accumulation of glycosylated sphingolipids may enhance the pathogenicity of αSyn [25, 121, 122]. In our studies, ambroxol did not prevent or reduce PD-associated manifestations of glucosyl-sphingolipid pathology.
Pharmacologically, lysosomal trapping and alkalinization is mostly considered as a disadvantage and may even causes phospholipidosis [60, 92, 123]. Lysosomal function is particularly critical in neurodegenerative diseases, which heavily rely on autophagolysosomal pathways for removal of waste such as αSyn oligomers [124,125,126]. However, paradoxically, chloroquine at high doses was shown to work as an anti-aging medication in mice [127, 128]. Also, alkaline compounds were able to preserve progranulin and improve the outcome in progranulin-deficiency-associated mouse dementia models [129], and alkaline tricyclic antidepressants provided therapeutic benefits in mouse models of major depression presumably by partial inhibition of lysosomal acidic sphingomyelinase [130]. Hence, the in vivo outcome is a function of the dose, pH dependent kinetics, microenvironment, cell—and disease-specific lysosomal biology. In GBA1-associated PD, ambroxol presumably binds to mutant GBA1 in the ER, preventing its premature degradation, hence allowing its transport to the lysosome where ambroxol detaches and releases the enzyme. However, in non-GBA1 associated PD, GCase activity is only mildly reduced likely owing to lysosomal deposits of αSyn oligomers. It appears unlikely that ambroxol trapped in lysosomes would alleviate the unhealthy reciprocal interaction of GCase and αSyn. But although theoretically rather unfavorable, ambroxol did improve the in vivo outcome in non-GBA1 dependent neurodegenerative models not only in our mice but also in transgenic mice overexpressing human α-synuclein [38], in hemi-PD like rats induced by intra-striatal 6-hydroxydopamine (6-OHDA) [131], and in a mouse model of familial amyotrophic lateral sclerosis (ALS) [132] suggesting that lysosomal exocytosis outweighs or counterbalances the observed sphingolipid trapping in lysosomes [82, 103]. It might be possible to take therapeutic advantage of these effects of ambroxol if it was combined with extracellular capture of synuclein for example, with active or passive immunization.
Data availability
All data that were analysed for the study are presented within the manuscript or supplementary files. The mass spectrometry proteomics data have been deposited to the ProteomeXchange Consortium via the PRIDE partner repository with the dataset identifier PXD039172. Data deposited to PRIDE; Project accession: PXD039172. Lipidomic and metabolomic studies of mouse brain and plasma and in wildtype and Pink1SNCA double mutant mice with/without ambroxol: (S-BSST1946), available at https://www.ebi.ac.uk/biostudies/studies/S-BSST1946?key=1b15a58b-643a-4357-a489-f1081592dad4, Targeted and untargeted lipidomic and metabolomic analyses in HT22 cells extracts with/without preformed fibrils: S-BSST1897, available at https://www.ebi.ac.uk/biostudies/studies/S-BSST1897?key=d2a20a65-a88f-44a4-9654-672a72f8a9ac.
References
Kalia LV, Lang AE. Parkinson’s disease. Lancet. 2015;386:896–912.
Darweesh SK, Verlinden VJ, Stricker BH, Hofman A, Koudstaal PJ, Ikram MA. Trajectories of prediagnostic functioning in Parkinson’s disease. Brain. 2017;140:429–41.
Dickson DW. Parkinson’s disease and parkinsonism: neuropathology. Cold Spring Harb Perspect Med. 2012;2:a009258.
Braak H, Del Tredici K, Rub U, de Vos RA, Jansen Steur EN, Braak E. Staging of brain pathology related to sporadic Parkinson’s disease. Neurobiol Aging. 2003;24:197–211.
Pont-Sunyer C, Hotter A, Gaig C, Seppi K, Compta Y, Katzenschlager R, et al. The onset of nonmotor symptoms in Parkinson’s disease (the ONSET PD study). Mov Disord. 2015;30:229–37.
Braak H, Braak E, Yilmazer D, Schultz C, de Vos RA, Jansen EN. Nigral and extranigral pathology in Parkinson’s disease. J Neural Transm. 1995;46:15–31.
Peelaerts W, Bousset L, Van der Perren A, Moskalyuk A, Pulizzi R, Giugliano M, et al. alpha-Synuclein strains cause distinct synucleinopathies after local and systemic administration. Nature. 2015;522:340–4.
Blanz J, Saftig P. Parkinson’s disease: acid-glucocerebrosidase activity and alpha-synuclein clearance. J Neurochem. 2016;139:198–215.
Bi M, Du X, Jiao Q, Chen X, Jiang H. Expanding the role of proteasome homeostasis in Parkinson’s disease: beyond protein breakdown. Cell Death Dis. 2021;12:154.
Thibaudeau TA, Anderson RT, Smith DM. A common mechanism of proteasome impairment by neurodegenerative disease-associated oligomers. Nat Commun. 2018;9:1097.
Magalhaes J, Gegg ME, Migdalska-Richards A, Doherty MK, Whitfield PD, Schapira AH. Autophagic lysosome reformation dysfunction in glucocerebrosidase deficient cells: relevance to Parkinson disease. Hum Mol Genet. 2016;25:3432–45.
Sepúlveda D, Cisternas-Olmedo M, Arcos J, Nassif M, Vidal RL. Contribution of autophagy-lysosomal pathway in the exosomal secretion of alpha-Synuclein and its impact in the progression of Parkinson’s disease. Front Mol Neurosci. 2022;15:805087.
Hattori N, Sato S. Mitochondrial dysfunction in Parkinson’s disease. J Neural Transm (Vienna). 2024;131:1415–28.
Osellame LD, Duchen MR. Defective quality control mechanisms and accumulation of damaged mitochondria link Gaucher and Parkinson diseases. Autophagy. 2013;9:1633–5.
Date Y, Sasazawa Y, Kitagawa M, Gejima K, Suzuki A, Saya H, et al. Novel autophagy inducers by accelerating lysosomal clustering against Parkinson’s disease. eLife. 2024;13:e98649.
Martinez-Vicente M, Vila M. Autophagy and protein quality control in Parkinson’s disease. Cold Spring Harb Perspect Med. 2024;a041619. https://doi.org/10.1101/cshperspect.a041619.
Szinyákovics J, Keresztes F, Kiss EA, Falcsik G, Vellai T, Kovács T. Potent new targets for autophagy enhancement to delay neuronal ageing. Cells. 2023;12:1753.
Alvarez-Erviti L, Seow Y, Schapira AH, Gardiner C, Sargent IL, Wood MJ, et al. Lysosomal dysfunction increases exosome-mediated alpha-synuclein release and transmission. Neurobiol Dis. 2011;42:360–7.
Izco M, Carlos E, Alvarez-Erviti L. The two faces of exosomes in Parkinson’s disease: from pathology to therapy. Neuroscientist. 2022;28:180–93.
Xie YX, Naseri NN, Fels J, Kharel P, Na Y, Lane D, et al. Lysosomal exocytosis releases pathogenic α-synuclein species from neurons in synucleinopathy models. Nat Commun. 2022;13:4918.
Paul A, Jacoby G, Laor Bar-Yosef D, Beck R, Gazit E, Segal D. Glucosylceramide associated with Gaucher disease forms amyloid-like twisted ribbon fibrils that induce α-synuclein aggregation. ACS Nano. 2021;15:11854–68.
Kurzawa-Akanbi M, Tammireddy S, Fabrik I, Gliaudelytė L, Doherty MK, Heap R, et al. Altered ceramide metabolism is a feature in the extracellular vesicle-mediated spread of alpha-synuclein in Lewy body disorders. Acta Neuropathol. 2021;142:961–84.
Henderson MX, Sedor S, McGeary I, Cornblath EJ, Peng C, Riddle DM, et al. Glucocerebrosidase activity modulates neuronal susceptibility to pathological α-synuclein insult. Neuron. 2020;105:822–36.e7.
Kim S, Yun SP, Lee S, Umanah GE, Bandaru VVR, Yin X, et al. GBA1 deficiency negatively affects physiological alpha-synuclein tetramers and related multimers. Proc Natl Acad Sci USA. 2018;115:798–803.
Zunke F, Moise AC, Belur NR, Gelyana E, Stojkovska I, Dzaferbegovic H, et al. Reversible conformational conversion of α-synuclein into toxic assemblies by glucosylceramide. Neuron. 2018;97:92–107.e10.
Murphy KE, Gysbers AM, Abbott SK, Tayebi N, Kim WS, Sidransky E, et al. Reduced glucocerebrosidase is associated with increased alpha-synuclein in sporadic Parkinson’s disease. Brain. 2014;137:834–48.
Horowitz M, Braunstein H, Zimran A, Revel-Vilk S, Goker-Alpan O. Lysosomal functions and dysfunctions: Molecular and cellular mechanisms underlying Gaucher disease and its association with Parkinson disease. Adv Drug Deliv Rev. 2022;187:114402.
Schulze H, Sandhoff K. Lysosomal lipid storage diseases. Cold Spring Harb Perspect Biol. 2011;3:a004804.
Tegeder I, Kögel D. When lipid homeostasis runs havoc: Lipotoxicity links lysosomal dysfunction to autophagy. Matrix Biol. 2021;100-101:99–117.
Clark LN, Ross BM, Wang Y, Mejia-Santana H, Harris J, Louis ED, et al. Mutations in the glucocerebrosidase gene are associated with early-onset Parkinson disease. Neurology. 2007;69:1270–7.
Sidransky E, Nalls MA, Aasly JO, Aharon-Peretz J, Annesi G, Barbosa ER, et al. Multicenter analysis of glucocerebrosidase mutations in Parkinson’s disease. N Engl J Med. 2009;361:1651–61.
Chiasserini D, Paciotti S, Eusebi P, Persichetti E, Tasegian A, Kurzawa-Akanbi M, et al. Selective loss of glucocerebrosidase activity in sporadic Parkinson’s disease and dementia with Lewy bodies. Mol Neurodegener. 2015;10:15.
Do J, McKinney C, Sharma P, Sidransky E. Glucocerebrosidase and its relevance to Parkinson disease. Mol Neurodegener. 2019;14:36.
Klatt-Schreiner K, Valek L, Kang JS, Khlebtovsky A, Trautmann S, Hahnefeld L, et al. High glucosylceramides and low anandamide contribute to sensory loss and pain in Parkinson’s disease. Mov Disord. 2020;35:1822–33.
Alcalay RN, Levy OA, Waters CC, Fahn S, Ford B, Kuo SH, et al. Glucocerebrosidase activity in Parkinson’s disease with and without GBA mutations. Brain. 2015;138:2648–58.
Valek L, Tran B, Wilken-Schmitz A, Trautmann S, Heidler J, Schmid T, et al. Prodromal sensory neuropathy in Pink1−/− SNCA(A53T) double mutant Parkinson mice. Neuropathol Appl Neurobiol. 2021;47:1060–79.
Williams D, Glasstetter LM, Jong TT, Chen T, Kapoor A, Zhu S, et al. High-throughput screening for small-molecule stabilizers of misfolded glucocerebrosidase in Gaucher disease and Parkinson’s disease. Proc Natl Acad Sci USA. 2024;121:e2406009121.
Migdalska-Richards A, Daly L, Bezard E, Schapira AH. Ambroxol effects in glucocerebrosidase and α-synuclein transgenic mice. Ann Neurol. 2016;80:766–75.
Migdalska-Richards A, Ko WKD, Li Q, Bezard E, Schapira AHV. Oral ambroxol increases brain glucocerebrosidase activity in a nonhuman primate. Synapse. 2017;71:e21967.
Ambrosi G, Ghezzi C, Zangaglia R, Levandis G, Pacchetti C, Blandini F. Ambroxol-induced rescue of defective glucocerebrosidase is associated with increased LIMP-2 and saposin C levels in GBA1 mutant Parkinson’s disease cells. Neurobiol Dis. 2015;82:235–42.
McNeill A, Magalhaes J, Shen C, Chau KY, Hughes D, Mehta A, et al. Ambroxol improves lysosomal biochemistry in glucocerebrosidase mutation-linked Parkinson disease cells. Brain. 2014;137:1481–95.
Mullin S, Smith L, Lee K, D’Souza G, Woodgate P, Elflein J, et al. Ambroxol for the treatment of patients with Parkinson disease with and without glucocerebrosidase gene mutations: a nonrandomized, noncontrolled trial. JAMA Neurol. 2020;77:427–34.
Zhao Y, Ren J, Padilla-Parra S, Fry EE, Stuart DI. Lysosome sorting of β-glucocerebrosidase by LIMP-2 is targeted by the mannose 6-phosphate receptor. Nat Commun. 2014;5:4321.
Dobert JP, Bub S, Mächtel R, Januliene D, Steger L, Regensburger M, et al. Activation and purification of ß-glucocerebrosidase by exploiting its transporter LIMP-2 - implications for novel treatment strategies in Gaucher’s and Parkinson’s disease. Adv Sci (Weinh). 2024;11:e2401641.
Pietrafesa D, Casamassa A, Benassi B, Santoro M, Marano M, Consales C, et al. Investigating the impact of the Parkinson’s-associated GBA1 E326K mutation on β-glucocerebrosidase dimerization and interactome dynamics through an in silico approach. Int J Mol Sci. 2024;25:11443.
Bendikov-Bar I, Maor G, Filocamo M, Horowitz M. Ambroxol as a pharmacological chaperone for mutant glucocerebrosidase. Blood Cells Mol Dis. 2013;50:141–5.
Siemeling O, Slingerland S, van der Zee S, van Laar T. Study protocol of the GRoningen early-PD Ambroxol treatment (GREAT) trial: a randomized, double-blind, placebo-controlled, single center trial with ambroxol in Parkinson patients with a GBA mutation. BMC Neurol. 2024;24:146.
Colucci F, Avenali M, De Micco R, Fusar Poli M, Cerri S, Stanziano M, et al. Ambroxol as a disease-modifying treatment to reduce the risk of cognitive impairment in GBA-associated Parkinson’s disease: a multicentre, randomised, double-blind, placebo-controlled, phase II trial. The AMBITIOUS study protocol. BMJ Neurol Open. 2023;5:e000535.
Han TU, Sam R, Sidransky E. Small molecule chaperones for the treatment of Gaucher disease and GBA1-associated Parkinson disease. Front Cell Dev Biol. 2020;8:271.
Narita A, Shirai K, Itamura S, Matsuda A, Ishihara A, Matsushita K, et al. Ambroxol chaperone therapy for neuronopathic Gaucher disease: a pilot study. Ann Clin Transl Neurol. 2016;3:200–15.
Chu SY, Chien CC, Hwu WL, Wang PJ, Chien YH. Early initiation of high-dose oral ambroxol in combination with enzyme replacement therapy in a neuropathic Gaucher infant. Blood Cells Mol Dis. 2020;81:102402.
Aflaki E, Westbroek W, Sidransky E. The complicated relationship between Gaucher disease and Parkinsonism: insights from a rare disease. Neuron. 2017;93:737–46.
Somerville EN, James A, Beetz C, Schwieger R, Barrel G, Kandaswamy KK, et al. Plasma glucosylceramide levels are regulated by ATP10D and are not involved in Parkinson’s disease pathogenesis. Ann Neurol. 2025;97:873–8.
Gispert S, Brehm N, Weil J, Seidel K, Rub U, Kern B, et al. Potentiation of neurotoxicity in double-mutant mice with Pink1 ablation and A53T-SNCA overexpression. Hum Mol Genet. 2015;24:1061–76.
Abou-Sleiman PM, Muqit MM, Wood NW. Expanding insights of mitochondrial dysfunction in Parkinson’s disease. Nat Rev Neurosci. 2006;7:207–19.
Maiti P, Manna J, Dunbar GL. Current understanding of the molecular mechanisms in Parkinson’s disease: Targets for potential treatments. Transl Neurodegener. 2017;6:28.
Plotegher N, Duchen MR. Crosstalk between Lysosomes and mitochondria in Parkinson’s disease. Front Cell Dev Biol. 2017;5:110.
Pan T, Kondo S, Le W, Jankovic J. The role of autophagy-lysosome pathway in neurodegeneration associated with Parkinson’s disease. Brain. 2008;131:1969–78.
Valek L, Auburger G, Tegeder I. Sensory neuropathy and nociception in rodent models of Parkinson’s disease. Dis Model Mech. 2019;12:dmm039396.
Breiden B, Sandhoff K. Emerging mechanisms of drug-induced phospholipidosis. Biol Chem. 2019;401:31–46.
Hinkovska-Galcheva V, Treadwell T, Shillingford JM, Lee A, Abe A, Tesmer JJG, et al. Inhibition of lysosomal phospholipase A2 predicts drug-induced phospholipidosis. J Lipid Res. 2021;62:100089.
Abe A, Hinkovska-Galcheva V, Bouchev P, Bouley R, Shayman JA. The role of lysosomal phospholipase A2 in the catabolism of bis(monoacylglycerol)phosphate and association with phospholipidosis. J Lipid Res. 2024;65:100574.
Kim YM, Yum MS, Heo SH, Kim T, Jin HK, Bae JS, et al. Pharmacologic properties of high-dose ambroxol in four patients with Gaucher disease and myoclonic epilepsy. J Med Genet. 2020;57:124–31.
Valek L, Tran BN, Tegeder I. Cold avoidance and heat pain hypersensitivity in neuronal nucleoredoxin knockout mice. Free Radic Biol Med. 2022;192:84–97.
Albuquerque B, Haussler A, Vannoni E, Wolfer DP, Tegeder I. Learning and memory with neuropathic pain: impact of old age and progranulin deficiency. Front Behav Neurosci. 2013;7:174.
Fischer C, Endle H, Schumann L, Wilken-Schmitz A, Kaiser J, Gerber S, et al. Prevention of age-associated neuronal hyperexcitability with improved learning and attention upon knockout or antagonism of LPAR2. Cell Mol Life Sci. 2021;78:1029–50.
Vogel A, Ueberbach T, Wilken-Schmitz A, Hahnefeld L, Franck L, Weyer MP, et al. Repetitive and compulsive behavior after early-life-pain associated with reduced long-chain sphingolipid species. Cell Biosci. 2023;13:155.
Sens A, Rischke S, Hahnefeld L, Dorochow E, Schäfer SMG, Thomas D, et al. Pre-analytical sample handling standardization for reliable measurement of metabolites and lipids in LC-MS-based clinical research. J Mass Spectrom Adv Clin Lab. 2023;28:35–46.
Seneviratne AK, Xu M, Henao JJA, Fajardo VA, Hao Z, Voisin V, et al. The mitochondrial transacylase, tafazzin, regulates for AML stemness by modulating intracellular levels of phospholipids. Cell Stem Cell. 2019;24:621–36.e16.
Brunkhorst-Kanaan N, Klatt-Schreiner K, Hackel J, Schroter K, Trautmann S, Hahnefeld L, et al. Targeted lipidomics reveal derangement of ceramides in major depression and bipolar disorder. Metabolism. 2019;95:65–76.
Gurke R, Thomas D, Schreiber Y, Schafer SMG, Fleck SC, Geisslinger G, et al. Determination of endocannabinoids and endocannabinoid-like substances in human K3EDTA plasma - LC-MS/MS method validation and pre-analytical characteristics. Talanta. 2019;204:386–94.
Kulak NA, Pichler G, Paron I, Nagaraj N, Mann M. Minimal, encapsulated proteomic-sample processing applied to copy-number estimation in eukaryotic cells. Nat Methods. 2014;11:319–24. Epub 014 Feb 2.
Rappsilber J, Mann M, Ishihama Y. Protocol for micro-purification, enrichment, pre-fractionation and storage of peptides for proteomics using StageTips. Nat Protoc. 2007;2:1896–906.
Cox J, Mann M. MaxQuant enables high peptide identification rates, individualized p.p.b.-range mass accuracies and proteome-wide protein quantification. Nat Biotechnol. 2008;26:1367–72.
Pang Z, Zhou G, Ewald J, Chang L, Hacariz O, Basu N, et al. Using MetaboAnalyst 5.0 for LC-HRMS spectra processing, multi-omics integration and covariate adjustment of global metabolomics data. Nat Protoc. 2022;17:1735–61.
Ahmed MR, Bychkov E, Kook S, Zurkovsky L, Dalby KN, Gurevich EV. Overexpression of GRK6 rescues L-DOPA-induced signaling abnormalities in the dopamine-depleted striatum of hemiparkinsonian rats. Exp Neurol. 2015;266:42–54.
Hara S, Arawaka S, Sato H, Machiya Y, Cui C, Sasaki A, et al. Serine 129 phosphorylation of membrane-associated alpha-synuclein modulates dopamine transporter function in a G protein-coupled receptor kinase-dependent manner. Mol Biol Cell. 2013;24:1649–60. s1-3.
Vogel A, Wilken-Schmitz A, Hummel R, Lang M, Gurke R, Schreiber Y, et al. Low brain endocannabinoids associated with persistent non-goal directed nighttime hyperactivity after traumatic brain injury in mice. Sci Rep. 2020;10:14929.
Tran BN, Valek L, Wilken-Schmitz A, Fuhrmann DC, Namgaladze D, Wittig I, et al. Reduced exploratory behavior in neuronal nucleoredoxin knockout mice. Redox Biol. 2021;45:102054.
Galper J, Dean NJ, Pickford R, Lewis SJG, Halliday GM, Kim WS, et al. Lipid pathway dysfunction is prevalent in patients with Parkinson’s disease. Brain. 2022;145:3472–87.
Torres-Odio S, Key J, Hoepken HH, Canet-Pons J, Valek L, Roller B, et al. Progression of pathology in PINK1-deficient mouse brain from splicing via ubiquitination, ER stress, and mitophagy changes to neuroinflammation. J Neuroinflammation. 2017;14:154.
Fois G, Hobi N, Felder E, Ziegler A, Miklavc P, Walther P, et al. A new role for an old drug: Ambroxol triggers lysosomal exocytosis via pH-dependent Ca2+ release from acidic Ca2+ stores. Cell Calcium. 2015;58:628–37.
Li Y, Wu Q, Hu E, Wang Y, Lu H. Quantitative mass spectrometry imaging of metabolomes and lipidomes for tracking changes and therapeutic response in traumatic brain injury surrounding injured area at chronic phase. ACS Chem Neurosci. 2021;12:1363–75.
Rubilar JC, Outeiro TF, Klein AD. The lysosomal β-glucocerebrosidase strikes mitochondria: implications for Parkinson’s therapeutics. Brain. 2024;147:2610–20.
Li H, Ham A, Ma TC, Kuo SH, Kanter E, Kim D, et al. Mitochondrial dysfunction and mitophagy defect triggered by heterozygous GBA mutations. Autophagy. 2019;15:113–30.
Vankoningsloo S, Piens M, Lecocq C, Gilson A, De Pauw A, Renard P, et al. Mitochondrial dysfunction induces triglyceride accumulation in 3T3-L1 cells: role of fatty acid beta-oxidation and glucose. J Lipid Res. 2005;46:1133–49.
Chauhan MZ, Chacko JG, Ghaffarieh A, Moulin CM, Pelaez D, Uwaydat SH, et al. Mitochondrial triglyceride dysregulation in optic nerves following indirect traumatic optic neuropathy. Biomolecules. 2022;12:1885.
Fernandes HJR, Kent JP, Bruntraeger M, Bassett AR, Koulman A, Metzakopian E, et al. Mitochondrial and endoplasmic reticulum stress trigger triglyceride accumulation in models of Parkinson’s disease independent of mutations in MAPT. Metabolites. 2023;13:112.
Mileykovskaya E, Dowhan W. Cardiolipin-dependent formation of mitochondrial respiratory supercomplexes. Chem Phys Lipids. 2014;179:42–8.
Meyer N, Henkel L, Linder B, Zielke S, Tascher G, Trautmann S, et al. Autophagy activation, lipotoxicity and lysosomal membrane permeabilization synergize to promote pimozide- and loperamide-induced glioma cell death. Autophagy. 2021;17:3424–43.
Brisson L, Bański P, Sboarina M, Dethier C, Danhier P, Fontenille MJ, et al. Lactate dehydrogenase B controls lysosome activity and autophagy in cancer. Cancer Cell. 2016;30:418–31.
Shayman JA, Tesmer JJG. Lysosomal phospholipase A2. Biochim Biophys Acta Mol Cell Biol Lipids. 2019;1864:932–40.
Sandhoff K. Metabolic and cellular bases of sphingolipidoses. Biochem Soc Trans. 2013;41:1562–8.
Kita N, Hamamoto A, Gowda SGB, Takatsu H, Nakayama K, Arita M, et al. Glucosylceramide flippases contribute to cellular glucosylceramide homeostasis. J Lipid Res. 2024;65:100508.
Martin S, Smolders S, Van den Haute C, Heeman B, van Veen S, Crosiers D, et al. Mutated ATP10B increases Parkinson’s disease risk by compromising lysosomal glucosylceramide export. Acta Neuropathol. 2020;139:1001–24.
Norris AC, Yazlovitskaya EM, Zhu L, Rose BS, May JC, Gibson-Corley KN, et al. Deficiency of the lipid flippase ATP10A causes diet-induced dyslipidemia in female mice. Sci Rep. 2024;14:343.
Snider SR, Fahn S, Isgreen WP, Cote LJ. Primary sensory symptoms in Parkinsonism. Neurology. 1976;26:423–9.
Wolpe N, Zhang J, Nombela C, Ingram JN, Wolpert DM, Rowe JB. Sensory attenuation in Parkinson’s disease is related to disease severity and dopamine dose. Sci Rep. 2018;8:15643.
Vos M, Geens A, Böhm C, Deaulmerie L, Swerts J, Rossi M, et al. Cardiolipin promotes electron transport between ubiquinone and complex I to rescue PINK1 deficiency. J Cell Biol. 2017;216:695–708.
Funk RS, Krise JP. Cationic amphiphilic drugs cause a marked expansion of apparent lysosomal volume: implications for an intracellular distribution-based drug interaction. Mol Pharmacol. 2012;9:1384–95.
Albright JM, Sydor MJ, Shannahan J, Ferreira CR, Holian A. Imipramine treatment alters sphingomyelin, cholesterol, and glycerophospholipid metabolism in isolated macrophage lysosomes. Biomolecules. 2023;13:1732.
Krogsaeter E, Rosato AS, Grimm C. TRPMLs and TPCs: Targets for lysosomal storage and neurodegenerative disease therapy? Cell Calcium. 2022;103:102553.
Tsunemi T, Perez-Rosello T, Ishiguro Y, Yoroisaka A, Jeon S, Hamada K, et al. Increased lysosomal exocytosis induced by lysosomal Ca2+ channel agonists protects human dopaminergic neurons from α-synuclein toxicity. J Neurosci. 2019;39:5760–72.
Miao Y, Li G, Zhang X, Xu H, Abraham SN. A TRP channel senses lysosome neutralization by pathogens to trigger their expulsion. Cell. 2015;161:1306–19.
Sagini K, Buratta S, Delo F, Pellegrino RM, Giovagnoli S, Urbanelli L, et al. Drug-induced lysosomal impairment is associated with the release of extracellular vesicles carrying autophagy markers. Int J Mol Sci. 2021;22:12922.
Rabia M, Leuzy V, Soulage C, Durand A, Fourmaux B, Errazuriz-Cerda E, et al. Bis(monoacylglycero)phosphate, a new lipid signature of endosome-derived extracellular vesicles. Biochimie. 2020;178:26–38.
Lin SC, Chang CC, Tsou SH, Chiu PY, Cheng JF, Hung HC, et al. Protective effects of ambroxol on Aβ and α-synuclein-induced neurotoxicity through glucocerebrosidase activation in HT-22 hippocampal neuronal cells. Int J Mol Sci. 2024;25:12114.
Pantoom S, Hules L, Schöll C, Petrosyan A, Monticelli M, Pospech J, et al. Mechanistic insight into the mode of action of acid β-glucosidase enhancer ambroxol. Int J Mol Sci. 2022;23:3536.
Poehler AM, Xiang W, Spitzer P, May VE, Meixner H, Rockenstein E, et al. Autophagy modulates SNCA/alpha-synuclein release, thereby generating a hostile microenvironment. Autophagy. 2014;10:2171–92.
Loria F, Vargas JY, Bousset L, Syan S, Salles A, Melki R, et al. alpha-Synuclein transfer between neurons and astrocytes indicates that astrocytes play a role in degradation rather than in spreading. Acta Neuropathol. 2017;134:789–808.
Gaspar R, Pallbo J, Weininger U, Linse S, Sparr E. Ganglioside lipids accelerate alpha-synuclein amyloid formation. Biochim Biophys Acta Proteins Proteom. 2018;1866:1062–72.
Uchihara T, Giasson BI. Propagation of alpha-synuclein pathology: hypotheses, discoveries, and yet unresolved questions from experimental and human brain studies. Acta Neuropathol. 2016;131:49–73.
Bernis ME, Babila JT, Breid S, Wusten KA, Wullner U, Tamguney G. Prion-like propagation of human brain-derived alpha-synuclein in transgenic mice expressing human wild-type alpha-synuclein. Acta Neuropathol Commun. 2015;3:75.
Chakraborty R, Nonaka T, Hasegawa M, Zurzolo C. Tunnelling nanotubes between neuronal and microglial cells allow bi-directional transfer of α-Synuclein and mitochondria. Cell Death Dis. 2023;14:329.
Abounit S, Bousset L, Loria F, Zhu S, de Chaumont F, Pieri L, et al. Tunneling nanotubes spread fibrillar α-synuclein by intercellular trafficking of lysosomes. EMBO J. 2016;35:2120–38.
Scheiblich H, Eikens F, Wischhof L, Opitz S, Jüngling K, Cserép C, et al. Microglia rescue neurons from aggregate-induced neuronal dysfunction and death through tunneling nanotubes. Neuron. 2024;112:3106–25.e8.
Danzer KM, Kranich LR, Ruf WP, Cagsal-Getkin O, Winslow AR, Zhu L, et al. Exosomal cell-to-cell transmission of alpha synuclein oligomers. Mol Neurodegener. 2012;7:42.
Guo M, Wang J, Zhao Y, Feng Y, Han S, Dong Q, et al. Microglial exosomes facilitate α-synuclein transmission in Parkinson’s disease. Brain. 2020;143:1476–97.
Scesa G, Moyano AL, Bongarzone ER, Givogri MI. Port-to-port delivery: mobilization of toxic sphingolipids via extracellular vesicles. J Neurosci Res. 2016;94:1333–40.
Stykel MG, Humphries KM, Kamski-Hennekam E, Buchner-Duby B, Porte-Trachsel N, Ryan T, et al. α-Synuclein mutation impairs processing of endomembrane compartments and promotes exocytosis and seeding of α-synuclein pathology. Cell Rep. 2021;35:109099.
Mazzulli JR, Xu YH, Sun Y, Knight AL, McLean PJ, Caldwell GA, et al. Gaucher disease glucocerebrosidase and alpha-synuclein form a bidirectional pathogenic loop in synucleinopathies. Cell. 2011;146:37–52.
Suzuki M, Fujikake N, Takeuchi T, Kohyama-Koganeya A, Nakajima K, Hirabayashi Y, et al. Glucocerebrosidase deficiency accelerates the accumulation of proteinase K-resistant alpha-synuclein and aggravates neurodegeneration in a Drosophila model of Parkinson’s disease. Hum Mol Genet. 2015;24:6675–86.
Abe A, Hiraoka M, Shayman JA. A role for lysosomal phospholipase A2 in drug induced phospholipidosis. Drug Metab Lett. 2007;1:49–53.
Xilouri M, Brekk OR, Stefanis L. Autophagy and alpha-synuclein: relevance to Parkinson’s disease and related synucleopathies. Mov Disord. 2016;31:178–92.
Watanabe Y, Tatebe H, Taguchi K, Endo Y, Tokuda T, Mizuno T, et al. p62/SQSTM1-dependent autophagy of Lewy body-like alpha-synuclein inclusions. PLoS One. 2012;7:e52868.
Cuervo AM, Stefanis L, Fredenburg R, Lansbury PT, Sulzer D. Impaired degradation of mutant alpha-synuclein by chaperone-mediated autophagy. Science. 2004;305:1292–5.
Doeppner TR, Coman C, Burdusel D, Ancuta DL, Brockmeier U, Pirici DN, et al. Long-term treatment with chloroquine increases lifespan in middle-aged male mice possibly via autophagy modulation, proteasome inhibition and glycogen metabolism. Aging. 2022;14:4195–210.
Varma VR, Desai RJ, Navakkode S, Wong LW, Anerillas C, Loeffler T, et al. Hydroxychloroquine lowers Alzheimer’s disease and related dementias risk and rescues molecular phenotypes related to Alzheimer’s disease. Mol Psychiatry. 2023;28:1312–26.
Capell A, Liebscher S, Fellerer K, Brouwers N, Willem M, Lammich S, et al. Rescue of progranulin deficiency associated with frontotemporal lobar degeneration by alkalizing reagents and inhibition of vacuolar ATPase. J Neurosci. 2011;31:1885–94.
Kornhuber J, Tripal P, Reichel M, Muhle C, Rhein C, Muehlbacher M, et al. Functional Inhibitors of Acid Sphingomyelinase (FIASMAs): a novel pharmacological group of drugs with broad clinical applications. Cell Physiol Biochem. 2010;26:9–20.
Mishra A, Krishnamurthy S. Neurorestorative effects of sub-chronic administration of ambroxol in rodent model of Parkinson’s disease. Naunyn Schmiedebergs Arch Pharmacol. 2020;393:429–44.
Bouscary A, Quessada C, Mosbach A, Callizot N, Spedding M, Loeffler JP, et al. Ambroxol hydrochloride improves motor functions and extends survival in a mouse model of familial amyotrophic lateral sclerosis. Front Pharmacol. 2019;10:883.
Percie du Sert N, Hurst V, Ahluwalia A, Alam S, Avey MT, Baker M, et al. The ARRIVE guidelines 2.0: updated guidelines for reporting animal research. BMJ Open Sci. 2020;4:e100115.
Acknowledgements
The study was supported by the Deutsche Forschungsgemeinschaft (CRC1039 A03 to IT; CRC1039 Z01 and 445757098 to GG; TE 322/11-1 to IT). The authors would like to thank Yannick Schreiber, Erika Dorochow, Alena Sens, Rahmat Mojaradfar and Pia Viktoria Wagner for technical support in performing the LC-HRMS and LC-MS/MS experiments.
Funding
Open Access funding enabled and organized by Projekt DEAL.
Author information
Authors and Affiliations
Contributions
LF and LV performed behavioural experiments in mice, collected tissue and blood samples, did protein, RNA, mitochondrial and histology studies; LF did experiments in HT22 cells and contributed to drafting the manuscript; LH established, performed and supervised untargeted lipidomic and metabolomic analyses; MPW contributed to live imaging studies; ST analysed sphingolipids; CA analysed endocannabinoids; DT and RG supervised lipid analyses; GG acquired funding and resources; IW did the proteomic analyses; IT conceived and designed the study, acquired funding and ethical approval, analysed IntelliCage behavioural data, compiled and analysed data, made the figures and tables and wrote the paper (draft and editing). All authors contributed to writing or editing parts of the manuscript and agree to the latest version of the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Ethical approval
Mouse behavioural studies were approved by the local Ethics Committee for animal research (Darmstadt, Germany) under FK1026, FK1059, and FK1096. The maintenance of the breeding colony of Pink1−/−SNCAA53T mice and observation up to onset of PD symptoms were approved under FK1032 (12.02.205-11.02.2020) and FK1131 (17.02.2020-31.12.2023). For primary cultures and tissue analyses of wild type mice, tissue was obtained after euthanization according to $4 of the German Tierschutzgesetz in compliance with the ARRIVE principles [133], and with European guidelines and GV-SOLAS recommendations for animal welfare in science.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Franck, L., Valek, L., Hahnefeld, L. et al. Modest improvement of metabolic and behavioral deficits with long-term ambroxol treatment in a Pink1−/−SNCAA53T double mutant mouse model of Parkinson’s disease. Acta Pharmacol Sin 47, 836–859 (2026). https://doi.org/10.1038/s41401-025-01690-9
Received:
Accepted:
Published:
Version of record:
Issue date:
DOI: https://doi.org/10.1038/s41401-025-01690-9
