
Abstract
Follicular lymphoma (FL) is the most common indolent type of B-cell non-Hodgkin lymphoma. Advances in treatment have improved overall survival, but early relapse or transformation to aggressive disease is associated with inferior outcome. To identify early genetic events and track tumor clonal evolution, we performed multi-omics analysis of 94 longitudinal biopsies from 44 FL patients; 22 with transformation (tFL) and 22 with relapse without transformation (nFL). Deep whole-exome sequencing confirmed recurrent mutations in genes encoding epigenetic regulators (CREBBP, KMT2D, EZH2, EP300), with similar mutational landscape in nFL and tFL patients. Calculation of genomic distances between longitudinal samples revealed complex evolutionary patterns in both subgroups. CREBBP and KMT2D mutations were identified as genetic events that occur early in the disease course, and cases with CREBBP KAT domain mutations had low risk of transformation. Gains in chromosomes 12 and 18 (TCF4), and loss in 6q were identified as early and stable copy number alterations. Identification of such early and stable genetic events may provide opportunities for early disease detection and disease monitoring. Integrative analysis revealed that tumors with EZH2 mutations exhibited reduced gene expression of numerous histone genes, including histone linker genes. This might contribute to the epigenetic dysregulation in FL.
Similar content being viewed by others


Introduction
Follicular lymphoma (FL) is the most common indolent type of B-cell non-Hodgkin lymphoma. The malignancy is characterized by multiple relapses, but advances in treatment has led to a median survival approaching 20 years. Still, a subset of patients experiences inferior outcomes. This includes those who progress within 24 months after chemo-immunotherapy (POD24), and those who experience transformation to aggressive disease, typically diffuse large B-cell lymphoma (DLBCL) [1, 2], occurring at an annual rate of 2-3% [3].
The initial event in FL pathogenesis is the immunoglobulin heavy chain (IgH) - BCL2 translocation t(14;18). This alteration can also be detected at a very low frequency in circulating B lymphocytes of healthy individuals [4, 5], and additional genetic alterations are required for overt FL formation. FL tumors harbor mutations in genes encoding epigenetic modifiers, of which CREBBP and KMT2D are the most frequently mutated genes (60–80%), while EZH2 and EP300 mutations are less frequent [6,7,8,9,10,11,12]. Common copy number alterations (CNAs) include gain of 2p, 5, 6p, 7, 12, 18 and 21, and loss on 1p, 6q and 17p [13,14,15]. Despite the progress in our understanding of the key genomic events in FL, the molecular alterations underlying transformation to DLBCL are less well characterized. DLBCL transformed tumors are clonally related to the original FL tumor as evidenced by conservation of the t(14;18) breakpoint region and the somatic hypermutation (SHM) pattern [13, 16, 17]. Activation of the MYC oncogene and inactivation of tumor suppressor genes such as CDKN2A/B and TP53 are often linked to transformation [9, 14, 18,19,20].
Identification of FL patients with high risk of early relapse and/or transformation at the time of diagnosis remains a key challenge. Several clinical and molecular risk stratification methods have been proposed, but not implemented in clinical practice. Two molecular predictors, m7-FLIPI and a mutation associated with progression (MAP) signature, incorporate the mutational status of seven genes of which only CREBBP was shared [21, 22]. In addition, gene expression-based signatures predicted outcome in the pre-rituximab era [23] and a 23-gene expression signature predicted progression-free survival for patients treated with R-chemotherapy [24]. However, neither of these methods were designed to predict transformation. Transformation predictive gene expression signatures include embryonic stem cell-like modules [25], and a BTK gene expression score based on a subset of NF-kB target genes, first developed in the pre-rituximab era [26], and later validated in a cohort of FL patients treated with rituximab [27]. Leveraging machine learning on whole-genome tumor sequencing data, a classifier was recently developed to stratify FL patients into DLBCL-like (dFL) or constrained FL (cFL) [28]. The classifier, based on the mutational status of 49 genes and 19 regions targeted by aberrant somatic hypermutation, demonstrated that dFL cases had increased rate of histologic transformation and a 10-year shorter median time to transformation than cFL [28].
Furthermore, whole exome sequencing (WES) studies of tumor clonal evolution in FL suggested that transformed tumor clones often evolve by divergent evolution from an inferred early common progenitor cell (CPC) pool [8, 9, 13, 17, 29]. A larger study based on targeted sequencing reported that linear evolution from an earlier major clone was more frequent than divergent evolution in cases with progression of low-grade FL as compared to cases with transformation [30]. These studies did not use any metrics to calculate the genomic diversity. Characterization of early genetic driver events may provide markers for transformation risk, and identify targets for therapeutic intervention [31]. However, there is extensive intra- and inter-individual clonal heterogeneity in FL [32], which may explain why it has been challenging to develop robust molecular predictors, based on a single biopsy per patient.
Here, we performed integrative analyses of deep whole-exome sequencing (WES) data, transcriptome sequencing (RNA-seq) and copy number alterations on multiple longitudinal biopsies from a larger cohort of FL patients, enriched for cases who experienced transformation. This facilitated a detailed exploration of the clonal evolution patterns between cases who experienced multiple relapses vs. those who experienced transformation, and enabled identification of genetic alterations that occur early in the disease course.
Material and methods
Patient samples
The discovery cohort consisted of n = 97 tumor biopsies and n = 42 matched blood samples from 44 FL patients diagnosed as FL1-3A. A total of 22 cases experienced transformation during the disease course, of whom 19 had confirmed histological transformation, whereas the remaining 22 patients experienced progression or relapse of FL (Supplementary Table 1). Tonsil specimens were obtained after written informed consent from healthy donors undergoing tonsillectomy. The study was performed according to the Declaration of Helsinki on Ethical Principles for Medical Research and all patients had given written informed consent. The study was approved by the South East Regional Committee for Medical and Health Research Ethics, Norway (IDs: 23192 (2014/127) and 2010/1147a).
Whole exome sequencing and variant detection
Genomic DNA from n = 97 fresh-frozen biopsies and 42 EDTA-treated blood samples were extracted using the Maxwell 16 DNA isolation automated magnetic bead instrument (Promega). Genomic libraries were prepared from genomic DNA using the SureSelectXT Human All Exon V6 + UTR kit (90 biopsies) or the SureSelectXT Human All Exon V5 kit (7 biopsies) (Agilent, California, USA), per the manufacturer’s instructions at the Oslo University Hospital Genomics core facility. Whole exome capture and paired-end sequencing was performed using an Illumina HiSeq 4000 or HiSeq 2500 instrument, at an average depth of 717X for the tumors and 725X for the matched normal samples (Supplementary Fig. 1). We used a benchmarked bioinformatics pipeline [33] to process the sequencing reads and perform somatic calling for tumor biopsies with matched blood samples as germline controls (Supplementary Fig. 2A), and performed more stringent filtering approaches for two patients lacking a matched blood sample (Supplementary Fig. 2B). Three biopsies were excluded due to tumor cell fraction below 10% as estimated by variant allelic fractions (VAF), giving a total cohort of WES data from n = 94 biopsies.
Driver gene discovery
Putative cancer driver genes were predicted from the mutational landscape of FL cases through multiple algorithms, specifically MutSig2CV, IntOGen, and 2020Plus [34,35,36]. Variants in serial biopsies were unified prior to analysis. For IntOGen and 2020Plus, we used silent and non-silent variants in coding regions as input, and otherwise default parameters. For MutSig2CV, all variants were used as input, with default parameters. We considered driver genes with a q-value < 0.1 as significant.
Clonal evolution and genomic distance analysis
Variant clustering was performed with PyClone [37], using variant read counts from the sequence alignments and copy number information inferred from SNP6.0 data. Clonal phylogenies were constructed for all tumors from the same patient, based on the clustering patterns and cluster means using ClonEvol [38]. To calculate genomic distances, we used all variants with VAF > 0.25 and determined the Euclidean distance of clonal mutations for each pair of tumors for a given patient, as previously described [39].
Copy number analysis
A total of n = 90 tumor biopsies from the discovery cohort were subjected to copy number analysis using the Affymetrix Genome Wide SNP6.0 Array platform (Gene Chip lot 4296976), as previously described [27]. Briefly, allele-specific CNAs were calculated using the R-package ASCAT (v2.4 30) and adjusted for tumor ploidy. The segmentation files were split into genomic region bins to produce comparable segments across all patients. For each patient, we selected the most severe copy number state across genomic regions as defined by the maximum absolute value of the log2 (raw copy state of both alleles) minus the tumor ploidy minus 1 for each patient. The resulting unified segment files were input to GISTIC 2.0 (v2.0.22) [40]. The copynumber R package was used for plotting the frequency of samples with aberrations.
Identification of early and stable copy number profiles
For detection of early and stable copy number events, we utilized cases with multiple biopsies, SNP6.0 availability and tumor purity >10% (n = 34). We harmonized genomic regions across patient samples by using all available CN segment break points from ASCAT analysis above. We then selected these harmonized regions in which the logR copy state stayed within a ± 0.3 range. Early and stable copy number profiles per patient were then defined by keeping the regions in which at the first available biopsy, there was an alteration above or below 0.3 logR state, and were defined recurrent if occurring in more than three cases (9% of all cases). To extract the cytoband information for each region, the binned segments were annotated with Bioconductor’s AnnotationHub package (v2.20.2) and the AHCytoBands R package.
Bulk RNA-sequencing and imputation of B-cell specific transcriptome
RNA was extracted from n = 64 fresh frozen FL biopsies from the WES discovery cohort and from FACS-sorted B cell populations from tonsils of healthy donors (Germinal Center B cells (n = 4), naive B cells (n = 4) and memory B cells (n = 4)). RNA libraries were prepared using the TruSeq RNA library kit and sequenced on Illumina HiSeq machines (2500/4000). High quality reads were mapped to the Ensembl GRCh37.p75 using Salmon [41] in order to quantify gene expression levels in the form of transcripts per million (TPM). CIBERSORTx [42] was run at B mode, using LM22 merged into four cell types [43] for deconvolution of bulk RNA-seq and for imputation of B-cell specific RNA expression, followed by stringent filtering. Significantly differentially expressed genes were identified by EdgeR, using the exact test by classic mode.
Validation cohorts
For validation, we obtained an independent cohort of FL patients included in two randomized Nordic clinical studies, with targeted DNA sequencing data (n = 92) [44], CNA and Affymetrix gene expression data (n = 82) [27]. An additional WGS validation cohort from BC Cancer (BCC), Canada, was obtained from Dreval et al. (n = 209) [28]. Single-cell RNA sequencing data were retrieved from Han et al. [45]. Additional expression and genomic targeting measurements are available for download as part of the full DepMap data release https://depmap.org/portal.
Statistical analysis
R (version 3.6.3) was used to perform statistical testing. The Kaplan-Meier method was used for survival analysis. The type of statistical test used (Student’s t-test, Fisher’s exact and non-parametric tests) is specified in the respective figure legends.
Results
Genetic alterations in longitudinal FL biopsies
To identify genomic alterations associated with POD24 and/or transformation, we performed deep WES, RNA-seq and SNP6.0 analysis of a unique retrospective cohort of 94 longitudinal biopsies from 44 FL patients (Fig. 1A, Supplementary Table 1). The cohort was enriched for patients who experienced clinical or histological transformation during the disease course (tFL group, 22 patients), whereas the remaining cases experienced progression or relapse of FL (nFL group, 22 patients). The tFL patients and patients who experienced POD24 had shorter overall survival (Supplementary Fig. 3). We identified a total of 6580 non-synonymous coding variants and found that post-treatment biopsies (n = 42) had significantly higher mutational burden than pre-treatment biopsies (n = 52) (Supplementary Fig. 4A). tFL patients had higher mutational burden than nFL patients (Supplementary Fig. 4B), suggesting that higher mutational burden is associated with an increased risk of transformation (p < 0.01, logistic regression). Analysis of mutational signatures in pre-treatment biopsies reflected known mutational mechanisms in lymphoid cancers including somatic hypermutation (SBS9) and activity of activation-induced cytidine deaminase (SBS84). Interestingly, the mutational signature patterns were commonly preserved in longitudinal samples (Supplementary Figs. 5A, 5B).

A Schematic overview of the FL multi-omics data cohort. The FL cohort was enriched for patients who experienced transformation (tFL group) as compared to FL patients who relapsed without transformation (nFL group). The longitudinal samples were subjected to multi-omics analysis by WES, SNP array, and RNA-seq. B Recurrent and significantly mutated genes in nFL and tFL patients as identified from WES (n = 94 samples) by at least one of the three applied tools (MutSig2CV, IntOgen pipeline and 2020Plus). Only cancer driver genes appearing at average VAF > 0.15 (adjusted for tumor content) are shown. The genes are ordered based on their mutational frequency. Mutation types are color-coded as indicated; for genes with multiple mutations, the most severe mutation with consequence for change in the protein structure is shown. The FL classification was performed as described in Dreval et al. [28]. C Forest plots of cancer driver gene mutations with odds ratio for association with nFL group vs. tFL group; n = 44, statistical testing by Fisher exact test. Left: the unified mutations per patient were used for the test. Right: the mutations in the pretreatment biopsies were used for the test. Mutant EZH2 was significantly enriched in the tFL group. D-E Kaplan–Meier curves showing D overall survival and E time to transformation between cases with CREBBP KAT domain mutation vs. the other cases in the WES discovery cohort.
We identified 77 cancer driver genes and focused on 51 genes with average VAFs > 0.15 after adjustment for tumor content (Fig. 1B, Supplementary Fig. 6, Supplementary Table 2, 3). As demonstrated previously [6,7,8,9,10,11,12], almost all tumors (96.4%) harbored at least one mutation in a chromatin modifying gene, including CREBBP, KMT2D, EZH2, EP300 and ARID1A, supporting the notion that dysregulation of epigenetic modifications plays an important role in FL pathogenesis. We detected potential novel driver genes in FL, including CBLB, VPS39, DIO2 and CPT1C (Fig. 1B, Supplementary Fig. 7). Of the 51 potential cancer driver genes, none were associated with POD24 status (data not shown), but EZH2 was significantly more often mutated in the tFL group compared to the nFL group when using unified data per patient or when using diagnostic biopsies (Fig. 1C, p < 0.016 for unified mutational data, and p < 0.02 for diagnostic biopsies, Fisher’s exact test). To further validate this observation, we used an independent Nordic cohort of FL patients treated with rituximab without chemotherapy [44], of whom 21 out of 84 patients (25%) had histologic transformation. We also used data from an external BCC cohort of 184 FL cases without transformation and 25 FL cases who later experienced transformation [28]. However, the frequency of EZH2 mutations was not significantly higher in cases who later experienced histologic transformation in these two cohorts (Supplementary Table 4).
We next performed the FL classification into cFL or dFL and the single gene approximation by CREBBP KAT domain mutation for all biopsies as described [28]. The FL class remained stable over time for 83% of the cases with multiple biopsies (Fig. 1B). The diagnostic biopsies from tFL cases and POD24 cases were mostly classified as dFL, although not statistically significant (p = 0.736 and p = 0.181, Fisher exact test, Supplementary Fig. 8A-B). The CREBBP KAT approximation was highly associated with the cFL/dFL classification (p < 0.00001, Fisher exact test; Supplementary Fig. 8C). In our dataset, the CREBBP KAT domain approximation had stronger predictive value than the full FL classifier, as cases with one CREBBP KAT domain mutation had significantly better outcome (p < 0.018, Fig. 1D, Supplementary Fig. 8D), and longer time to transformation (median time to transformation not reached vs. 6.2 years in the other cases (p < 0.033, Fig. 1E, Supplementary Fig. 8E). The CREBBP KAT domain approximation to classify samples in the Nordic validation cohort with targeted sequencing data was however not predictive of time to transformation (Supplementary Fig. 8F).
Inter- and intra-patient variability in genomic distances and tumor clonal evolution
Clonality analysis of WES data coupled with allele-specific copy number data identified the tumor clonal evolutionary trajectory in serial biopsies from 30 patients. We identified cases with tumor(s) evolving independently from an inferred CPC (i.e. divergent evolution) and cases in which a later biopsy evolved directly from a major clone in the first biopsy or that the major clone was shared (i.e. linear evolution) (Fig. 2A, B). To quantitative measure the genetic divergence of serial biopsies from individual patients, we calculated genomic distances as the Euclidean distance of clonal mutations for each pair of tumors for a given patient. The genomic distance between FL and tFL biopsies exhibited a shallower increase in slope with increasing time than between FL and FL biopsies (Fig. 2C). We also observed that the genomic distance varied substantially over time for individual patients, indicating prominent intra-patient heterogeneity (Fig. 2D). As expected, biopsy comparisons classified as linear evolution had significantly smaller genomic distances compared to biopsy comparisons classified as divergent evolution (Fig. 2E, Supplementary Fig. 9A). The overall distribution of genomic distances did not differ between the nFL and tFL groups, or between FL-to-FL biopsy comparison within the nFL group vs. FL-to-tFL/DLBCL biopsy comparison within the tFL group (Fig. 2F). In contrast, POD24 patients had smaller genomic distance between biopsies than non-POD24 patients (Fig. 2F). The type of treatment did not significantly affect the genomic distance (Supplementary Fig. 9B). In conclusion, genomic distances varied greatly, and differed between POD24 and non-POD24 patients but not between nFL and tFL patients.

Tumor genome evolution was inferred with PyClone from ultra-deep WES of longitudinal samples, and clonal phylogenies were constructed for all tumors from the same patient, based on the clustering patterns and cluster means using ClonEvol. Genomic distances were calculated on the variants with cellular fraction above 0.25 as inferred by PyClone. A The nFL patient P21 and tFL patient P32 are examples of divergent evolution: The two biopsies of P21 are represented by disjoint sets of clones. The subclonal composition of all three biopsies from tFL patient P32 suggests divergent evolution from a CPC as the most prevalent clone of each biopsy was absent from the other biopsies. B The nFL patient P14 and the tFL patient P29 are examples of linear evolution. For P14, the relapsed tumor evolved directly from the major clone in the pretreatment biopsy (clone 1). For P29, the two biopsies shared the dominant clone (clone 1). C Genomic distances between any two biopsies of each patient were calculated on the variants with cellular fraction above 0.25 as inferred by PyClone. Shown is genomic distance vs. time interval of the compared serial biopsies and D Genomic distance ordered on the x-axis based on the largest genomic distance between any two biopsies of a patient. E Comparison of the genomic distances in nFL cases (left) or tFL cases (right) classified as divergent or linear evolution. F Comparison of the genomic distances when comparing nFL vs tFL patients (left), or comparing two FL biopsies vs. FL and DLBCL biopsies within individual patients (middle), or when comparing biopsies from non-POD24 patients vs. biopsies from POD24 patients (right). Statistical difference by Student t-test, p < 0.05.
CREBBP and KMT2D are early mutational events in FL
We next hypothesized that mutations present at VAF = 0.5 in all biopsies from a given patient must have arisen in the CPC, and therefore considered to be early dominant clone mutations (DCMs), whereas mutations present at VAF = 0.5 in some, but not all patient’s biopsies were considered to be late DCMs (Fig. 3A). In total, we identified 24 genes that were targeted by DCM in more than 10% of the patients (Fig. 3B). Of these, CREBBP and KMT2D were the genes that most frequently harbored early DCMs, occurring in 56 and 71% of nFL and tFL patients (CREBBP) and in 44 and 50% of nFL and tFL patients (KMT2D). In six patients, BCL2 was targeted by both early and late DCMs, indicative of repeated rounds of aberrant somatic hypermutation (Fig. 3B). TNFRSF14 mutations occurred in 40% of FL cases and were mostly classified as late DCMs, suggesting that these mutations may provide a growth advantage rather than being necessary for cancer initiation. Genes with early DCM that occurred at lower recurrence included PCLO, STAT6, ARID1A and ATP6V1B2, whereas TBL1XR1, TP53, and HIST1H1D/H1-5 more often harbored late DCM (Fig. 3B).

Tumor genome evolution was inferred with PyClone and CloneEvol as in Fig. 2. A Illustration of the mapping of Dominant Clonal Mutations (DCMs) to divergent phylogenetic trees from a common progenitor cell (CPC). Mutations found in all tumor clones, such as “A”, are assumed to have arisen in CPC, and were defined as early DCMs. Other mutations found in one or more (but not all) dominant clones were defined as late DCMs, such as “B” and “C”. B Genes harboring dominant clone mutations, DCM genes. DCM genes mutated in at least four patients out of 32 are shown.
Early and stable genomic alterations in FL pathogenesis
To identify significant CNAs, GISTIC2 analysis was applied to unified copy number segmentation data generated from multiple biopsies. We validated several known recurrent focal CNAs in FL, including loss of 1p36.33 (TNFRSF14), 16p13.3 (CREBBP), 6q23.3 (TNFAIP3, PBOVI and PERP) and gain of 2p11.2 (REL, BCL11) (Supplementary Fig. 10-11, Supplementary Table 5). Novel focal CNAs were also revealed, including loss of 4q21.22 and 13q14.3 and gain of 1q23.1 and 11q11.22. Several genes belonging to the Fc receptor-like family, FCRLA, FCRLB and FCRL5 were shown to have copy number gains in FL. FCRL5 has a regulatory role in B cell receptor function [46]. We found only weak evidence of associations between potential driver genes and CNAs (Supplementary Fig. 12).
To infer genomic alterations occurring in CPC, we developed a novel computational pipeline to identify early copy number alterations that remained stable throughout progression and treatment (Supplementary Fig. 13). For each patient, we first identified segmented regions in which the logR copy state stayed within ± 0.3 range across all available longitudinal biopsies. Next, regions with an alteration (adjusted logR changes below −0.3 or above 0.3 in the primary biopsy) were identified as early and stable copy number profiles for a given patient (Supplementary Fig. 13 and 14). Our analysis provided results from 34 cases and the majority of the stable and early CNAs were also identified in the broader GISTIC analysis, either as focal alterations or overlapped by arm level changes. However, we also discovered additional regions of interest, including loss of 10q21 and 2p16.3 (Fig. 4A). Early and stable CNAs that occurred in at least five patients include gains on 7p22.3-q36.3, 8q11.21-24.3, 12p11.23-q21.33, and 18p11.32-q23 and loss of 2p16.3, 6q11.1-25.3 and 14q11.2, suggesting that these CNAs may drive early FL initiation and are maintained during disease progression (Fig. 4A, B). In addition, a few CNAs were seen more frequent in the tFL group, such as gain of 7p22.3-q36.3, 12q24.12-31 and loss of 19p13.3-2 (Fig. 4A, B). The most recurrent early and stable CNA was gain of 18p11.32-q23, occurring in 21% of the patients.

A Bubble plot identifying all genomic regions, by genomic location, in which an amplification was identified at the first available biopsy and the copy number state remained within ±0.3 logR or alterations above 0.3 logR increased in magnitude across all available biopsies of a patient. B Bubble plot identifying all genomic regions, plotted by genomic location, in which a deletion was identified at the first available biopsy and the copy state stayed within ±0.3 logR or alterations below −0.3 logR increased in magnitude across all available biopsies of a patient. The y-axis represents the number of cases in which that genomic region was identified as an early and stable copy number region. For A–B, the bubble plot size represents the size of the genomic region identified and color indicates the ratio of tFL cases over nFL cases identified as containing the region as an early stable copy state. SNP6.0 data was available for 34 patients with serial biopsies. C 2D plot showing the Pearson correlations for cis-genes in the discovery and the Nordic validation cohort. In addition, significantly differentially expressed cis-genes with a Pearson correlation above 0.4 in both cohorts were highlighted in orange. D–F Correlation of omics data and gene dependency data for TCF4, using DepMap portal [68]. D TCF4 RNA expression and protein expression. E TCF4 RNA expression with RNA interference data, showing dependency of cell lines on gene presence. Negative values below −0.5 correlate with depletion of TCF4 with decreasing cell proliferation rates. F TCF4 RNA expression with CRISPR dependency scores (Chronos). Chronos scores below −0.5 correlate with depletion of TCF4 with decreasing cell proliferation rates. For E-F, the linear regression was correlated for the 3 highlighted lineage types, the R squared and p-value for the linear fit is calculated.
To validate these findings in an independent FL cohort, we analyzed SNP6.0 data from our Nordic validation cohort which included 10 cases with serial samples [27]. Despite the small cohort size, we were able to confirm recurrent (n > 2) loss of 14q11.2 and gains of 12q12-21 and 18p11.32-q23 as early and stable alterations (Supplementary Fig. 15).
Novel cis-regulated genes within early and stable copy number regions in FL
To explore the functional consequence of early and stable copy number changes, we utilized RNA sequencing of the remaining tissue from the WES cohort. Application of CIBERSORTx enumerated various cell types in each bulk RNA-seq sample and demonstrated high variability in the abundance of B cells (range 20–78%), with the estimated fractions being highly concordant with the frequencies of tumor cell VAFs (Supplementary Fig. S16, Spearman correlation, r = 0.72). Therefore, to overcome gene expression biases due to variability in tumor cell content across the bulk RNA cohort, B-cell transcriptomes inferred from CIBERSORTx were used for downstream analyses. Identification of cis-correlated genes was performed by obtaining Pearson’s correlation between the CNA and the B-cell specific gene expression levels. Additionally, for genes with a Pearson’s correlation of 0.5 or higher, we performed differential gene expression (DEG) analysis on samples with and without the identified CNA. We detected 46 cis-regulated genes that were differentially expressed compared to cases without the CNA (Fig. 4C).
Some known oncogenes were included in the list of significant cis-correlated genes, yet the majority were genes with less known roles in FL oncogenesis. For validation of these cis-correlated genes, we utilized available microarray and SNP6.0 data from 82 cases from our Nordic validation cohort. Applying the previously described analysis, we discovered 32 differentially expressed cis-genes with a Pearson’s correlation above 0.4. Eight of these genes (HAUS1, TXNL1, MED30, VAPA, YWHAZ, TCF4, MBD1 and TRAPPC8) overlapped with the differentially expressed cis-genes in the discovery cohort (Pearson correlation, r > 0.6, Fig. 4C), suggesting that these genes may contribute to early FL pathogenesis. Analysis of the contribution to lymphoma fitness through the cancer cell encyclopedia dependency map revealed that HAUS1 and MED30 are pan-cancer essential genes, while TCF4 tended to have lymphoid specific dependencies (Supplementary Fig. 17). Further investigation into TCF4 revealed increased RNA and protein expression in lymphoid derived cell lines including mature B-cell malignancy-based cell lines compared to pan-cancer cell lines (Fig. 4D). Furthermore, depletion of TCF4 by both RNAi and CRISPR correlated with a decrease in cell fitness particularly in lymphoid and B-cell malignancy-based cell lines (Fig. 4E, F).
Downregulated expression of histone genes is common in FL and is associated with EZH2 mutation
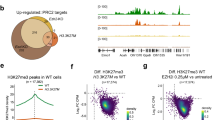
To evaluate the impact of mutations associated with tFL, we next used B-cell imputed transcriptional profiles to determine genes impacted by the mutational status of KMT2D, CREBBP or EZH2. Tumors with EZH2 hotspot mutations (Y646, A682 and A692) had upregulated gene expression of PTPN22, LMO2 and LIMS1 relative to tumors with WT EZH2. Moreover, we demonstrated a remarkable downregulation of numerous histone genes, including histone linker genes (H1-2/HIST1H1C, H1-3/HIST1H1D and H1-5/HIST1H1B), and core histone genes H2-H4 (Fig. 5A, Supplementary Table 6). This association between EZH2 mutation and downregulation of histone genes was not found when identifying DEGs between biopsies with or without mutations in either CREBBP or KMT2D (Supplementary Fig. S18A, B, Supplementary Table 7, 8). Probing the Nordic validation dataset [27], we imputed B-cell transcriptomes and validated the upregulation of PTPN22 and LIMS1 and the downregulation of specific histone genes, including H1-2, H2AC6, H4C8 and H4C14 (Fig. 5A).

Association between EZH2 mutation and downregulation of histone genes. B-cell transcriptomes were inferred by CIBERSORTx from RNA-seq data (this study, n = 64) and from the Nordic cohort with gene expression data described in Steen et al. [27] (n = 82). Volcano plot displaying significant differentially expressed genes (DEG) in cases with EZH2 mutations vs. wild-type in A left plot: the RNA-seq cohort (this study), middle plot: the Nordic cohort [27]. In the right plot, analysis of scRNA-seq data from Han et al. [45] is showing DEG in malignant B cells from EZH2 mutant cases vs. wild-type cases. Significant DEG were defined as −0.35 < FC(log2)>0.35 and p < 0.05. B Unsupervised hierarchical clustering analysis of the expression level of histone genes in the RNA-seq cohort of FL patients (this study) and in FACS-sorted B-cell populations from healthy donor tonsils. Three clusters with generally higher (HL-H), intermediate (HL-M) and lower (HL-L) histone gene expression patterns were identified.
To gain further support for a relationship between EZH2 mutations and downregulation of histone genes in malignant B cells, we re-analyzed the single-cell RNA-sequencing dataset from Han et al. [45], which contained data from four cases with EZH2 mutations and 18 cases with EZH2 WT. Also in this dataset, H1-2 was significantly downregulated in the malignant B cells with EZH2 mutations (Fig. 5A).
To characterize lymphoma cell expression of histone genes in relation to normal B-cell development, we included RNA-seq data from naive, memory and germinal center (GC) B cells purified from tonsillar tissue from normal donors. Using unsupervised clustering of FL samples and normal B-cell populations, we identified three main clusters: Cluster 1 had overall high expression of histone genes and contained normal GC B cells, cluster 2 had intermediate expression and cluster 3 had intermediate to low expression and contained naïve and memory B cells (Fig. 5B). FL samples were found in all clusters, but most FL samples had downregulated several histone genes compared to cell-of-origin GC B cells.
Discussion
Here, we report one of the largest longitudinal multi-omics studies on FL enriched for cases with transformation to DLBCL. Our comprehensive discovery cohort analysis validated genes previously implicated in FL and identified several novel driver genes. The longitudinal sampling enabled us to determine the sequential order of genetic events in FL and revealed that mutations in CREBBP and KMT2D and copy number gains in 7p, 8q, 12p and 18p and loss of 6q are genetic events that occur early in the disease course. By integrative analysis of WES data and B-cell transcriptome data, we discovered that samples containing EZH2 mutations had reduced expression of several histone genes including histone linker gene H1-2, which might contribute to the epigenetic deregulation in FL.
FL cells are dependent on epigenetic alterations, and mutations in at least one of the genes CREBBP, KMT2D, EZH2, EP300 and ARIDA were found in 96% of the biopsies in our discovery cohort, in agreement with earlier studies that have explored the genetic landscape of FL [6,7,8,9,10,11,12]. Clonal evolution reconstructions from multiple biopsies provide an opportunity to characterize early and late genetic events during FL development. This is of particular importance in FL as it may give clues to improved prognostication and treatment stratification. We confirmed that mutations in CREBBP and KMT2D are among the earliest genetic events in FL pathogenesis. Support for KMT2D mutations being early events comes from studying genetic ablation of the gene in mice overexpressing Bcl-2, which led to an increased incidence of GC-derived lymphomas resembling human tumors [47, 48]. Accumulating evidence, including our study, suggest that CREBBP mutations also arise as early events in common progenitor cells giving rise to FL, and later propagate in disease relapse or transformation [8, 11, 12, 47, 49,50,51]. This hypothesis is further strengthened as mutations in CREBBP could be detected by ultra-deep sequencing of peripheral blood samples obtained several years before the development of overt FL [52]. It is worth noting that many of the genes harboring DCMs have been identified as significantly mutated genes or as potential driver genes, emphasizing their putative driver roles in FL.
Additionally, we found a striking downregulation of histone gene expression, including the linker histone H1-2/HIST1H1C and several isoforms of core histones H2, H3 and H4 in malignant B-cells that were associated with the presence of EZH2 mutations. This was validated in a separate cohort with bulk RNA-seq data, and in an external scRNA-seq cohort. In addition, HIST1H1D/H1-5 was identified as a driver gene with 10% recurrence in our WES cohort. Mutations in HIST1H1B-E were reported in 27% of FL patients and were largely restricted to the C-terminus of the proteins [53]. Linker histone genes affect chromatin compaction [54]. Strikingly, in conditional KO mice, loss of HIST1H1C and HIST1H1E conferred enhanced fitness and self-renewal properties to GC B cells, leading to aggressive lymphomas with an increased repopulating potential [55], suggesting histone linker genes as bona fide tumor suppressor genes. Further work is needed to confirm a role for mutant EZH2 in regulation of H1-2 expression.
EZH2 mutations were enriched in FL cases who experienced transformation to DLBCL in our WES discovery cohort, but this finding was not validated in two independent and external FL cohorts. Our initial finding also seemed counterintuitive to the m7-FLIPI score, in which EZH2 mutation contributes to lower risk in the m7-FLIPI predictor, and EZH2 WT is associated with shorter failure free survival in the high FLIPI patient group [21]. Of note, the m7-FLIPI was not prognostic in our validation cohort of FL patients that were treated with rituximab without chemotherapy [44]. Similar findings were obtained in a large retrospective analysis of 590 relapsed or refractory FL patients, in which EZH2 mutation was not associated with longer overall survival [56]. Collectively, the presence of EZH2 mutation is not enough to predict risk of transformation, suggesting that broader approaches are needed to identify robust biomarkers with predictive power of transformation. We therefore tested the recently developed FL classification [28]. In our WES discovery cohort, the CREBBP KAT domain approximation had stronger predictive value than the full FL classifier, and cases with CREBBP KAT domain mutation had much longer time to transformation. Although our WES cohort is small, these results confirm the findings of Dreval et al. [28] that cFL/cFL-like cases are restrained from transformation. We were however not able to validate this in a separate cohort of targeted sequencing data from FL patients treated with rituximab without chemotherapy. Future larger studies with multi-omics data and therapy annotations might further refine genetic subtypes of FL.
Through the utilization of microdissection of early stage disease, including follicular lymphoma in situ and duodenal follicular lymphoma, a few early copy number alterations have been identified [57]. Given our extensive cohort of longitudinally sampled FL, we were able to expand the landscape of genetic copy number events in CPCs by using a novel exploratory analysis to identify copy state lesions that remain present from diagnosis to transformation despite varying treatment schedules. We similarly identified deletion of 6q and gains of 7p, 8, 12 and 18 as overly recurrent. Chromosome 7 gains have been linked to EZH2 alterations in FL [58], although we did not see a correlation between this gain and EZH2 gene expression. However, we did reveal differential expression and a correlation that pointed to the anti-apoptotic gene YWHAZ (also referred to as 14-3-3ζ) and TCF4 (E2-2) transcription factor as genes of interest. Furthermore, TCF4 depletion correlated with a decrease in cell fitness in lymphoid and B-cell malignancy-based cell lines. TCF4 was previously recognized as associated with ABC-like DLBCL and a driver of IgM expression in B-cells [59] and is influencing the B-cell decision to produce follicular versus marginal zone populations [60]. Further investigations into targeting vulnerabilities of 18q gain in DLBCL, revealed that TCF4 could be directly targeted by bromodomain and extra-terminal motif (BET) inhibitors/degraders. BET inhibitors have shown efficacy in lymphoma [61] and our results may suggest benefit in a subset of FL CPCs.
Through the calculation of genomic distance between the longitudinal biopsies of each patient, we identified complex patterns of evolution from a putative CPC at similar levels in nFL and tFL patients. A larger genomic distance was observed between two longitudinal biopsies acquired before any treatment, indicating divergent evolution over time at the different spatial locations. Araf et al. [32] demonstrated the existence of spatial tumor genome heterogeneity within the same patient by analyzing biopsies that were acquired from different locations at the same time. Similarly, Haebe et al. investigated site-to-site heterogeneity in FL biopsies by single-cell mRNA- and VDJ-sequencing [62]. In most patients, they found clear evidence for divergent disease evolution between the tumor sites. Thus, given the prominent spatial heterogeneity in FL, a single biopsy will not capture the full mutational spectrum.
Moving forward, identifying early and stable alterations that may correspond to CPCs, could guide design of rationale risk stratification. Identification of early genetic events by utilization of ctDNA may provide opportunities for early diagnosis and monitoring of disease and might predict transformation before clinical onset despite the known spatial heterogeneities in FL [63]. With this perspective, it would be interesting to test how the FL classifier or the CREBBP approximation performs using ctDNA sequencing of longitudinal plasma samples in prospective FL studies. Immunotherapy with CAR T cells or bispecific antibodies/T cell engagers are promising choices of therapies for patients with early relapse or transformed disease, with CR responses ranging from 60–79% [64,65,66,67]. As these therapies move into earlier lines of treatment, development of robust biomarkers to identify the high-risk patients at time of diagnosis will be of key importance for better implementation of effective treatment.
Data availability
RNA-sequencing data are available at Gene Expression Omnibus (accession no. GSE272403), whereas patient sensitive DNA sequencing data can be made available upon request.
Code availability
All code used for analyses are available through our GitHub repository, https://github.com/myklebustlab/ LymphomaFLserial.
References
Casulo C, Byrtek M, Dawson KL, Zhou X, Farber CM, Flowers CR, et al. Early relapse of follicular lymphoma after rituximab plus cyclophosphamide, doxorubicin, vincristine, and prednisone defines patients at high risk for death: an analysis from the national lymphocare study. J Clin Oncol. 2015;33:2516–22.
Sarkozy C, Trneny M, Xerri L, Wickham N, Feugier P, Leppa S, et al. Risk factors and outcomes for patients with follicular lymphoma who had histologic transformation after response to first-line immunochemotherapy in the PRIMA trial. J Clin Oncol. 2016;34:2575–82.
Link BK, Maurer MJ, Nowakowski GS, Ansell SM, Macon WR, Syrbu SI, et al. Rates and outcomes of follicular lymphoma transformation in the immunochemotherapy era: a report from the University of Iowa/MayoClinic Specialized Program of Research Excellence Molecular Epidemiology Resource. J Clin Oncol. 2013;31:3272–8.
Roulland S, Navarro JM, Grenot P, Milili M, Agopian J, Montpellier B, et al. Follicular lymphoma-like B cells in healthy individuals: a novel intermediate step in early lymphomagenesis. J Exp Med. 2006;203:2425–31.
Roulland S, Kelly RS, Morgado E, Sungalee S, Solal-Celigny P, Colombat P, et al. t(14;18) Translocation: A predictive blood biomarker for follicular lymphoma. J Clin Oncol. 2014;32:1347–55.
Morin RD, Mendez-Lago M, Mungall AJ, Goya R, Mungall KL, Corbett RD, et al. Frequent mutation of histone-modifying genes in non-Hodgkin lymphoma. Nature. 2011;476:298–303.
Bödör C, Grossmann V, Popov N, Okosun J, O’Riain C, Tan K, et al. EZH2 mutations are frequent and represent an early event in follicular lymphoma. Blood. 2013;122:3165–8.
Okosun J, Bodor C, Wang J, Araf S, Yang CY, Pan C, et al. Integrated genomic analysis identifies recurrent mutations and evolution patterns driving the initiation and progression of follicular lymphoma. Nat Genet. 2014;46:176–81.
Pasqualucci L, Khiabanian H, Fangazio M, Vasishtha M, Messina M, Holmes A-Á, et al. Genetics of Follicular Lymphoma Transformation. Cell Rep. 2014;6:130–40.
Krysiak K, Gomez F, White BS, Matlock M, Miller CA, Trani L, et al. Recurrent somatic mutations affecting B-cell receptor signaling pathway genes in follicular lymphoma. Blood. 2017;129:473–83.
Green MR, Gentles AJ, Nair RV, Irish JM, Kihira S, Liu CL, et al. Hierarchy in somatic mutations arising during genomic evolution and progression of follicular lymphoma. Blood. 2013;121:1604–11.
Green MR, Kihira S, Liu CL, Nair RV, Salari R, Gentles AJ, et al. Mutations in early follicular lymphoma progenitors are associated with suppressed antigen presentation. Proc Natl Acad Sci. 2015;112:E1116–E1125.
Eide MB, Liestøl K, Lingjærde OC, Hystad ME, Kresse SH, Meza-Zepeda L, et al. Genomic alterations reveal potential for higher grade transformation in follicular lymphoma and confirm parallel evolution of tumor cell clones. Blood. 2010;116:1489–97.
Bouska A, Zhang W, Gong Q, Iqbal J, Scuto A, Vose J, et al. Combined copy number and mutation analysis identifies oncogenic pathways associated with transformation of follicular lymphoma. Leukemia. 2017;31:83–91.
Ma MCJ, Tadros S, Bouska A, Heavican T, Yang H, Deng Q, et al. Subtype-specific and co-occurring genetic alterations in B-cell non-Hodgkin lymphoma. Haematologica. 2022;107:690–701.
Zelenetz AD, Chen TT, Levy R. Clonal expansion in follicular lymphoma occurs subsequent to antigenic selection. J Exp Med. 1992;176:1137–48.
Carlotti E, Wrench D, Matthews J, Iqbal S, Davies A, Norton A, et al. Transformation of follicular lymphoma to diffuse large B-cell lymphoma may occur by divergent evolution from a common progenitor cell or by direct evolution from the follicular lymphoma clone. Blood. 2009;113:3553–7.
Lo Coco F, Gaidano G, Louie DC, Offit K, Chaganti RS, Dalla-Favera R. p53 mutations are associated with histologic transformation of follicular lymphoma. Blood. 1993;82:2289–95.
Sander CA, Yano T, Clark HM, Harris C, Longo DL, Jaffe ES, et al. p53 mutation is associated with progression in follicular lymphomas. Blood. 1993;82:1994–2004.
Lossos IS, Alizadeh AA, Diehn M, Warnke R, Thorstenson Y, Oefner PJ, et al. Transformation of follicular lymphoma to diffuse large-cell lymphoma: alternative patterns with increased or decreased expression of c-myc and its regulated genes. Proc Natl Acad Sci USA. 2002;99:8886–91.
Pastore A, Jurinovic V, Kridel R, Hoster E, Staiger AM, Szczepanowski M, et al. Integration of gene mutations in risk prognostication for patients receiving first-line immunochemotherapy for follicular lymphoma: a retrospective analysis of a prospective clinical trial and validation in a population-based registry. Lancet Oncol. 2015;16:1111–22.
Russler-Germain DA, Krysiak K, Ramirez C, Mosior M, Watkins MP, Gomez F, et al. Mutations associated with progression in follicular lymphoma predict inferior outcomes at diagnosis: Alliance A151303. Blood Adv. 2023;7:5524–39.
Dave SS, Wright G, Tan B, Rosenwald A, Gascoyne RD, Chan WC, et al. Prediction of Survival in Follicular Lymphoma Based on Molecular Features of Tumor-Infiltrating Immune Cells. N. Engl J Med. 2004;351:2159–69.
Huet S, Tesson B, Jais JP, Feldman AL, Magnano L, Thomas E, et al. A gene-expression profiling score for prediction of outcome in patients with follicular lymphoma: a retrospective training and validation analysis in three international cohorts. Lancet Oncol. 2018;19:549–61.
Gentles AJ, Alizadeh AA, Lee S, Myklebust J, Shachaf C, Shahbaba B, et al. A pluripotency signature predicts histologic transformation and influences survival in follicular lymphoma patients. Blood. 2009;114:3158–66.
Brodtkorb M, Lingjærde OC, Huse K, Trøen G, Hystad ME, Hilden VI, et al. Whole-genome integrative analysis reveals expression signatures predicting transformation in follicular lymphoma. Blood. 2014;123:1051–4.
Steen CB, Leich E, Myklebust JH, Lockmer S, Wise JF, Wahlin BE, et al. A clinico-molecular predictor identifies follicular lymphoma patients at risk of early transformation after first-line immunotherapy. Haematologica. 2019;104:e460–e464.
Dreval K, Hilton LK, Cruz M, Shaalan H, Ben-Neriah S, Boyle M, et al. Genetic subdivisions of follicular lymphoma defined by distinct coding and noncoding mutation patterns. Blood. 2023;142:561–73.
Fitzgibbon J, Iqbal S, Davies A, O’Shea D, Carlotti E, Chaplin T, et al. Genome-wide detection of recurring sites of uniparental disomy in follicular and transformed follicular lymphoma. Leukemia. 2007;21:1514–20.
Kridel R, Chan FC, Mottok A, Boyle M, Farinha P, Tan K, et al. Histological Transformation and Progression in Follicular Lymphoma: A Clonal Evolution Study. PLoS Med. 2016;13:e1002197.
Heitzer E, Haque IS, Roberts CES, Speicher MR. Current and future perspectives of liquid biopsies in genomics-driven oncology. Nat Rev Genet. 2019;20:71–88.
Araf S, Wang J, Korfi K, Pangault C, Kotsiou E, Rio-Machin A, et al. Genomic profiling reveals spatial intra-tumor heterogeneity in follicular lymphoma. Leukemia. 2018;32:1261–5.
Alioto TS, Buchhalter I, Derdak S, Hutter B, Eldridge MD, Hovig E, et al. A comprehensive assessment of somatic mutation detection in cancer using whole-genome sequencing. Nat Commun. 2015;6:10001.
Gonzalez-Perez A, Perez-Llamas C, Deu-Pons J, Tamborero D, Schroeder MP, Jene-Sanz A, et al. IntOGen-mutations identifies cancer drivers across tumor types. Nat Methods. 2013;10:1081–2.
Tokheim CJ, Papadopoulos N, Kinzler KW, Vogelstein B, Karchin R. Evaluating the evaluation of cancer driver genes. Proc Natl Acad Sci USA. 2016;113:14330–5.
Lawrence MS, Stojanov P, Polak P, Kryukov GV, Cibulskis K, Sivachenko A, et al. Mutational heterogeneity in cancer and the search for new cancer-associated genes. Nature. 2013;499:214–8.
Roth A, Khattra J, Yap D, Wan A, Laks E, Biele J, et al. PyClone: statistical inference of clonal population structure in cancer. Nat Methods. 2014;11:396–8.
Dang HX, White BS, Foltz SM, Miller CA, Luo J, Fields RC, et al. ClonEvol: clonal ordering and visualization in cancer sequencing. Ann Oncol. 2017;28:3076–82.
Rosenthal R, Cadieux EL, Salgado R, Bakir MA, Moore DA, Hiley CT, et al. Neoantigen-directed immune escape in lung cancer evolution. Nature. 2019;567:479–85.
Mermel CH, Schumacher SE, Hill B, Meyerson ML, Beroukhim R, Getz G. GISTIC2.0 facilitates sensitive and confident localization of the targets of focal somatic copy-number alteration in human cancers. Genome Biol. 2011;12:R41.
Patro R, Duggal G, Love MI, Irizarry RA, Kingsford C. Salmon provides fast and bias-aware quantification of transcript expression. Nat Methods. 2017;14:417–9.
Newman AMSC, Gentles AJ, Chaudhuri AA, Khodadoust MS, Esfahani MS, Steiner D, et al. High-throughput tissue dissection and cell purification with digital cytometry. Nat Biotechnol. 2019;37:773–82.
Newman AM, Liu CL, Green MR, Gentles AJ, Feng W, Xu Y, et al. Robust enumeration of cell subsets from tissue expression profiles. Nat Methods. 2015;12:453–7.
Lockmer S, Ren W, Brodtkorb M, Østenstad B, Wahlin BE, Pan-Hammarström Q, et al. M7-FLIPI is not prognostic in follicular lymphoma patients with first-line rituximab chemo-free therapy. Br J Haematol. 2020;188:259–67.
Han G, Deng Q, Marques-Piubelli ML, Dai E, Dang M, Ma MCJ, et al. Follicular Lymphoma Microenvironment Characteristics Associated with Tumor Cell Mutations and MHC Class II Expression. Blood Cancer Discov. 2022;3:428–43.
Zhu Z, Li R, Li H, Zhou T, Davis RS. FCRL5 exerts binary and compartment-specific influence on innate-like B-cell receptor signaling. Proc Natl Acad Sci USA. 2013;110:E1282–1290.
Zhang J, Dominguez-Sola D, Hussein S, Lee JE, Holmes AB, Bansal M, et al. Disruption of KMT2D perturbs germinal center B cell development and promotes lymphomagenesis. Nat Med. 2015;21:1190–8.
Ortega-Molina A, Boss IW, Canela A, Pan H, Jiang Y, Zhao C, et al. The histone lysine methyltransferase KMT2D sustains a gene expression program that represses B cell lymphoma development. Nat Med. 2015;21:1199–208.
Horton SJ, Giotopoulos G, Yun H, Vohra S, Sheppard O, Bashford-Rogers R, et al. Early loss of Crebbp confers malignant stem cell properties on lymphoid progenitors. Nat Cell Biol. 2017;19:1093–104.
García-Ramírez I, Tadros S, González-Herrero I, Martín-Lorenzo A, Rodríguez-Hernández G, Moore D, et al. Crebbp loss cooperates with Bcl2 overexpression to promote lymphoma in mice. Blood. 2017;129:2645–56.
Zhang J, Vlasevska S, Wells VA, Nataraj S, Holmes AB, Duval R, et al. The CREBBP Acetyltransferase Is a Haploinsufficient Tumor Suppressor in B-cell Lymphoma. Cancer Discov. 2017;7:322–37.
Schroers-Martin JG, Soo J, Brisou G, Scherer F, Kurtz DM, Sworder BJ, et al. Tracing Founder Mutations in Circulating and Tissue-Resident Follicular Lymphoma Precursors. Cancer Discov. 2023;13:1310–23.
Li H, Kaminski MS, Li Y, Yildiz M, Ouillette P, Jones S, et al. Mutations in linker histone genes HIST1H1 B, C, D, and E; OCT2 (POU2F2); IRF8; and ARID1A underlying the pathogenesis of follicular lymphoma. Blood. 2014;123:1487–98.
Fyodorov DV, Zhou BR, Skoultchi AI, Bai Y. Emerging roles of linker histones in regulating chromatin structure and function. Nat Rev Mol Cell Biol. 2018;19:192–206.
Yusufova N, Kloetgen A, Teater M, Osunsade A, Camarillo JM, Chin CR, et al. Histone H1 loss drives lymphoma by disrupting 3D chromatin architecture. Nature. 2021;589:299–305.
Okosun J, Bödör C, Batlevi C, N. Nagy N, Michot J, Schneider T, et al. EZH2 gain-of-function mutations are not associated with more favorable prognosis in relapsed/refractory follicualr lymphoma (FL): A preliminary analysis of 590 patients. Hematological Oncol. 2019;S2:192–3.
Mamessier E, Song JY, Eberle FC, Pack S, Drevet C, Chetaille B, et al. Early lesions of follicular lymphoma: a genetic perspective. Haematologica. 2014;99:481–8.
Huet S, Xerri L, Tesson B, Mareschal S, Taix S, Mescam-Mancini L, et al. EZH2 alterations in follicular lymphoma: biological and clinical correlations. Blood Cancer J. 2017;7:e555.
Jain N, Hartert K, Tadros S, Fiskus W, Havranek O, Ma MCJ, et al. Targetable genetic alterations of TCF4 (E2-2) drive immunoglobulin expression in diffuse large B cell lymphoma. Sci Transl Med. 2019;11:eaav5599.
Wikström I, Forssell J, Goncalves M, Colucci F, Holmberg D. E2-2 regulates the expansion of pro-B cells and follicular versus marginal zone decisions. J Immunol. 2006;177:6723–9.
Blum KA, Abramson J, Maris M, Flinn I, Goy A, Mertz J, et al. A phase I study of CPI-0610, a bromodomain and extra terminal protein (BET) inhibitor in patients with relapsed or refractory lymphoma. Ann Oncol. 2018;29:iii7–iii9.
Haebe S, Shree T, Sathe A, Day G, Czerwinski DK, Grimes SM, et al. Single-cell analysis can define distinct evolution of tumor sites in follicular lymphoma. Blood. 2021;137:2869–80.
Scherer F, Kurtz DM, Newman AM, Stehr H, Craig AF, Esfahani MS, et al. Distinct biological subtypes and patterns of genome evolution in lymphoma revealed by circulating tumor DNA. Sci Transl Med. 2016;8:364ra155.
Fowler NH, Chavez JC, Riedell PA. Moving T-Cell Therapies into the Standard of Care for Patients with Relapsed or Refractory Follicular Lymphoma: A Review. Target Oncol. 2024;4:495–510.
Jacobson CA, Chavez JC, Sehgal AR, William BM, Munoz J, Salles G, et al. Axicabtagene ciloleucel in relapsed or refractory indolent non-Hodgkin lymphoma (ZUMA-5): a single-arm, multicentre, phase 2 trial. Lancet Oncol. 2022;23:91–103.
Morschhauser F, Dahiya S, Palomba ML, Martin Garcia-Sancho A, Reguera Ortega JL, Kuruvilla J et al. Lisocabtagene maraleucel in follicular lymphoma: the phase 2 TRANSCEND FL study. Nat Med, 2024. Epub ahead of print.
Budde LE, Sehn LH, Matasar M, Schuster SJ, Assouline S, Giri P, et al. Safety and efficacy of mosunetuzumab, a bispecific antibody, in patients with relapsed or refractory follicular lymphoma: a single-arm, multicentre, phase 2 study. Lancet Oncol. 2022;23:1055–65.
Nusinow DP, Szpyt J, Ghandi M, Rose CM, McDonald ER 3rd, et al. Quantitative Proteomics of the Cancer Cell Line Encyclopedia. Cell. 2020;180:387–402.
Acknowledgements
We acknowledge Merete Hektoen and Lars Birger Aasheim for technical assistance, and the support staff at the Genomics Core Facility and the Flow Cytometry Core Facility at Institute for Cancer Research, Oslo University Hospital. This project was funded by the Norwegian Cancer Society (Grant No. 182694), South-Eastern Norway Regional Health Authority (Grant No. 21521), the foundation KG Jebsen (#19, Centre for B-cell malignancies), the Norwegian Research Council (grants no 218241 and 221580 to Norwegian Cancer Genomics Consortium and grant number 332727 through the Centres of Excellence scheme), the Radium Hospital Foundation and the Swedish Cancer Society, Radiumhemmets, the Center for Innovative Medicine (CIMED), and a Mayo Clinic and KI collaborative grant.
Author information
Authors and Affiliations
Contributions
B.B., J.F.W., E.H., O.M., E.B.S., H.H., and J.H.M. conceptualized and designed the study. B.B., G.T., V.H., J.F.W., and H.H. collected samples and B.B., J.F.W. and V.H. isolated DNA and RNA. B.B., J.F.W, D.V., S.N., A.S., C.B.S., and O.C.L. analyzed data and performed bioinformatics analyses, and J.F.W developed the pipeline for early and stable CNAs. M.S.L. provided TCGA panel of normal data. Y.N.B., M.B., and H.H. collected and reviewed clinical data. L.M.Z and S.L. provided sequencing technologies and expertise, as well as generation of the sequencing data. K.B. provided pathology review. B.B., D.V., J.F.W., S.N., A.S., M.B., O.C.L., E.H., E.B.S., H.H. and J.H.M provided data interpretation. W.R., E.K. and Q.P.H provided targeted DNA sequencing data, gene expression data and clinical data for the Nordic validation cohort [44]. B.B., J.F.W and J.H.M. drafted the article and all authors critically revised the article. All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Competing interests
EK has served on the advisory boards of Celgene, Janssen and AbbVie, and has provided educational lectures for Celgene, Janssen, AbbVie and Astra Zeneca. OCL has served as a statistical advisor for Novartis and provided educational lectures for Nykode Therapeutics. HH has served on the advisory boards of Roche, Celgene, Nordic Nanovector, Novartis and Takeda and has provided educational lectures for Novartis. EBS owns stock in Nordic Nanovector and is a named inventor on a patent filed by the National Cancer Institute: “Methods for selecting and treating lymphoma types” licensed to NanoString Technologies; named inventor on a patent: “Evaluation of mantle cell lymphoma and methods related thereof.” All other authors declare no competing interest.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Bai, B., Wise, J.F., Vodák, D. et al. Multi-omics profiling of longitudinal samples reveals early genomic changes in follicular lymphoma. Blood Cancer J. 14, 147 (2024). https://doi.org/10.1038/s41408-024-01124-5
Received:
Revised:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41408-024-01124-5
This article is cited by
-
Molecular landscape of distinct follicular lymphoma histologic grades: insights from genomic and transcriptome analyses
Leukemia (2025)
-
Exposomics: a review of methodologies, applications, and future directions in molecular medicine
EMBO Molecular Medicine (2025)
-
Effect of POD24 on the prognosis of follicular and other indolent B-cell non-hodgkin lymphomas
Annals of Hematology (2025)
